
On the Potential Energy Surface of the Pyrene Dimer


Abstract
1. Introduction
2. Results
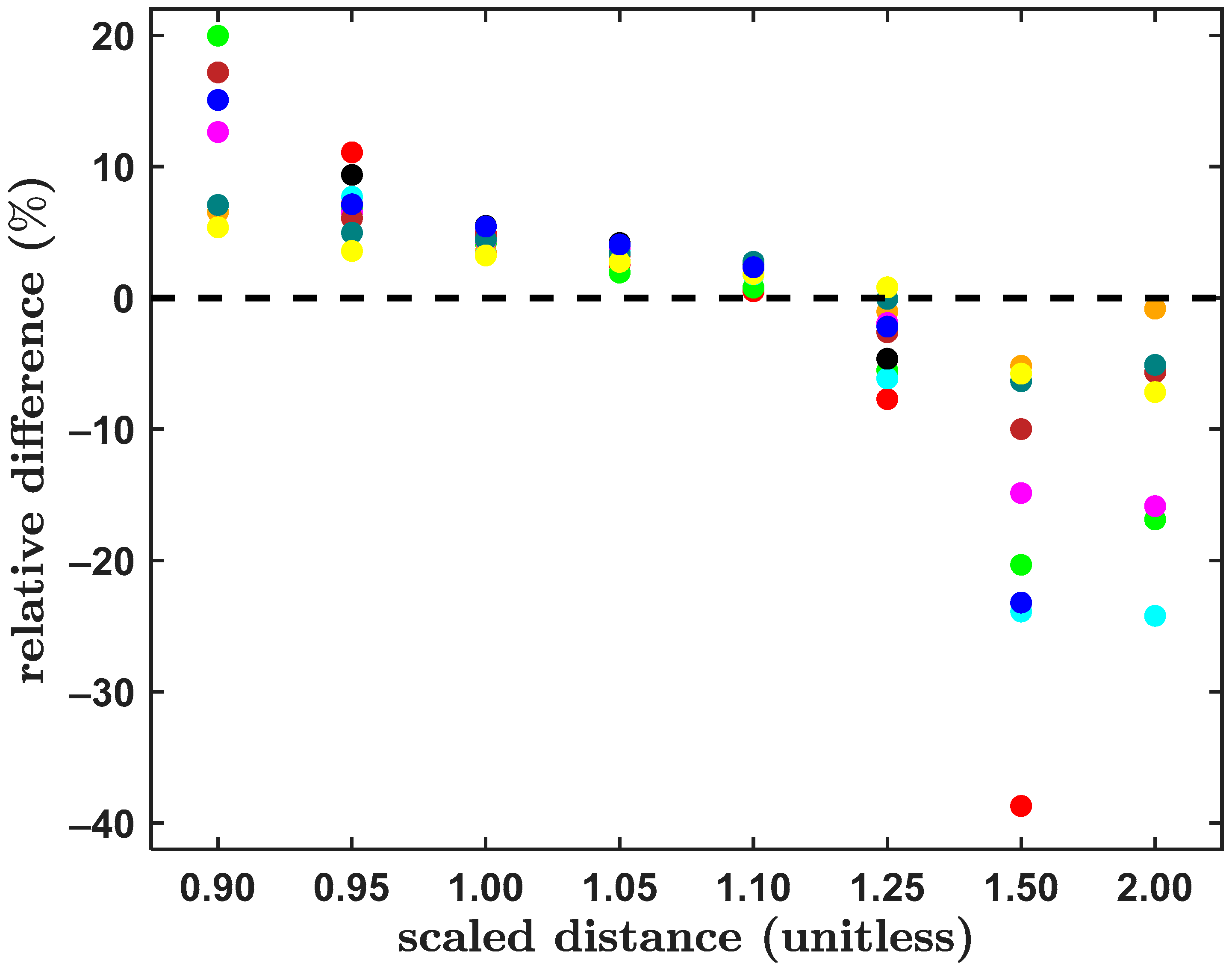
2.1. Systems from the S66x8 Set
2.2. The Benzene Dimer Structures
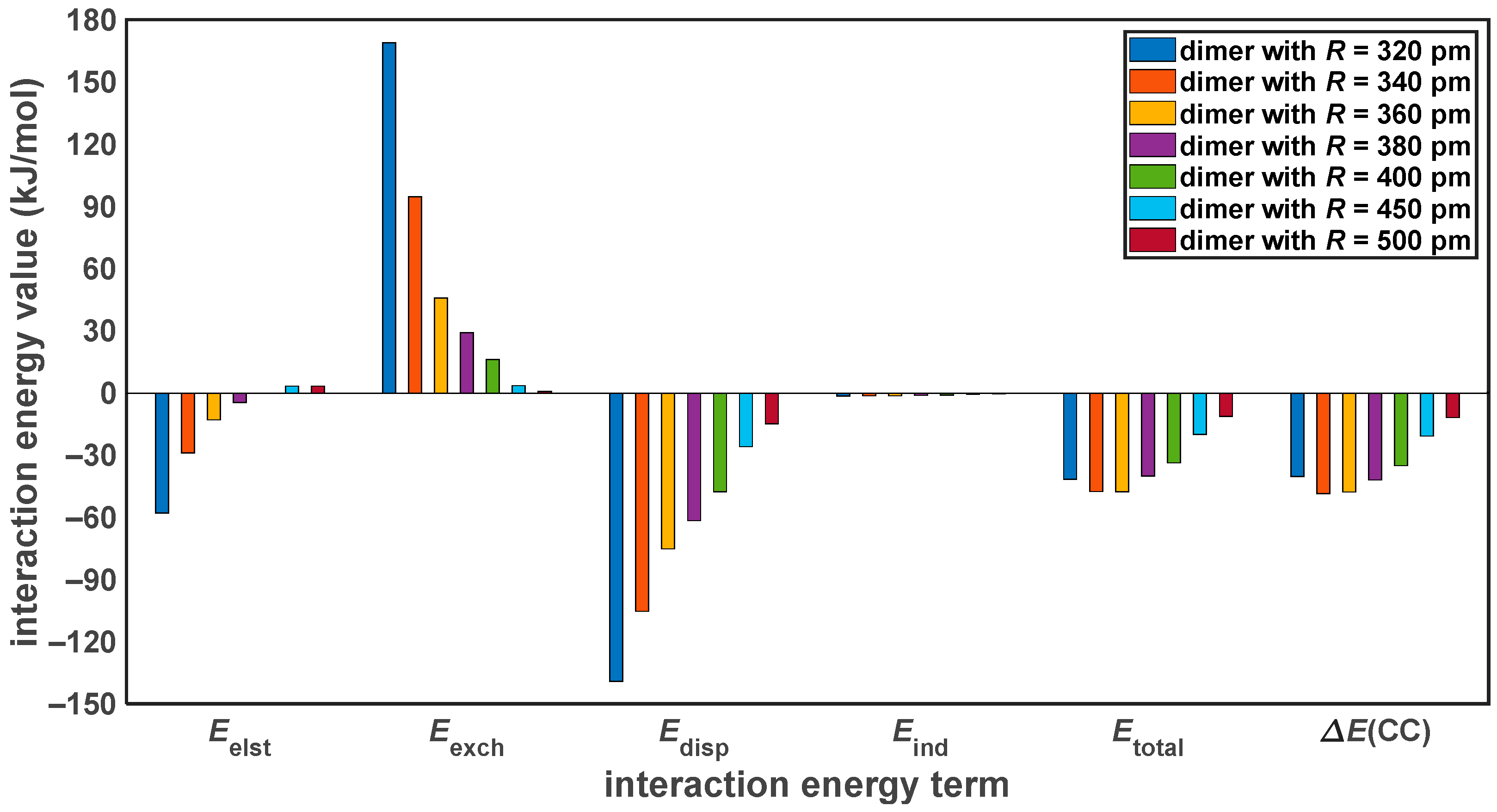
2.3. Dissociation of the Pyrene Dimer
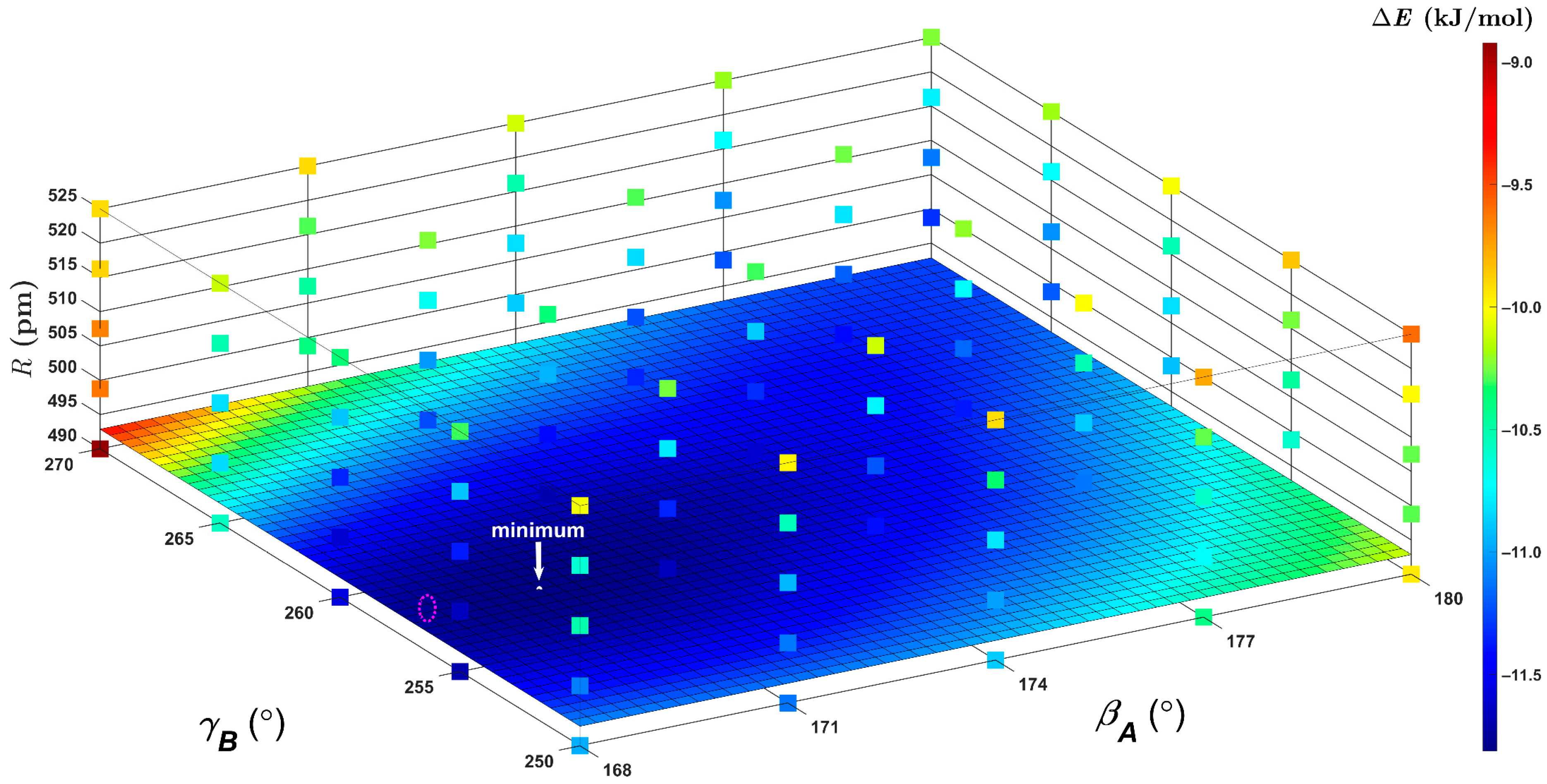
2.4. Minima of the Pyrene Ddimer
3. Discussion
3.1. Discrepancies between the Canonical and DLPNO CCSD(T)/CBS Results
3.2. Stacking Preferences of the Pyrene Dimers
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, X.; Ge, W.; Guo, S.; Bai, J.; Hong, W. Characterization and Application of Supramolecular Junctions. Angew. Chem. 2023, 62, 202216819. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Fu, Y.; Wang, M.; Qiu, R.; Wang, Y.; Stoddart, J.F.; Wang, Y.; Chen, H. Robust Single-Supermolecule Switches Operating in Response to Two Different Noncovalent Interaction. J. Am. Chem. Soc. 2023, 145, 18800–18811. [Google Scholar] [CrossRef] [PubMed]
- Poriel, C.; Rault-Berthelot, J. Dihydroindenofluorenes as building units in organic semiconductors for organic electronics. Chem. Soc. Rev. 2023, 52, 6754–6805. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Brouillac, C.; Jacques, E.; Quiton, C.; Poriel, C. π-Conjugated Nanohoops: A New Generation of Curved Materials for Organic Electronics. Angew. Chem. 2024, 63, e202402608. [Google Scholar] [CrossRef]
- Zhao, W.; Ding, Z.; Yang, Z.; Lu, T.; Yang, B.; Jiang, S. Remarkable Off–On Tunable Solid-State Luminescence by the Regulation of Pyrene Dimer. Chem. Eur. J. 2024, 30, e202303202. [Google Scholar] [CrossRef]
- Liao, Q.; Huang, A.; Wang, J.; Chang, K.; Li, H.; Yao, P.; Zhong, C.; Xie, P.; Wang, J.; Li, Z.; et al. Controllable π–π coupling of intramolecular dimer models in aggregated states. Chem. Sci. 2024, 15, 4364–4373. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Zheng, Y.; Wang, X.; Lei, J.; Wang, H.; Tian, X.; Zou, S.; Bloino, J.; Gou, Q.; Caminati, W.; et al. Scissor-like Face to Face π−π Stacking: A Surprising Preference Induced by the Isocyano Group in the Self-Assembled Dimer of Phenyl Isocyanide. J. Phys. Chem. Lett. 2022, 13, 9934–9940. [Google Scholar] [CrossRef]
- Germer, S.; Bauer, M.; Hübner, O.; Marten, R.; Dreuw, A.; Himmel, H.-J. Isolated Dimers Versus Solid-State Dimers of N-Heteropolycycles: Matrix-Isolation Spectroscopy in Concert with Quantum Chemistry. Chem. Eur. J. 2023, 29, e202302296. [Google Scholar] [CrossRef]
- Miao, X.; Preitschopf, T.; Sturm, F.; Fischer, F.; Fischer, I.; Lemmens, A.K.; Limbacher, M.; Mitric, R. Stacking Is Favored over Hydrogen Bonding in Azaphenanthrene Dimers. J. Phys. Chem. Lett. 2022, 13, 8939–8944. [Google Scholar] [CrossRef]
- Torres-Hernández, F.; Pinillos, P.; Li, W.; Saragi, R.T.; Camiruaga, A.; Juanes, M.; Usabiaga, I.; Lessari, A.; Fernández, J.A. Competition between O−H and S−H Intermolecular Interactions in Conformationally Complex Systems: The 2-Phenylethanethiol and 2-Phenylethanol Dimers. J. Phys. Chem. Lett. 2024, 15, 5674–5680. [Google Scholar] [CrossRef]
- Goerigk, L.; Hansen, A.; Bauer, C.; Ehrlich, S.; Najibi, A.; Grimme, S. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. [Google Scholar] [CrossRef] [PubMed]
- Hübner, O.; Thusek, J.; Himmel, H.-J. Pyridine Dimers and Their Low-Temperature Isomerization: A High-Resolution Matrix-Isolation Spectroscopy Study. Angew. Chem. 2023, 62, e202218042. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Werner, H.-J. Explicitly correlated local coupled-cluster methods using pair natural orbitals. WIREs Comput. Mol. Sci. 2018, 8, e1371. [Google Scholar] [CrossRef]
- Calvin, J.A.; Peng, C.; Rishi, V.; Kumar, A.; Valeev, E.F. Many-Body Quantum Chemistry on Massively Parallel Computers. Chem. Rev. 2021, 121, 1203–1231. [Google Scholar] [CrossRef]
- Saragi, R.T.; Calabrese, C.; Juanes, M.; Pinacho, R.; Rubio, J.E.; Pérez, C.; Lessari, A. π-Stacking Isomerism in Polycyclic Aromatic Hydrocarbons: The 2-Naphthalenethiol Dimer. J. Phys. Chem. Lett. 2023, 14, 207–231. [Google Scholar] [CrossRef]
- Riplinger, C.; Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys. 2013, 138, 034106. [Google Scholar] [CrossRef] [PubMed]
- Riplinger, C.; Pinski, P.; Becker, U.; Valeev, E.F.; Neese, F. Sparse maps—A systematic infrastructure for reduced-scaling electronic structure methods. II. Linear scaling domain based pair natural orbital coupled cluster theory. J. Chem. Phys. 2016, 144, 024109. [Google Scholar] [CrossRef]
- Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139, 134101. [Google Scholar] [CrossRef]
- Santra, G.; Semidalas, E.; Mehta, N.; Karton, A.; Martin, J.M.L. S66x8 noncovalent interactions revisited: New benchmark and performance of composite localized coupled-cluster methods. Phys. Chem. Chem. Phys. 2022, 24, 25555–25570. [Google Scholar] [CrossRef]
- Nagy, P.R.; Gyevi-Nagy, L.; Lőrincz, B.D.; Kállay, M. Pursuing the basis set limit of CCSD(T) non-covalent interaction energies for medium-sized complexes: Case study on the S66 compilation. Mol. Phys. 2022, 121, e2109526. [Google Scholar] [CrossRef]
- Donchev, A.G.; Taube, A.G.; Decolvenaere, E.; Hargus, C.; McGibbon, R.T.; Law, K.-H.; Gregersen, B.A.; Li, J.-L.; Palmo, K.; Siva, K.; et al. Quantum chemical benchmark databases of gold-standard dimer interaction energies. Sci. Data 2021, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Al-Hamdani, Y.S.; Nagy, P.R.; Zen, A.; Barton, D.; Kállay, M.; Bradenburg, J.G.; Tkatchenko, A. Interactions between large molecules pose a puzzle for reference quantum mechanical methods. Nat. Commun. 2021, 12, 3927. [Google Scholar] [CrossRef] [PubMed]
- Czernek, J.; Brus, J. Reliable Dimerization Energies for Modeling of Supramolecular Junctions. Int. J. Mol. Sci. 2024, 25, 602. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Riplinger, C.; Becker, U.; Liakos, D.G.; Minenkov, Y.; Cavallo, L.; Neese, F. Communication: An improved linear scaling perturbative triples correction for the domain based local pair-natural orbital based singles and doubles coupled cluster method [DLPNO-CCSD(T)]. J. Chem. Phys. 2018, 148, 011101. [Google Scholar] [CrossRef] [PubMed]
- Řezáč, J.; Riley, K.E.; Hobza, P. S66: A Well-balanced Database of Benchmark Interaction Energies Relevant to Biomolecular Structures. J. Chem. Theory Comput. 2011, 7, 2427–2438. [Google Scholar] [CrossRef]
- Czernek, J.; Brus, J. Revisiting the Most Stable Structures of the Benzene Dimer. Int. J. Mol. Sci. 2024, 25, 8272. [Google Scholar] [CrossRef]
- Zhang, X.; Fu, Z.; Jiang, X.; Yang, Z.; Wang, S.; Wang, K.; Wu, Z.; Zhang, S.-T.; Liu, H.; Yang, B. Robust formation of discrete non-covalent pyrene dimers in an amorphous film by strong π-π interaction. Chem. Commun. 2022, 58, 8250–8253. [Google Scholar] [CrossRef]
- Shao, C.; Zhai, Y.; Cardenas-Salvarez, A.; Zhang, W.; Grajales-Gonzales, E.; Bai, X.; Li, Y.; Monge-Palacios, M.; Sarathy, S.M. High-resolution mass spectrometry of pyrene dimers formed in a jet-stirred reactor. Combust. Flame 2023, 255, 112886. [Google Scholar] [CrossRef]
- Feng, X.; Wang, X.; Redshaw, C.; Tang, B.Z. Aggregation behaviour of pyrene-based luminescent materials, from molecular design and optical properties to application. Chem. Soc. Rev. 2023, 52, 6715–6753. [Google Scholar] [CrossRef]
- Leboucher, H.; Simon, A.; Rapacioli, M. Structures and stabilities of PAH clusters solvated by water aggregates: The case of the pyrene dimer. J. Chem. Phys. 2023, 158, 114308. [Google Scholar] [CrossRef]
- Xia, Z.-A.; Yao, M.; Wang, S.; Yang, D.; Wang, Z.; Wu, R.; Zhang, S.-T.; Liu, H.; Yang, B. Tailoring pyrene excimer luminescence via controlled sulfur oxidation. J. Mater. Chem. C 2024, 12, 9305–9311. [Google Scholar] [CrossRef]
- Shao, C.; Wang, Q.; Zhang, W.; Bennett, A.; Li, Y.; Guo, J.; Im, H.G.; Roberts, W.L.; Violi, A.; Sarathy, M. Elucidating the polycyclic aromatic hydrocarbons involved in soot inception. Commun. Chem. 2023, 6, 223. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.-B.; Zhang, P.; Gu, Y.; Wang, J.-Q.; Han, M.-M.; Chen, C.; Zhan, X.-J.; Xie, Z.-L.; Zou, B.; Peng, Q.; et al. The Influence of Molecular Packing on the Emissive Behavior of Pyrene Derivatives: Mechanoluminiscence and Mechanochromism. Adv. Opt. Mater. 2018, 6, 1800198. [Google Scholar] [CrossRef]
- Gray, M.; Herbert, J.M. Assessing the domain-based local pair natural orbital (DLPNO) approximation for non-covalent interactions in sizable supramolecular complexes. J. Chem. Phys. 2024, 161, 054114. [Google Scholar] [CrossRef] [PubMed]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Peterson, K.A.; Woon, D.E.; Dunnig, T.J., Jr. Benchmark calculations with correlated wave functions. J. Chem. Phys. 1994, 100, 7410–7415. [Google Scholar] [CrossRef]
- The Benchmark Energy & Geometry Database (BEGDB). Available online: http://www.begdb.org/ (accessed on 28 August 2024).
- Kesharwani, M.K.; Karton, M.; Sylvetsky, N.; Martin, J.M.L. The S66 Non-Covalent Interactions Benchmark Reconsidered Using Explicitly Correlated Methods Near the Basis Set Limit. Austr. J. Chem. 2018, 71, 238–248. [Google Scholar] [CrossRef]
- Podeszwa, R.; Bukowski, R.; Szalewicz, K. Potential Energy Surface for the Benzene Dimer and Perturbational Analysis of π−π Interactions. J. Phys. Chem. A 2006, 110, 10345–10354. [Google Scholar] [CrossRef]
- Schnell, M.; Erlekam, U.; Bunker, P.R.; von Helden, G.; Grabow, J.-U.; Meijer, G.; van der Avoird, A. Structure of the Benzene Dimer—Governed by Dynamics. Angew. Chem. Int. Ed. 2013, 52, 5180–5183. [Google Scholar] [CrossRef] [PubMed]
- Herman, K.M.; Aprà, E.; Xantheas, S.S. A critical comparison of CH···π versus π···π interactions in the benzene dimer: Obtaining benchmarks at the CCSD(T) level and assessing the accuracy of lower scaling methods. Phys. Chem. Chem. Phys. 2023, 25, 4824–4838. [Google Scholar] [CrossRef] [PubMed]
- Podeszwa, R.; Szalewicz, K. Physical origins of interactions in dimers of polycyclic aromatic hydrocarbons. Phys. Chem. Chem. Phys. 2008, 10, 2735–2746. [Google Scholar] [CrossRef] [PubMed]
- Marshall, M.S.; Burns, L.A.; Sherrill, C.D. Basis set convergence of the coupled-cluster correction: Best practices for benchmarking non-covalent interactions and the attendant revision of the S22, NBC10, HBC6, and HSG databases. J. Chem. Phys. 2011, 135, 194102. [Google Scholar] [CrossRef]
- Patkowski, K. Recent developments in symmetry-adapted perturbation theory. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 10, e1452. [Google Scholar] [CrossRef]
- Shahbaz, M.; Szalewicz, K. Evaluation of methods for obtaining dispersion energies used in density functional calculations of intermolecular interactions. Theor. Chem. Acc. 2019, 138, 25. [Google Scholar] [CrossRef]
- Czernek, J.; Brus, J.; Czerneková, V.; Kobera, L. Quantifying the Intrinsic Strength of C–H⋯O Intermolecular Interactions. Molecules 2023, 28, 4478. [Google Scholar] [CrossRef]
- Henrichsmeyer, J.; Thelen, M.; Bröckel, M.; Fadel, M.; Behnle, S.; Sekkal-Rahal, M.; Fink, R.F. Rationalizing Aggregate Structures with Orbital Contributions to the Exchange-Repulsion Energy. ChemPhysChem 2023, 24, e202300097. [Google Scholar] [CrossRef]
- Gray, M.; Herbert, J.M. Origins of Offset-Stacking in Porous Frameworks. J. Phys. Chem. C 2023, 127, 2675–2686. [Google Scholar] [CrossRef]
- Rapacioli, M.; Spiegelman, F.; Talbi, D.; Mineva, T.; Goursot, A.; Heine, T.; Seifert, G. Correction for dispersion and Coulombic interactions in molecular clusters with density functional derived methods: Application to polycyclic aromatic hydrocarbon clusters. J. Chem. Phys. 2009, 130, 244304. [Google Scholar] [CrossRef]
- Baba, M.; Saitoh, M.; Kowaka, Y.; Taguma, K.; Yoshida, K.; Semba, Y.; Kasahara, S.; Yamanaka, T.; Ohshima, Y.; Hsu, Y.-C.; et al. Vibrational and rotational structure and excited-state dynamics of pyrene. J. Chem. Phys. 2009, 131, 224318. [Google Scholar] [CrossRef]
- Hoche, J.; Schmitt, H.-C.; Humeniuk, A.; Fischer, I.; Mitrić, R.; Röhr, M.I.S. The mechanism of excimer formation: An experimental and theoretical study on the pyrene dimer. Phys. Chem. Chem. Phys. 2017, 19, 25002–25015. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.; Biennier, L.; Klippenstein, S.J.; Sims, I.R.; Rowe, B.R. Exploring the Role of PAHs in the Formation of Soot: Pyrene Dimerization. J. Phys. Chem. Lett. 2010, 1, 2962–2967. [Google Scholar] [CrossRef]
- Sandler, I.; Chen, J.; Taylor, M.; Sharma, S.; Ho, J. Accuracy of DLPNO-CCSD(T): Effect of Basis Set and System Size. J. Phys. Chem. A 2021, 125, 1553–1563. [Google Scholar] [CrossRef] [PubMed]
- Carter-Fenk, K.; Herbert, J.M. Reinterpreting π-stacking. Phys. Chem. Chem. Phys. 2020, 22, 24870–24886. [Google Scholar] [CrossRef]
- Carter-Fenk, K.; Herbert, J.M. Electrostatics does not dictate the slip-stacked arrangement of aromatic π–π interactions. Chem. Sci. 2020, 11, 6758–6765. [Google Scholar] [CrossRef]
- Cabaleiro-Lago, E.M.; Rodríguez-Otero, J.; Vázquez, S.A. Electrostatic penetration effects stand at the heart of aromatic π interactions. Phys. Chem. Chem. Phys. 2022, 24, 8979–8991. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.J.; Knowles, P.J.; Manby, F.R.; Black, J.A.; Doll, K.; Hesselmann, A.; Kats, D.; Kohn, A.; Korona, T.; Kreplin, D.A.; et al. The Molpro quantum chemistry package. J. Chem. Phys. 2020, 152, 144107. [Google Scholar] [CrossRef]
- Balasubramani, S.G.; Chen, G.P.; Coriani, S.; Diedenhofen, M.; Frank, M.S.; Franzke, Y.J.; Furche, F.; Grotjahn, R.; Harding, M.E.; Hättig, C.; et al. TURBOMOLE: Modular program suite for ab initio quantum-chemical and condensed-matter simulations. J. Chem. Phys. 2020, 152, 184107. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M. RI-MP2: First derivatives and global consistency. Theor. Chem. Acc. 1997, 97, 331–340. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M.; Patzelt, H.; Ahlrichs, R. RI-MP2: Optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143–152. [Google Scholar] [CrossRef]
- Czernek, J.; Brus, J.; Czerneková, V. A Cost Effective Scheme for the Highly Accurate Description of Intermolecular Binding in Large Complexes. Int. J. Mol. Sci. 2022, 23, 15773. [Google Scholar] [CrossRef]
- Halkier, A.; Helgaker, T.; Jørgensen, P.; Klopper, W.; Koch, H.; Olsen, J.; Wilson, A.K. Basis-set convergence in correlated calculations on Ne, N2, and H2O. Chem. Phys. Lett. 1998, 286, 243–252. [Google Scholar] [CrossRef]
- Pinski, P.; Riplinger, C.; Valeev, E.F.; Neese, F. Sparse maps–A systematic infrastructure for reduced-scaling electronic structure methods. I. An efficient and simple linear scaling local MP2 method that uses an intermediate basis of pair natural orbitals. J. Chem. Phys. 2015, 143, 034108. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Heßelmann, A.; Jansen, G. Density-functional theory-symmetry-adapted intermolecular perturbation theory with density fitting: A new efficient method to study intermolecular interaction energies. J. Chem. Phys. 2005, 122, 014103. [Google Scholar] [CrossRef] [PubMed]
- Czernek, J.; Brus, J.; Czerneková, V. A computational inspection of the dissociation energy of mid-sized organic dimers. J. Chem. Phys. 2022, 156, 204303. [Google Scholar] [CrossRef]
- Heßelmann, A.; Jansen, G. First-order intermolecular interaction energies from Kohn–Sham orbitals. Chem. Phys. Lett. 2002, 357, 464–470. [Google Scholar] [CrossRef]
- Heßelmann, A.; Jansen, G. Intermolecular dispersion energies from time-dependent density functional theory. Chem. Phys. Lett. 2003, 367, 778–784. [Google Scholar] [CrossRef]
- Heßelmann, A.; Jansen, G. Intermolecular induction and exchange-induction energies from coupled-perturbed Kohn–Sham density functional theory. Chem. Phys. Lett. 2002, 362, 319–325. [Google Scholar] [CrossRef]
- Moszynski, R.; Heijmen, T.G.A.; Jeziorski, B. Symmetry-adapted perturbation theory for the calculation of Hartree–Fock interaction energies. Mol. Phys. 1996, 88, 741–758. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Frish, M.J.; Trucks, J.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Dai, Y.; Rambaldi, F.; Negri, F. Eclipsed and Twisted Excimers of Pyrene and 2-Azapyrene: How Nitrogen Substitution Impacts Excimer Emission. Molecules 2024, 29, 507. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System (Designation in S66 Set) | CCSD(T)/CBS |ΔE| Estimate/kJ/mol | ||||
|---|---|---|---|---|---|
| From Equation (1) | From Equation (2) | From Ref. [22] (a) | From Ref. [20] (b) | From Ref. [19] (c) | |
| benzene–benzene (S24) | 10.926 | 10.762 | 11.171 ± 0.293 | 11.238 | 10.548 |
| pyridine–pyridine (S25) | 15.462 | 15.340 | 15.481 ± 0.335 | 15.731 | 15.104 |
| uracil–uracil (S26) | 40.578 | 40.593 | 40.203 ± 0.418 | 40.652 | 40.246 |
| benzene–pyridine (S27) | 13.561 | 13.428 | 13.724 ± 0.293 | 13.824 | 13.205 |
| benzene–uracil (S28) | 23.239 | 23.101 | 22.928 ± 0.460 | 23.200 | 22.866 |
| pyridine–uracil (S29) | 27.905 | 27.774 | 27.656 ± 0.377 | 27.870 | 27.514 |
| benzene–ethene (S30) | 5.570 | 5.482 | — | 5.619 | 5.310 |
| uracil–ethene (S31) | 13.860 | 13.800 | — | 13.823 | 13.627 |
| uracil–ethyne (S32) | 15.362 | 15.333 | — | 15.376 | 15.008 |
| pyridine–ethene (S33) | 7.455 | 7.378 | — | 7.464 | 7.201 |
| Parameter | Configuration (Symmetry) | |||
|---|---|---|---|---|
| L (C2h) | G (Cs) | S (C2h) | X (D2h) | |
| /pm | 344 | 347 | 350 | 355 |
| ΔE/kJ/mol | –52.1 | –50.3 | –49.1 | –48.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czernek, J.; Brus, J. On the Potential Energy Surface of the Pyrene Dimer. Int. J. Mol. Sci. 2024, 25, 10762. https://doi.org/10.3390/ijms251910762
Czernek J, Brus J. On the Potential Energy Surface of the Pyrene Dimer. International Journal of Molecular Sciences. 2024; 25(19):10762. https://doi.org/10.3390/ijms251910762
Chicago/Turabian StyleCzernek, Jiří, and Jiří Brus. 2024. "On the Potential Energy Surface of the Pyrene Dimer" International Journal of Molecular Sciences 25, no. 19: 10762. https://doi.org/10.3390/ijms251910762
APA StyleCzernek, J., & Brus, J. (2024). On the Potential Energy Surface of the Pyrene Dimer. International Journal of Molecular Sciences, 25(19), 10762. https://doi.org/10.3390/ijms251910762