
Potential Mechanisms of Tunneling Nanotube Formation and Their Role in Pathology Spread in Alzheimer’s Disease and Other Proteinopathies

 , , , , , , ,
, , , , , , ,
Abstract
1. Introduction
2. Pathologic Aggregation and Spreading of Misfolded Aβ and Tau Protein in AD Pathogenesis
2.1. Ab and Tau Aggregation and Formation of Pathogenic Species
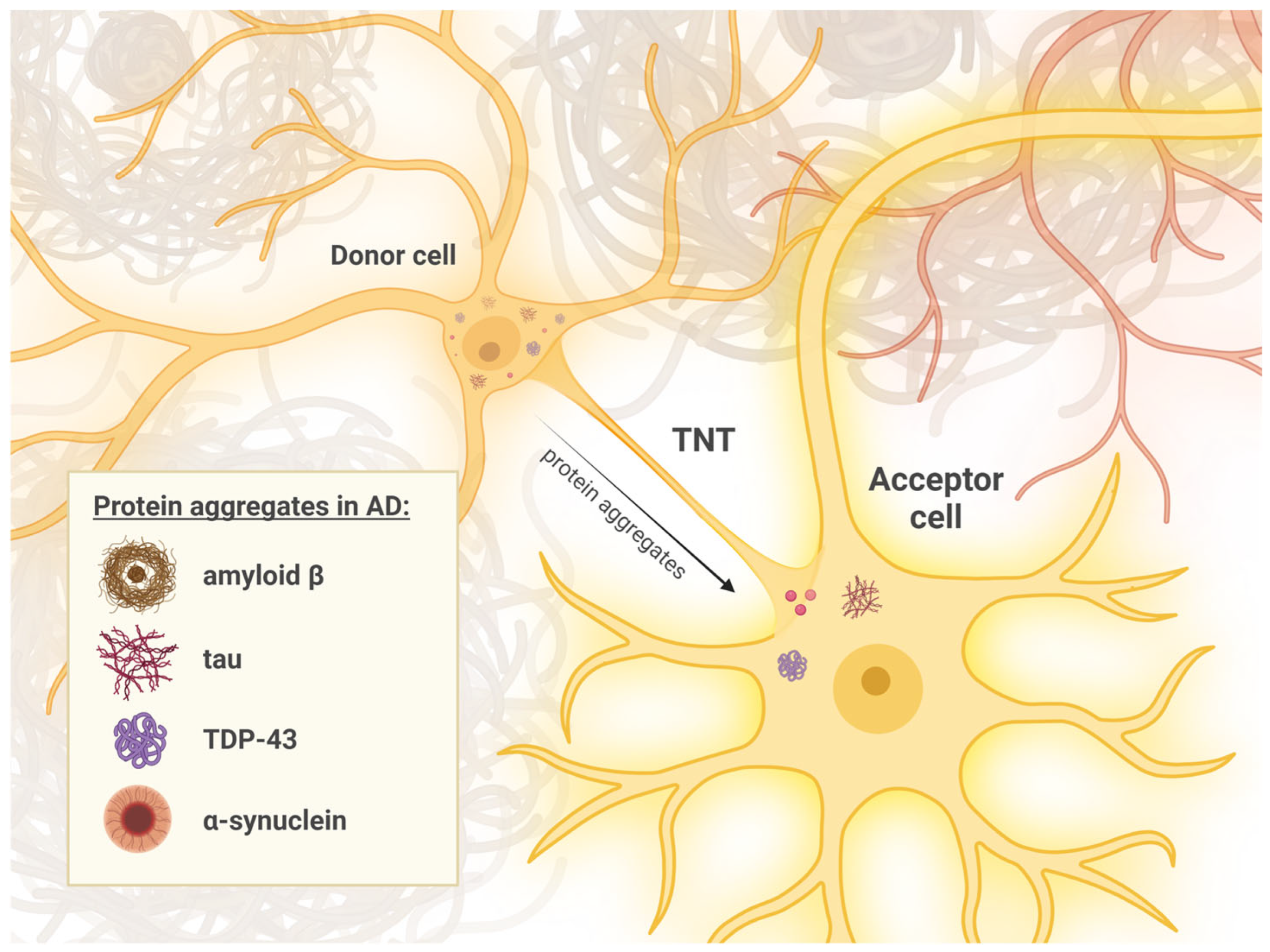
2.2. Cellular Mechanisms of Pathology Transmission
3. TNT Structure, Biogenesis, and Significance in Health and Disease
3.1. Potential Mechanisms of TNT Formation
3.2. TNTs—Role in Physiology and Pathology
4. TNTs’ Role in AD
4.1. Amyloid-Beta
4.2. Tau
4.3. Other Proteins
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Atri, A. The Alzheimer’s Disease Clinical Spectrum: Diagnosis and Management. Med. Clin. N. Am. 2019, 103, 263–293. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Action Plan on the Public Health Response to Dementia 2017–2025; World Health Organization: Geneva, Switzerland, 2017; ISBN 978-92-4-151348-7. [Google Scholar]
- GBD 2019 Dementia Forecasting Collaborators. Estimation of the Global Prevalence of Dementia in 2019 and Forecasted Prevalence in 2050: An Analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- Waldemar, G.; Dubois, B.; Emre, M.; Georges, J.; McKeith, I.G.; Rossor, M.; Scheltens, P.; Tariska, P.; Winblad, B. EFNS Recommendations for the Diagnosis and Management of Alzheimer’s Disease and Other Disorders Associated with Dementia: EFNS Guideline. Eur. J. Neurol. 2007, 14, e1–e26. [Google Scholar] [CrossRef] [PubMed]
- Tabert, M.H.; Liu, X.; Doty, R.L.; Serby, M.; Zamora, D.; Pelton, G.H.; Marder, K.; Albers, M.W.; Stern, Y.; Devanand, D.P. A 10-Item Smell Identification Scale Related to Risk for Alzheimer’s Disease. Ann. Neurol. 2005, 58, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The Cholinergic System in the Pathophysiology and Treatment of Alzheimer’s Disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Mielke, M.L.; Gómez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. Reactive Glia Not Only Associates with Plaques but Also Parallels Tangles in Alzheimer’s Disease. Am. J. Pathol. 2011, 179, 1373–1384. [Google Scholar] [CrossRef]
- Liu, X.; Che, R.; Liang, W.; Zhang, Y.; Wu, L.; Han, C.; Lu, H.; Song, W.; Wu, Y.; Wang, Z. Clusterin Transduces Alzheimer-Risk Signals to Amyloidogenesis. Signal Transduct. Target Ther. 2022, 7, 1–4. [Google Scholar] [CrossRef]
- Depp, C.; Sun, T.; Sasmita, A.O.; Spieth, L.; Berghoff, S.A.; Nazarenko, T.; Overhoff, K.; Steixner-Kumar, A.A.; Subramanian, S.; Arinrad, S.; et al. Myelin Dysfunction Drives Amyloid-β Deposition in Models of Alzheimer’s Disease. Nature 2023, 618, 349–357. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Bateman, R.J.; Munsell, L.Y.; Morris, J.C.; Swarm, R.; Yarasheski, K.E.; Holtzman, D.M. Human Amyloid-β Synthesis and Clearance Rates as Measured in Cerebrospinal Fluid in Vivo. Nat. Med. 2006, 12, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wang, Z.; Wang, R.; Zhang, X.; Zhang, S.; Wu, Y.; Staufenbiel, M.; Cai, F.; Song, W. Amyloid-β Protein (Aβ) Glu11 Is the Major β-Secretase Site of β-Site Amyloid-β Precursor Protein-Cleaving Enzyme 1(BACE1), and Shifting the Cleavage Site to Aβ Asp1 Contributes to Alzheimer Pathogenesis. Eur. J. Neurosci. 2013, 37, 1962–1969. [Google Scholar] [CrossRef] [PubMed]
- Alexander, G.C.; Emerson, S.; Kesselheim, A.S. Evaluation of Aducanumab for Alzheimer Disease: Scientific Evidence and Regulatory Review Involving Efficacy, Safety, and Futility. JAMA 2021, 325, 1717–1718. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Viola, K.L.; Bicca, M.A.; Bebenek, A.M.; Kranz, D.L.; Nandwana, V.; Waters, E.A.; Haney, C.R.; Lee, M.; Gupta, A.; Brahmbhatt, Z.; et al. The Therapeutic and Diagnostic Potential of Amyloid β Oligomers Selective Antibodies to Treat Alzheimer’s Disease. Front. Neurosci. 2022, 15, 768646. [Google Scholar] [CrossRef]
- Wang, A.-Y.; Hu, H.-Y.; Huang, L.-Y.; Xiao, C.-Y.; Li, Q.-Y.; Tan, L.; Hu, H. Risk Factors for Cognitive Decline in Non-Demented Elders with Amyloid-Beta Positivity. Alzheimer’s Res. Ther. 2024, 16, 189. [Google Scholar] [CrossRef]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-Associated Protein Tau (Tau) Is a Major Antigenic Component of Paired Helical Filaments in Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef]
- Vossel, K.A.; Zhang, K.; Brodbeck, J.; Daub, A.C.; Sharma, P.; Finkbeiner, S.; Cui, B.; Mucke, L. Tau Reduction Prevents Abeta-Induced Defects in Axonal Transport. Science 2010, 330, 198. [Google Scholar] [CrossRef]
- Vossel, K.A.; Xu, J.C.; Fomenko, V.; Miyamoto, T.; Suberbielle, E.; Knox, J.A.; Ho, K.; Kim, D.H.; Yu, G.-Q.; Mucke, L. Tau Reduction Prevents Aβ-Induced Axonal Transport Deficits by Blocking Activation of GSK3β. J. Cell Biol. 2015, 209, 419–433. [Google Scholar] [CrossRef]
- Baker, S.; Polanco, J.C.; Götz, J. Extracellular Vesicles Containing P301L Mutant Tau Accelerate Pathological Tau Phosphorylation and Oligomer Formation but Do Not Seed Mature Neurofibrillary Tangles in ALZ17 Mice. J. Alzheimer’s Dis. 2016, 54, 1207–1217. [Google Scholar] [CrossRef]
- Dujardin, S.; Commins, C.; Lathuiliere, A.; Beerepoot, P.; Fernandes, A.R.; Kamath, T.V.; De Los Santos, M.B.; Klickstein, N.; Corjuc, D.L.; Corjuc, B.T.; et al. Tau Molecular Diversity Contributes to Clinical Heterogeneity in Alzheimer’s Disease. Nat. Med. 2020, 26, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary Tangles but Not Senile Plaques Parallel Duration and Severity of Alzheimer’s Disease. Neurology 1992, 42, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Kon, T.; Tanji, K.; Mori, F.; Kimura, A.; Kakita, A.; Wakabayashi, K. Immunoreactivity of Myelin-Associated Oligodendrocytic Basic Protein in Lewy Bodies. Neuropathology 2019, 39, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein Strains Cause Distinct Synucleinopathies after Local and Systemic Administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Buciuc, M.; Whitwell, J.L.; Boeve, B.F.; Ferman, T.J.; Graff-Radford, J.; Savica, R.; Kantarci, K.; Fields, J.A.; Knopman, D.S.; Petersen, R.C.; et al. TDP-43 Is Associated with a Reduced Likelihood of Rendering a Clinical Diagnosis of Dementia with Lewy Bodies in Autopsy-Confirmed Cases of Transitional/Diffuse Lewy Body Disease. J. Neurol. 2020, 267, 1444–1453. [Google Scholar] [CrossRef]
- Arai, Y.; Yamazaki, M.; Mori, O.; Muramatsu, H.; Asano, G.; Katayama, Y. α-Synuclein-Positive Structures in Cases with Sporadic Alzheimer’s Disease: Morphology and Its Relationship to Tau Aggregation. Brain Res. 2001, 888, 287–296. [Google Scholar] [CrossRef]
- Larson, M.E.; Sherman, M.A.; Greimel, S.; Kuskowski, M.; Schneider, J.A.; Bennett, D.A.; Lesné, S.E. Soluble α-Synuclein Is a Novel Modulator of Alzheimer’s Disease Pathophysiology. J. Neurosci. 2012, 32, 10253–10266. [Google Scholar] [CrossRef]
- Shi, M.; Tang, L.; Toledo, J.B.; Ginghina, C.; Wang, H.; Aro, P.; Jensen, P.H.; Weintraub, D.; Chen-Plotkin, A.S.; Irwin, D.J.; et al. Cerebrospinal Fluid α-Synuclein Contributes to the Differential Diagnosis of Alzheimer’s Disease. Alzheimer’s Dement. 2018, 14, 1052–1062. [Google Scholar] [CrossRef]
- Berge, G.; Sando, S.B.; Albrektsen, G.; Lauridsen, C.; Møller, I.; Grøntvedt, G.R.; Bråthen, G.; White, L.R. Alpha-Synuclein Measured in Cerebrospinal Fluid from Patients with Alzheimer’s Disease, Mild Cognitive Impairment, or Healthy Controls: A Two Year Follow-up Study. BMC Neurol. 2016, 16, 180. [Google Scholar] [CrossRef]
- Toledo, J.B.; Korff, A.; Shaw, L.M.; Trojanowski, J.Q.; Zhang, J. CSF α-Synuclein Improves Diagnostic and Prognostic Performance of CSF Tau and Aβ in Alzheimer’s Disease. Acta Neuropathol. 2013, 126, 683–697. [Google Scholar] [CrossRef]
- Oikawa, T.; Nonaka, T.; Terada, M.; Tamaoka, A.; Hisanaga, S.; Hasegawa, M. α-Synuclein Fibrils Exhibit Gain of Toxic Function, Promoting Tau Aggregation and Inhibiting Microtubule Assembly. J. Biol. Chem. 2016, 291, 15046–15056. [Google Scholar] [CrossRef] [PubMed]
- Toledo, J.B.; Gopal, P.; Raible, K.; Irwin, D.J.; Brettschneider, J.; Sedor, S.; Waits, K.; Boluda, S.; Grossman, M.; Van Deerlin, V.M.; et al. Pathological α-Synuclein Distribution in Subjects with Coincident Alzheimer’s and Lewy Body Pathology. Acta Neuropathol. 2016, 131, 393–409. [Google Scholar] [CrossRef] [PubMed]
- Buerger, K.; Ewers, M.; Pirttilä, T.; Zinkowski, R.; Alafuzoff, I.; Teipel, S.J.; DeBernardis, J.; Kerkman, D.; McCulloch, C.; Soininen, H.; et al. CSF Phosphorylated Tau Protein Correlates with Neocortical Neurofibrillary Pathology in Alzheimer’s Disease. Brain 2006, 129, 3035–3041. [Google Scholar] [CrossRef] [PubMed]
- Twohig, D.; Rodriguez-Vieitez, E.; Sando, S.B.; Berge, G.; Lauridsen, C.; Møller, I.; Grøntvedt, G.R.; Bråthen, G.; Patra, K.; Bu, G.; et al. The Relevance of Cerebrospinal Fluid α-Synuclein Levels to Sporadic and Familial Alzheimer’s Disease. Acta Neuropathol. Commun. 2018, 6, 130. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Whitwell, J.L.; Weigand, S.D.; Murray, M.E.; Tosakulwong, N.; Liesinger, A.M.; Petrucelli, L.; Senjem, M.L.; Knopman, D.S.; Boeve, B.F.; et al. TDP-43 Is a Key Player in the Clinical Features Associated with Alzheimer’s Disease. Acta Neuropathol. 2014, 127, 811–824. [Google Scholar] [CrossRef]
- Katsumata, Y.; Fardo, D.W.; Kukull, W.A.; Nelson, P.T. Dichotomous Scoring of TDP-43 Proteinopathy from Specific Brain Regions in 27 Academic Research Centers: Associations with Alzheimer’s Disease and Cerebrovascular Disease Pathologies. Acta Neuropathol. Commun. 2018, 6, 142. [Google Scholar] [CrossRef]
- Josephs, K.A.; Whitwell, J.L.; Tosakulwong, N.; Weigand, S.D.; Murray, M.E.; Liesinger, A.M.; Petrucelli, L.; Senjem, M.L.; Ivnik, R.J.; Parisi, J.E.; et al. TAR DNA-Binding Protein 43 and Pathological Subtype of Alzheimer’s Disease Impact Clinical Features. Ann. Neurol. 2015, 78, 697–709. [Google Scholar] [CrossRef]
- Pergu, R.; Dagar, S.; Kumar, H.; Kumar, R.; Bhattacharya, J.; Mylavarapu, S.V.S. The Chaperone ERp29 Is Required for Tunneling Nanotube Formation by Stabilizing MSec. J. Biol. Chem. 2019, 294, 7177–7193. [Google Scholar] [CrossRef]
- Li, A.; Han, X.; Deng, L.; Wang, X. Mechanical Properties of Tunneling Nanotube and Its Mechanical Stability in Human Embryonic Kidney Cells. Front. Cell Dev. Biol. 2022, 10, 955676. [Google Scholar] [CrossRef]
- Zaccard, C.R.; Watkins, S.C.; Kalinski, P.; Fecek, R.J.; Yates, A.L.; Salter, R.D.; Ayyavoo, V.; Rinaldo, C.R.; Mailliard, R.B. CD40L Induces Functional Tunneling Nanotube Networks Exclusively in Dendritic Cells Programmed by Mediators of Type 1 Immunity. J. Immunol. 2015, 194, 1047–1056. [Google Scholar] [CrossRef]
- Martínez, A.D.; Eugenín, E.A.; Brañes, M.C.; Bennett, M.V.L.; Sáez, J.C. Identification of Second Messengers That Induce Expression of Functional Gap Junctions in Microglia Cultured from Newborn Rats. Brain Res. 2002, 943, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, K.A.; Yerkovich, S.T.; Hopkins, P.M.-A.; Chambers, D.C. Characterization of Intercellular Communication and Mitochondrial Donation by Mesenchymal Stromal Cells Derived from the Human Lung. Stem Cell Res. Ther. 2016, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.V.; Morrison, T.J.; Doherty, D.F.; McAuley, D.F.; Matthay, M.A.; Kissenpfennig, A.; O’Kane, C.M.; Krasnodembskaya, A.D. Mitochondrial Transfer via Tunneling Nanotubes Is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the In Vitro and In Vivo Models of ARDS. Stem Cells 2016, 34, 2210–2223. [Google Scholar] [CrossRef] [PubMed]
- Dupont, M.; Souriant, S.; Lugo-Villarino, G.; Maridonneau-Parini, I.; Vérollet, C. Tunneling Nanotubes: Intimate Communication between Myeloid Cells. Front. Immunol. 2018, 9, 43. [Google Scholar] [CrossRef]
- Caneparo, L.; Pantazis, P.; Dempsey, W.; Fraser, S.E. Intercellular Bridges in Vertebrate Gastrulation. PLoS ONE 2011, 6, e20230. [Google Scholar] [CrossRef]
- Cordero Cervantes, D.; Khare, H.; Wilson, A.M.; Mendoza, N.D.; Coulon-Mahdi, O.; Lichtman, J.W.; Zurzolo, C. 3D Reconstruction of the Cerebellar Germinal Layer Reveals Tunneling Connections between Developing Granule Cells. Sci. Adv. 2023, 9, eadf3471. [Google Scholar] [CrossRef]
- Wang, X.; Bukoreshtliev, N.V.; Gerdes, H.-H. Developing Neurons Form Transient Nanotubes Facilitating Electrical Coupling and Calcium Signaling with Distant Astrocytes. PLoS ONE 2012, 7, e47429. [Google Scholar] [CrossRef]
- Wang, X.; Veruki, M.L.; Bukoreshtliev, N.V.; Hartveit, E.; Gerdes, H.-H. Animal Cells Connected by Nanotubes Can Be Electrically Coupled through Interposed Gap-Junction Channels. Proc. Natl. Acad. Sci. USA 2010, 107, 17194–17199. [Google Scholar] [CrossRef]
- Dubey, G.P.; Ben-Yehuda, S. Intercellular Nanotubes Mediate Bacterial Communication. Cell 2011, 144, 590–600. [Google Scholar] [CrossRef]
- Gousset, K.; Schiff, E.; Langevin, C.; Marijanovic, Z.; Caputo, A.; Browman, D.T.; Chenouard, N.; de Chaumont, F.; Martino, A.; Enninga, J.; et al. Prions Hijack Tunnelling Nanotubes for Intercellular Spread. Nat. Cell Biol. 2009, 11, 328–336. [Google Scholar] [CrossRef]
- Dilna, A.; Deepak, K.V.; Damodaran, N.; Kielkopf, C.S.; Kagedal, K.; Ollinger, K.; Nath, S. Amyloid-β Induced Membrane Damage Instigates Tunneling Nanotube-like Conduits by P21-Activated Kinase Dependent Actin Remodulation. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166246. [Google Scholar] [CrossRef] [PubMed]
- Rostami, J.; Holmqvist, S.; Lindström, V.; Sigvardson, J.; Westermark, G.T.; Ingelsson, M.; Bergström, J.; Roybon, L.; Erlandsson, A. Human Astrocytes Transfer Aggregated Alpha-Synuclein via Tunneling Nanotubes. J. Neurosci. 2017, 37, 11835–11853. [Google Scholar] [CrossRef] [PubMed]
- Tardivel, M.; Bégard, S.; Bousset, L.; Dujardin, S.; Coens, A.; Melki, R.; Buée, L.; Colin, M. Tunneling Nanotube (TNT)-Mediated Neuron-to Neuron Transfer of Pathological Tau Protein Assemblies. Acta Neuropathol. Commun. 2016, 4, 117. [Google Scholar] [CrossRef] [PubMed]
- Abounit, S.; Bousset, L.; Loria, F.; Zhu, S.; de Chaumont, F.; Pieri, L.; Olivo-Marin, J.-C.; Melki, R.; Zurzolo, C. Tunneling Nanotubes Spread Fibrillar α-Synuclein by Intercellular Trafficking of Lysosomes. EMBO J. 2016, 35, 2120–2138. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Fu, H.; Hardy, J.; Duff, K.E. Selective Vulnerability in Neurodegenerative Diseases. Nat. Neurosci. 2018, 21, 1350–1358. [Google Scholar] [CrossRef]
- Ansari, M.A.; Scheff, S.W. Oxidative Stress in the Progression of Alzheimer Disease in the Frontal Cortex. J. Neuropathol. Exp. Neurol. 2010, 69, 155–167. [Google Scholar] [CrossRef]
- Du, L.; Zhang, X.; Han, Y.Y.; Burke, N.A.; Kochanek, P.M.; Watkins, S.C.; Graham, S.H.; Carcillo, J.A.; Szabó, C.; Clark, R.S.B. Intra-Mitochondrial Poly(ADP-Ribosylation) Contributes to NAD+ Depletion and Cell Death Induced by Oxidative Stress. J. Biol. Chem. 2003, 278, 18426–18433. [Google Scholar] [CrossRef]
- Agarwal, M.; Alam, M.R.; Haider, M.K.; Malik, M.Z.; Kim, D.-K. Alzheimer’s Disease: An Overview of Major Hypotheses and Therapeutic Options in Nanotechnology. Nanomaterials 2020, 11, 59. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Sabbagh, M.; Cohen, S. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 1631–1632. [Google Scholar] [CrossRef] [PubMed]
- Tucker, S.; Möller, C.; Tegerstedt, K.; Lord, A.; Laudon, H.; Sjödahl, J.; Söderberg, L.; Spens, E.; Sahlin, C.; Waara, E.R.; et al. The Murine Version of BAN2401 (mAb158) Selectively Reduces Amyloid-β Protofibrils in Brain and Cerebrospinal Fluid of Tg-ArcSwe Mice. J. Alzheimers Dis. 2015, 43, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Söderberg, L.; Johannesson, M.; Nygren, P.; Laudon, H.; Eriksson, F.; Osswald, G.; Möller, C.; Lannfelt, L. Lecanemab, Aducanumab, and Gantenerumab—Binding Profiles to Different Forms of Amyloid-Beta Might Explain Efficacy and Side Effects in Clinical Trials for Alzheimer’s Disease. Neurotherapeutics 2023, 20, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Farlow, M.; Anand, R.; Messina, J., Jr.; Hartman, R.; Veach, J. A 52-Week Study of the Efficacy of Rivastigmine in Patients with Mild to Moderately Severe Alzheimer’s Disease. Eur. Neurol. 2000, 44, 236–241. [Google Scholar] [CrossRef]
- Rountree, S.D.; Atri, A.; Lopez, O.L.; Doody, R.S. Effectiveness of Antidementia Drugs in Delaying Alzheimer’s Disease Progression. Alzheimer’s Dement. 2013, 9, 338–345. [Google Scholar] [CrossRef]
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-Synaptic Spread of Tau Pathology In Vivo. PLoS ONE 2012, 7, e31302. [Google Scholar] [CrossRef]
- Ulusoy, A.; Rusconi, R.; Pérez-Revuelta, B.I.; Musgrove, R.E.; Helwig, M.; Winzen-Reichert, B.; Monte, D.A.D. Caudo-rostral Brain Spreading of A-synuclein through Vagal Connections. EMBO Mol. Med. 2013, 5, 1119–1127. [Google Scholar] [CrossRef]
- de Calignon, A.; Polydoro, M.; Suárez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of Tau Pathology in a Model of Early Alzheimer’s Disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef]
- Yetman, M.J.; Lillehaug, S.; Bjaalie, J.G.; Leergaard, T.B.; Jankowsky, J.L. Transgene Expression in the Nop-tTA Driver Line Is Not Inherently Restricted to the Entorhinal Cortex. Brain Struct. Funct. 2016, 221, 2231–2249. [Google Scholar] [CrossRef]
- Fritschi, S.K.; Langer, F.; Kaeser, S.A.; Maia, L.F.; Portelius, E.; Pinotsi, D.; Kaminski, C.F.; Winkler, D.T.; Maetzler, W.; Keyvani, K.; et al. Highly Potent Soluble Amyloid-β Seeds in Human Alzheimer Brain but Not Cerebrospinal Fluid. Brain 2014, 137, 2909–2915. [Google Scholar] [CrossRef]
- Eisele, Y.S.; Obermüller, U.; Heilbronner, G.; Baumann, F.; Kaeser, S.A.; Wolburg, H.; Walker, L.C.; Staufenbiel, M.; Heikenwalder, M.; Jucker, M. Peripherally Applied Abeta-Containing Inoculates Induce Cerebral Beta-Amyloidosis. Science 2010, 330, 980–982. [Google Scholar] [CrossRef] [PubMed]
- Domert, J.; Rao, S.B.; Agholme, L.; Brorsson, A.-C.; Marcusson, J.; Hallbeck, M.; Nath, S. Spreading of Amyloid-β Peptides via Neuritic Cell-to-Cell Transfer Is Dependent on Insufficient Cellular Clearance. Neurobiol. Dis. 2014, 65, 82–92. [Google Scholar] [CrossRef]
- Marzesco, A.-M.; Flötenmeyer, M.; Bühler, A.; Obermüller, U.; Staufenbiel, M.; Jucker, M.; Baumann, F. Highly Potent Intracellular Membrane-Associated Aβ Seeds. Sci. Rep. 2016, 6, 28125. [Google Scholar] [CrossRef]
- He, Z.; Guo, J.L.; McBride, J.D.; Narasimhan, S.; Kim, H.; Changolkar, L.; Zhang, B.; Gathagan, R.J.; Yue, C.; Dengler, C.; et al. Amyloid-β Plaques Enhance Alzheimer’s Brain Tau-Seeded Pathologies by Facilitating Neuritic Plaque Tau Aggregation. Nat. Med. 2018, 24, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Petralia, R.S.; Kurushima, H.; Patel, H.; Jung, M.; Volk, L.; Chowdhury, S.; Shepherd, J.D.; Dehoff, M.; Li, Y.; et al. Arc/Arg3.1 Regulates an Endosomal Pathway Essential for Activity-Dependent β-Amyloid Generation. Cell 2011, 147, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, J.; Thériault, C.; Lessard-Beaudoin, M.; Marcil, A.; Dahan, S.; Lavoie, C. LDLR-Related Protein 10 (LRP10) Regulates Amyloid Precursor Protein (APP) Trafficking and Processing: Evidence for a Role in Alzheimer’s Disease. Mol. Neurodegener. 2012, 7, 31. [Google Scholar] [CrossRef]
- Fol, R.; Braudeau, J.; Ludewig, S.; Abel, T.; Weyer, S.W.; Roederer, J.-P.; Brod, F.; Audrain, M.; Bemelmans, A.-P.; Buchholz, C.J.; et al. Viral Gene Transfer of APPsα Rescues Synaptic Failure in an Alzheimer’s Disease Mouse Model. Acta Neuropathol. 2016, 131, 247–266. [Google Scholar] [CrossRef]
- Feng, T.; Tammineni, P.; Agrawal, C.; Jeong, Y.Y.; Cai, Q. Autophagy-Mediated Regulation of BACE1 Protein Trafficking and Degradation. J. Biol. Chem. 2017, 292, 1679–1690. [Google Scholar] [CrossRef]
- Sadleir, K.R.; Kandalepas, P.C.; Buggia-Prévot, V.; Nicholson, D.A.; Thinakaran, G.; Vassar, R. Presynaptic Dystrophic Neurites Surrounding Amyloid Plaques Are Sites of Microtubule Disruption, BACE1 Elevation, and Increased Aβ Generation in Alzheimer’s Disease. Acta Neuropathol. 2016, 132, 235–256. [Google Scholar] [CrossRef]
- Zhao, J.; Fu, Y.; Yasvoina, M.; Shao, P.; Hitt, B.; O’Connor, T.; Logan, S.; Maus, E.; Citron, M.; Berry, R.; et al. β-Site Amyloid Precursor Protein Cleaving Enzyme 1 Levels Become Elevated in Neurons around Amyloid Plaques: Implications for Alzheimer’s Disease Pathogenesis. J. Neurosci. 2007, 27, 3639–3649. [Google Scholar] [CrossRef]
- Kandalepas, P.C.; Sadleir, K.R.; Eimer, W.A.; Zhao, J.; Nicholson, D.A.; Vassar, R. The Alzheimer’s β-Secretase BACE1 Localizes to Normal Presynaptic Terminals and to Dystrophic Presynaptic Terminals Surrounding Amyloid Plaques. Acta Neuropathol. 2013, 126, 329–352. [Google Scholar] [CrossRef] [PubMed]
- Huse, J.T.; Liu, K.; Pijak, D.S.; Carlin, D.; Lee, V.M.-Y.; Doms, R.W. Beta-Secretase Processing in the Trans-Golgi Network Preferentially Generates Truncated Amyloid Species That Accumulate in Alzheimer’s Disease Brain. J. Biol. Chem. 2002, 277, 16278–16284. [Google Scholar] [CrossRef] [PubMed]
- Choy, R.W.-Y.; Cheng, Z.; Schekman, R. Amyloid Precursor Protein (APP) Traffics from the Cell Surface via Endosomes for Amyloid β (Aβ) Production in the Trans-Golgi Network. Proc. Natl. Acad. Sci. USA 2012, 109, E2077–E2082. [Google Scholar] [CrossRef] [PubMed]
- Frieg, B.; Han, M.; Giller, K.; Dienemann, C.; Riedel, D.; Becker, S.; Andreas, L.B.; Griesinger, C.; Schröder, G.F. Cryo-EM Structures of Lipidic Fibrils of Amyloid-β (1–40). Nat. Commun. 2024, 15, 1297. [Google Scholar] [CrossRef] [PubMed]
- Bruinsma, I.B.; te Riet, L.; Gevers, T.; ten Dam, G.B.; van Kuppevelt, T.H.; David, G.; Küsters, B.; de Waal, R.M.W.; Verbeek, M.M. Sulfation of Heparan Sulfate Associated with Amyloid-Beta Plaques in Patients with Alzheimer’s Disease. Acta Neuropathol. 2010, 119, 211–220. [Google Scholar] [CrossRef]
- Jana, A.K.; Batkulwar, K.B.; Kulkarni, M.J.; Sengupta, N. Glycation Induces Conformational Changes in the Amyloid-β Peptide and Enhances Its Aggregation Propensity: Molecular Insights. Phys. Chem. Chem. Phys. 2016, 18, 31446–31458. [Google Scholar] [CrossRef]
- Arosio, P.; Knowles, T.P.J.; Linse, S. On the Lag Phase in Amyloid Fibril Formation. Phys. Chem. Chem. Phys. 2015, 17, 7606–7618. [Google Scholar] [CrossRef]
- Frankel, R.; Törnquist, M.; Meisl, G.; Hansson, O.; Andreasson, U.; Zetterberg, H.; Blennow, K.; Frohm, B.; Cedervall, T.; Knowles, T.P.J.; et al. Autocatalytic Amplification of Alzheimer-Associated Aβ42 Peptide Aggregation in Human Cerebrospinal Fluid. Commun. Biol. 2019, 2, 365. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-Beta Protein Dimers Isolated Directly from Alzheimer’s Brains Impair Synaptic Plasticity and Memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef]
- Brinkmalm, G.; Hong, W.; Wang, Z.; Liu, W.; O’Malley, T.T.; Sun, X.; Frosch, M.P.; Selkoe, D.J.; Portelius, E.; Zetterberg, H.; et al. Identification of Neurotoxic Cross-Linked Amyloid-β Dimers in the Alzheimer’s Brain. Brain 2019, 142, 1441–1457. [Google Scholar] [CrossRef]
- Nguyen, H.L.; Linh, H.Q.; Krupa, P.; La Penna, G.; Li, M.S. Amyloid β Dodecamer Disrupts the Neuronal Membrane More Strongly than the Mature Fibril: Understanding the Role of Oligomers in Neurotoxicity. J. Phys. Chem. B 2022, 126, 3659–3672. [Google Scholar] [CrossRef] [PubMed]
- Sideris, D.I.; Danial, J.S.H.; Emin, D.; Ruggeri, F.S.; Xia, Z.; Zhang, Y.P.; Lobanova, E.; Dakin, H.; De, S.; Miller, A.; et al. Soluble Amyloid Beta-Containing Aggregates Are Present throughout the Brain at Early Stages of Alzheimer’s Disease. Brain Commun. 2021, 3, fcab147. [Google Scholar] [CrossRef] [PubMed]
- Roos, T.T.; Garcia, M.G.; Martinsson, I.; Mabrouk, R.; Israelsson, B.; Deierborg, T.; Kobro-Flatmoen, A.; Tanila, H.; Gouras, G.K. Neuronal Spreading and Plaque Induction of Intracellular Aβ and Its Disruption of Aβ Homeostasis. Acta Neuropathol. 2021, 142, 669–687. [Google Scholar] [CrossRef] [PubMed]
- Hefti, M.M.; Farrell, K.; Kim, S.; Bowles, K.R.; Fowkes, M.E.; Raj, T.; Crary, J.F. High-Resolution Temporal and Regional Mapping of MAPT Expression and Splicing in Human Brain Development. PLoS ONE 2018, 13, e0195771. [Google Scholar] [CrossRef] [PubMed]
- Trabzuni, D.; Wray, S.; Vandrovcova, J.; Ramasamy, A.; Walker, R.; Smith, C.; Luk, C.; Gibbs, J.R.; Dillman, A.; Hernandez, D.G.; et al. MAPT Expression and Splicing Is Differentially Regulated by Brain Region: Relation to Genotype and Implication for Tauopathies. Hum. Mol. Genet. 2012, 21, 4094–4103. [Google Scholar] [CrossRef]
- Majounie, E.; Cross, W.; Newsway, V.; Dillman, A.; Vandrovcova, J.; Morris, C.M.; Nalls, M.A.; Ferrucci, L.; Owen, M.J.; O’Donovan, M.C.; et al. Variation in Tau Isoform Expression in Different Brain Regions and Disease States. Neurobiol. Aging 2013, 34, 1922.e7–1922.e12. [Google Scholar] [CrossRef]
- Liu, C.; Götz, J. Profiling Murine Tau with 0N, 1N and 2N Isoform-Specific Antibodies in Brain and Peripheral Organs Reveals Distinct Subcellular Localization, with the 1N Isoform Being Enriched in the Nucleus. PLoS ONE 2013, 8, e84849. [Google Scholar] [CrossRef]
- Sündermann, F.; Fernandez, M.-P.; Morgan, R.O. An Evolutionary Roadmap to the Microtubule-Associated Protein MAP Tau. BMC Genom. 2016, 17, 264. [Google Scholar] [CrossRef]
- Sotiropoulos, I.; Galas, M.-C.; Silva, J.M.; Skoulakis, E.; Wegmann, S.; Maina, M.B.; Blum, D.; Sayas, C.L.; Mandelkow, E.-M.; Mandelkow, E.; et al. Atypical, Non-Standard Functions of the Microtubule Associated Tau Protein. Acta Neuropathol. Commun. 2017, 5, 91. [Google Scholar] [CrossRef]
- Busciglio, J.; Lorenzo, A.; Yeh, J.; Yankner, B.A. Beta-Amyloid Fibrils Induce Tau Phosphorylation and Loss of Microtubule Binding. Neuron 1995, 14, 879–888. [Google Scholar] [CrossRef]
- Hernandez, P.; Lee, G.; Sjoberg, M.; Maccioni, R.B. Tau Phosphorylation by Cdk5 and Fyn in Response to Amyloid Peptide Abeta (25–35): Involvement of Lipid Rafts. J. Alzheimers Dis. 2009, 16, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Terwel, D.; Muyllaert, D.; Dewachter, I.; Borghgraef, P.; Croes, S.; Devijver, H.; Van Leuven, F. Amyloid Activates GSK-3beta to Aggravate Neuronal Tauopathy in Bigenic Mice. Am. J. Pathol. 2008, 172, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.R.; Wiedau-Pazos, M.; Sang, T.-K.; Wagle, N.; Brown, C.A.; Massachi, S.; Geschwind, D.H. Human Wild-Type Tau Interacts with Wingless Pathway Components and Produces Neurofibrillary Pathology in Drosophila. Neuron 2002, 34, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, A.; Kabat, J.; Novak, M.; Wu, Q.; Grundke-Iqbal, I.; Iqbal, K. Phosphorylation of Tau at Both Thr 231 and Ser 262 Is Required for Maximal Inhibition of Its Binding to Microtubules. Arch. Biochem. Biophys. 1998, 357, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive Microglia Drive Tau Pathology and Contribute to the Spreading of Pathological Tau in the Brain. Brain 2015, 138, 1738–1755. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, K.; Konerth, M.; Kokiko-Cochran, O.N.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T. Regulation of Tau Pathology by the Microglial Fractalkine Receptor. Neuron 2010, 68, 19–31. [Google Scholar] [CrossRef]
- Puangmalai, N.; Sengupta, U.; Bhatt, N.; Gaikwad, S.; Montalbano, M.; Bhuyan, A.; Garcia, S.; McAllen, S.; Sonawane, M.; Jerez, C.; et al. Lysine 63-Linked Ubiquitination of Tau Oligomers Contributes to the Pathogenesis of Alzheimer’s Disease. J. Biol. Chem. 2022, 298, 101766. [Google Scholar] [CrossRef]
- Song, H.-L.; Kim, N.-Y.; Park, J.; Kim, M.I.; Jeon, Y.-N.; Lee, S.-J.; Cho, K.; Shim, Y.-L.; Lee, K.-H.; Mun, Y.-S.; et al. Monoclonal Antibody Y01 Prevents Tauopathy Progression Induced by Lysine 280-Acetylated Tau in Cell and Mouse Models. J. Clin. Investig. 2023, 133, e156537. [Google Scholar] [CrossRef]
- Gu, J.; Xu, W.; Jin, N.; Li, L.; Zhou, Y.; Chu, D.; Gong, C.-X.; Iqbal, K.; Liu, F. Truncation of Tau Selectively Facilitates Its Pathological Activities. J. Biol. Chem. 2020, 295, 13812–13828. [Google Scholar] [CrossRef]
- Kyalu Ngoie Zola, N.; Balty, C.; Pyr Dit Ruys, S.; Vanparys, A.A.T.; Huyghe, N.D.G.; Herinckx, G.; Johanns, M.; Boyer, E.; Kienlen-Campard, P.; Rider, M.H.; et al. Specific Post-Translational Modifications of Soluble Tau Protein Distinguishes Alzheimer’s Disease and Primary Tauopathies. Nat. Commun. 2023, 14, 3706. [Google Scholar] [CrossRef]
- Rodriguez Camargo, D.C.; Sileikis, E.; Chia, S.; Axell, E.; Bernfur, K.; Cataldi, R.L.; Cohen, S.I.A.; Meisl, G.; Habchi, J.; Knowles, T.P.J.; et al. Proliferation of Tau 304–380 Fragment Aggregates through Autocatalytic Secondary Nucleation. ACS Chem. Neurosci. 2021, 12, 4406–4415. [Google Scholar] [CrossRef] [PubMed]
- Patterson, K.R.; Remmers, C.; Fu, Y.; Brooker, S.; Kanaan, N.M.; Vana, L.; Ward, S.; Reyes, J.F.; Philibert, K.; Glucksman, M.J.; et al. Characterization of Prefibrillar Tau Oligomers in Vitro and in Alzheimer Disease. J. Biol. Chem. 2011, 286, 23063–23076. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Awasthi, S.; Bryan, L.; Ehrlich, R.S.; Tonali, N.; Balog, S.; Yang, J.; Sewald, N.; Mayer, M. Fluorescence-Based Monitoring of Early-Stage Aggregation of Amyloid-β, Amylin Peptide, Tau, and α-Synuclein Proteins. ACS Chem. Neurosci. 2024, 15, 3113–3123. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D.J. Soluble Amyloid Beta-Protein Dimers Isolated from Alzheimer Cortex Directly Induce Tau Hyperphosphorylation and Neuritic Degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.A.; Hipp, S.A.; Rijal Upadhaya, A.; Balakrishnan, K.; Ospitalieri, S.; Koper, M.J.; Largo-Barrientos, P.; Uytterhoeven, V.; Reichwald, J.; Rabe, S.; et al. Aβ-Induced Acceleration of Alzheimer-Related τ-Pathology Spreading and Its Association with Prion Protein. Acta Neuropathol. 2019, 138, 913–941. [Google Scholar] [CrossRef]
- Pooler, A.M.; Polydoro, M.; Maury, E.A.; Nicholls, S.B.; Reddy, S.M.; Wegmann, S.; William, C.; Saqran, L.; Cagsal-Getkin, O.; Pitstick, R.; et al. Amyloid Accelerates Tau Propagation and Toxicity in a Model of Early Alzheimer’s Disease. Acta Neuropathol. Commun. 2015, 3, 14. [Google Scholar] [CrossRef]
- Dai, C.; Chen, X.; Kazim, S.F.; Liu, F.; Gong, C.-X.; Grundke-Iqbal, I.; Iqbal, K. Passive Immunization Targeting the N-Terminal Projection Domain of Tau Decreases Tau Pathology and Improves Cognition in a Transgenic Mouse Model of Alzheimer Disease and Tauopathies. J. Neural Transm. 2015, 122, 607–617. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. Amyloid-β May Be Released from Non-Junctional Varicosities of Axons Generated from Abnormal Tau-Containing Brainstem Nuclei in Sporadic Alzheimer’s Disease: A Hypothesis. Acta Neuropathol. 2013, 126, 303–306. [Google Scholar] [CrossRef]
- Vasconcelos, B.; Stancu, I.-C.; Buist, A.; Bird, M.; Wang, P.; Vanoosthuyse, A.; Van Kolen, K.; Verheyen, A.; Kienlen-Campard, P.; Octave, J.-N.; et al. Heterotypic Seeding of Tau Fibrillization by Pre-Aggregated Abeta Provides Potent Seeds for Prion-like Seeding and Propagation of Tau-Pathology in Vivo. Acta Neuropathol. 2016, 131, 549–569. [Google Scholar] [CrossRef]
- Lee, W.J.; Brown, J.A.; Kim, H.R.; La Joie, R.; Cho, H.; Lyoo, C.H.; Rabinovici, G.D.; Seong, J.-K.; Seeley, W.W. Alzheimer’s Disease Neuroimaging Initiative Regional Aβ-Tau Interactions Promote Onset and Acceleration of Alzheimer’s Disease Tau Spreading. Neuron 2022, 110, 1932–1943.e5. [Google Scholar] [CrossRef]
- Bejanin, A.; Schonhaut, D.R.; La Joie, R.; Kramer, J.H.; Baker, S.L.; Sosa, N.; Ayakta, N.; Cantwell, A.; Janabi, M.; Lauriola, M.; et al. Tau Pathology and Neurodegeneration Contribute to Cognitive Impairment in Alzheimer’s Disease. Brain 2017, 140, 3286–3300. [Google Scholar] [CrossRef] [PubMed]
- Savage, M.J.; Kalinina, J.; Wolfe, A.; Tugusheva, K.; Korn, R.; Cash-Mason, T.; Maxwell, J.W.; Hatcher, N.G.; Haugabook, S.J.; Wu, G.; et al. A Sensitive Aβ Oligomer Assay Discriminates Alzheimer’s and Aged Control Cerebrospinal Fluid. J. Neurosci. 2014, 34, 2884–2897. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, U.; Portelius, E.; Hansson, O.; Farmer, K.; Castillo-Carranza, D.; Woltjer, R.; Zetterberg, H.; Galasko, D.; Blennow, K.; Kayed, R. Tau Oligomers in Cerebrospinal Fluid in Alzheimer’s Disease. Ann. Clin. Transl. Neurol. 2017, 4, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.; Schröder, J.; Blomberg, M.; Engvall, B.; Pantel, J.; Ida, N.; Basun, H.; Wahlund, L.-O.; Werle, E.; Jauss, M.; et al. Cerebrospinal Fluid Aβ42 Is Increased Early in Sporadic Alzheimer’s Disease and Declines with Disease Progression. Ann. Neurol. 1999, 45, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Calafate, S.; Buist, A.; Miskiewicz, K.; Vijayan, V.; Daneels, G.; de Strooper, B.; de Wit, J.; Verstreken, P.; Moechars, D. Synaptic Contacts Enhance Cell-to-Cell Tau Pathology Propagation. Cell Rep. 2015, 11, 1176–1183. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Kang, J.-E.; Lee, J.; Stewart, F.R.; Verges, D.K.; Silverio, L.M.; Bu, G.; Mennerick, S.; Holtzman, D.M. Endocytosis Is Required for Synaptic Activity-Dependent Release of Amyloid-Beta in Vivo. Neuron 2008, 58, 42–51. [Google Scholar] [CrossRef]
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A.C.M.; et al. Neuronal Activity Enhances Tau Propagation and Tau Pathology in Vivo. Nat. Neurosci. 2016, 19, 1085–1092. [Google Scholar] [CrossRef]
- Lazarov, O.; Lee, M.; Peterson, D.A.; Sisodia, S.S. Evidence That Synaptically Released Beta-Amyloid Accumulates as Extracellular Deposits in the Hippocampus of Transgenic Mice. J. Neurosci. 2002, 22, 9785–9793. [Google Scholar] [CrossRef]
- Dujardin, S.; Bégard, S.; Caillierez, R.; Lachaud, C.; Delattre, L.; Carrier, S.; Loyens, A.; Galas, M.-C.; Bousset, L.; Melki, R.; et al. Ectosomes: A New Mechanism for Non-Exosomal Secretion of Tau Protein. PLoS ONE 2014, 9, e100760. [Google Scholar] [CrossRef]
- Lachenal, G.; Pernet-Gallay, K.; Chivet, M.; Hemming, F.J.; Belly, A.; Bodon, G.; Blot, B.; Haase, G.; Goldberg, Y.; Sadoul, R. Release of Exosomes from Differentiated Neurons and Its Regulation by Synaptic Glutamatergic Activity. Mol. Cell. Neurosci. 2011, 46, 409–418. [Google Scholar] [CrossRef]
- Hamlett, E.D.; Goetzl, E.J.; Ledreux, A.; Vasilevko, V.; Boger, H.A.; LaRosa, A.; Clark, D.; Carroll, S.L.; Carmona-Iragui, M.; Fortea, J.; et al. Neuronal Exosomes Reveal Alzheimer’s Disease Biomarkers in Down Syndrome. Alzheimer’s Dement. 2017, 13, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of Preclinical Alzheimer’s Disease by a Profile of Pathogenic Proteins in Neurally Derived Blood Exosomes: A Case-Control Study. Alzheimer’s Dement. 2015, 11, 600–607.e1. [Google Scholar] [CrossRef] [PubMed]
- Abner, E.L.; Jicha, G.A.; Shaw, L.M.; Trojanowski, J.Q.; Goetzl, E.J. Plasma Neuronal Exosomal Levels of Alzheimer’s Disease Biomarkers in Normal Aging. Ann. Clin. Transl. Neurol. 2016, 3, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Kapogiannis, D.; Schwartz, J.B.; Lobach, I.V.; Goetzl, L.; Abner, E.L.; Jicha, G.A.; Karydas, A.M.; Boxer, A.; Miller, B.L. Decreased Synaptic Proteins in Neuronal Exosomes of Frontotemporal Dementia and Alzheimer’s Disease. FASEB J. 2016, 30, 4141–4148. [Google Scholar] [CrossRef]
- Sardar Sinha, M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s Disease Pathology Propagation by Exosomes Containing Toxic Amyloid-Beta Oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The Release and Trans-Synaptic Transmission of Tau via Exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Yamada, K.A.; Finn, M.B.; Sloviter, R.S.; Bales, K.R.; May, P.C.; Schoepp, D.D.; Paul, S.M.; Mennerick, S.; Holtzman, D.M. Synaptic Activity Regulates Interstitial Fluid Amyloid-Beta Levels in Vivo. Neuron 2005, 48, 913–922. [Google Scholar] [CrossRef]
- Katsinelos, T.; Zeitler, M.; Dimou, E.; Karakatsani, A.; Müller, H.-M.; Nachman, E.; Steringer, J.P.; Ruiz de Almodovar, C.; Nickel, W.; Jahn, T.R. Unconventional Secretion Mediates the Trans-Cellular Spreading of Tau. Cell Rep. 2018, 23, 2039–2055. [Google Scholar] [CrossRef]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE Influences Amyloid-β (Aβ) Clearance despite Minimal apoE/Aβ Association in Physiological Conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Zhang, J.; Liu, Q.; Liu, C.-C.; Zhang, L.; Bu, G. Heparan Sulphate Proteoglycan and the Low-Density Lipoprotein Receptor-Related Protein 1 Constitute Major Pathways for Neuronal Amyloid-Beta Uptake. J. Neurosci. 2011, 31, 1644–1651. [Google Scholar] [CrossRef]
- Yu, C.; Nwabuisi-Heath, E.; Laxton, K.; Ladu, M.J. Endocytic Pathways Mediating Oligomeric Abeta42 Neurotoxicity. Mol. Neurodegener. 2010, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Nazere, K.; Takahashi, T.; Hara, N.; Muguruma, K.; Nakamori, M.; Yamazaki, Y.; Morino, H.; Maruyama, H. Amyloid Beta Is Internalized via Macropinocytosis, an HSPG- and Lipid Raft-Dependent and Rac1-Mediated Process. Front. Mol. Neurosci. 2022, 15, 804702. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Herman, M.; Liu, L.; Simoes, S.; Acker, C.M.; Figueroa, H.; Steinberg, J.I.; Margittai, M.; Kayed, R.; Zurzolo, C.; et al. Small Misfolded Tau Species Are Internalized via Bulk Endocytosis and Anterogradely and Retrogradely Transported in Neurons. J. Biol. Chem. 2013, 288, 1856–1870. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan Sulfate Proteoglycans Mediate Internalization and Propagation of Specific Proteopathic Seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [PubMed]
- Marshall, K.E.; Vadukul, D.M.; Staras, K.; Serpell, L.C. Misfolded Amyloid-β-42 Impairs the Endosomal-Lysosomal Pathway. Cell Mol. Life Sci. 2020, 77, 5031–5043. [Google Scholar] [CrossRef]
- Rose, K.; Jepson, T.; Shukla, S.; Maya-Romero, A.; Kampmann, M.; Xu, K.; Hurley, J.H. Tau Fibrils Induce Nanoscale Membrane Damage and Nucleate Cytosolic Tau at Lysosomes. Proc. Natl. Acad. Sci. USA 2024, 121, e2315690121. [Google Scholar] [CrossRef]
- d’Errico, P.; Ziegler-Waldkirch, S.; Aires, V.; Hoffmann, P.; Mezö, C.; Erny, D.; Monasor, L.S.; Liebscher, S.; Ravi, V.M.; Joseph, K.; et al. Microglia Contribute to the Propagation of Aβ into Unaffected Brain Tissue. Nat. Neurosci. 2022, 25, 20–25. [Google Scholar] [CrossRef]
- Jiwaji, Z.; Tiwari, S.S.; Avilés-Reyes, R.X.; Hooley, M.; Hampton, D.; Torvell, M.; Johnson, D.A.; McQueen, J.; Baxter, P.; Sabari-Sankar, K.; et al. Reactive Astrocytes Acquire Neuroprotective as Well as Deleterious Signatures in Response to Tau and Aß Pathology. Nat. Commun. 2022, 13, 135. [Google Scholar] [CrossRef]
- Tamboli, I.Y.; Barth, E.; Christian, L.; Siepmann, M.; Kumar, S.; Singh, S.; Tolksdorf, K.; Heneka, M.T.; Lütjohann, D.; Wunderlich, P.; et al. Statins Promote the Degradation of Extracellular Amyloid β-Peptide by Microglia via Stimulation of Exosome-Associated Insulin-Degrading Enzyme (IDE) Secretion. J. Biol. Chem. 2010, 285, 37405–37414. [Google Scholar] [CrossRef]
- Pacheco-Quinto, J.; Eckman, E.A. Endothelin-Converting Enzymes Degrade Intracellular β-Amyloid Produced within the Endosomal/Lysosomal Pathway and Autophagosomes. J. Biol. Chem. 2013, 288, 5606–5615. [Google Scholar] [CrossRef]
- Pomilio, C.; Pavia, P.; Gorojod, R.M.; Vinuesa, A.; Alaimo, A.; Galvan, V.; Kotler, M.L.; Beauquis, J.; Saravia, F. Glial Alterations from Early to Late Stages in a Model of Alzheimer’s Disease: Evidence of Autophagy Involvement in Aβ Internalization. Hippocampus 2016, 26, 194–210. [Google Scholar] [CrossRef] [PubMed]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.-H. Nanotubular Highways for Intercellular Organelle Transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Onfelt, B.; Nedvetzki, S.; Benninger, R.K.P.; Purbhoo, M.A.; Sowinski, S.; Hume, A.N.; Seabra, M.C.; Neil, M.A.A.; French, P.M.W.; Davis, D.M. Structurally Distinct Membrane Nanotubes between Human Macrophages Support Long-Distance Vesicular Traffic or Surfing of Bacteria. J. Immunol. 2006, 177, 8476–8483. [Google Scholar] [CrossRef] [PubMed]
- Domhan, S.; Ma, L.; Tai, A.; Anaya, Z.; Beheshti, A.; Zeier, M.; Hlatky, L.; Abdollahi, A. Intercellular Communication by Exchange of Cytoplasmic Material via Tunneling Nano-Tube Like Structures in Primary Human Renal Epithelial Cells. PLoS ONE 2011, 6, e21283. [Google Scholar] [CrossRef]
- Wittig, D.; Wang, X.; Walter, C.; Gerdes, H.-H.; Funk, R.H.W.; Roehlecke, C. Multi-Level Communication of Human Retinal Pigment Epithelial Cells via Tunneling Nanotubes. PLoS ONE 2012, 7, e33195. [Google Scholar] [CrossRef]
- Nasoni, M.G.; Carloni, S.; Canonico, B.; Burattini, S.; Cesarini, E.; Papa, S.; Pagliarini, M.; Ambrogini, P.; Balduini, W.; Luchetti, F. Melatonin Reshapes the Mitochondrial Network and Promotes Intercellular Mitochondrial Transfer via Tunneling Nanotubes after Ischemic-like Injury in Hippocampal HT22 Cells. J. Pineal Res. 2021, 71, e12747. [Google Scholar] [CrossRef]
- Sartori-Rupp, A.; Cordero Cervantes, D.; Pepe, A.; Gousset, K.; Delage, E.; Corroyer-Dulmont, S.; Schmitt, C.; Krijnse-Locker, J.; Zurzolo, C. Correlative Cryo-Electron Microscopy Reveals the Structure of TNTs in Neuronal Cells. Nat. Commun. 2019, 10, 342. [Google Scholar] [CrossRef]
- Davis, D.M.; Sowinski, S. Membrane Nanotubes: Dynamic Long-Distance Connections between Animal Cells. Nat. Rev. Mol. Cell Biol. 2008, 9, 431–436. [Google Scholar] [CrossRef]
- Polak, R.; de Rooij, B.; Pieters, R.; den Boer, M.L. B-Cell Precursor Acute Lymphoblastic Leukemia Cells Use Tunneling Nanotubes to Orchestrate Their Microenvironment. Blood 2015, 126, 2404–2414. [Google Scholar] [CrossRef]
- Panasiuk, M.; Rychłowski, M.; Derewońko, N.; Bieńkowska-Szewczyk, K. Tunneling Nanotubes as a Novel Route of Cell-to-Cell Spread of Herpesviruses. J. Virol. 2018, 92, e00090-18. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef] [PubMed]
- Sergeant, N.; David, J.P.; Lefranc, D.; Vermersch, P.; Wattez, A.; Delacourte, A. Different Distribution of Phosphorylated Tau Protein Isoforms in Alzheimer’s and Pick’s Diseases. FEBS Lett. 1997, 412, 578–582. [Google Scholar] [CrossRef] [PubMed]
- McCully, J.D.; Cowan, D.B.; Pacak, C.A.; Toumpoulis, I.K.; Dayalan, H.; Levitsky, S. Injection of Isolated Mitochondria during Early Reperfusion for Cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H94–H105. [Google Scholar] [CrossRef] [PubMed]
- Bukoreshtliev, N.V.; Wang, X.; Hodneland, E.; Gurke, S.; Barroso, J.F.V.; Gerdes, H.-H. Selective Block of Tunneling Nanotube (TNT) Formation Inhibits Intercellular Organelle Transfer between PC12 Cells. FEBS Lett. 2009, 583, 1481–1488. [Google Scholar] [CrossRef]
- Barutta, F.; Kimura, S.; Hase, K.; Bellini, S.; Corbetta, B.; Corbelli, A.; Fiordaliso, F.; Barreca, A.; Papotti, M.G.; Ghiggeri, G.M.; et al. Protective Role of the M-Sec-Tunneling Nanotube System in Podocytes. J. Am. Soc. Nephrol. 2021, 32, 1114–1130. [Google Scholar] [CrossRef]
- Dilsizoglu Senol, A.; Pepe, A.; Grudina, C.; Sassoon, N.; Reiko, U.; Bousset, L.; Melki, R.; Piel, J.; Gugger, M.; Zurzolo, C. Effect of Tolytoxin on Tunneling Nanotube Formation and Function. Sci. Rep. 2019, 9, 5741. [Google Scholar] [CrossRef]
- Wang, F.; Chen, X.; Cheng, H.; Song, L.; Liu, J.; Caplan, S.; Zhu, L.; Wu, J.Y. MICAL2PV Suppresses the Formation of Tunneling Nanotubes and Modulates Mitochondrial Trafficking. EMBO Rep. 2021, 22, e52006. [Google Scholar] [CrossRef]
- Zhu, D.; Tan, K.S.; Zhang, X.; Sun, A.Y.; Sun, G.Y.; Lee, J.C.-M. Hydrogen Peroxide Alters Membrane and Cytoskeleton Properties and Increases Intercellular Connections in Astrocytes. J. Cell Sci. 2005, 118, 3695–3703. [Google Scholar] [CrossRef]
- Lin, X.; Wang, W.; Chang, X.; Chen, C.; Guo, Z.; Yu, G.; Shao, W.; Wu, S.; Zhang, Q.; Zheng, F.; et al. ROS/mtROS Promotes TNTs Formation via the PI3K/AKT/mTOR Pathway to Protect against Mitochondrial Damages in Glial Cells Induced by Engineered Nanomaterials. Part. Fibre Toxicol. 2024, 21, 1. [Google Scholar] [CrossRef]
- Hase, K.; Kimura, S.; Takatsu, H.; Ohmae, M.; Kawano, S.; Kitamura, H.; Ito, M.; Watarai, H.; Hazelett, C.C.; Yeaman, C.; et al. M-Sec Promotes Membrane Nanotube Formation by Interacting with Ral and the Exocyst Complex. Nat. Cell Biol. 2009, 11, 1427–1432. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, J.; Sun, X.; Zhang, Y. Tunneling-Nanotube Development in Astrocytes Depends on P53 Activation. Cell Death Differ. 2011, 18, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Andresen, V.; Wang, X.; Ghimire, S.; Omsland, M.; Gjertsen, B.T.; Gerdes, H.H. Tunneling Nanotube (TNT) Formation Is Independent of P53 Expression. Cell Death Differ. 2013, 20, 1124. [Google Scholar] [CrossRef] [PubMed]
- Sowinski, S.; Jolly, C.; Berninghausen, O.; Purbhoo, M.A.; Chauveau, A.; Köhler, K.; Oddos, S.; Eissmann, P.; Brodsky, F.M.; Hopkins, C.; et al. Membrane Nanotubes Physically Connect T Cells over Long Distances Presenting a Novel Route for HIV-1 Transmission. Nat. Cell Biol. 2008, 10, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Lou, E.; Zhai, E.; Sarkari, A.; Desir, S.; Wong, P.; Iizuka, Y.; Yang, J.; Subramanian, S.; McCarthy, J.; Bazzaro, M.; et al. Cellular and Molecular Networking Within the Ecosystem of Cancer Cell Communication via Tunneling Nanotubes. Front. Cell Dev. Biol. 2018, 6, 95. [Google Scholar] [CrossRef]
- Guo, L.; Zhang, Y.; Yang, Z.; Peng, H.; Wei, R.; Wang, C.; Feng, M. Tunneling Nanotubular Expressways for Ultrafast and Accurate M1 Macrophage Delivery of Anticancer Drugs to Metastatic Ovarian Carcinoma. ACS Nano 2019, 13, 1078–1096. [Google Scholar] [CrossRef]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential Transfer of Mitochondria from Endothelial to Cancer Cells through Tunneling Nanotubes Modulates Chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef]
- Pasquier, J.; Galas, L.; Boulangé-Lecomte, C.; Rioult, D.; Bultelle, F.; Magal, P.; Webb, G.; Foll, F.L. Different Modalities of Intercellular Membrane Exchanges Mediate Cell-to-Cell P-Glycoprotein Transfers in MCF-7 Breast Cancer Cells. J. Biol. Chem. 2012, 287, 7374–7387. [Google Scholar] [CrossRef]
- Spitzer, N.C. Electrical Activity in Early Neuronal Development. Nature 2006, 444, 707–712. [Google Scholar] [CrossRef]
- Chauveau, A.; Aucher, A.; Eissmann, P.; Vivier, E.; Davis, D.M. Membrane Nanotubes Facilitate Long-Distance Interactions between Natural Killer Cells and Target Cells. Proc. Natl. Acad. Sci. USA 2010, 107, 5545–5550. [Google Scholar] [CrossRef]
- Dupont, M.; Souriant, S.; Balboa, L.; Vu Manh, T.-P.; Pingris, K.; Rousset, S.; Cougoule, C.; Rombouts, Y.; Poincloux, R.; Ben Neji, M.; et al. Tuberculosis-Associated IFN-I Induces Siglec-1 on Tunneling Nanotubes and Favors HIV-1 Spread in Macrophages. Elife 2020, 9, e52535. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Y.; Zhang, J.; Tu, J.; Wang, X.-J.; Su, X.-D.; Wang, L.; Zhang, Y. Tunneling-Nanotube Direction Determination in Neurons and Astrocytes. Cell Death Dis. 2012, 3, e438. [Google Scholar] [CrossRef] [PubMed]
- Djurkovic, M.A.; Leavitt, C.G.; Arnett, E.; Kriachun, V.; Martínez-Sobrido, L.; Titone, R.; Sherwood, L.J.; Hayhurst, A.; Schlesinger, L.S.; Shtanko, O. Ebola Virus Uses Tunneling Nanotubes as an Alternate Route of Dissemination. J. Infect. Dis. 2023, 228, S522–S535. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Katz, B.B.; Tomich, J.M.; Gallagher, T.; Fang, Y. Porcine Reproductive and Respiratory Syndrome Virus Utilizes Nanotubes for Intercellular Spread. J. Virol. 2016, 90, 5163–5175. [Google Scholar] [CrossRef] [PubMed]
- Pepe, A.; Pietropaoli, S.; Vos, M.; Barba-Spaeth, G.; Zurzolo, C. Tunneling Nanotubes Provide a Route for SARS-CoV-2 Spreading. Sci. Adv. 2022, 8, eabo0171. [Google Scholar] [CrossRef]
- Li, X.; Zhao, Y.; Teng, Q.-Y.; Zhang, X.-H.; Xue, J.; Zhang, G.-Z. Methyltransferase K-D-K-E Motif Influences the Intercellular Transmission of Newcastle Disease Virus. Virulence 2023, 14, 2186336. [Google Scholar] [CrossRef]
- Okafo, G.; Prevedel, L.; Eugenin, E. Tunneling Nanotubes (TNT) Mediate Long-Range Gap Junctional Communication: Implications for HIV Cell to Cell Spread. Sci. Rep. 2017, 7, 16660. [Google Scholar] [CrossRef]
- Roberts, K.L.; Manicassamy, B.; Lamb, R.A. Influenza A Virus Uses Intercellular Connections to Spread to Neighboring Cells. J. Virol. 2015, 89, 1537–1549. [Google Scholar] [CrossRef]
- Pinto, G.; Brou, C.; Zurzolo, C. Tunneling Nanotubes: The Fuel of Tumor Progression? Trends Cancer 2020, 6, 874–888. [Google Scholar] [CrossRef]
- Kolba, M.D.; Dudka, W.; Zaręba-Kozioł, M.; Kominek, A.; Ronchi, P.; Turos, L.; Chroscicki, P.; Wlodarczyk, J.; Schwab, Y.; Klejman, A.; et al. Tunneling Nanotube-Mediated Intercellular Vesicle and Protein Transfer in the Stroma-Provided Imatinib Resistance in Chronic Myeloid Leukemia Cells. Cell Death Dis. 2019, 10, 817. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Qiu, Y.; Shi, Y.; Cai, J.; Wang, B.; Wei, X.; Ke, Q.; Sui, X.; Wang, Y.; et al. Cell Adhesion-Mediated Mitochondria Transfer Contributes to Mesenchymal Stem Cell-Induced Chemoresistance on T Cell Acute Lymphoblastic Leukemia Cells. J. Hematol. Oncol. 2018, 11, 11. [Google Scholar] [CrossRef]
- Omsland, M.; Andresen, V.; Gullaksen, S.-E.; Ayuda-Durán, P.; Popa, M.; Hovland, R.; Brendehaug, A.; Enserink, J.; McCormack, E.; Gjertsen, B.T. Tyrosine Kinase Inhibitors and Interferon-α Increase Tunneling Nanotube (TNT) Formation and Cell Adhesion in Chronic Myeloid Leukemia (CML) Cell Lines. FASEB J. 2020, 34, 3773–3791. [Google Scholar] [CrossRef] [PubMed]
- Valdebenito, S.; Malik, S.; Luu, R.; Loudig, O.; Mitchell, M.; Okafo, G.; Bhat, K.; Prideaux, B.; Eugenin, E.A. Tunneling Nanotubes, TNT, Communicate Glioblastoma with Surrounding Non-Tumor Astrocytes to Adapt Them to Hypoxic and Metabolic Tumor Conditions. Sci. Rep. 2021, 11, 14556. [Google Scholar] [CrossRef] [PubMed]
- Delage, E.; Cervantes, D.C.; Pénard, E.; Schmitt, C.; Syan, S.; Disanza, A.; Scita, G.; Zurzolo, C. Differential Identity of Filopodia and Tunneling Nanotubes Revealed by the Opposite Functions of Actin Regulatory Complexes. Sci. Rep. 2016, 6, 39632. [Google Scholar] [CrossRef] [PubMed]
- Jansens, R.J.J.; Van den Broeck, W.; De Pelsmaeker, S.; Lamote, J.A.S.; Van Waesberghe, C.; Couck, L.; Favoreel, H.W. Pseudorabies Virus US3-Induced Tunneling Nanotubes Contain Stabilized Microtubules, Interact with Neighboring Cells via Cadherins, and Allow Intercellular Molecular Communication. J. Virol. 2017, 91, e00749-17. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Subramaniam, S. Rhes Travels from Cell to Cell and Transports Huntington Disease Protein via TNT-like Protrusion. J. Cell Biol. 2019, 218, 1972–1993. [Google Scholar] [CrossRef]
- Ramírez-Jarquín, U.N.; Sharma, M.; Shahani, N.; Li, Y.; Boregowda, S.; Subramaniam, S. Rhes Protein Transits from Neuron to Neuron and Facilitates Mutant Huntingtin Spreading in the Brain. Sci. Adv. 2022, 8, eabm3877. [Google Scholar] [CrossRef]
- Grudina, C.; Kouroupi, G.; Nonaka, T.; Hasegawa, M.; Matsas, R.; Zurzolo, C. Human NPCs Can Degrade α-Syn Fibrils and Transfer Them Preferentially in a Cell Contact-Dependent Manner Possibly through TNT-like Structures. Neurobiol. Dis. 2019, 132, 104609. [Google Scholar] [CrossRef]
- Chakraborty, R.; Nonaka, T.; Hasegawa, M.; Zurzolo, C. Tunnelling Nanotubes between Neuronal and Microglial Cells Allow Bi-Directional Transfer of α-Synuclein and Mitochondria. Cell Death Dis. 2023, 14, 329. [Google Scholar] [CrossRef]
- Padmanabhan, S.; Manjithaya, R. Leaderless Secretory Proteins of the Neurodegenerative Diseases via TNTs: A Structure-Function Perspective. Front. Mol. Neurosci. 2023, 16, 983108. [Google Scholar] [CrossRef]
- Abounit, S.; Wu, J.W.; Duff, K.; Victoria, G.S.; Zurzolo, C. Tunneling Nanotubes: A Possible Highway in the Spreading of Tau and Other Prion-like Proteins in Neurodegenerative Diseases. Prion 2016, 10, 344–351. [Google Scholar] [CrossRef]
- Dilsizoglu Senol, A.; Samarani, M.; Syan, S.; Guardia, C.M.; Nonaka, T.; Liv, N.; Latour-Lambert, P.; Hasegawa, M.; Klumperman, J.; Bonifacino, J.S.; et al. α-Synuclein Fibrils Subvert Lysosome Structure and Function for the Propagation of Protein Misfolding between Cells through Tunneling Nanotubes. PLoS Biol. 2021, 19, e3001287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Sun, Z.; Chen, X.; Zhang, Y.; Guo, A.; Zhang, Y. Intercellular Transport of Tau Protein and β-Amyloid Mediated by Tunneling Nanotubes. Am. J. Transl. Res. 2021, 13, 12509–12522. [Google Scholar] [PubMed]
- Konstantinidis, E.; Dakhel, A.; Beretta, C.; Erlandsson, A. Long-Term Effects of Amyloid-Beta Deposits in Human iPSC-Derived Astrocytes. Mol. Cell Neurosci. 2023, 125, 103839. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; O’Connor, T.; Vassar, R. The Contribution of Activated Astrocytes to Aβ Production: Implications for Alzheimer’s Disease Pathogenesis. J. Neuroinflamm. 2011, 8, 150. [Google Scholar] [CrossRef]
- Sriram, K.; Benkovic, S.A.; Hebert, M.A.; Miller, D.B.; O’Callaghan, J.P. Induction of Gp130-Related Cytokines and Activation of JAK2/STAT3 Pathway in Astrocytes Precedes Up-Regulation of Glial Fibrillary Acidic Protein in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Model of Neurodegeneration: Key signaling pathway for astrogliosis in vivo? J. Biol. Chem. 2004, 279, 19936–19947. [Google Scholar] [CrossRef]
- Gu, X.-L.; Long, C.-X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic Expression of Parkinson’s Disease-Related A53T α-Synuclein Causes Neurodegeneration in Mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Amyloid β Oligomers (AβOs) in Alzheimer’s Disease. J. Neural Transm. 2018, 125, 177–191. [Google Scholar] [CrossRef]
- Davis, N.; Mota, B.C.; Stead, L.; Palmer, E.O.C.; Lombardero, L.; Rodríguez-Puertas, R.; de Paola, V.; Barnes, S.J.; Sastre, M. Pharmacological Ablation of Astrocytes Reduces Aβ Degradation and Synaptic Connectivity in an Ex Vivo Model of Alzheimer’s Disease. J. Neuroinflamm. 2021, 18, 73. [Google Scholar] [CrossRef]
- Liu, M.; Sui, D.; Dexheimer, T.; Hovde, S.; Deng, X.; Wang, K.-W.; Lin, H.L.; Chien, H.-T.; Kweon, H.K.; Kuo, N.S.; et al. Hyperphosphorylation Renders Tau Prone to Aggregate and to Cause Cell Death. Mol. Neurobiol. 2020, 57, 4704–4719. [Google Scholar] [CrossRef]
- Chastagner, P.; Loria, F.; Vargas, J.Y.; Tois, J.; I Diamond, M.; Okafo, G.; Brou, C.; Zurzolo, C. Fate and Propagation of Endogenously Formed Tau Aggregates in Neuronal Cells. EMBO Mol. Med. 2020, 12, e12025. [Google Scholar] [CrossRef]
- Swaney, K.F.; Li, R. Function and Regulation of the Arp2/3 Complex during Cell Migration in Diverse Environments. Curr. Opin. Cell Biol. 2016, 42, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Keller, K.E.; Bradley, J.M.; Sun, Y.Y.; Yang, Y.-F.; Acott, T.S. Tunneling Nanotubes Are Novel Cellular Structures That Communicate Signals Between Trabecular Meshwork Cells. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5298–5307. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Bhat, S.; Syan, S.; Kuchitsu, Y.; Fukuda, M.; Zurzolo, C. Rab11a-Rab8a Cascade Regulates the Formation of Tunneling Nanotubes through Vesicle Recycling. J. Cell Sci. 2018, 131, jcs215889. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia Clear Neuron-Released α-Synuclein via Selective Autophagy and Prevent Neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef]
- Scheiblich, H.; Eikens, F.; Wischhof, L.; Opitz, S.; Jüngling, K.; Cserép, C.; Schmidt, S.V.; Lambertz, J.; Bellande, T.; Pósfai, B.; et al. Microglia Rescue Neurons from Aggregate-Induced Neuronal Dysfunction and Death through Tunneling Nanotubes. Neuron 2024, 112, 3106–3125.e8. [Google Scholar] [CrossRef]
- Shim, K.H.; Kang, M.J.; Youn, Y.C.; An, S.S.A.; Kim, S. Alpha-Synuclein: A Pathological Factor with Aβ and Tau and Biomarker in Alzheimer’s Disease. Alzheimers Res. Ther. 2022, 14, 201. [Google Scholar] [CrossRef]
- Giusti, V.; Kaur, G.; Giusto, E.; Civiero, L. Brain Clearance of Protein Aggregates: A Close-up on Astrocytes. Mol. Neurodegener. 2024, 19, 5. [Google Scholar] [CrossRef]
- Scheiblich, H.; Dansokho, C.; Mercan, D.; Schmidt, S.V.; Bousset, L.; Wischhof, L.; Eikens, F.; Odainic, A.; Spitzer, J.; Griep, A.; et al. Microglia Jointly Degrade Fibrillar Alpha-Synuclein Cargo by Distribution through Tunneling Nanotubes. Cell 2021, 184, 5089–5106.e21. [Google Scholar] [CrossRef]
- Meneses, A.; Koga, S.; O’Leary, J.; Dickson, D.W.; Bu, G.; Zhao, N. TDP-43 Pathology in Alzheimer’s Disease. Mol. Neurodegener. 2021, 16, 84. [Google Scholar] [CrossRef]
- Šušnjar, U.; Škrabar, N.; Brown, A.-L.; Abbassi, Y.; Phatnani, H.; Cortese, A.; Cereda, C.; Bugiardini, E.; Cardani, R.; Meola, G.; et al. Cell Environment Shapes TDP-43 Function with Implications in Neuronal and Muscle Disease. Commun. Biol. 2022, 5, 1–17. [Google Scholar] [CrossRef]
- Conicella, A.E.; Dignon, G.L.; Zerze, G.H.; Schmidt, H.B.; D’Ordine, A.M.; Kim, Y.C.; Rohatgi, R.; Ayala, Y.M.; Mittal, J.; Fawzi, N.L. TDP-43 α-Helical Structure Tunes Liquid-Liquid Phase Separation and Function. Proc. Natl. Acad. Sci. USA 2020, 117, 5883–5894. [Google Scholar] [CrossRef] [PubMed]
- van Eersel, J.; Ke, Y.D.; Gladbach, A.; Bi, M.; Götz, J.; Kril, J.J.; Ittner, L.M. Cytoplasmic Accumulation and Aggregation of TDP-43 upon Proteasome Inhibition in Cultured Neurons. PLoS ONE 2011, 6, e22850. [Google Scholar] [CrossRef] [PubMed]
- Latimer, C.S.; Stair, J.G.; Hincks, J.C.; Currey, H.N.; Bird, T.D.; Keene, C.D.; Kraemer, B.C.; Liachko, N.F. TDP-43 Promotes Tau Accumulation and Selective Neurotoxicity in Bigenic Caenorhabditis Elegans. Dis. Models Mech. 2022, 15, dmm049323. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, L.; Gao, C.; Arakawa, H.; Perry, G.; Wang, X. TDP-43 Inhibitory Peptide Alleviates Neurodegeneration and Memory Loss in an APP Transgenic Mouse Model for Alzheimer’s Disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165580. [Google Scholar] [CrossRef]
- Davis, S.A.; Gan, K.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 Expression Influences Amyloidβ Plaque Deposition and Tau Aggregation. Neurobiol. Dis. 2017, 103, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Ederle, H.; Funk, C.; Abou-Ajram, C.; Hutten, S.; Funk, E.B.E.; Kehlenbach, R.H.; Bailer, S.M.; Dormann, D. Nuclear Egress of TDP-43 and FUS Occurs Independently of Exportin-1/CRM1. Sci. Rep. 2018, 8, 7084. [Google Scholar] [CrossRef]
- Pinarbasi, E.S.; Cağatay, T.; Fung, H.Y.J.; Li, Y.C.; Chook, Y.M.; Thomas, P.J. Active Nuclear Import and Passive Nuclear Export Are the Primary Determinants of TDP-43 Localization. Sci. Rep. 2018, 8, 7083. [Google Scholar] [CrossRef]
- Archbold, H.C.; Jackson, K.L.; Arora, A.; Weskamp, K.; Tank, E.M.-H.; Li, X.; Miguez, R.; Dayton, R.D.; Tamir, S.; Klein, R.L.; et al. TDP43 Nuclear Export and Neurodegeneration in Models of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Sci. Rep. 2018, 8, 4606. [Google Scholar] [CrossRef]
- Martinez-Gonzalez, L.; Cuevas, E.P.; Tosat-Bitrián, C.; Nozal, V.; Gil, C.; Palomo, V.; Martín-Requero, Á.; Martinez, A. TTBK1 and CK1 Inhibitors Restore TDP-43 Pathology and Avoid Disease Propagation in Lymphoblast from Alzheimer’s Disease Patients. Front. Mol. Neurosci. 2023, 16, 1243277. [Google Scholar] [CrossRef]
- Barton, S.K.; Lau, C.L.; Chiam, M.D.F.; Tomas, D.; Muyderman, H.; Beart, P.M.; Turner, B.J. Mutant TDP-43 Expression Triggers TDP-43 Pathology and Cell Autonomous Effects on Primary Astrocytes: Implications for Non-Cell Autonomous Pathology in ALS. Neurochem. Res. 2020, 45, 1451–1459. [Google Scholar] [CrossRef]
- Lee, S.; Kim, S.; Kang, H.-Y.; Lim, H.R.; Kwon, Y.; Jo, M.; Jeon, Y.-M.; Kim, S.R.; Kim, K.; Ha, C.M.; et al. The Overexpression of TDP-43 in Astrocytes Causes Neurodegeneration via a PTP1B-Mediated Inflammatory Response. J. Neuroinflamm. 2020, 17, 299. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, E.; Audrain, M.; Ratnam, M.; Ollier, R.; Fuchs, A.; Piorkowska, K.; Pfeifer, A.; Kosco-Vilbois, M.; Seredenina, T.; Afroz, T. Targeting the TDP-43 Low Complexity Domain Blocks Spreading of Pathology in a Mouse Model of ALS/FTD. Acta Neuropathol. Commun. 2024, 12, 156. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Kawakami, E.; Endo, K.; Misawa, H.; Watabe, K. Formation and Spreading of TDP-43 Aggregates in Cultured Neuronal and Glial Cells Demonstrated by Time-Lapse Imaging. PLoS ONE 2017, 12, e0179375. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Type of TNTs | Description | Mechanism of AD Pathology |
|---|---|---|
| “thin” TNTs | Cell junctions made of actin with a maximum diameter of 700 nm. They can only transport small molecules with a maximum mass of 1.2 kDa. | Not fully known. |
| “thick” TNTs | Cell junctions made of actin and microtubules with a diameter exceeding 700 nm. They are significantly more stable and can transport large molecules with a mass greater than 1.2 kDa, organelles, and viruses. | TNTs facilitates the transport of molecules such as tau, α-syn, and Aβ to spread neuronal damage. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotarba, S.; Kozłowska, M.; Scios, M.; Saramowicz, K.; Barczuk, J.; Granek, Z.; Siwecka, N.; Wiese, W.; Golberg, M.; Galita, G.; et al. Potential Mechanisms of Tunneling Nanotube Formation and Their Role in Pathology Spread in Alzheimer’s Disease and Other Proteinopathies. Int. J. Mol. Sci. 2024, 25, 10797. https://doi.org/10.3390/ijms251910797
Kotarba S, Kozłowska M, Scios M, Saramowicz K, Barczuk J, Granek Z, Siwecka N, Wiese W, Golberg M, Galita G, et al. Potential Mechanisms of Tunneling Nanotube Formation and Their Role in Pathology Spread in Alzheimer’s Disease and Other Proteinopathies. International Journal of Molecular Sciences. 2024; 25(19):10797. https://doi.org/10.3390/ijms251910797
Chicago/Turabian StyleKotarba, Szymon, Marta Kozłowska, Małgorzata Scios, Kamil Saramowicz, Julia Barczuk, Zuzanna Granek, Natalia Siwecka, Wojciech Wiese, Michał Golberg, Grzegorz Galita, and et al. 2024. "Potential Mechanisms of Tunneling Nanotube Formation and Their Role in Pathology Spread in Alzheimer’s Disease and Other Proteinopathies" International Journal of Molecular Sciences 25, no. 19: 10797. https://doi.org/10.3390/ijms251910797
APA StyleKotarba, S., Kozłowska, M., Scios, M., Saramowicz, K., Barczuk, J., Granek, Z., Siwecka, N., Wiese, W., Golberg, M., Galita, G., Sychowski, G., Majsterek, I., & Rozpędek-Kamińska, W. (2024). Potential Mechanisms of Tunneling Nanotube Formation and Their Role in Pathology Spread in Alzheimer’s Disease and Other Proteinopathies. International Journal of Molecular Sciences, 25(19), 10797. https://doi.org/10.3390/ijms251910797