
The Complex Interplay of TGF-β and Notch Signaling in the Pathogenesis of Fibrosis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Fibrosis Development
1.1. Cellular and Molecular Mechanisms
1.2. TGF-β Pathway and Its Role in Fibrosis
1.3. Notch Signaling and Fibrogenesis
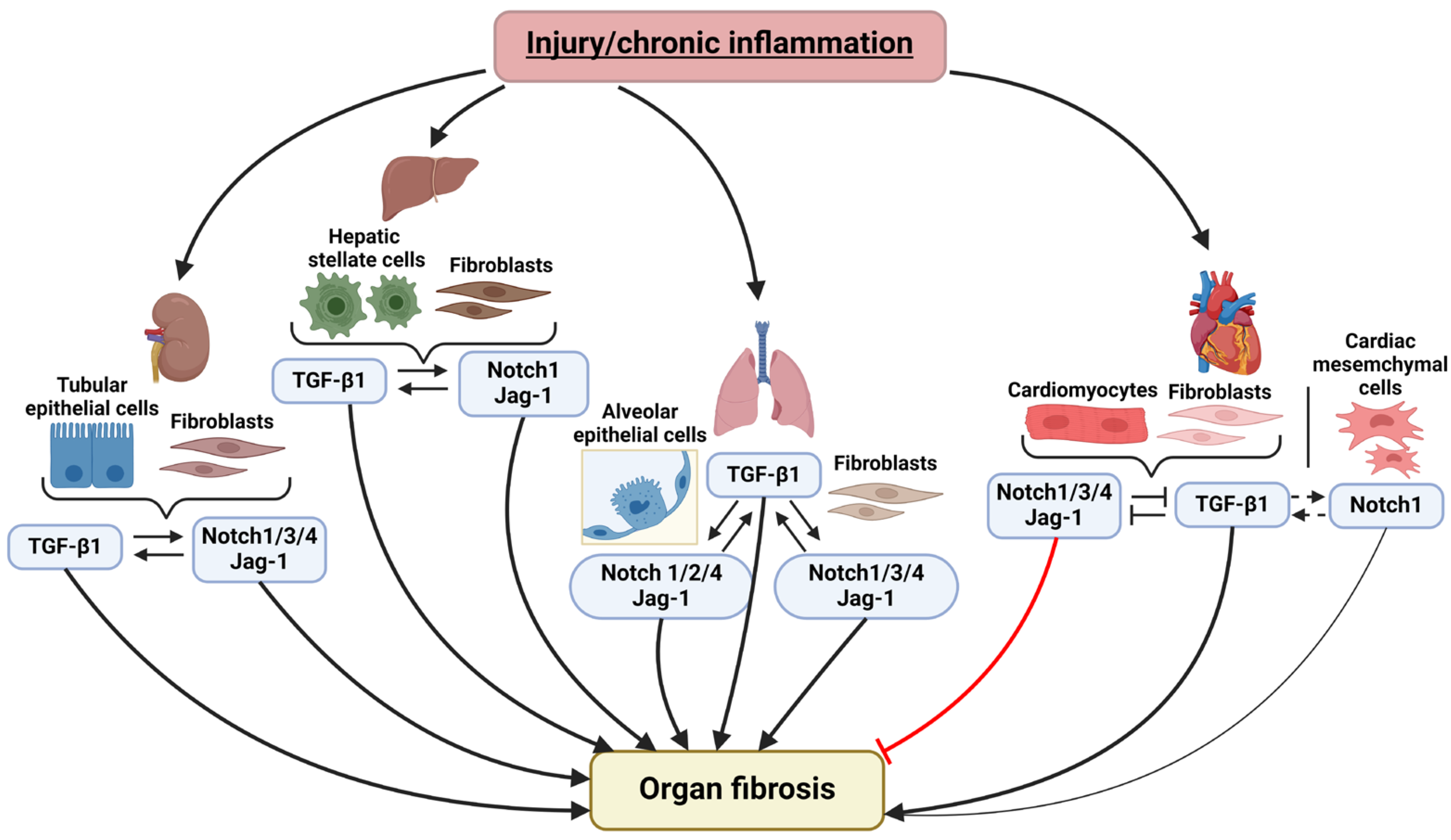
2. Notch and TGF-β Signaling Pathways Interplay in the Fibrosis of Various Organs
2.1. Notch Signaling and TGF-β Pathway Crosstalk in Pulmonary Fibrosis
2.2. Notch and TGF-β in Liver Fibrosis
2.3. Notch and TGF-β Interaction in Kidney Fibrosis
2.4. Crosstalk between Notch and TGF-β in Cardiac Fibrosis
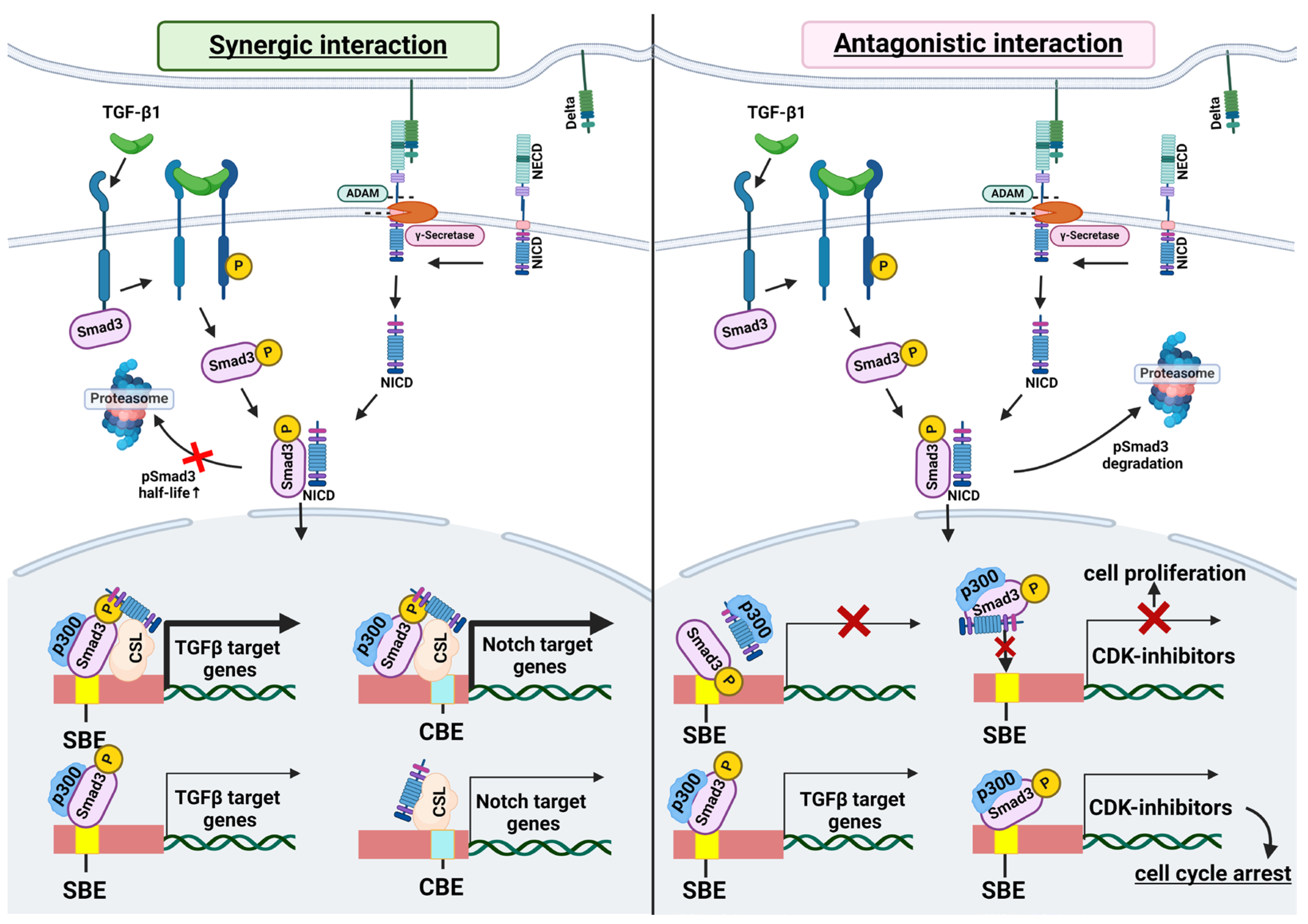
3. Molecular Mechanisms of the Interplay between TGF-β and Notch Signaling
3.1. Interaction of SMAD3 and NICD
3.2. Shared Transcriptional Targets
3.3. Interactions Mediated by Reactive Oxygen Species (ROS)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meng, X.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Wilson, M.D. Fibrogenesis: Mechanisms, Dynamics and Clinical Implications. Iran. J. Pathol. 2015, 10, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Pei, Q.; Yi, Q.; Tang, L. Liver Fibrosis Resolution: From Molecular Mechanisms to Therapeutic Opportunities. Int. J. Mol. Sci. 2023, 24, 9671. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Fu, P.; Ma, L. Kidney fibrosis: From mechanisms to therapeutic medicines. Signal. Transduct. Target. Ther. 2023, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, M.L.; Wijsenbeek, M.S. Management of idiopathic pulmonary fibrosis. Clin. Chest Med. 2021, 42, 275–285. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef]
- Do, N.N.; Eming, S.A. Skin fibrosis: Models and mechanisms. Curr. Res. Transl. Med. 2016, 64, 185–193. [Google Scholar] [CrossRef]
- Borrelli, M.R.; Shen, A.H.; Lee, G.K.; Momeni, A.; Longaker, M.T.; Wan, D.C. Radiation-induced skin fibrosis. Ann. Plast. Surg. 2019, 83, S59–S64. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.H.; Li, J.; Sun, L.Q. Molecular Mechanisms and Treatment of Radiation-Induced Lung Fibrosis. Curr. Drug. Targets 2013, 14, 1347–1356. [Google Scholar] [CrossRef]
- Duong-Quy, S.; Vo-Pham-Minh, T.; Tran-Xuan, Q.; Huynh-Anh, T.; Vo-Van, T.; Vu-Tran-Thien, Q.; Nguyen-Nhu, V. Post-COVID-19 Pulmonary Fibrosis: Facts-Challenges and Futures: A Narrative Review. Pulm. Ther. 2023, 9, 295–307. [Google Scholar] [CrossRef]
- Herrick, S.E.; Wilm, B. Post-Surgical Peritoneal Scarring and Key Molecular Mechanisms. Biomolecules 2021, 11, 692. [Google Scholar] [CrossRef] [PubMed]
- Savin, I.A.; Zenkova, M.A.; Sen’kova, A.V. Bronchial Asthma, Airway Remodeling and Lung Fibrosis as Successive Steps of One Process. Int. J. Mol. Sci. 2023, 24, 16042. [Google Scholar] [CrossRef]
- Hu, B.; Phan, S.H. Notch in fibrosis and as a target of anti-fibrotic therapy. Pharmacol. Res. 2016, 108, 57–64. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Mack, M. Inflammation and fibrosis. Matrix Biol. 2018, 68–69, 106–121. [Google Scholar] [CrossRef]
- Liu, T.; Hu, B.; Choi, Y.Y.; Chung, M.; Ullenbruch, M.; Yu, H.; Lowe, J.B.; Phan, S.H. Notch1 Signaling in FIZZ1 Induction of Myofibroblast Differentiation. Am. J. Pathol. 2009, 174, 1745–1755. [Google Scholar] [CrossRef] [PubMed]
- Baum, J.; Duffy, H.S. Fibroblasts and Myofibroblasts: What Are We Talking About? J. Cardiovasc. Pharmacol. 2011, 57, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi-Ikeda, K.; Maeno, T.; Matsui, H.; Ueno, M.; Hara, K.; Aoki, Y.; Aoki, F.; Shimizu, T.; Doi, H.; Kawai-Kowase, K.; et al. Notch Induces Myofibroblast Differentiation of Alveolar Epithelial Cells via Transforming Growth Factor-β-Smad3 Pathway. Am. J. Respir. Cell Mol. Biol. 2011, 45, 136–144. [Google Scholar] [CrossRef]
- Sun, Y.B.Y.; Qu, X.; Caruana, G.; Li, J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation 2016, 92, 102–107. [Google Scholar] [CrossRef]
- Kuppe, C.; Ibrahim, M.M.; Kranz, J.; Zhang, X.; Ziegler, S.; Perales-Patón, J.; Jansen, J.; Reimer, K.C.; Smith, J.R.; Dobie, R.; et al. Decoding myofibroblast origins in human kidney fibrosis. Nature 2021, 589, 281–286. [Google Scholar] [CrossRef]
- Lee, J.H.; Massagué, J. TGF-β in developmental and fibrogenic EMTs. Semin. Cancer Biol. 2022, 86, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; You, Y.; Jiang, H.; Wang, Z.Z. Epithelial–mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J. Cell. Physiol. 2017, 232, 3261–3272. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef]
- Marconi, G.D.; Fonticoli, L.; Rajan, T.S.; Pierdomenico, S.D.; Trubiani, O.; Pizzicannella, J.; Diomede, F. Epithelial-Mesenchymal Transition (EMT): The Type-2 EMT in Wound Healing, Tissue Regeneration and Organ Fibrosis. Cells 2021, 10, 1587. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Zhang, Y.; Jin, D.; Kang, X.; Zhou, R.; Sun, Y.; Lian, F.; Tong, X. Signaling Pathways Involved in Diabetic Renal Fibrosis. Front. Cell Dev. Biol. 2021, 9, 696542. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Transforming growth factor–β in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Hu, H.H.; Cao, G.; Wu, X.Q.; Vaziri, N.D.; Zhao, Y.Y. Wnt signaling pathway in aging-related tissue fibrosis and therapies. Ageing Res. Rev. 2020, 60, 101063. [Google Scholar] [CrossRef]
- Yue, X.; Shan, B.; Lasky, J.A. TGF-β: Titan of Lung Fibrogenesis. Curr. Enzym. Inhib. 2010, 6, 67–77. [Google Scholar] [CrossRef]
- Tzavlaki, K.; Moustakas, A. TGF-β Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef] [PubMed]
- Dituri, F.; Cossu, C.; Mancarella, S.; Giannelli, G. The Interactivity between TGFβ and BMP Signaling in Organogenesis, Fibrosis, and Cancer. Cells 2019, 8, 1130. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Border, W.A.; Huang, Y.; Noble, N.A. TGF-β isoforms in renal fibrogenesis. Kidney Int. 2003, 64, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.H.; Chen, D.Q.; Wang, Y.N.; Feng, Y.L.; Cao, G.; Vaziri, N.D.; Zhao, Y.Y. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem. Biol. Interact. 2018, 292, 76–83. [Google Scholar] [CrossRef]
- Hu, B.; Wu, Z.; Phan, S.H. Smad3 Mediates Transforming Growth Factor-β–Induced α-Smooth Muscle Actin Expression. Am. J. Respir. Cell. Mol. Biol. 2003, 29, 397–404. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef]
- Choi, S.Y.; Ryu, Y.; Kee, H.J.; Cho, S.-N.; Kim, G.R.; Cho, J.Y.; Kim, H.S.; Kim, I.K.; Jeong, M.H. Tubastatin A suppresses renal fibrosis via regulation of epigenetic histone modification and Smad3-dependent fibrotic genes. Vascul. Pharmacol. 2015, 72, 130–140. [Google Scholar] [CrossRef]
- Pan, X.; Chen, Z.; Huang, R.; Yao, Y.; Ma, G. Transforming Growth Factor β1 Induces the Expression of Collagen Type I by DNA Methylation in Cardiac Fibroblasts. PLoS ONE 2013, 8, e60335. [Google Scholar] [CrossRef]
- Sutariya, B.; Jhonsa, D.; Saraf, M.N. TGF-β: The connecting link between nephropathy and fibrosis. Immunopharmacol. Immunotoxicol. 2016, 38, 39–49. [Google Scholar] [CrossRef]
- Lai, J.M.; Zhang, X.; Liu, F.F.; Yang, R.; Li, S.Y.; Zhu, L.B.; Zou, M.; Cheng, W.H.; Zhu, J.H. Redox-sensitive MAPK and Notch3 regulate fibroblast differentiation and activation: A dual role of ERK1/2. Oncotarget 2016, 7, 43731–43745. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-β/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem.Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.C. Notch signaling: Control of cell communication and cell fate. Development 2004, 131, 965–973. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Gozlan, O.; Sprinzak, D. Notch signaling in development and homeostasis. Development 2023, 150, dev201138. [Google Scholar] [CrossRef]
- Wasnick, R.; Korfei, M.; Piskulak, K.; Henneke, I.; Wilhelm, J.; Mahavadi, P.; Dartsch, R.C.; von der Beck, D.; Koch, M.; Shalashova, I.; et al. Notch1 Induces Defective Epithelial Surfactant Processing and Pulmonary Fibrosis. Am. J. Respir. Crit. Care. Med. 2023, 207, 283–299. [Google Scholar] [CrossRef]
- Yuan, X.; Wu, H.; Han, N.; Xu, H.; Chu, Q.; Yu, S.; Chen, Y.; Wu, K. Notch signaling and EMT in non-small cell lung cancer: Biological significance and therapeutic application. J. Hematol. Oncol. 2014, 7, 87. [Google Scholar] [CrossRef]
- Hu, B.; Phan, S.H. Myofibroblasts. Curr. Opin. Rheumatol. 2013, 25, 71–77. [Google Scholar] [CrossRef]
- Wang, J.; Yang, L.; Mei, J.; Li, Z.; Huang, Y.; Sun, H.; Zheng, K.; Kuang, H.; Luo, W. Knockdown of Notch Suppresses Epithelial-mesenchymal Transition and Induces Angiogenesis in Oral Submucous Fibrosis by Regulating TGF-β1. Biochem. Genet. 2024, 62, 1055–1069. [Google Scholar] [CrossRef]
- Chistyakova, I.V.; Bakalenko, N.I.; Malashicheva, A.B.; Atyukov, M.A.; Petrov, A.S. The role of Notch-dependent differentiation of resident fibroblasts in the development of pulmonary fibrosis. Transl. Med. 2022, 9, 96–104. [Google Scholar] [CrossRef]
- Yuan, C.; Ni, L.; Zhang, C.; Wu, X. The Role of Notch3 Signaling in Kidney Disease. Oxid. Med. Cell Longev. 2020, 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.X. Notch3 regulates the activation of hepatic stellate cells. World J. Gastroenterol. 2012, 18, 1397. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, J.; Guo, G.; Moridaira, K.; Fitzgerald, M.; McCracken, R.; Tolley, T.; Klahr, S. Transforming Growth Factor-β Induces Renal Epithelial Jagged-1 Expression in Fibrotic Disease. J. Am. Soc. Nephrol. 2002, 13, 1499–1508. [Google Scholar] [CrossRef]
- Kachanova, O.; Lobov, A.; Malashicheva, A. The Role of the Notch Signaling Pathway in Recovery of Cardiac Function after Myocardial Infarction. Int. J. Mol. Sci. 2022, 23, 12509. [Google Scholar] [CrossRef] [PubMed]
- Nemir, M.; Metrich, M.; Plaisance, I.; Lepore, M.; Cruchet, S.; Berthonneche, C.; Sarre, A.; Radtke, F.; Pedrazzini, T. The Notch pathway controls fibrotic and regenerative repair in the adult heart. Eur. Heart J. 2014, 35, 2174–2185. [Google Scholar] [CrossRef]
- Fan, Y.H.; Dong, H.; Pan, Q.; Cao, Y.J.; Li, H.; Wang, H.C. Notch signaling may negatively regulate neonatal rat cardiac fibroblast-myofibroblast transformation. Physiol. Res. 2011, 60, 739–748. [Google Scholar] [CrossRef]
- Zhou, X.; Fang, Y.; Wan, L.; Xu, Q.; Huang, H.; Zhu, R.; Wu, Q.; Liu, J. Notch signaling inhibits cardiac fibroblast to myofibroblast transformation by antagonizing TGF-β1/Smad3 signaling. J. Cell Physiol. 2018, 234, 8834–8845. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, L.; Wang, S.; Cheng, H.; Xu, L.; Pei, G.; Wang, Y.; Fu, C.; Jiang, Y.; He, C.; et al. Signaling pathways and targeted therapy for myocardial infarction. Sig. Transduct. Target. Ther. 2022, 7, 78. [Google Scholar] [CrossRef]
- Russell, J.L.; Goetsch, S.C.; Gaiano, N.R.; Hill, J.A.; Olson, E.N.; Schneider, J.W. A Dynamic Notch Injury Response Activates Epicardium and Contributes to Fibrosis Repair. Circ. Res. 2011, 108, 51–59. [Google Scholar] [CrossRef]
- Docshin, P.; Bairqdar, A.; Malashicheva, A. Interplay between BMP2 and Notch signaling in endothelial-mesenchymal transition: Implications for cardiac fibrosis. Stem Cell Investig. 2023, 10, 18. [Google Scholar] [CrossRef]
- Saito, A.; Horie, M.; Nagase, T. TGF-β Signaling in Lung Health and Disease. Int. J. Mol. Sci. 2018, 19, 2460. [Google Scholar] [CrossRef] [PubMed]
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Asp. Med. 2019, 65, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Zong, D.; Ouyang, R.; Li, J.; Chen, Y.; Chen, P. Notch signaling in lung diseases: Focus on Notch1 and Notch3. Ther. Adv. Respir. Dis. 2016, 10, 468–484. [Google Scholar] [CrossRef] [PubMed]
- Vera, L.; Garcia-Olloqui, P.; Petri, E.; Viñado, A.C.; Valera, P.S.; Blasco-Iturri, Z.; Calvo, I.A.; Cenzano, I.; Ruppert, C.; Zulueta, J.J.; et al. Notch3 Deficiency Attenuates Pulmonary Fibrosis and Impedes Lung-Function Decline. Am. J. Respir. Cell Mol. Biol. 2021, 64, 465–476. [Google Scholar] [CrossRef]
- Matsuno, Y.; Coelho, A.L.; Jarai, G.; Westwick, J.; Hogaboam, C.M. Notch signaling mediates TGF-β1-induced epithelial–mesenchymal transition through the induction of Snai1. Int. J. Biochem. Cell Biol. 2012, 44, 776–789. [Google Scholar] [CrossRef]
- Bakalenko, N.; Smirnova, D.; Gaifullina, L.; Kuchur, P.; Ian, D.; Atyukov, M.; Liu, J.; Malashicheva, A. NOTCH4 Is a New Player in the Development of Pulmonary Fibrosis. Gene Expr. 2024, 23, 273–281. [Google Scholar] [CrossRef]
- Zavadil, J.; Böttinger, E.P. TGF-β and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef]
- Zavadil, J.; Cermak, L.; Soto-Nieves, N.; Böttinger, E.P. Integration of TGF-β/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004, 23, 1155–1165. [Google Scholar] [CrossRef]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef]
- D’Amico, G.; Morabito, A.; D’Amico, M.; Pasta, L.; Malizia, G.; Rebora, P.; Valsecchi, M.G. Clinical states of cirrhosis and competing risks. J. Hepatol. 2018, 68, 563–576. [Google Scholar] [CrossRef]
- Elpek, G.Ö. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J. Gastroenterol. 2014, 20, 7260. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Aimaiti, Y.; Yusufukadier, M.; Li, W.; Tuerhongjiang, T.; Shadike, A.; Meiheriayi, A.; Gulisitan; Abudusalamu, A.; Wang, H.; Tuerganaili, A.; et al. TGF-β1 signaling activates hepatic stellate cells through Notch pathway. Cytotechnology 2019, 71, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Genz, B.; Coleman, M.A.; Irvine, K.M.; Kutasovic, J.R.; Miranda, M.; Gratte, F.D.; Tirnitz-Parker, J.E.E.; Olynyk, J.K.; Calvopina, D.A.; Weis, A.; et al. Overexpression of miRNA-25-3p inhibits Notch1 signaling and TGF-β-induced collagen expression in hepatic stellate cells. Sci. Rep. 2019, 9, 8541. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shen, R.W.; Han, B.; Li, Z.; Xiong, L.; Zhang, F.Y.; Cong, B.B.; Zhang, B. Notch signaling mediated by TGF-β/Smad pathway in concanavalin A-induced liver fibrosis in rats. World J. Gastroenterol. 2017, 23, 2330. [Google Scholar] [CrossRef]
- Mancarella, S.; Gigante, I.; Serino, G.; Pizzuto, E.; Dituri, F.; Valentini, M.F.; Wang, J.; Chen, X.; Armentano, R.; Calvisi, D.F.; et al. Crenigacestat blocking notch pathway reduces liver fibrosis in the surrounding ecosystem of intrahepatic CCA viaTGF-β inhibition. J. Exp. Clin. Cancer Res. 2022, 41, 331. [Google Scholar] [CrossRef]
- Yu, C.; Xiong, C.; Tang, J.; Hou, X.; Liu, N.; Bayliss, G.; Zhuang, S. Histone demethylase JMJD3 protects against renal fibrosis by suppressing TGFβ and Notch signaling and preserving PTEN expression. Theranostics 2021, 11, 2706–2721. [Google Scholar] [CrossRef]
- Xiao, Z.; Zhang, J.; Peng, X.; Dong, Y.; Jia, L.; Li, H.; Du, J. The Notch γ-secretase inhibitor ameliorates kidney fibrosis via inhibition of TGF-β/Smad2/3 signaling pathway activation. Int. J. Biochem. Cell Biol. 2014, 55, 65–71. [Google Scholar] [CrossRef]
- Kriz, W.; Kaissling, B.; Le Hir, M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: Fact or fantasy? J. Clin. Investig. 2011, 121, 468–474. [Google Scholar] [CrossRef]
- Hong, W.; Zhang, G.; Lu, H.; Guo, Y.; Zheng, S.; Zhu, H.; Xiao, Y.; Papa, A.P.D.; Wu, C.; Sun, L.; et al. Epithelial and interstitial Notch1 activity contributes to the myofibroblastic phenotype and fibrosis. Cell Commun. Signal. 2019, 17, 145. [Google Scholar] [CrossRef]
- Tang, R.; Xiao, X.; Lu, Y.; Li, H.; Zhou, Q.; Kwadwo Nuro-Gyina, P.; Li, X. Interleukin-22 attenuates renal tubular cells inflammation and fibrosis induced by TGF-β1 through Notch1 signaling pathway. Ren. Fail. 2020, 42, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Gao, C.; Chen, G.; Li, X.; Li, J.; Wan, Q.; Xu, Y. Notch Signaling Molecules Activate TGF- β in Rat Mesangial Cells under High Glucose Conditions. J. Diabetes Res. 2013, 2013, 979702. [Google Scholar] [CrossRef] [PubMed]
- Grabias, B.M.; Konstantopoulos, K. Notch4-dependent antagonism of canonical TGF-β1 signaling defines unique temporal fluctuations of SMAD3 activity in sheared proximal tubular epithelial cells. Am. J. Physiol. Renal Physiol. 2013, 305, F123–F133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Pan, X.; Zou, Q.; Xia, Y.; Chen, J.; Hao, Q.; Wang, H.; Sun, D. Notch3 Ameliorates Cardiac Fibrosis After Myocardial Infarction by Inhibiting the TGF-β1/Smad3 Pathway. Cardiovasc. Toxicol. 2015, 16, 316–324. [Google Scholar] [CrossRef]
- Li, Z.; Nie, M.; Yu, L.; Tao, D.; Wang, Q.; He, Y.; Liu, Y.; Zhang, Y.; Han, H.; Wang, H. Blockade of the Notch Signaling Pathway Promotes M2 Macrophage Polarization to Suppress Cardiac Fibrosis Remodeling in Mice With Myocardial Infarction. Front. Cardiovasc. Med. 2022, 8, 639476. [Google Scholar] [CrossRef]
- Zhang, K.; Han, X.; Zhang, Z.; Zheng, L.; Hu, Z.; Yao, Q.; Cui, H.; Shu, G.; Si, M.; Li, C.; et al. The liver-enriched lnc-LFAR1 promotes liver fibrosis by activating TGFβ and Notch pathways. Nat. Commun. 2017, 8, 144. [Google Scholar] [CrossRef]
- Blokzijl, A.; Dahlqvist, C.; Reissmann, E.; Falk, A.; Moliner, A.; Lendahl, U.; Ibáñez, C.F. Cross-talk between the Notch and TGF-β signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J. Cell Biol. 2003, 163, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chang, A.; Chang, L.; Niessen, K.; Eapen, S.; Setiadi, A.; Karsan, A. Differential Regulation of Transforming Growth Factor β Signaling Pathways by Notch in Human Endothelial Cells. J. Biol. Chem. 2009, 284, 19452–19462. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lowther, W.; Kato, K.; Bianco, C.; Kenney, N.; Strizzi, L.; Raafat, D.; Hirota, M.; Khan, N.I.; Bargo, S.; et al. Notch4 intracellular domain binding to Smad3 and inhibition of the TGF-β signaling. Oncogene 2005, 24, 5365–5374. [Google Scholar] [CrossRef]
- Carlson, M.E.; Hsu, M.; Conboy, I.M. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature 2008, 454, 528–532. [Google Scholar] [CrossRef]
- Samon, J.B.; Champhekar, A.; Minter, L.M.; Telfer, J.C.; Miele, L.; Fauq, A.; Das, P.; Golde, T.E.; Osborne, B.A. Notch1 and TGFβ1 cooperatively regulate Foxp3 expression and the maintenance of peripheral regulatory T cells. Blood 2008, 112, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Masuda, S.; Kumano, K.; Shimizu, K.; Imai, Y.; Kurokawa, M.; Ogawa, S.; Miyagishi, M.; Taira, K.; Hirai, H.; Chiba, S. Notch1 oncoprotein antagonizes TGF-β/Smad-mediated cell growth suppression via sequestration of coactivator p300. Cancer Sci. 2005, 96, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef] [PubMed]
- Niessen, K.; Fu, Y.; Chang, L.; Hoodless, P.A.; McFadden, D.; Karsan, A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J. Cell Biol. 2008, 182, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef]
- Zhao, Y.; Fan, X.; Wang, Q.; Zhen, J.; Li, X.; Zhou, P.; Lang, Y.; Sheng, Q.; Zhang, T.; Huang, T.; et al. ROS promote hyper-methylation of NDRG2 promoters in a DNMTS-dependent manner: Contributes to the progression of renal fibrosis. Redox Biol. 2023, 62, 102674. [Google Scholar] [CrossRef]
- Latella, G. Redox Imbalance in Intestinal Fibrosis: Beware of the TGFβ-1, ROS, and Nrf2 Connection. Dig. Dis. Sci. 2018, 63, 312–320. [Google Scholar] [CrossRef]
- Yazaki, K.; Matsuno, Y.; Yoshida, K.; Sherpa, M.; Nakajima, M.; Matsuyama, M.; Kiwamoto, T.; Morishima, Y.; Ishii, Y.; Hizawa, N. ROS-Nrf2 pathway mediates the development of TGF-β1-induced epithelial-mesenchymal transition through the activation of Notch signaling. Eur. J. Cell Biol. 2021, 100, 151181. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Shin, S.; Slocum, S.L.; Agoston, E.S.; Wakabayashi, J.; Kwak, M.K.; Misra, V.; Biswal, S.; Yamamoto, M.; Kensler, T.W. Regulation of Notch1 Signaling by Nrf2: Implications for Tissue Regeneration. Sci. Signal. 2010, 3, ra52. [Google Scholar] [CrossRef]
- Edeling, M.; Ragi, G.; Huang, S.; Pavenstädt, H.; Susztak, K. Developmental signalling pathways in renal fibrosis: The roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 2016, 12, 426–439. [Google Scholar] [CrossRef]
- Distler, J.H.W.; Györfi, A.H.; Ramanujam, M.; Whitfield, M.L.; Königshoff, M.; Lafyatis, R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [CrossRef] [PubMed]
- Mia, M.M.; Singh, M.K. New Insights into Hippo/YAP Signaling in Fibrotic Diseases. Cells 2022, 11, 2065. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakalenko, N.; Kuznetsova, E.; Malashicheva, A. The Complex Interplay of TGF-β and Notch Signaling in the Pathogenesis of Fibrosis. Int. J. Mol. Sci. 2024, 25, 10803. https://doi.org/10.3390/ijms251910803
Bakalenko N, Kuznetsova E, Malashicheva A. The Complex Interplay of TGF-β and Notch Signaling in the Pathogenesis of Fibrosis. International Journal of Molecular Sciences. 2024; 25(19):10803. https://doi.org/10.3390/ijms251910803
Chicago/Turabian StyleBakalenko, Nadezhda, Evdokiya Kuznetsova, and Anna Malashicheva. 2024. "The Complex Interplay of TGF-β and Notch Signaling in the Pathogenesis of Fibrosis" International Journal of Molecular Sciences 25, no. 19: 10803. https://doi.org/10.3390/ijms251910803
APA StyleBakalenko, N., Kuznetsova, E., & Malashicheva, A. (2024). The Complex Interplay of TGF-β and Notch Signaling in the Pathogenesis of Fibrosis. International Journal of Molecular Sciences, 25(19), 10803. https://doi.org/10.3390/ijms251910803