Abstract
In classical amyloidoses, amyloid fibres form through the nucleation and accretion of protein monomers, with protofibrils and fibrils exhibiting a cross-β motif of parallel or antiparallel β-sheets oriented perpendicular to the fibre direction. These protofibrils and fibrils can intertwine to form mature amyloid fibres. Similar phenomena can occur in blood from individuals with circulating inflammatory molecules (and also some originating from viruses and bacteria). Such pathological clotting can result in an anomalous amyloid form termed fibrinaloid microclots. Previous proteomic analyses of these microclots have shown the presence of non-fibrin(ogen) proteins, suggesting a more complex mechanism than simple entrapment. We thus provide evidence against such a simple entrapment model, noting that clot pores are too large and centrifugation would have removed weakly bound proteins. Instead, we explore whether co-aggregation into amyloid fibres may involve axial (multiple proteins within the same fibril), lateral (single-protein fibrils contributing to a fibre), or both types of integration. Our analysis of proteomic data from fibrinaloid microclots in different diseases shows no significant quantitative overlap with the normal plasma proteome and no correlation between plasma protein abundance and their presence in fibrinaloid microclots. Notably, abundant plasma proteins like α-2-macroglobulin, fibronectin, and transthyretin are absent from microclots, while less abundant proteins such as adiponectin, periostin, and von Willebrand factor are well represented. Using bioinformatic tools, including AmyloGram and AnuPP, we found that proteins entrapped in fibrinaloid microclots exhibit high amyloidogenic tendencies, suggesting their integration as cross-β elements into amyloid structures. This integration likely contributes to the microclots’ resistance to proteolysis. Our findings underscore the role of cross-seeding in fibrinaloid microclot formation and highlight the need for further investigation into their structural properties and implications in thrombotic and amyloid diseases. These insights provide a foundation for developing novel diagnostic and therapeutic strategies targeting amyloidogenic cross-seeding in blood clotting disorders.
1. Introduction
Blood homeostasis is a finely tuned process involving a complex series of reactions known as the blood clotting cascade. Central to this process is the conversion of fibrinogen to fibrin, catalysed by the serine protease thrombin, resulting in the formation of polymeric fibrin clots. Fibrinogen, a major plasma protein, is pivotal in this cascade, undergoing a remarkable transformation from a soluble protein to an insoluble fibrin matrix. While the mechanisms of normal clot formation are well understood, recent studies have unveiled the formation of pathological clot structures, termed fibrinaloid microclots, in the presence of inflammatory molecules, including those with viral and bacterial origins. These fibrinaloid microclots display unique proteomic profiles and amyloid-like properties, suggesting a complex interplay between protein misfolding and aggregation beyond simple entrapment. This paper explores the proteomic characteristics of fibrinaloid microclots, investigates the mechanisms of their formation, and highlights their potential implications in thrombotic and amyloid diseases.
An important part of blood homeostasis involves the blood clotting cascade. This is well known (Figure 1A) and our focus is on the catalysis by the serine protease thrombin of the conversion of fibrinogen to make polymeric fibrin. Fibrinogen is one of the most abundant plasma proteins, present at some 2–4 g.L−1 (e.g., [1,2,3,4]). It is a cigar-shaped molecule of ca 5 × 45 nm, and consists of several chains (e.g., [5,6,7,8]). The action of thrombin leads to the serial removal of two fibrinopeptides, which sets in motion a remarkable self-organisation by which the fibrinogen molecules interact to form protofibrils, fibrils, and then fibres of some 50–100 nm diameter (Figure 1B), implying some hundreds of fibrinogen molecules in each length element of the typical fibrin fibre. In normal clots, the direction of the fibrinogen molecules and fibrin protofibrils is parallel to that of the fibres.
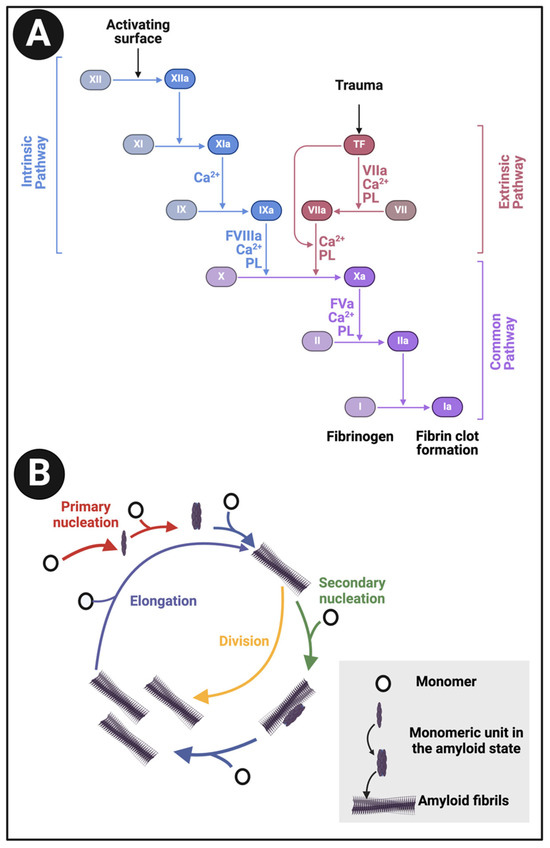
Figure 1.
(A): The clotting cascade and (B) fibrinogen conversion to fibrin. The clotting cascade involves the intrinsic, extrinsic, and common pathways, each comprising various clotting factors. The intrinsic pathway includes factors I (fibrinogen), II (prothrombin), IX (Christmas factor), X (Stuart-Prower factor), XI (plasma thromboplastin), and XII (Hageman factor). The extrinsic pathway consists of factors I, II, VII (stable factor), and X. The common pathway involves factors I, II, V, VIII, and X. These factors circulate in the bloodstream as zymogens and are activated into serine proteases, which catalyse the cleavage of subsequent zymogens into more serine proteases, ultimately activating fibrinogen. The serine proteases include factors II, VII, IX, X, XI, and XII, while factors V, VIII, and XIII are not serine proteases. The intrinsic pathway is activated by exposed endothelial collagen, whereas the extrinsic pathway is triggered by tissue factor released by endothelial cells after external damage. Drawn using Biorender.com.
The pore sizes of typical clots are of the order 0.5–5 μm when fibrinogen is at its physiological concentrations (e.g., [9,10,11,12,13]) (the pore diameters can be far lower at massively extraphysiological fibrinogen concentrations [14]), so without specific binding of some kind they are clearly incapable of simply entrapping molecules of globular proteins (with diameters in the low nm range). A complex set of reactions also contribute to normal clot degradation (fibrinolysis) [15].
1.1. Proteins of Identical Sequence Can Adopt Alternative, Stable Macrostates
The famous ‘unboiling an egg’ experiment of Christian Anfinsen [16,17] involved the chemical denaturation of ribonuclease followed, upon removal by dialysis of the chemical denaturant, by its refolding into what was considered to be the same native form as the original made naturally following ribosomal synthesis. Importantly, this led to the conclusion that the information necessary for a protein’s tertiary structure could be encoded solely in its primary amino acid sequence. Unfortunately, it was also widely assumed that this structure was thus the thermodynamically most stable under the conditions of interest. This latter assumption could only be just that (an assumption) because of the astronomical number of conformations that a string of n amino acids might adopt even as a single molecule [18,19], let alone when ensembles of a given protein form inclusion bodies. Indeed, we now know of many classes of example in which proteins can adopt very different conformations despite having the identical primary structure. Prions, amyloids, and prionoids are three such classes [20,21,22,23]. In fact, the existence of very different macrostates or folds, between which proteins can in fact switch physiologically, is surprisingly common [24,25,26,27]. Such proteins have been referred to as ‘metamorphic’ [28,29,30,31,32]. While the present versions of Alphafold cannot (and were not designed to) predict such a multiplicity of macroscopic conformations effectively [33,34] (though see their utility for isoenergetic microstates [35]), the primary sequences do in fact contain such information [36,37]. There is also evidence that proteins capable of adopting multiple macrostates were in fact selected adaptively [38,39,40,41] and can be designed accordingly [42].
Given the above, and that our focus here is on amyloidogenic proteins, it is of special interest that α-helix-to-β-sheet transitions are a noteworthy feature of such proteins [36], and particularly, for our present purposes, some in SARS-CoV-2 [43], where several proteins are amyloidogenic [44,45]. We note too that some alleles of the fibrinogen Aα chain may produce highly amyloidogenic proteolytic fragments [46], and that fibrinogen can bind to well-established amyloids such as Aβ [47,48,49,50].
1.2. Structure and Interconversion of Amyloid(ogenic) Proteins
In contrast to the expectations of the Anfinsen experiment, it has become well established that many (and maybe even most) proteins can fold into states that are considerably more stable than the one natively or most commonly adopted as they leave the ribosome, but that this thermodynamically favoured conformation (or, more accurately, the set of isoenergetic conformations) is normally kinetically inaccessible due to a massive energy barrier of some 36–38 kcal.mol−1 [21,51]. A particular class of these more stable conformations involve a cross(ed)-β-sheet motif [52,53,54,55,56,57,58], and they become insoluble because they tend to aggregate and self-assemble; following their discovery by Virchow in 1854 (reviewed by Sipe and Cohen [59]), they are referred to as amyloids (see, e.g., [60,61,62,63,64,65]). As is well known, they are intimately (if at best only partially) involved in a variety of diseases, including Alzheimer’s [66] and Parkinson’s. Such syndromes are collectively referred to as amyloidoses (e.g., [67,68,69,70,71,72,73]). As phrased by Burdukiewicz et al. [74], “Despite their diversity, all amyloid proteins can undergo aggregation initiated by short segments called hot spots”. This is a massive field, so our focus is on those parts that most reflect the present core question, which is around their self-assembly. This—commonly the transition from an α-helix structure to a β-sheet one [75]—necessarily involves partial unfolding [76,77,78,79,80], and this plausibly underpins the mechanism of cross-seeding.
1.3. Rules for Amyloidogenesis and Cross-β Formation
In contrast to the classical secondary structure of β-sheets, where the rules for their formation in terms of amino acid sequence are broadly polar–apolar–polar–apolar–(etc.) [81], the sequence rules for amyloidogenic potential generally, and for cross-β sheet formation [82] in particular, are rather more obscure. This is not helped by the relative paucity of data on (sub)sequences that can encode amyloidogenicity, but experimental and computational progress is being made (e.g., [83,84,85] and Table 1) and databases of amyloidogenic hexapeptides [86] and amyloid–amyloid interactions exist [87]. It is indeed reasonable that the most predictive properties for residue amyloidogenicity are “hydrophobicity index [88], average flexibility indices (a normalized fluctuational displacement of an amino acid residue) [89], polarizability parameter [90] and thermodynamic β-sheet propensity [74,91].

Table 1.
Some computational resources for predicting amyloidogenic regions in proteins.
Figure 2A shows a prediction from AmyloGram [74] of a well-established amyloidogenic protein in the form of the human prion protein PrPc, when a significant run of residues towards the C-terminus has an amyloidogenicity score (referred to as a ‘probability of self-assembly’) exceeding 0.75 (and see later). Figure 2B also shows the predictions for the fibrinogen A/α chain, which shares some of these features. Figure 2C shows the predictions for the fibrinogen A/α chain at AnuPP, giving a broadly similar picture.
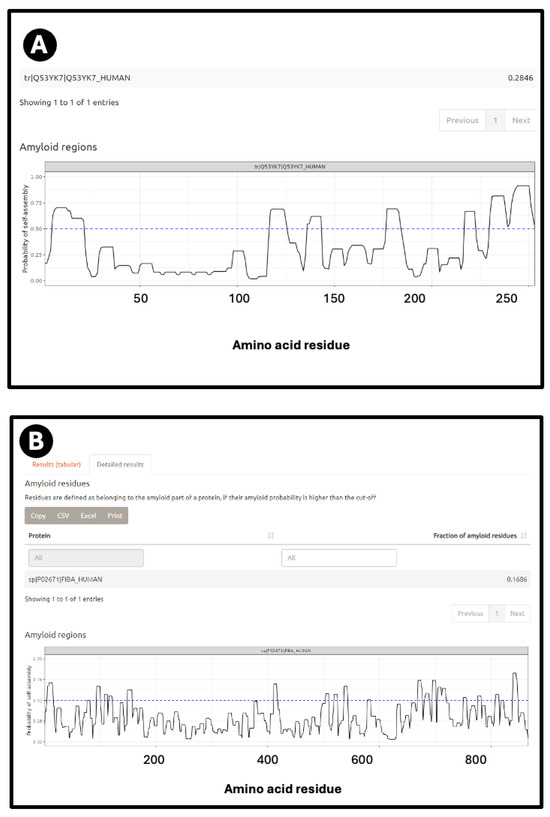

Figure 2.
Prediction of amyloidogenic regions of (A) human prion protein and (B) fibrinogen α chain on AmyloGram [74,99], and (C) fibrinogen α chain on AnuPP [104]. In the latter case, amyloidogenic regions are shown in green. Blue columns indicate residue numbers.
1.4. Prevalence of Amyloidogenicity
As can be established by testing various sequences on the above servers (we focused on AmyloGram [74,99], see later), as well as the classical amyloidoses, a very great many [123,124,125] (and possibly most [126]) proteins can exhibit amyloid formation under certain conditions [65,127]. Examples include insulin [128,129,130], lysozyme [131,132,133,134,135,136,137,138,139], proteins providing structure/texture in various processed foods and other gels [140,141,142], and even certain yeast [143,144,145] and bacterial [146] proteins, including some that can be ‘inherited’.
1.5. Amyloid Structures
Historically, establishing the structures of amyloid fibres formed even by single proteins or peptides was difficult because of their insoluble nature, but this is being changed by techniques such as solid-state NMR (e.g., [147,148,149,150,151]) and nowadays, in particular, cryoEM (e.g., [152,153,154,155,156,157]). These make it clear that amyloidogenic stretches of proteins can be responsible for cross-β fibril and fibre formation. An example is given in Figure 3, reproduced from an open-access paper [157].
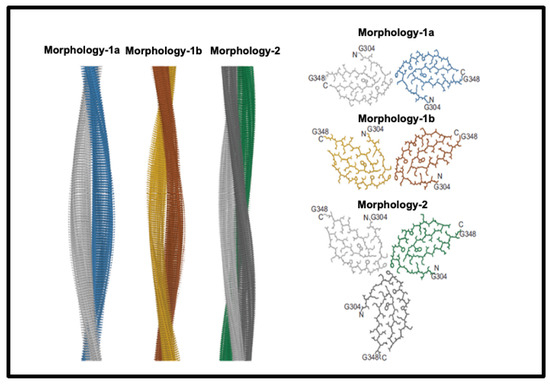
Figure 3.
Structures of amyloid fibres: Fibril formation by cross-ß elements. Reproduced from an open-access paper [157].
1.6. Amyloid Detection with Thioflavin T and Other Stains
A continuing and historically important discovery is the fact that the dye thioflavin T (ThT; https://pubchem.ncbi.nlm.nih.gov/compound/Thioflavin-T, accessed on 1 October 2024) binds to a whole series of amyloids, with a concomitant increase in its fluorescence [158]. This occurs because rotation of the normally rotatable single bond between the benzothiazole and dimethylaniline rings allows for fluorescence from an excited state to be dissipated and hence quenched. When the ThT is bound appropriately to a macromolecule, no such rotation is possible, fluorescence occurs, and thus ThT is a fluorogenic stain for amyloids (e.g., [159,160,161,162,163,164,165,166]). Note too that in the absence of amyloid target, ThT forms micelles with a critical micelle concentration of some 4 μM [159]. As phrased by Biancalana and Koide [161], “ThT binds to diverse fibrils, despite their distinct amino acid sequences, strongly suggesting that ThT recognizes a structural feature common among fibrils. Because amyloid fibrils share the cross-β architecture, it is generally accepted that the surfaces of cross-β structures form the ThT-binding sites”. Indeed, a number of such crystal structures have been solved, e.g., [167], or the relevant structures determined by other means (e.g., [168,169,170,171]), binding being seen as perpendicular to the cross-β elements and thus parallel to the fibrils themselves [52,147,169]. Figure 4 gives an example from an open-access publication [172].
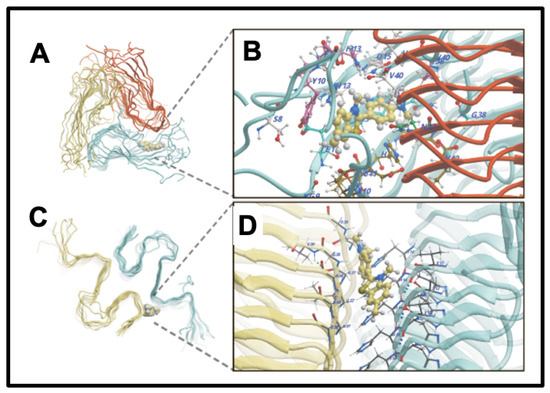
Figure 4.
Thioflavin T (ThT) binds to amyloid fibrils by recognizing a structural feature common among them. Amyloid fibrils share a cross-β architecture, where the β-sheets are oriented perpendicular to the fibril axis. The surfaces of these cross-β structures form the binding sites for ThT, which results in a characteristic increase in fluorescence upon binding. This property makes ThT a widely used fluorescent stain for detecting and studying amyloid fibrils. ThT binding sites illustrated are of Aβ40 (A,B) and Aβ42 (C,D) fibrils [172].
Note, however, that there can be quite subtle differences in the binding modes of ThT to specific amyloids, leading to changes in fluorescence intensity [172,173,174]. All of this said, and noting that ThT can in some cases bind to non-amyloid structures (e.g., [175]), ThT certainly remains the most popular amyloidogenic dye (e.g., [21,160,161,164,165,176,177]).
This difference in the precise mode of binding of a given dye can be observed in the studies of oligothiophene dyes (marketed as ‘Amytrackers’) by Nilsson and colleagues, where spectral as well as intensity changes can be observed between different amyloid structures (e.g., [178,179,180,181,182,183]).
Many other fluorogenic dyes that bind amyloid also exist, some with desirable spectral properties. Examples include NIAD-4 [184] that has an enhanced Stokes shift [166,185,186,187,188,189], and others that excite and emit towards the (near infra)red end of the spectrum (e.g., [187,190,191,192,193]).
1.7. Alternative Blood Clotting
In the same way that many proteins can adopt an amyloid form, as described above, it was discovered [194,195] that fibrinogen can polymerise into an anomalous amyloid-like form, that stained very strongly with thioflavin T and indeed other stains such as Amytracker stains (see Figure 5 of confocal micrographs where ThT and Amytrackers were exposed to plasma from healthy participants and those with type 2 diabetes [196]. The typical size of these clots, which were termed microclots (e.g., [197,198,199,200,201]), is in the range 2–200 μm (e.g., [194,198,201,202,203,204,205]).
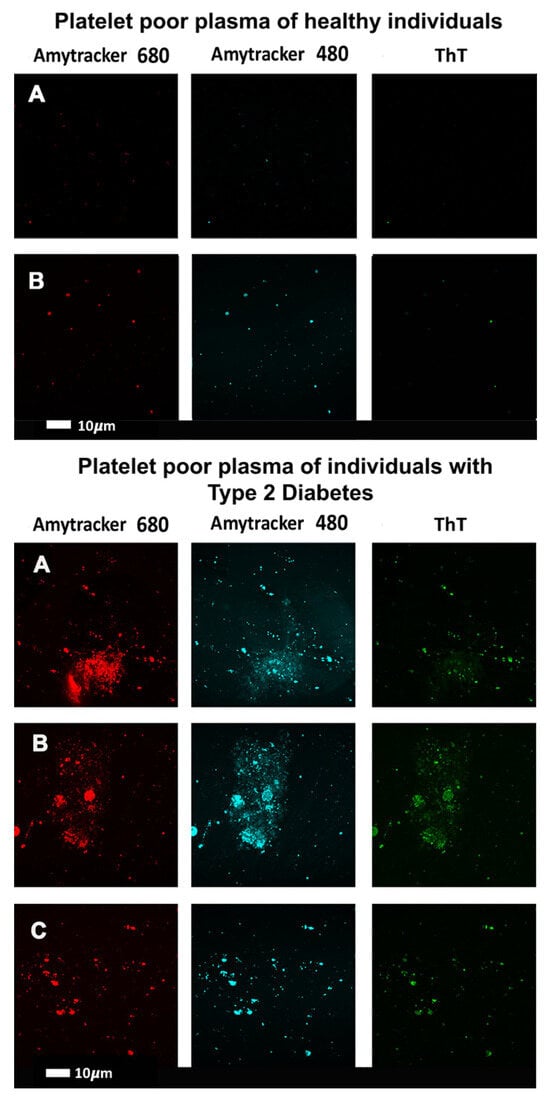
Figure 5.
Confocal micrographs where thioflavin T (T) and Amytrackers were used to stain plasma from healthy participants (upper two rows) and those with type 2 diabetes (lower three rows) [196]. Stains used are as indicated.
According to AmyPro [101], the most amyloidogenic region of fibrinogen α chain encompasses residues 148–160 (KRLEVDIDIKIRS), though Amylogram implies a second region nearer the C-terminus is even greater (Figure 2A).
It is interesting to note that fibrinogen is itself able to interact with other small amyloids, actually inhibiting their extension into larger fibrils [206,207,208], indicating that while it is well capable of binding amyloidogenic sequences, its own amyloidogenicity is only normally manifest during clotting itself, under the action of thrombin, though certain amyloidogenic alleles can lead to a fibrin amyloidosis (e.g., [209,210,211]). Other peptides can bind preferentially to (at least normal forms of) fibrin but not fibrinogen [212,213,214].
1.8. Size of Fibres in Classical Amyloidoses and in Normal and Fibrinaloid Clotting
To assist in understanding the nature of the fibres and how they may differ between ‘normal’ and fibrinaloid microclots, it is worth rehearsing the diameter of a typical monomer fibril of a cross-β element in a fibril. This depends, of course, on the length of the amyloidogenic run of amino acids that forms it, but is typically 1–2 nm or so. A protofibril consisting of 2–4 intertwined monomer fibrils may be 4–11 nm for molecules such as Aβ [215], 7 nm for tau [216], 11 nm for the prion protein in its amyloid PrPSc form [217], 6–15 nm for α-synuclein [218,219], and 7–13 nm for transthyretin [220].
By contrast (though cf. [99] for artificial super-amyloidogenic hexapeptides), the diameter of individual clot fibres is roughly 100 nm for amyloid clots (e.g., [194]) and is similar in many cases for normal ones [221], but can be as much as 400 nm or even more for normal, non-amyloid ones [222,223,224,225,226]. For normal non-amyloid clots, this would require several hundred elements, and for the amyloid version, very long runs of crossed-β features (that run in a criss-cross manner perpendicular to the long axis of the fibre), and many, many protofibrils intertwining around each other by lateral co-aggregation.
Normal clots are far better studied, and their diameter, for instance, depends on the fibrinogen concentration, consistent with general chemical kinetics. However, what the exact structures are, especially for the fibrinaloid ones, and what eventually stops them increasing in both length and diameter indefinitely, is not yet known. The ability of normal fibrinogen [227] and other proteins [228] to convert into a β-sheet-rich form is probably highly relevant. These observations also depend, of course, on a variety of factors such as the degree of hydration; initial fibrinogen [229] and thrombin [230] concentrations; levels of small molecules [194,231], of metal, and other ions [232,233,234,235]; and so on. However, the point of this paragraph is that these are clearly very much larger numbers for the fibre diameters in fibrinaloid microclots than are those seen in classical amyloidoses.
1.9. Inclusion Bodies, Compared with the Growth and Aggregation of Classical Amyloid Fibrils
Inclusion body formation is a well-known feature of recombinant protein expression (e.g., [236,237,238,239,240,241,242,243]) and is usually considered to occur due to the protein it is desired to fold being unable to keep up with the rates of its synthesis. Inclusion body formation largely involves a somewhat random or amorphous type of aggregation driven by interactions between hydrophobic residues of proteins that have failed to fold properly, even if they may sometimes contain or induce amyloid-like structures [127,244,245,246,247,248,249,250]. They mostly consist of the same polypeptide (so are sometimes considered a useful means of recombinant protein purification) but can certainly entrap other proteins via non-covalent interactions [248,251].
This contrasts with the type of ordered self-organisation seen in amyloid fibrils where multiple copies of the same protein also come together but into much more regular or structured shapes (e.g., [61,76,77,252,253,254,255,256,257]). The hallmark is one of various parallel or antiparallel cross-β sheet motifs [46,258,259,260,261] that run perpendicular to the fibril axis. They provide for a very characteristic X-ray diffraction peak reflecting a spacing of some 4.7 Å [262,263,264].
A given protein can even adopt various amyloid forms, known here as polymorphisms (e.g., [52,265,266,267,268,269,270,271,272,273,274,275,276,277,278]). The same is true of prions (e.g., [279,280,281,282,283]), arguably the most ‘extreme’ forms of amyloid(ogenic) proteins, and indeed the coinage of the term ‘prionoid’ (e.g., [284,285,286,287,288,289]) reflects this kind of overlap or continuum. In one sense [21], it is obvious that there must be parallels between the kinds of fibril formation that are seen in classical amyloidogenesis (commonly in the range 2–25 nm diameter [255,290,291,292,293]) and that seen in both normal and pathological blood clotting (although those fibrils are commonly at least 10x larger in diameter [198], see above), since in both cases fibrils are an observable result. Lengths of fibrils in classical amyloidoses can be 1 μm or so [294]. Consequently, in this section, we review what is known of amyloid fibril formation in the classical amyloidoses [255].
1.10. General Phases of Amyloid Fibril Formation
Extensive kinetic and imaging studies in vitro (e.g., [65,255,295,296,297,298,299,300,301,302,303]), often using ThT, have recognised several stages of amyloid fibre formation [263], starting with a lag [166,304] or nucleation phase creating oligomers (the most cytotoxic forms [305,306]); then an elongation phase in which protofibrils then fibrils are formed, the latter via protofibrils twisting round each other; and ending with a stationary phase in which fibril and plaque formation is complete (or, more accurately, any elongation and fragmentation or inhibition are occuring at the same rates [255,298,307,308]). Such sigmoidal curves are very similar to those of the batch growth of microbes [309]. It is the fibrils in which the cross-β structures are manifest, implying a structural transition whose detailed mechanism is far from clear.
1.11. Protein Entrapment in Microclots; Cross-Seeding
Our own work on the proteomics of fibrinaloid microclots has referred to ‘entrapment’ of non-fibrin proteins in the microclots. However, this cannot be a simple entrapment like a fish in a in a mesh net; the pores are far too big and in any case the centrifugation would have washed soluble proteins away from any weak binding or entrapment. Consequently, the ‘entrapment’ must actually be a forcing of the other proteins to become insoluble, likely by making cross-β sheets and thus joining the tightly-bound-but-noncovalent party and be incorporated into the growing amyloid fibrils. In the terminology of Bondarev and colleagues (Figure 6) and see below), these could be either or both of axial and lateral co-aggregation. Evidence for this includes the fact that there is no relationship between what is ‘entrapped’ in the fibrinaloid microclots and the normal plasma abundance of proteins (e.g., albumins and transferrin are pretty well the most abundant and mostly do not appear). Specifically, i there is an amyloidogenic transition such that different proteins line up to make the amyloids (axial co-aggregation in Figure 6), only those proteins capable of doing this will be entrapped, and others will be excluded. In some cases, anti-proteolytic substances such as antiplasmin [310] and α1 antitrypsin (SERPINA1) [205] may also be present in some abundance.
In a similar vein, many proteins besides α-synuclein are found in the Lewy bodies that can occur in dementia [311,312], while there is considerable experimental evidence for the co-incorporation of different amyloidogenic proteins into the same fibrils [87,313,314,315]. The same is true in transthyretin amyloidosis [316]. This is sometimes referred to as ‘cross-talk’ or ‘cross-feeding’, heterotypic interaction [317,318], and—perhaps most commonly—‘cross-seeding’ [79,172,319,320,321,322,323,324,325,326,327,328,329,330,331,332,333,334,335,336,337,338,339,340,341,342,343,344,345,346,347,348,349,350,351,352,353,354,355,356,357,358]. Note that even simple polyQ features (occurring as C-terminal ‘tails’ in various diseases) can do this [331,359,360,361,362,363], possibly also by incorporating transition metals [364]. Equally importantly, not all amyloids (‘donors’) can cross-seed other ones [142] (‘acceptors’) and hence be entrapped in the fibrils of the acceptors; there is significant selectivity, whose sequence/structural basis remains unknown.

Figure 6.
Different classes or types of protein co-aggregation: (A) Titration; (B) Sequestration; (C) Axial and (D) Lateral. Adapted from [365]. Different colours indicate different protein types.
The occurrence of multiple amyloid proteins within the same fibril is reviewed by Bondarev and colleagues [98,365], who refer to it as axial co-aggregation (Figure 6). The server AmyloComp [98] (Table 1) also predicts the likelihood of proteins forming axial co-aggregates; that for SERPINA1 and the fibrinogen alpha chain is especially high (unpublished). As phrased by them [98], “The core of these amyloid fibrils is a columnar structure [79,172,319,320,321,322,323,324,325,326,327,328,329,330,331,332,333,334,335,336,337,338,339,340,341,342,343,344,345,346,347,348,349,350,351,352,353,354,355,356,357,358] produced by axial stacking of β-strand-loop-β-strand motifs called ‘β-arches’ [366,367,368,369,370]”. Well-established examples include RIP1/RIP3, which can induce necroptosis [371], and the HET-s protein, which also contains the Rip homotopic interaction motif (RHIM) [372]. Clearly, any protein capable of forming these β-arches can then do so, so as to make a hetero-fibril, which is what we suggest is the main means of ‘entrapment’ of other proteins in fibrinaloid microclots (provided the amyloidogenic regions are of sufficient length [373]). The idea we develop here, with considerable evidence, is that while normal clotting plausibly binds proteins by titration and sequestration in the terminology of [98,365] (Figure 6), the amyloid-containing fibrinaloid microclots involve axial and lateral co-aggregation. That fibrinogen can interact with a variety of known amyloidogenic proteins is beyond dispute [374,375]; causing them thereby to create new epitopes can even account for autoantibody generation [22]. A preprint has been lodged in bioRxiv [376].
2. Results
2.1. Absence of Relationship between Microclot Proteome and Plasma Concentration
At least two lines of evidence indicate the lack of relationship between the amount of a protein in plasma and its appearance in fibrinaloid microclots. First, the only overlap between the proteomic data of Kruger and colleagues [377] (who did not report on fibrinogen) and those of Schofield et al. [205] was the protein apolipoprotein A2 (marked in blue in Figure 7). The data were taken from two quite different diseases (acute sepsis [205] vs. long COVID [377]), with ‘normal’ proteome levels spanning several orders of magnitude, and so while the content of these proteins in the average proteomes will not have differed by more than a factor of two at most, their appearance in the microclots differed massively. Secondly, we extracted the ‘top 20’ data from the pie chart representing the average of three individuals in Figure 3 of [205] and related those (where available) to the average plasma protein concentration [378], indicating that there was no such relation (r2 = 0.1 for the data in Figure 7). We also assessed some of the most abundant plasma proteins as tabulated in [379] for their presence or otherwise in the microclots in either study [205,377] (Figure 8); it is obvious that many of the most abundant proteins are not notably entrapped in the microclots, so those that are clearly selected, presumably by integration into the amyloid mixtures. (The data underpinning all these analyses are given in a spreadsheet in Supplementary Materials.)
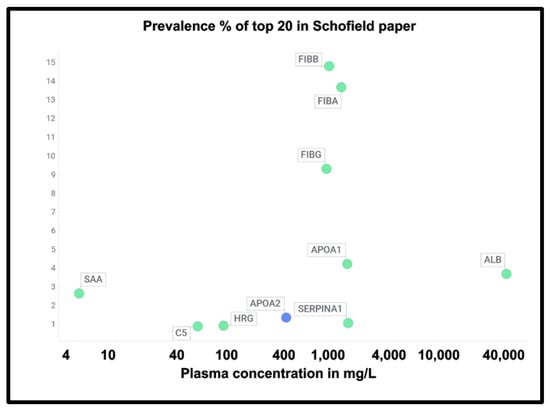
Figure 7.
Prevalence of proteins (y-axis) in fibrinaloid microclots in the Schofield ‘top 20’ (green) and the one example also seen in the Kruger study (blue) versus average plasma concentrations that are taken from [378] except for TGFB1 [380] and periostin [381]. Abbreviations as in the list of abbreviations. The line of ‘best fit’ is not shown as it has a correlation coefficient r2 of only 0.1.
A chief source of protein abundances in plasma is [378]. In addition, we used other sources for high-abundance and other detected proteins including C9 [382,383], complement factor H [384], thyroxine-binding globulin [385], retinol-binding protein [386], TGFβ1 [380], periostin [381], CXCL7 (PFA4) [387], (pre)kallikrein [388], galectin-3-binding protein (LG3BP) [389], thrombospondin-1 [390], and ITIH1/2 [391]. LG3BP is of interest, as it is substantially lowered in the plasma of those with mild cognitive impairment or Alzheimer’s [392], arguably because it has been removed in amyloid microclots. Similarly, thrombospondin-1 also interacts with Aβ [393]. Reference [391] is very valuable in its own right, since while its coverage lacks some of the low-abundance proteins of interest here, it does list quantitative values for 197 plasma proteins; where both are available, the concentrations are well correlated (Figure 9) (slope = 1.06, r2 = 0.87), taking as the values for the Heck study [391] the averages of six data points for the controls of two patients at the first three time points. Consequently, we use the Heck dataset in most of what follows.

Figure 8.
Good correlation between the plasma proteome concentration data in [391] by Heck and colleagues compared with other measurements of the proteome cited in the text and in Supplementary Information. The slope of the line is 0.95 and the correlation coefficient 0.83. Colours encode the datasets in which fibrinaloid proteins were or were not observed, as in Figure 7 and Figure 9, viz. blue both, green Schofield, red Kruger, and yellow neither. Abbreviations as in the list of abbreviations.


Figure 9.
Relationship between the standard plasma proteome concentrations (taken from [391]) and their detection in the Kruger (K), Schofield (S), both (B) studies, or neither (N). Size of symbol encodes protein length in residues. Abbreviations as in list of abbreviations. (A) protein concentrations from other studies delineated in the text and the supplementary spreadsheet. (B) Proteins from the study of Heck and colleagues [391].
Using both sets of plasma proteome data (since some important markers do not appear in both), we again see large number of proteins that are high in abundance in the plasma proteome that nevertheless are not ‘entrapped’ in the fibrinaloid microclots, and similarly others in low abundance in the proteome nonetheless appear in the fibrinaloid microclots. The conclusion is very clear: there is a significant selectivity with regard to proteins that are entrapped within fibrinaloid microclots. We note too that atomic force microscopy [394] has provided evidence for both axial [395] and lateral [396] co-aggregation, and that cross-seeding can also explain the comorbidities seen with protein misfolding diseases [397].
What is not always clear is whether these heterotypic interactions tend to promote or to inhibit amyloidogenesis (or even both at different stages, as can occur with the curli protein CsgA and fibrinogen [398]; the same is true for fibrinogen and phenol-soluble modulins [399]).
SERPINA1 (α1 antitrypsin), as found by Schofield et al. [205] as a major constituent of microclots, contains four β-sheets and is also able to interact with the amyloidogenic transthyretin [400,401,402].
Some amyloidogenic proteins, such as apolipoprotein B-100, which contains a massive crossed-β (‘β-belt’) structure [403], were not detected; however, this is plausible because they were fully embedded within lipoproteins and thus not in plasma. (They are, however, capable of becoming embedded in neurofibrillary tangles [404].)
2.2. Amyloidogenicity of Proteins ‘Entrapped’ in Microclots
If axial or lateral co-aggregation is responsible for the ‘entrapment’ of proteins in fibrinaloid microclots, one would suppose that all the proteins involved would themselves be amyloidogenic. This can be tested using amyloidogenicity prediction programs of the type given in Table X. We chose AmyloGram [74], available at http://biongram.biotech.uni.wroc.pl/AmyloGram/ (accessed on 1 October 2024). For the proteins entrapped within microclots, we took the proteomics data from Table 2 from the long COVID study of Kruger and colleagues (who did not report on fibrinogen) and the Table 3 of the study of microclots in intensive care patients of Toh and colleagues [205]. The conclusion is very clear: every single one of the proteins detected in the microclots is highly amyloidogenic, and the microclots evidently involve cross-seeding. However, there was little correlation between amyloidogenicity and protein length (Figure 10) (r2 = 0.275). However, all but three of the Kruger proteins and all but four of the Schofield ‘top 20’ had an amyloidogenicity score exceeding 0.8 (Figure 10).
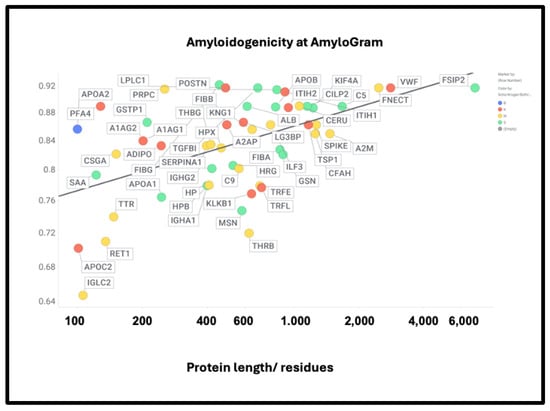
Figure 10.
High amyloidogenicity of proteins in the Kruger (red) and Schofield ‘top 20’ (green) studies and in both (blue) plus amyloidogenicity of proteins seen in neither (yellow), and its broad independence from protein length. The line of best fit indicated has an r2 of just 0.23.
However, there was no correlation with the overall amyloidogenicity as calculated using AmyloGram and either the plasma proteome abundance or whether the proteins were in the fibrinaloid microclots (Figure 11) (r2 = 0.02):
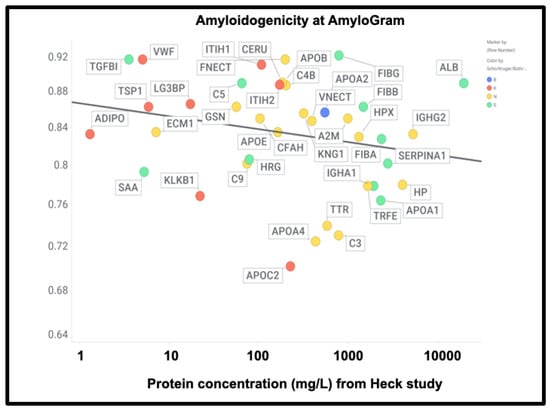
Figure 11.
High amyloidogenicity of proteins in the Kruger (red) and Schofield (green) studies and in both (blue) plus amyloidogenicity of proteins seen in neither (yellow), and its independence from plasma protein concentrations slope =0.01, (r2 = 0.05) as recorded in the Heck study [391] (means of first three time points averaged over two controls).
The presence of von Willebrand factor and adiponectin in the fibrinaloid microclots is very interesting, despite their comparatively low plasma concentrations (Figure 11). The former is among the most amyloidogenic proteins in the list (Figure 11) and is notably entrapped and removed by microclots in SARS-CoV-2 infection [405], while the latter is correlated with amyloid Aβ deposition [406] and may be protective [407]. LPLC1 (Figure 10, so low in concentration it does not appear in Figure 11; the human protein atlas https://www.proteinatlas.org/ENSG00000125999-BPIFB1/blood+protein (accessed on 1 October 2024 ) estimates its plasma concentration by mass spectrometry to be 2.7 μg/L) is also of interest. LPLC1 stands for “Long palate, lung and nasal epithelium carcinoma-associated protein 1” or also “BPI fold-containing family B member 1” (BNIB1), and to be clear, it is Uniprot Q8TDL5; again, it has a very high amyloidogenicity (~0.91). Finally, thrombospondin-1 is also very over-represented, and it too was neuroprotective against Aβ [393,408,409]. A clear pattern emerges.
The precise nature and extent of the amyloidogenicity necessary to induce or be entrapped in fibrinaloid microclots is as yet unclear, but inspection of the detailed data from the analyses at AmyloGram (unpublished) showed that each of the proteins involved possessed a segment of amyloidogenicity (referred to on its website and in the subset of figures displayed here as a ‘probability of self-assembly’) that exceed 0.75 in the data that could be acquired at the AmyloGram website. Figure 12, Figure 13, Figure 14 and Figure 15 show four examples of results from such an analysis at the Amylogram website: the first (Figure 12) is α-2-antiplasmin (prominent in the findings of [310]), where there is an initial run plus two further prominent peaks. α-2-antiplasmin is of course well known as an inhibitor of fibrinolysis [410].
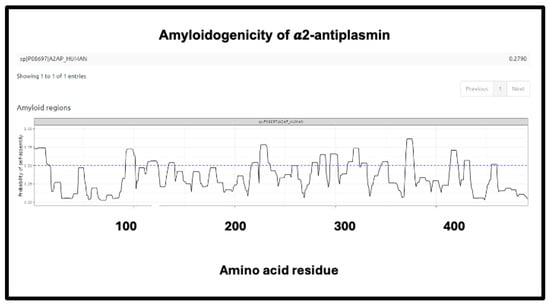
Figure 12.
Amyloidogenicity of α-2-antiplasmin. The FASTA sequence was obtained from the Uniprot site given in Supplementary Table S1 and run on the Amylogram website http://biongram.biotech.uni.wroc.pl/AmyloGram/ (accessed on 1 October 2024) with the results as indicated.
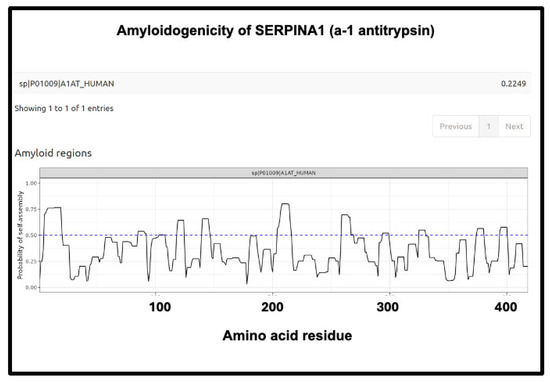
Figure 13.
Amyloidogenicity of SERPINA1 (α1-antitrypsin). The FASTA sequence was obtained from the Uniprot site given in Supplementary Table S1 and run on the Amylogram website http://biongram.biotech.uni.wroc.pl/AmyloGram/ (accessed on 1 October 2024) with the results as indicated.
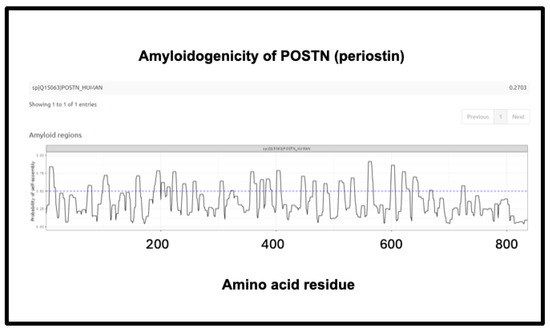
Figure 14.
Amyloidogenicity of POSTN (periostin). The FASTA sequence was obtained from the Uniprot site given in Supplementary Table S1 and run on the Amylogram website http://biongram.biotech.uni.wroc.pl/AmyloGram/ (accessed on 1 October 2024) with the results as indicated.
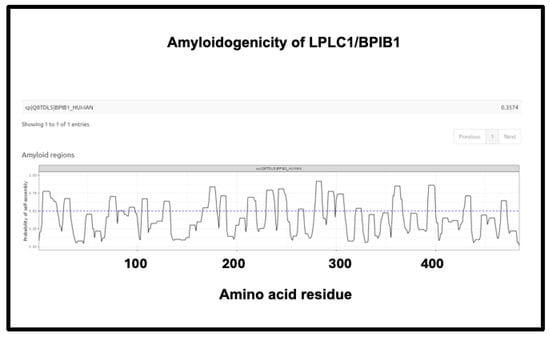
Figure 15.
Amyloidogenicity of LPLC1/BPIB1. The FASTA sequence was obtained from the Uniprot site given in Supplementary Table S1 and run on the Amylogram website http://biongram.biotech.uni.wroc.pl/AmyloGram/ (accessed on 1 October 2024) with the results as indicated.
The next (Figure 13) is SERPINA1 (α1-antitrypsin), where there is a long run at the beginning just exceeding 0.75. SERPINA1 is the most abundant anti-protease in plasma (see also Figure 11) and has several parallel–antiparallel β-sheets in its ground-state conformation [411,412,413,414,415], which, interestingly, is metastable [416,417,418]. It can also interact with amyloidogenic transthyretin [401] and is associated with the severity and progression of SARS-CoV-2 [419], consistent with its role in assisting fibrinaloid microclot formation. It is thus entirely plausible that it could participate in amyloid formation, and fragments of it certainly do [420,421].
The third is the inflammatory marker periostin, representing 1.17% of the ‘top 20’ proteins in Schofield et al. [205], and containing no fewer than eight regions of amyloidogenicity with a score exceeding 0.75. It features in these fibrinaloid microclots despite being one of the least abundant plasma proteins of those under consideration (372 ng/mL according to [381], 98 ng/mL in [422] and just 10 ng/mL according to [423,424]) (209 ng/mL is stated for serum [425]). Notably, however, as well as being among the most amyloidogenic of those surveyed (Figure 10), it is highly predictive of Aβ deposition [392,426] and is also involved in lung fibrosis [427,428].
Finally, the fourth is LPLC1 (also known as BPIB1 or BPIFB1). This protein has a very high amyloidogenic propensity of 0.9176, Figure 10) and no fewer than eight regions in which the amyloidogenicity score exceeds 0.75 (Figure 15), despite a minuscule concentration in normal plasma. Interestingly, it is involved in innate immunity [429], especially in mucosa [430], and bears similarities to lipopolysaccharide-binding protein (LPS being a molecule that can trigger fibrinaloid formation [194,195,431]). BPIB1 can also inhibit Epstein–Barr virus proliferation [432,433,434] (something of major potential relevance in long COVID [435,436,437]). Overall, the fact that it is so concentrated in fibrinaloid microclots in the case of long COVID is thus very notable.
2.3. Comparison with the Normal Clot Proteome
Undas and colleagues [438] provided (their Table 2) a quantitative list of protein-bound proteins in normal clots (that were presumably non-amyloid, though that was not in fact tested). Figure 16 compares the prevalence of proteins in their clots with that of plasma proteins, showing a reasonable correlation (slope = 0.67, r2 = 0.29) between the two. This contrasts with that for the fibrinaloid microclot proteome (Figure 7), where the value for r2 was just 0.1.
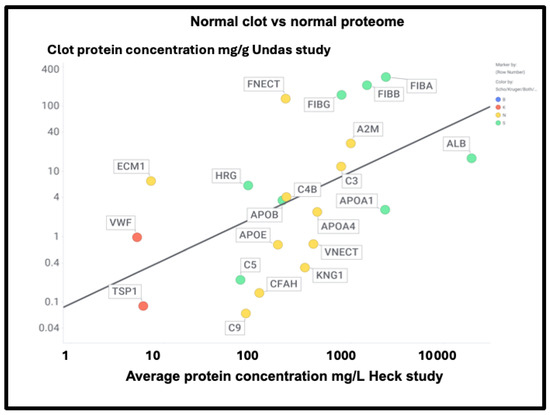
Figure 16.
Comparison of the proteome content of normal (non-amyloid) clots vs the standard plasma proteome in controls in the Heck study (average of two controls over first three time points). Colour encoding is as in Figure 11.
Figure 17 shows those that were also assessed in the Kruger and Schofield studies, along with their amyloidogenicity. There is no correlation whatsoever (r2 ~0.02), again showing how very different the composition of fibrinaloid microclots is from that of normal clots.
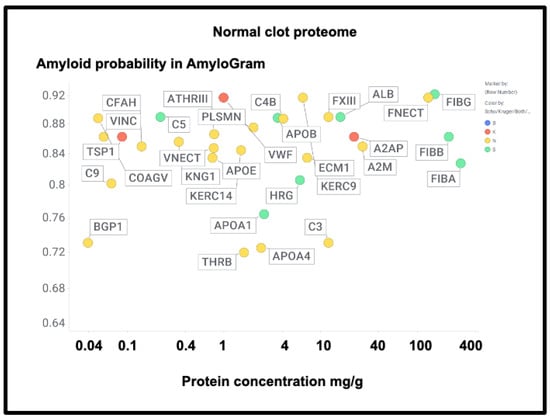
Figure 17.
The proteome in normal clots. Data are taken from [438] and also coded as to whether the proteins were observed in the fibrinaloid microclots observed by Schofield (green), Kruger (red), both (blue), or neither (yellow).
A particularly noteworthy observation here (Figure 17) is the relatively high amounts of fibronectin seen in normal clots (130 mg/g protein [438], fibronectin typically being present in plasma at 300–400 mg/L [439,440,441]), as it was not seen in the fibrinaloid microclots. While fibrin is highly amyloidogenic (Figure 10 and Figure 17) and in vivo can produce insoluble fibrillar components that may be incorporated in the extracellular matrix [442,443,444], fibronectin is somewhat unusual for two reasons: first, it is large (2477 residues). Secondly, it is relatively thermostable [445], especially in some of its domains [446]. Together, these features can plausibly account for the difficulty of unfolding and incorporating it into an amyloid clot compared to a normal one (see also the next paragraph for amyloidogenicity vs thermostability). Similar comments relate to α-2-macroglobulin (27 mg/g, 1474 residues, Figure 17) and to Factor XIII (12 mg/g, 732 residues), which is in fact inhibited by α-2-macroglobulin. Factor XIII is a transglutaminase (linking glutamate and lysine residues) that is responsible for stiffening normal clots by crosslinking them [447,448,449]—mainly via the γ but also partly the α chains [450]—yet does not appear in the fibrinaloid microclot proteomes. This would be entirely consistent with their completely different structures relative to that of normal clots. Lastly, complement factor 3 is fairly well represented in both plasma (Figure 16; 778 mg/L in the Heck study) and in normal clots (Figure 17; 12 mg/g) yet is not found in fibrinaloid microclots; consistent with the thrust of our arguments, its total amyloidogenic propensity at just 0.73 is among the lowest of those evinced in this study.
2.4. Amyloidogenicity vs. Thermostability
As noted, amyloidogenesis of a non-amyloid form of a protein necessarily requires a significant unfolding [76,77,78,79,80], and in general terms, the resistance to a protein unfolding is reflected in its thermostability. The point about the experimental lack of amyloidogenicity in a thermostable protein thus leads to the question of whether experimental amyloidogenic proteins are in fact normally relatively non-thermostable in their non-amyloid forms. This turns out to be strongly supported by substantial evidence [252,261,451,452,453,454,455,456,457].
3. Discussion
An important question about amyloid(ogenic) protein fibres in general, and fibrinaloid microclots (our main focus) in particular, is the nature and location of the proteins that they contain. A variety of studies have provided data on both normal clots and fibrinaloid microclots, as well as the normal plasma proteome. With occasional exceptions (such as C-reactive protein—not involved here) their concentrations are fairly constant, and since (i) they cover four orders of magnitude in the proteins considered here, and (ii) they were well correlated in two studies (Figure 8), we consider their standard concentrations to be a good guide as to the likelihood of non-fibrinogen proteins being entrapped in a clot if entrapment simply reflects their plasma concentrations. In normal clots, that expectation is broadly borne out (Figure 15). However, this is far from being the case with the fibrinaloid microclots that form in certain diseases, stain strongly with thioflavin T, and are far more resistant than are normal clots to fibrinolysis (Figure 18). Seemingly, as with prion proteins, the presence of a small amount of the thermodynamically stabler amyloid form is enough to trigger conversion of a very large number of monomers in the amyloid polymer form in almost an ‘all-or-nothing’ manner (Figure 19).
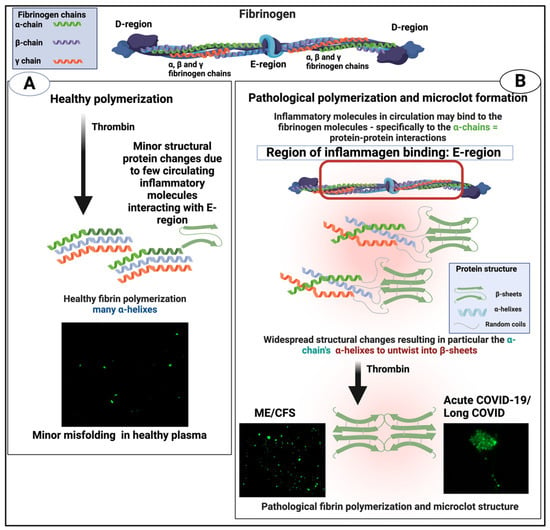
Figure 18.
Healthy versus pathological (amyloid) clotting. Taken from [22]. Created with Biorender.com. Microclots fluoresce green in the presence of thioflavin T.
First, their proteome varies strongly with the disease (Figure 7), in a way that cannot reflect changes in the bulk plasma proteome. Secondly, their proteome constitution is far from being related to the concentrations of bulk plasma proteins, with some being excluded and others being highly concentrated (Figure 9, Figure 10, Figure 11, and Figure 16; twelve are summarised for convenience in Table 2). Clearly, there must be special mechanisms at work, the most obvious, given their strikingly high amyloidogenicity scores, being a cross-seeding where the various proteins are actually incorporated into the cross-β elements of the fibrils themselves. This said, there is a consonance in Table 2 between the proteins highlighted as being in fibrinaloid microclots and biological explanations based on their known roles. Most of those that are higher are in the Kruger [377] long COVID study (Table 2). Long COVID is of course a chronic disease, and very different from the acute conditions characterising individual in an ICU such as those in the Toh study [205]. Long COVID exhibits similarities with myalgic encephalopathy/chronic fatigue syndrome (ME/CFS), however [435,458,459,460,461,462,463,464], so it is of interest that thrombospondin and platelet factor 4 are both raised in the plasma of individuals with ME/CFS [463].

Figure 19.
Individual fibrinogen molecules upon polymerisation either polymerise into a normal clot form, which is relatively easily removed by fibrinolysis, or into an anomalous amyloid form or forms, which are not (Taken from [465]). Created with Biorender.com.
Clot Fibrinaloids vs. Classical Amyloid Fibrils
Much is known about classical amyloids, and plausibly this knowledge will contribute to our emerging understanding of fibrinaloids, but there is one crucial difference, and that is the size of the fibres. A typical oligomer made up of a standard cross-β amyloid chain crisscrossing in a direction perpendicular to the fibril direction has a diameter in the range 2 to 5 nm [466], and these can aggregate to make protofibrils in the range 4–11 nm diameter [467]. The amyloid fibrils themselves typically involve a few intertwined protofibrils per unit length [276], and thus have a diameter of the order of 6 to 12 nm [468,469,470], although in principle they could become larger [276]. This means that generally most fibrils consist of only a few intertwined protofibrils per unit length. By contrast, the fibrils observed in fibrinaloid microclots in plasma samples have a diameter that is often in the range 50 to 100 nm. This means that they must contain many more lateral fibres per unit length, likely of the order of 100–300. This ability to increase numbers laterally obviously bears strongly on the potential ability of microclots to aggregate further to become macroclots, and maybe ultimately to lead to the kinds of occlusions involved in stroke and myocardial infarctions.
It has been suggested [471,472] that at least some types of classical amyloidoses have a ‘proteome signature’ that would include serum amyloid P-component (SAP) and heparan sulphate proteoglycans (HSPG), as well as apolipoprotein A4, apolipoprotein E, and vitronectin (see also [62,473,474,475,476]), consistent with some kind of cross-seeding; interestingly, none of these was reported in either the Schofield or Kruger studies, though the latter three proteins were easily observed in normal clots (Figure 17). This adds to the evidence that while amyloid in character, the proteomic contents of fibrinaloid microclots are different in many other ways (besides diameter) from the amyloid fibrils in classical amyloidoses.
Another particular feature of the fibrinaloid microclots is their resistance to fibrinolysis (see [21,22,198,310,477,478]), most easily seen in our proteome studies, where a double trypsin digestion was required for successful peptide-based mass spectrometric proteome analysis [310,377]. The presence of molecules such as α-2-antiplasmin [310] and SERPINA1 will certainly have contributed, but it is of course well known that amyloid forms of protein are far more resistant to proteolysis than their native unfolded or globular versions, especially among prion proteins where PrPSc is even resistant to proteolysis by proteinase K (e.g., [479,480,481,482,483]).
Some molecules such as bacterial cell wall compounds can clearly serve as triggers for amyloidogenesis in both fibrinaloid microclots [194,195,196,431,484,485,486,487,488] and other cases [489,490,491]. What we propose here is that the massive changes in fibrinogen structure necessary for its conversion to an amyloid form, as also observed by others [204,205,492], must then involve cross-seeding, as this provides a simple mechanism that at once accounts for (i) the proteomics, (ii) the resistance to proteolysis, and (iii) the amyloid nature of the mixed clots. At the same time, we recognise that while our analysis is both clear and robust, future studies would benefit from comparing the proteomes of fibrinaloid and normal clots at the same time on the same instrument.
Overall, however, while the present findings make it clear that cross-seeded proteins must be involved in fibrinaloid formation, we are far from knowing their specific locations and whether and how they co-aggregate axially and/or laterally (as per Figure 6). Such studies would require the resolution of an atomic force microscope (e.g., [226,394,493,494]), which, on the basis of the present findings, it now seems worthwhile to pursue.
4. Materials and Methods
Data were downloaded from the sources indicated and were not otherwise transformed save to average the first three timepoints and two individuals representing the controls in [391]. The values from Figure 3 of Schofield et al. [205] were determined digitally from the averaged pie chart therein. An Excel file provided as Supplementary Information summarises the data, that are commonly displayed in this paper using the Spotfire program (https://www.spotfire.com/ (accessed on 1 October 2024), version 12.0). Other methods run on websites for analysing the proteomic datasets for amyloidogenicity were as follows: Amylogram was run on its website http://biongram.biotech.uni.wroc.pl/AmyloGram/ (accessed on 1 October 2024) using the FASTA sequences of the relevant proteins obtained from Uniprot (addresses as set down in the Supplementary data). Fibrinogen α was also run on the AnuPP website https://web.iitm.ac.in/bioinfo2/ANuPP/homeseq1/ (accessed on 1 October 2024).
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms251910809/s1.
Author Contributions
D.B.K. and E.P. contributed to the conceptualisation, analyses, funding acquisition, drafting, and final editing. All authors have read and agreed to the published version of the manuscript.
Funding
DBK thanks the Balvi Foundation (grant 18) and the Novo Nordisk Foundation for funding (grant NNF20CC0035580). EP: Funding was provided by NRF of South Africa (grant number 142142), SA MRC (self-initiated research (SIR) grant), and the Balvi Foundation. The content and findings reported and illustrated are the sole deduction, view, and responsibility of the researchers and do not reflect the official position and sentiments of the funders. The APC was funded by the journal.
Data Availability Statement
Data are given in Supplementary Table S1.
Conflicts of Interest
E.P. is a named inventor on a patent application covering the use of fluorescence methods for microclot detection in long COVID. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Meade, T.W.; Chakrabarti, R.; Haines, A.P.; North, W.R.; Stirling, Y. Characteristics affecting fibrinolytic activity and plasma fibrinogen concentrations. Br. Med. J. 1979, 1, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.M.; Howard, S.C.; Power, D.A.; Gutnikov, S.A.; Algra, A.; van Gijn, J.; Clark, T.G.; Murphy, M.F.; Warlow, C.P. Fibrinogen concentration and risk of ischemic stroke and acute coronary events in 5113 patients with transient ischemic attack and minor ischemic stroke. Stroke 2004, 35, 2300–2305. [Google Scholar] [CrossRef] [PubMed]
- Kattula, S.; Byrnes, J.R.; Wolberg, A.S. Fibrinogen and Fibrin in Hemostasis and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e13–e21. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.Y.; Sun, H.L.; Sun, P.Y.; Jian, J.M.; Li, W.W.; Shen, Y.Y.; Zeng, F.; Wang, Y.J.; Bu, X.L. The Correlations between Plasma Fibrinogen with Amyloid-Beta and Tau Levels in Patients with Alzheimer’s Disease. Front. Neurosci. 2020, 14, 625844. [Google Scholar] [CrossRef]
- Weisel, J.W. Fibrinogen and fibrin. Adv. Protein Chem. 2005, 70, 247–299. [Google Scholar] [CrossRef]
- Weisel, J.W.; Litvinov, R.I. Fibrin Formation, Structure and Properties. Subcell. Biochem. 2017, 82, 405–456. [Google Scholar] [CrossRef]
- Göbel, K.; Eichler, S.; Wiendl, H.; Chavakis, T.; Kleinschnitz, C.; Meuth, S.G. The Coagulation Factors Fibrinogen, Thrombin, and Factor XII in Inflammatory Disorders—A Systematic Review. Front. Immunol. 2018, 9, 1731. [Google Scholar] [CrossRef]
- Litvinov, R.I.; Pieters, M.; de Lange-Loots, Z.; Weisel, J.W. Fibrinogen and Fibrin. Subcell. Biochem. 2021, 96, 471–501. [Google Scholar] [CrossRef]
- Carr, M.E., Jr.; Hardin, C.L. Fibrin has larger pores when formed in the presence of erythrocytes. Am. J. Physiol. 1987, 253, H1069–H1073. [Google Scholar] [CrossRef]
- Collet, J.P.; Lesty, C.; Montalescot, G.; Weisel, J.W. Dynamic changes of fibrin architecture during fibrin formation and intrinsic fibrinolysis of fibrin-rich clots. J. Biol. Chem. 2003, 278, 21331–21335. [Google Scholar] [CrossRef]
- Weigandt, K.M.; White, N.; Chung, D.; Ellingson, E.; Wang, Y.; Fu, X.; Pozzo, D.C. Fibrin clot structure and mechanics associated with specific oxidation of methionine residues in fibrinogen. Biophys. J. 2012, 103, 2399–2407. [Google Scholar] [CrossRef] [PubMed]
- Eyisoylu, H.; Hazekamp, E.D.; Cruts, J.; Koenderink, G.H.; de Maat, M.P.M. Flow affects the structural and mechanical properties of the fibrin network in plasma clots. J. Mater. Sci. Mater. Med. 2024, 35, 8. [Google Scholar] [CrossRef] [PubMed]
- Zakharov, A.; Awan, M.; Cheng, T.; Gopinath, A.; Lee, S.J.; Ramasubramanian, A.K.; Dasbiswas, K. Clots reveal anomalous elastic behavior of fiber networks. Sci. Adv. 2024, 10, eadh1265. [Google Scholar] [CrossRef] [PubMed]
- Wufsus, A.R.; Macera, N.E.; Neeves, K.B. The hydraulic permeability of blood clots as a function of fibrin and platelet density. Biophys. J. 2013, 104, 1812–1823. [Google Scholar] [CrossRef] [PubMed]
- Risman, R.A.; Kirby, N.C.; Bannish, B.E.; Hudson, N.E.; Tutwiler, V. Fibrinolysis: An illustrated review. Res. Pract. Thromb. Haemost. 2023, 7, 100081. [Google Scholar] [CrossRef]
- Anfinsen, C.B.; Haber, E.; Sela, M.; White, F.H. The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain. Proc. Natl. Acad. Sci. USA 1961, 47, 1309–1314. [Google Scholar] [CrossRef]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef]
- Levinthal, C. How to Fold Graciously. In Proceedings of the Mossbauer Spectroscopy in Biological Systems, Monticello, IL, USA, 17–18 March 1969; pp. 22–24. [Google Scholar]
- Martínez, L. Introducing the Levinthal’s Protein Folding Paradox and Its Solution. J. Chem. Educ. 2014, 91, 1918–1923. [Google Scholar] [CrossRef]
- Aguzzi, A.; Lakkaraju, A.K.K. Cell Biology of Prions and Prionoids: A Status Report. Trends Cell Biol. 2016, 26, 40–51. [Google Scholar] [CrossRef]
- Kell, D.B.; Pretorius, E. Proteins behaving badly. Substoichiometric molecular control and amplification of the initiation and nature of amyloid fibril formation: Lessons from and for blood clotting. Prog. Biophys. Mol. Biol. 2017, 123, 16–41. [Google Scholar] [CrossRef]
- Kell, D.B.; Pretorius, E. Are fibrinaloid microclots a cause of autoimmunity in Long Covid and other post-infection diseases? Biochem. J. 2023, 480, 1217–1240. [Google Scholar] [CrossRef] [PubMed]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Porter, L.L.; Looger, L.L. Extant fold-switching proteins are widespread. Proc. Natl. Acad. Sci. USA 2018, 115, 5968–5973. [Google Scholar] [CrossRef] [PubMed]
- LiWang, A.; Porter, L.L.; Wang, L.P. Fold-switching proteins. Biopolymers 2021, 112, e23478. [Google Scholar] [CrossRef]
- Porter, L.L. Fluid protein fold space and its implications. Bioessays 2023, 45, e2300057. [Google Scholar] [CrossRef]
- Kim, A.K.; Porter, L.L. Functional and Regulatory Roles of Fold-Switching Proteins. Structure 2021, 29, 6–14. [Google Scholar] [CrossRef]
- Chen, N.; Das, M.; LiWang, A.; Wang, L.P. Sequence-Based Prediction of Metamorphic Behavior in Proteins. Biophys. J. 2020, 119, 1380–1390. [Google Scholar] [CrossRef]
- Das, M.; Chen, N.; LiWang, A.; Wang, L.P. Identification and characterization of metamorphic proteins: Current and future perspectives. Biopolymers 2021, 112, e23473. [Google Scholar] [CrossRef]
- Artsimovitch, I.; Ramirez-Sarmiento, C.A. Metamorphic proteins under a computational microscope: Lessons from a fold-switching RfaH protein. Comput. Struct. Biotechnol. J. 2022, 20, 5824–5837. [Google Scholar] [CrossRef]
- Retamal-Farfán, I.; González-Higueras, J.; Galaz-Davison, P.; Rivera, M.; Ramírez-Sarmiento, C.A. Exploring the structural acrobatics of fold-switching proteins using simplified structure-based models. Biophys. Rev. 2023, 15, 787–799. [Google Scholar] [CrossRef]
- Porter, L.L.; Artsimovitch, I.; Ramirez-Sarmiento, C.A. Metamorphic proteins and how to find them. Curr. Opin. Struct. Biol. 2024, 86, 102807. [Google Scholar] [CrossRef] [PubMed]
- Strodel, B. Energy Landscapes of Protein Aggregation and Conformation Switching in Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167182. [Google Scholar] [CrossRef] [PubMed]
- Schafer, J.W.; Chakravarty, D.; Chen, E.A.; Porter, L.L. Sequence clustering confounds AlphaFold2. bioRxiv 2024. [Google Scholar] [CrossRef]
- Casadevall, G.; Duran, C.; Estévez-Gay, M.; Osuna, S. Estimating conformational heterogeneity of tryptophan synthase with a template-based Alphafold2 approach. Protein Sci. 2022, 31, e4426. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Looger, L.L.; Porter, L.L. A sequence-based method for predicting extant fold switchers that undergo alpha-helix <--> beta-strand transitions. Biopolymers 2021, 112, e23471. [Google Scholar] [CrossRef]
- Porter, L.L.; Chakravarty, D.; Schafer, J.W.; Chen, E.A. ColabFold predicts alternative protein structures from single sequences, coevolution unnecessary for AF-cluster. bioRxiv 2023. [Google Scholar] [CrossRef]
- Chang, Y.G.; Cohen, S.E.; Phong, C.; Myers, W.K.; Kim, Y.I.; Tseng, R.; Lin, J.; Zhang, L.; Boyd, J.S.; Lee, Y.; et al. Circadian rhythms. A protein fold switch joins the circadian oscillator to clock output in cyanobacteria. Science 2015, 349, 324–328. [Google Scholar] [CrossRef]
- Chakravarty, D.; Sreenivasan, S.; Swint-Kruse, L.; Porter, L.L. Identification of a covert evolutionary pathway between two protein folds. Nat. Commun. 2023, 14, 3177. [Google Scholar] [CrossRef]
- Schafer, J.W.; Porter, L.L. Evolutionary selection of proteins with two folds. Nat. Commun. 2023, 14, 5478. [Google Scholar] [CrossRef]
- Siemer, A.B. What makes functional amyloids work? Crit. Rev. Biochem. Mol. Biol. 2022, 57, 399–411. [Google Scholar] [CrossRef]
- Porter, L.L.; He, Y.; Chen, Y.; Orban, J.; Bryan, P.N. Subdomain interactions foster the design of two protein pairs with approximately 80% sequence identity but different folds. Biophys. J. 2015, 108, 154–162. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Porter, L.L. Predictable fold switching by the SARS-CoV-2 protein ORF9b. Protein Sci. 2021, 30, 1723–1729. [Google Scholar] [CrossRef] [PubMed]
- Nyström, S.; Hammarström, P. Amyloidogenesis of SARS-CoV-2 Spike Protein. J. Am. Chem. Soc. 2022, 144, 8945–8950. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, T.; Gadhave, K.; Kapuganti, S.K.; Kumar, P.; Brotzakis, Z.F.; Saumya, K.U.; Nayak, N.; Kumar, A.; Joshi, R.; Mukherjee, B.; et al. Amyloidogenic proteins in the SARS-CoV and SARS-CoV-2 proteomes. Nat. Commun. 2023, 14, 945. [Google Scholar] [CrossRef] [PubMed]
- Serpell, L.C.; Benson, M.; Liepnieks, J.J.; Fraser, P.E. Structural analyses of fibrinogen amyloid fibrils. Amyloid 2007, 14, 199–203. [Google Scholar] [CrossRef]
- Zamolodchikov, D.; Berk-Rauch, H.E.; Oren, D.A.; Stor, D.S.; Singh, P.K.; Kawasaki, M.; Aso, K.; Strickland, S.; Ahn, H.J. Biochemical and structural analysis of the interaction between beta-amyloid and fibrinogen. Blood 2016, 128, 1144–1151. [Google Scholar] [CrossRef]
- Ahn, H.J.; Chen, Z.L.; Zamolodchikov, D.; Norris, E.H.; Strickland, S. Interactions of beta-amyloid peptide with fibrinogen and coagulation factor XII may contribute to Alzheimer’s disease. Curr. Opin. Hematol. 2017, 24, 427–431. [Google Scholar] [CrossRef]
- Singh, P.K.; Berk-Rauch, H.E.; Soplop, N.; Uryu, K.; Strickland, S.; Ahn, H.J. Analysis of beta-Amyloid-induced Abnormalities on Fibrin Clot Structure by Spectroscopy and Scanning Electron Microscopy. J. Vis. Exp. 2018, e58475. [Google Scholar] [CrossRef]
- Cajamarca, S.A.; Norris, E.H.; van der Weerd, L.; Strickland, S.; Ahn, H.J. Cerebral amyloid angiopathy-linked beta-amyloid mutations promote cerebral fibrin deposits via increased binding affinity for fibrinogen. Proc. Natl. Acad. Sci. USA 2020, 117, 14482–14492. [Google Scholar] [CrossRef]
- Cohen, F.E.; Prusiner, S.B. Pathologic conformations of prion proteins. Annu. Rev. Biochem. 1998, 67, 793–819. [Google Scholar] [CrossRef]
- Matiiv, A.B.; Trubitsina, N.P.; Matveenko, A.G.; Barbitoff, Y.A.; Zhouravleva, G.A.; Bondarev, S.A. Structure and Polymorphism of Amyloid and Amyloid-Like Aggregates. Biochemistry 2022, 87, 450–463. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.O.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-beta spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Sabaté, R.; Ventura, S. Cross-beta-sheet supersecondary structure in amyloid folds: Techniques for detection and characterization. Methods Mol. Biol. 2013, 932, 237–257. [Google Scholar] [CrossRef] [PubMed]
- Saiki, M.; Shiba, K.; Okumura, M. Structural stability of amyloid fibrils depends on the existence of the peripheral sequence near the core cross-beta region. FEBS Lett. 2015, 589, 3541–3547. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, R.; Ranson, N.A.; Radford, S.E. Amyloid structures: Much more than just a cross-beta fold. Curr. Opin. Struct. Biol. 2020, 60, 7–16. [Google Scholar] [CrossRef]
- Gonay, V.; Dunne, M.P.; Caceres-Delpiano, J.; Kajava, A.V. Developing machine-learning-based amyloid predictors with Cross-Beta DB. bioRxiv 2024. [Google Scholar] [CrossRef]
- Jahn, T.R.; Makin, O.S.; Morris, K.L.; Marshall, K.E.; Tian, P.; Sikorski, P.; Serpell, L.C. The common architecture of cross-beta amyloid. J. Mol. Biol. 2010, 395, 717–727. [Google Scholar] [CrossRef]
- Sipe, J.D.; Cohen, A.S. Review: History of the amyloid fibril. J. Struct. Biol. 2000, 130, 88–98. [Google Scholar] [CrossRef]
- Upadhyay, A.; Mishra, A. Amyloids of multiple species: Are they helpful in survival? Biol. Rev. Camb. Philos. Soc. 2018, 93, 1363–1386. [Google Scholar] [CrossRef]
- Willbold, D.; Strodel, B.; Schroder, G.F.; Hoyer, W.; Heise, H. Amyloid-type Protein Aggregation and Prion-like Properties of Amyloids. Chem. Rev. 2021, 121, 8285–8307. [Google Scholar] [CrossRef]
- Gottwald, J.; Röcken, C. The amyloid proteome: A systematic review and proposal of a protein classification system. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 526–542. [Google Scholar] [CrossRef] [PubMed]
- Louros, N.; Schymkowitz, J.; Rousseau, F. Mechanisms and pathology of protein misfolding and aggregation. Nat. Rev. Mol. Cell Biol. 2023, 24, 912–933. [Google Scholar] [CrossRef] [PubMed]
- Michaels, T.C.T.; Qian, D.; Šarić, A.; Vendruscolo, M.; Linse, S.; Knowles, T.P.J. Amyloid formation as a protein phase transition. Nat. Phys. 2023, 5, 379–397. [Google Scholar] [CrossRef]
- Wei, G.; Su, Z.; Reynolds, N.P.; Arosio, P.; Hamley, I.W.; Gazit, E.; Mezzenga, R. Self-assembling peptide and protein amyloids: From structure to tailored function in nanotechnology. Chem. Soc. Rev. 2017, 46, 4661–4708. [Google Scholar] [CrossRef]
- Grobler, C.; van Tongeren, M.; Gettemans, J.; Kell, D.; Pretorius, E. Alzheimer-type dementia: A systems view provides a unifying explanation of its development. J. Alzheimer’s Dis. 2023, 91, 43–70. [Google Scholar] [CrossRef]
- Blancas-Mejía, L.M.; Ramirez-Alvarado, M. Systemic amyloidoses. Annu. Rev. Biochem. 2013, 82, 745–774. [Google Scholar] [CrossRef]
- Buxbaum, J.N.; Linke, R.P. A molecular history of the amyloidoses. J. Mol. Biol. 2012, 421, 142–159. [Google Scholar] [CrossRef]
- Lavatelli, F.; di Fonzo, A.; Palladini, G.; Merlini, G. Systemic amyloidoses and proteomics: The state of the art. EuPA Open Proteom. 2016, 11, 4–10. [Google Scholar] [CrossRef][Green Version]
- Palladini, G.; Merlini, G. Systemic amyloidoses: What an internist should know. Eur. J. Intern. Med. 2013, 24, 729–739. [Google Scholar] [CrossRef]
- Nevone, A.; Merlini, G.; Nuvolone, M. Treating Protein Misfolding Diseases: Therapeutic Successes Against Systemic Amyloidoses. Front. Pharmacol. 2020, 11, 1024. [Google Scholar] [CrossRef]
- Nevzglyadova, O.V.; Mikhailova, E.V.; Soidla, T.R. Yeast red pigment, protein aggregates, and amyloidoses: A review. Cell Tissue Res. 2022, 388, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; Nath, A.K.; Pal, I.; Dey, S.G. Second Sphere Interactions in Amyloidogenic Diseases. Chem. Rev. 2022, 122, 12132–12206. [Google Scholar] [CrossRef] [PubMed]
- Burdukiewicz, M.; Sobczyk, P.; Rödiger, S.; Duda-Madej, A.; Mackiewicz, P.; Kotulska, M. Amyloidogenic motifs revealed by n-gram analysis. Sci. Rep. 2017, 7, 12961. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; LaRocque, J.J.; Dokholyan, N.V. Direct observation of protein folding, aggregation, and a prion-like conformational conversion. J. Biol. Chem. 2005, 280, 40235–40240. [Google Scholar] [CrossRef] [PubMed]
- Sulatskaya, A.I.; Kosolapova, A.O.; Bobylev, A.G.; Belousov, M.V.; Antonets, K.S.; Sulatsky, M.I.; Kuznetsova, I.M.; Turoverov, K.K.; Stepanenko, O.V.; Nizhnikov, A.A. beta-Barrels and Amyloids: Structural Transitions, Biological Functions, and Pathogenesis. Int. J. Mol. Sci. 2021, 22, 11316. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 2009, 5, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Sun, Y.; Ding, F. Structures and dynamics of beta-barrel oligomer intermediates of amyloid-beta16-22 aggregation. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1687–1697. [Google Scholar] [CrossRef]
- Ge, X.; Yang, Y.; Sun, Y.; Cao, W.; Ding, F. Islet Amyloid Polypeptide Promotes Amyloid-Beta Aggregation by Binding-Induced Helix-Unfolding of the Amyloidogenic Core. ACS Chem. Neurosci. 2018, 9, 967–975. [Google Scholar] [CrossRef]
- Sun, Y.; Ge, X.; Xing, Y.; Wang, B.; Ding, F. beta-barrel Oligomers as Common Intermediates of Peptides Self-Assembling into Cross-beta Aggregates. Sci. Rep. 2018, 8, 10353. [Google Scholar] [CrossRef]
- Bradley, L.H.; Wei, Y.; Thumfort, P.; Wurth, C.; Hecht, M.H. Protein design by binary patterning of polar and nonpolar amino acids. Methods Mol. Biol. 2007, 352, 155–166. [Google Scholar]
- Fändrich, M. On the structural definition of amyloid fibrils and other polypeptide aggregates. Cell. Mol. Life Sci. 2007, 64, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Keresztes, L.; Szogi, E.; Varga, B.; Farkas, V.; Perczel, A.; Grolmusz, V. Succinct Amyloid and Nonamyloid Patterns in Hexapeptides. ACS Omega 2022, 7, 35532–35537. [Google Scholar] [CrossRef] [PubMed]
- Kotulska, M.; Wojciechowski, J.W. Bioinformatics Methods in Predicting Amyloid Propensity of Peptides and Proteins. Methods Mol. Biol. 2022, 2340, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fatafta, H.; Khaled, M.; Kav, B.; Olubiyi, O.O.; Strodel, B. A brief history of amyloid aggregation simulations. Wires Comput. Mol. Sci. 2024, 14, e1703. [Google Scholar] [CrossRef]
- Beerten, J.; Van Durme, J.; Gallardo, R.; Capriotti, E.; Serpell, L.; Rousseau, F.; Schymkowitz, J. WALTZ-DB: A benchmark database of amyloidogenic hexapeptides. Bioinformatics 2015, 31, 1698–1700. [Google Scholar] [CrossRef]
- Burdukiewicz, M.; Rafacz, D.; Barbach, A.; Hubicka, K.; Bąkaaronla, L.; Lassota, A.; Stecko, J.; Szymańska, N.; Wojciechowski, J.W.; Kozakiewicz, D.; et al. AmyloGraph: A comprehensive database of amyloid-amyloid interactions. Nucleic Acids Res. 2023, 51, D352–D357. [Google Scholar] [CrossRef]
- Argos, P.; Rao, J.K.; Hargrave, P.A. Structural prediction of membrane-bound proteins. Eur. J. Biochem. 1982, 128, 565–575. [Google Scholar] [CrossRef]
- Bhaskaran, R.; Ponnuswamy, P. Positional flexibilities of amino acid residues in globular proteins. Int. J. Pept. Protein Res. 1988, 32, 241–255. [Google Scholar] [CrossRef]
- Charton, M.; Charton, B.I. The structural dependence of amino acid hydrophobicity parameters. J. Theor. Biol. 1982, 99, 629–644. [Google Scholar] [CrossRef]
- Kim, C.A.; Berg, J.M. Thermodynamic beta-sheet propensities measured using a zinc-finger host peptide. Nature 1993, 362, 267–270. [Google Scholar] [CrossRef]
- Planas-Iglesias, J.; Borko, S.; Swiatkowski, J.; Elias, M.; Havlasek, M.; Salamon, O.; Grakova, E.; Kunka, A.; Martinovic, T.; Damborsky, J.; et al. AggreProt: A web server for predicting and engineering aggregation prone regions in proteins. Nucleic Acids Res. 2024, 52, W159–W169. [Google Scholar] [CrossRef] [PubMed]
- Conchillo-Solé, O.; de Groot, N.S.; Avilés, F.X.; Vendrell, J.; Daura, X.; Ventura, S. AGGRESCAN: A server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinform. 2007, 8, 65. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, R.; Jamroz, M.; Szczasiuk, A.; Pujols, J.; Kmiecik, S.; Ventura, S. AGGRESCAN3D (A3D): Server for prediction of aggregation properties of protein structures. Nucleic Acids Res. 2015, 43, W306–W313. [Google Scholar] [CrossRef]
- Bárcenas, O.; Kuriata, A.; Zalewski, M.; Iglesias, V.; Pintado-Grima, C.; Firlik, G.; Burdukiewicz, M.; Kmiecik, S.; Ventura, S. Aggrescan4D: Structure-informed analysis of pH-dependent protein aggregation. Nucleic Acids Res. 2024, 52, W170–W175. [Google Scholar] [CrossRef]
- Yang, Z.; Wu, Y.; Liu, H.; He, L.; Deng, X. AMYGNN: A Graph Convolutional Neural Network-Based Approach for Predicting Amyloid Formation from Polypeptides. J. Chem. Inf. Model. 2024, 64, 1751–1762. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, P.P.; Kotulska, M. AmyLoad: Website dedicated to amyloidogenic protein fragments. Bioinformatics 2015, 31, 3395–3397. [Google Scholar] [CrossRef]
- Bondarev, S.A.; Uspenskaya, M.V.; Leclercq, J.; Falgarone, T.; Zhouravleva, G.A.; Kajava, A.V. AmyloComp: A Bioinformatic Tool for Prediction of Amyloid Co-aggregation. J. Mol. Biol. 2024, 436, 168437. [Google Scholar] [CrossRef] [PubMed]
- Szulc, N.; Burdukiewicz, M.; Gąsior-Głogowska, M.; Wojciechowski, J.W.; Chilimoniuk, J.; Mackiewicz, P.; Šneideris, T.; Smirnovas, V.; Kotulska, M. Bioinformatics methods for identification of amyloidogenic peptides show robustness to misannotated training data. Sci. Rep. 2021, 11, 8934. [Google Scholar] [CrossRef] [PubMed]
- Charoenkwan, P.; Ahmed, S.; Nantasenamat, C.; Quinn, J.M.W.; Moni, M.A.; Lio, P.; Shoombuatong, W. AMYPred-FRL is a novel approach for accurate prediction of amyloid proteins by using feature representation learning. Sci. Rep. 2022, 12, 7697. [Google Scholar] [CrossRef]
- Varadi, M.; De Baets, G.; Vranken, W.F.; Tompa, P.; Pancsa, R. AmyPro: A database of proteins with validated amyloidogenic regions. Nucleic Acids Res. 2018, 46, D387–D392. [Google Scholar] [CrossRef]
- Ahmed, A.B.; Znassi, N.; Chateau, M.T.; Kajava, A.V. A structure-based approach to predict predisposition to amyloidosis. Alzheimer’s Dement. 2015, 11, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Falgarone, T.; Villain, E.; Guettaf, A.; Leclercq, J.; Kajava, A.V. TAPASS: Tool for annotation of protein amyloidogenicity in the context of other structural states. J. Struct. Biol. 2022, 214, 107840. [Google Scholar] [CrossRef] [PubMed]
- Prabakaran, R.; Rawat, P.; Kumar, S.; Michael Gromiha, M. ANuPP: A Versatile Tool to Predict Aggregation Nucleating Regions in Peptides and Proteins. J. Mol. Biol. 2021, 433, 166707. [Google Scholar] [CrossRef] [PubMed]
- Bryan, A.W., Jr.; Menke, M.; Cowen, L.J.; Lindquist, S.L.; Berger, B. BETASCAN: Probable beta-amyloids identified by pairwise probabilistic analysis. PLoS Comput. Biol. 2009, 5, e1000333. [Google Scholar] [CrossRef] [PubMed]
- Bondarev, S.A.; Bondareva, O.V.; Zhouravleva, G.A.; Kajava, A.V. BetaSerpentine: A bioinformatics tool for reconstruction of amyloid structures. Bioinformatics 2018, 34, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Keresztes, L.; Szogi, E.; Varga, B.; Farkas, V.; Perczel, A.; Grolmusz, V. The Budapest Amyloid Predictor and Its Applications. Biomolecules 2021, 11, 500. [Google Scholar] [CrossRef]
- Louros, N.; Rousseau, F.; Schymkowitz, J. CORDAX web server: An online platform for the prediction and 3D visualization of aggregation motifs in protein sequences. Bioinformatics 2024, 40, btae279. [Google Scholar] [CrossRef]
- Thangakani, A.M.; Nagarajan, R.; Kumar, S.; Sakthivel, R.; Velmurugan, D.; Gromiha, M.M. CPAD, Curated Protein Aggregation Database: A Repository of Manually Curated Experimental Data on Protein and Peptide Aggregation. PLoS ONE 2016, 11, e0152949. [Google Scholar] [CrossRef]
- Citarella, A.A.; Di Biasi, L.; De Marco, F.; Tortora, G. ENTAIL: yEt aNoTher amyloid fIbrils cLassifier. BMC Bioinform. 2022, 23, 517. [Google Scholar] [CrossRef]
- Gasior, P.; Kotulska, M. FISH Amyloid—A new method for finding amyloidogenic segments in proteins based on site specific co-occurrence of aminoacids. BMC Bioinform. 2014, 15, 54. [Google Scholar] [CrossRef]
- Garbuzynskiy, S.O.; Lobanov, M.Y.; Galzitskaya, O.V. FoldAmyloid: A method of prediction of amyloidogenic regions from protein sequence. Bioinformatics 2010, 26, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Thangakani, A.M.; Kumar, S.; Nagarajan, R.; Velmurugan, D.; Gromiha, M.M. GAP: Towards almost 100 percent prediction for beta-strand-mediated aggregating peptides with distinct morphologies. Bioinformatics 2014, 30, 1983–1990. [Google Scholar] [CrossRef] [PubMed]
- Munir, F.; Gul, S.; Asif, A.; Minhas, F.A. MILAMP: Multiple Instance Prediction of Amyloid Proteins. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 18, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowski, J.W.; Szczurek, W.; Szulc, N.; Szefczyk, M.; Kotulska, M. PACT—Prediction of amyloid cross-interaction by threading. Sci. Rep. 2023, 13, 22268. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Escamilla, A.M.; Rousseau, F.; Schymkowitz, J.; Serrano, L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 2004, 22, 1302–1306. [Google Scholar] [CrossRef]
- Walsh, I.; Seno, F.; Tosatto, S.C.; Trovato, A. PASTA 2.0: An improved server for protein aggregation prediction. Nucleic Acids Res. 2014, 42, W301–W307. [Google Scholar] [CrossRef]
- Teng, Z.; Zhang, Z.; Tian, Z.; Li, Y.; Wang, G. ReRF-Pred: Predicting amyloidogenic regions of proteins based on their pseudo amino acid composition and tripeptide composition. BMC Bioinform. 2021, 22, 545. [Google Scholar] [CrossRef]
- Niu, M.; Li, Y.; Wang, C.; Han, K. RFAmyloid: A Web Server for Predicting Amyloid Proteins. Int. J. Mol. Sci. 2018, 19, 2071. [Google Scholar] [CrossRef]
- Maurer-Stroh, S.; Debulpaep, M.; Kuemmerer, N.; Lopez de la Paz, M.; Martins, I.C.; Reumers, J.; Morris, K.L.; Copland, A.; Serpell, L.; Serrano, L.; et al. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 2010, 7, 237–242. [Google Scholar] [CrossRef]
- Louros, N.; Konstantoulea, K.; De Vleeschouwer, M.; Ramakers, M.; Schymkowitz, J.; Rousseau, F. WALTZ-DB 2.0: An updated database containing structural information of experimentally determined amyloid-forming peptides. Nucleic Acids Res. 2020, 48, D389–D393. [Google Scholar] [CrossRef]
- Goldschmidt, L.; Teng, P.K.; Riek, R.; Eisenberg, D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2010, 107, 3487–3492. [Google Scholar] [CrossRef] [PubMed]
- Prabakaran, R.; Goel, D.; Kumar, S.; Gromiha, M.M. Aggregation prone regions in human proteome: Insights from large-scale data analyses. Proteins 2017, 85, 1099–1118. [Google Scholar] [CrossRef] [PubMed]
- Danilov, L.G.; Sukhanova, X.V.; Rogoza, T.M.; Antonova, E.Y.; Trubitsina, N.P.; Zhouravleva, G.A.; Bondarev, S.A. Identification of New FG-Repeat Nucleoporins with Amyloid Properties. Int. J. Mol. Sci. 2023, 24, 8571. [Google Scholar] [CrossRef] [PubMed]
- Falgarone, T.; Villain, E.; Richard, F.; Osmanli, Z.; Kajava, A.V. Census of exposed aggregation-prone regions in proteomes. Brief. Bioinform. 2023, 24, bbad183. [Google Scholar] [CrossRef] [PubMed]
- Nizhnikov, A.A.; Antonets, K.S.; Inge-Vechtomov, S.G. Amyloids: From Pathogenesis to Function. Biochemistry 2015, 80, 1127–1144. [Google Scholar] [CrossRef]
- Carrió, M.; González-Montalbán, N.; Vera, A.; Villaverde, A.; Ventura, S. Amyloid-like properties of bacterial inclusion bodies. J. Mol. Biol. 2005, 347, 1025–1037. [Google Scholar] [CrossRef]
- Fagihi, M.H.A.; Bhattacharjee, S. Amyloid Fibrillation of Insulin: Amelioration Strategies and Implications for Translation. ACS Pharmacol. Transl. Sci. 2022, 5, 1050–1061. [Google Scholar] [CrossRef]
- Miller, Y. Advancements and future directions in research of the roles of insulin in amyloid diseases. Biophys. Chem. 2022, 281, 106720. [Google Scholar] [CrossRef]
- Hua, Q.X.; Weiss, M.A. Mechanism of insulin fibrillation: The structure of insulin under amyloidogenic conditions resembles a protein-folding intermediate. J. Biol. Chem. 2004, 279, 21449–21460. [Google Scholar] [CrossRef]
- Kuo, C.T.; Chen, Y.L.; Hsu, W.T.; How, S.C.; Cheng, Y.H.; Hsueh, S.S.; Liu, H.S.; Lin, T.H.; Wu, J.W.; Wang, S.S. Investigating the effects of erythrosine B on amyloid fibril formation derived from lysozyme. Int. J. Biol. Macromol. 2017, 98, 159–168. [Google Scholar] [CrossRef]
- How, S.C.; Hsin, A.; Chen, G.Y.; Hsu, W.T.; Yang, S.M.; Chou, W.L.; Chou, S.H.; Wang, S.S. Exploring the influence of brilliant blue G on amyloid fibril formation of lysozyme. Int. J. Biol. Macromol. 2019, 38, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.; Miti, T.; Hasecke, F.; Meisl, G.; Hoyer, W.; Muschol, M.; Ullah, G. Mechanism of Fibril and Soluble Oligomer Formation in Amyloid Beta and Hen Egg White Lysozyme Proteins. J. Phys. Chem. B 2019, 123, 5678–5689. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xing, L.; Li, X.; Chen, N.; Liu, L.; Wang, J.; Zhou, X.; Liu, S. Manganese Ion-Induced Amyloid Fibrillation Kinetics of Hen Egg White-Lysozyme in Thermal and Acidic Conditions. ACS Omega 2023, 8, 16439–16449. [Google Scholar] [CrossRef] [PubMed]
- Gancar, M.; Kurin, E.; Bednarikova, Z.; Marek, J.; Mucaji, P.; Nagy, M.; Gazova, Z. Green tea leaf constituents inhibit the formation of lysozyme amyloid aggregates: An effect of mutual interactions. Int. J. Biol. Macromol. 2023, 242, 124856. [Google Scholar] [CrossRef]
- Khan, A.N.; Nabi, F.; Khan, R.H. Mechanistic and biophysical insight into the inhibitory and disaggregase role of antibiotic moxifloxacin on human lysozyme amyloid formation. Biophys. Chem. 2023, 298, 107029. [Google Scholar] [CrossRef]
- Swaminathan, R.; Ravi, V.K.; Kumar, S.; Kumar, M.V.; Chandra, N. Lysozyme: A model protein for amyloid research. Adv. Protein Chem. Struct. Biol. 2011, 84, 63–111. [Google Scholar] [CrossRef]
- Roy, S.; Srinivasan, V.R.; Arunagiri, S.; Mishra, N.; Bhatia, A.; Shejale, K.P.; Prajapati, K.P.; Kar, K.; Anand, B.G. Molecular insights into the phase transition of lysozyme into amyloid nanostructures: Implications of therapeutic strategies in diverse pathological conditions. Adv. Colloid. Interface Sci. 2024, 331, 103205. [Google Scholar] [CrossRef] [PubMed]
- Muthu, S.A.; Qureshi, A.; Sharma, R.; Bisaria, I.; Parvez, S.; Grover, S.; Ahmad, B. Redesigning the kinetics of lysozyme amyloid aggregation by cephalosporin molecules. J. Biomol. Struct. Dyn. 2024, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kamada, A.; Herneke, A.; Lopez-Sanchez, P.; Harder, C.; Ornithopoulou, E.; Wu, Q.; Wei, X.; Schwartzkopf, M.; Muller-Buschbaum, P.; Roth, S.V.; et al. Hierarchical propagation of structural features in protein nanomaterials. Nanoscale 2022, 14, 2502–2510. [Google Scholar] [CrossRef]
- Cao, Y.; Mezzenga, R. Design principles of food gels. Nat. Food 2020, 1, 106–118. [Google Scholar] [CrossRef]
- Rahman, M.M.; Pires, R.S.; Herneke, A.; Gowda, V.; Langton, M.; Biverstal, H.; Lendel, C. Food protein-derived amyloids do not accelerate amyloid beta aggregation. Sci. Rep. 2023, 13, 985. [Google Scholar] [CrossRef]
- Wickner, R.B.; Edskes, H.K.; Bateman, D.A.; Kelly, A.C.; Gorkovskiy, A.; Dayani, Y.; Zhou, A. Amyloid diseases of yeast: Prions are proteins acting as genes. Essays Biochem. 2014, 56, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Edskes, H.K.; Gorkovskiy, A.; Bezsonov, E.E.; Stroobant, E.E. Yeast and Fungal Prions: Amyloid-Handling Systems, Amyloid Structure, and Prion Biology. Adv. Genet. 2016, 93, 191–236. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Shen, H.C.; Komi, Y.; Sugiyama, S.; Kurinomaru, T.; Tomabechi, Y.; Krayukhina, E.; Okamoto, K.; Yokoyama, T.; Shirouzu, M.; et al. Amyloid conformation-dependent disaggregation in a reconstituted yeast prion system. Nat. Chem. Biol. 2022, 18, 321–331. [Google Scholar] [CrossRef]
- Rouse, S.L.; Matthews, S.J.; Dueholm, M.S. Ecology and Biogenesis of Functional Amyloids in Pseudomonas. J. Mol. Biol. 2018, 430, 3685–3695. [Google Scholar] [CrossRef]
- Ivancic, V.; Ekanayake, O.; Lazo, N. Binding Modes of Thioflavin T on the Surface of Amyloid Fibrils by NMR. ChemPhysChem 2016, 17, 2461–2464. [Google Scholar] [CrossRef]
- Tycko, R. Solid-state NMR studies of amyloid fibril structure. Annu. Rev. Phys. Chem. 2011, 62, 279–299. [Google Scholar] [CrossRef]
- Artikis, E.; Kraus, A.; Caughey, B. Structural biology of ex vivo mammalian prions. J. Biol. Chem. 2022, 298, 102181. [Google Scholar] [CrossRef]
- Martial, B.; Lefèvre, T.; Auger, M. Understanding amyloid fibril formation using protein fragments: Structural investigations via vibrational spectroscopy and solid-state NMR. Biophys. Rev. 2018, 10, 1133–1149. [Google Scholar] [CrossRef]
- Baek, Y.; Lee, M. Solid-state NMR spectroscopic analysis for structure determination of a zinc-bound catalytic amyloid fibril. Methods Enzymol. 2024, 697, 435–471. [Google Scholar] [CrossRef]
- Lövestam, S.; Scheres, S.H.W. High-throughput cryo-EM structure determination of amyloids. Faraday Discuss. 2022, 240, 243–260. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Arseni, D.; Zhang, W.; Huang, M.; Lovestam, S.; Schweighauser, M.; Kotecha, A.; Murzin, A.G.; Peak-Chew, S.Y.; Macdonald, J.; et al. Cryo-EM structures of amyloid-beta 42 filaments from human brains. Science 2022, 375, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Bücker, R.; Seuring, C.; Cazey, C.; Veith, K.; Garcia-Alai, M.; Grünewald, K.; Landau, M. The Cryo-EM structures of two amphibian antimicrobial cross-beta amyloid fibrils. Nat. Commun. 2022, 13, 4356. [Google Scholar] [CrossRef] [PubMed]
- Heerde, T.; Bansal, A.; Schmidt, M.; Fändrich, M. Cryo-EM structure of a catalytic amyloid fibril. Sci. Rep. 2023, 13, 4070. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.; Hoq, M.R.; Hallinan, G.I.; Li, D.; Bharath, S.R.; Vago, F.S.; Zhang, X.; Ozcan, K.A.; Newell, K.L.; Garringer, H.J.; et al. Cryo-EM structures of amyloid-beta and tau filaments in Down syndrome. Nat. Struct. Mol. Biol. 2024, 31, 903–909. [Google Scholar] [CrossRef]
- Sharma, K.; Stockert, F.; Shenoy, J.; Berbon, M.; Abdul-Shukkoor, M.B.; Habenstein, B.; Loquet, A.; Schmidt, M.; Fändrich, M. Cryo-EM observation of the amyloid key structure of polymorphic TDP-43 amyloid fibrils. Nat. Commun. 2024, 15, 486. [Google Scholar] [CrossRef]
- LeVine, H., 3rd. Thioflavine T interaction with synthetic Alzheimer’s disease beta-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci. 1993, 2, 404–410. [Google Scholar] [CrossRef]
- Khurana, R.; Coleman, C.; Ionescu-Zanetti, C.; Carter, S.A.; Krishna, V.; Grover, R.K.; Roy, R.; Singh, S. Mechanism of thioflavin T binding to amyloid fibrils. J. Struct. Biol. 2005, 151, 229–238. [Google Scholar] [CrossRef]
- Hawe, A.; Sutter, M.; Jiskoot, W. Extrinsic fluorescent dyes as tools for protein characterization. Pharm. Res. 2008, 25, 1487–1499. [Google Scholar] [CrossRef]
- Biancalana, M.; Koide, S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta 2010, 1804, 1405–1412. [Google Scholar] [CrossRef]
- Amdursky, N.; Erez, Y.; Huppert, D. Molecular rotors: What lies behind the high sensitivity of the thioflavin-T fluorescent marker. Acc. Chem. Res. 2012, 45, 1548–1557. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, I.M.; Sulatskaya, A.I.; Maskevich, A.A.; Uversky, V.N.; Turoverov, K.K. High Fluorescence Anisotropy of Thioflavin T in Aqueous Solution Resulting from Its Molecular Rotor Nature. Anal. Chem. 2016, 88, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Gade Malmos, K.; Blancas-Mejia, L.M.; Weber, B.; Buchner, J.; Ramirez-Alvarado, M.; Naiki, H.; Otzen, D. ThT 101: A primer on the use of thioflavin T to investigate amyloid formation. Amyloid 2017, 24, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Lin, T.Y.; Chang, D.; Guo, Z. Thioflavin T as an amyloid dye: Fibril quantification, optimal concentration and effect on aggregation. R. Soc. Open Sci. 2017, 4, 160696. [Google Scholar] [CrossRef] [PubMed]
- Aliyan, A.; Cook, N.P.; Marti, A.A. Interrogating Amyloid Aggregates using Fluorescent Probes. Chem. Rev. 2019, 119, 11819–11856. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, C.; Rimola, A.; Rodríguez-Santiago, L.; Ugliengo, P.; Álvarez-Larena, Á.; Gutiérrez-de-Terán, H.; Sodupe, M.; González-Duarte, P. Crystal structure of thioflavin-T and its binding to amyloid fibrils: Insights at the molecular level. Chem. Commun. 2010, 46, 1156–1158. [Google Scholar] [CrossRef]
- Ali-Torres, J.; Rimola, A.; Rodríguez-Rodríguez, C.; Rodríguez-Santiago, L.; Sodupe, M. Insights on the binding of Thioflavin derivative markers to amyloid-like fibril models from quantum chemical calculations. J. Phys. Chem. B 2013, 117, 6674–6680. [Google Scholar] [CrossRef]
- Kuang, G.; Murugan, N.A.; Tu, Y.; Nordberg, A.; Ågren, H. Investigation of the Binding Profiles of AZD2184 and Thioflavin T with Amyloid-beta(1-42) Fibril by Molecular Docking and Molecular Dynamics Methods. J. Phys. Chem. B 2015, 119, 11560–11567. [Google Scholar] [CrossRef]
- Shaban, H.A.; Valades-Cruz, C.A.; Savatier, J.; Brasselet, S. Polarized super-resolution structural imaging inside amyloid fibrils using Thioflavine T. Sci. Rep. 2017, 7, 12482. [Google Scholar] [CrossRef]
- Frieg, B.; Gremer, L.; Heise, H.; Willbold, D.; Gohlke, H. Binding modes of thioflavin T and Congo red to the fibril structure of amyloid-beta(1-42). Chem. Commun. 2020, 56, 7589–7592. [Google Scholar] [CrossRef]
- Sulatskaya, A.I.; Rychkov, G.N.; Sulatsky, M.I.; Mikhailova, E.V.; Melnikova, N.M.; Andozhskaya, V.S.; Kuznetsova, I.M.; Turoverov, K.K. New Evidence on a Distinction between A beta 40 and A beta 42 Amyloids: Thioflavin T Binding Modes, Clustering Tendency, Degradation Resistance, and Cross-Seeding. Int. J. Mol. Sci. 2022, 23, 5513. [Google Scholar] [CrossRef] [PubMed]
- Sulatskaya, A.I.; Kuznetsova, I.M.; Belousov, M.V.; Bondarev, S.A.; Zhouravleva, G.A.; Turoverov, K.K. Stoichiometry and Affinity of Thioflavin T Binding to Sup35p Amyloid Fibrils. PLoS ONE 2016, 11, e0156314. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, T.S.; Hunter, C.A. A closer look at amyloid ligands, and what they tell us about protein aggregates. Chem. Soc. Rev. 2024, 53, 1354–1374. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Sonoda, L.K.; Silman, I.; Sussman, J.L.; Rosenberry, T.L. Crystal structure of thioflavin T bound to the peripheral site of Torpedo californica acetylcholinesterase reveals how thioflavin T acts as a sensitive fluorescent reporter of ligand binding to the acylation site. J. Am. Chem. Soc. 2008, 130, 7856–7861. [Google Scholar] [CrossRef] [PubMed]
- Hudson, S.A.; Ecroyd, H.; Kee, T.W.; Carver, J.A. The thioflavin T fluorescence assay for amyloid fibril detection can be biased by the presence of exogenous compounds. FEBS J. 2009, 276, 5960–5972. [Google Scholar] [CrossRef]
- Lindberg, D.J.; Wranne, M.S.; Gilbert Gatty, M.; Westerlund, F.; Esbjörner, E.K. Steady-state and time-resolved Thioflavin-T fluorescence can report on morphological differences in amyloid fibrils formed by Abeta(1-40) and Abeta(1-42). Biochem. Biophys. Res. Commun. 2015, 458, 418–423. [Google Scholar] [CrossRef]
- Nilsson, K.P.R.; Åslund, A.; Berg, I.; Nyström, S.; Konradsson, P.; Herland, A.; Inganäs, O.; Stabo-Eeg, F.; Lindgren, M.; Westermark, G.T.; et al. Imaging distinct conformational states of amyloid-beta fibrils in Alzheimer’s disease using novel luminescent probes. ACS Chem. Biol. 2007, 2, 553–560. [Google Scholar] [CrossRef]
- Nilsson, K.P.R.; Ikenberg, K.; Åslund, A.; Fransson, S.; Konradsson, P.; Röcken, C.; Moch, H.; Aguzzi, A. Structural typing of systemic amyloidoses by luminescent-conjugated polymer spectroscopy. Am. J. Pathol. 2010, 176, 563–574. [Google Scholar] [CrossRef]
- Åslund, A.; Herland, A.; Hammarström, P.; Nilsson, K.P.R.; Jonsson, B.H.; Inganäs, O.; Konradsson, P. Studies of luminescent conjugated polythiophene derivatives: Enhanced spectral discrimination of protein conformational states. Bioconjug. Chem. 2007, 18, 1860–1868. [Google Scholar] [CrossRef]
- Klingstedt, T.; Åslund, A.; Simon, R.A.; Johansson, L.B.G.; Mason, J.J.; Nyström, S.; Hammarström, P.; Nilsson, K.P.R. Synthesis of a library of oligothiophenes and their utilization as fluorescent ligands for spectral assignment of protein aggregates. Org. Biomol. Chem. 2011, 9, 8356–8370. [Google Scholar] [CrossRef]
- Klingstedt, T.; Shirani, H.; Åslund, K.O.A.; Cairns, N.J.; Sigurdson, C.J.; Goedert, M.; Nilsson, K.P.R. The structural basis for optimal performance of oligothiophene-based fluorescent amyloid ligands: Conformational flexibility is essential for spectral assignment of a diversity of protein aggregates. Chemistry 2013, 19, 10179–10192. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.A.; Shirani, H.; Åslund, K.O.A.; Bäck, M.; Haroutunian, V.; Gandy, S.; Nilsson, K.P.R. Pentameric thiophene-based ligands that spectrally discriminate amyloid-beta and tau aggregates display distinct solvatochromism and viscosity-induced spectral shifts. Chemistry 2014, 20, 12537–12543. [Google Scholar] [CrossRef] [PubMed]
- Nesterov, E.E.; Skoch, J.; Hyman, B.T.; Klunk, W.E.; Bacskai, B.J.; Swager, T.M. In vivo optical Imaging of amyloid aggregates in brain: Design of fluorescent markers. Angew. Chem. Int. Ed. Engl. 2005, 44, 5452–5456. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Lim, E.; Hwang, D.; Huh, H.; Kim, S.K. Torsion-dependent fluorescence switching of amyloid-binding dye NIAD-4. Chem. Phys. Lett. 2015, 633, 109–113. [Google Scholar] [CrossRef]
- Brandenburg, E.; von Berlepsch, H.; Koksch, B. Specific in situ discrimination of amyloid fibrils versus alpha-helical fibres by the fluorophore NIAD-4. Mol. Biosyst. 2012, 8, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Staderini, M.; Martin, M.A.; Bolognesi, M.L.; Menéndez, J.C. Imaging of beta-amyloid plaques by near infrared fluorescent tracers: A new frontier for chemical neuroscience. Chem. Soc. Rev. 2015, 44, 1807–1819. [Google Scholar] [CrossRef]
- Peccati, F.; Pantaleone, S.; Solans-Monfort, X.; Sodupe, M. Fluorescent Markers for Amyloid-beta Detection: Computational Insights. Isr. J. Chem. 2017, 57, 686–698. [Google Scholar] [CrossRef]
- Li, D.; Yang, Y.; Li, C.; Liu, Y. The influence of hydrogen bonds on NIAD-4 for use in the optical imaging of amyloid fibrils. Phys. Chem. Chem. Phys. 2017, 19, 15849–15855. [Google Scholar] [CrossRef]
- Fu, H.; Cui, M. Fluorescent Imaging of Amyloid-beta Deposits in Brain: An Overview of Probe Development and a Highlight of the Applications for In Vivo Imaging. Curr. Med. Chem. 2018, 25, 2736–2759. [Google Scholar] [CrossRef]
- Gyasi, Y.I.; Pang, Y.P.; Li, X.R.; Gu, J.X.; Cheng, X.J.; Liu, J.; Xu, T.; Liu, Y. Biological applications of near infrared fluorescence dye probes in monitoring Alzheimer’s disease. Eur. J. Med. Chem. 2020, 187, 111982. [Google Scholar] [CrossRef]
- Peng, C.; Wang, X.; Li, Y.; Li, H.-W.; Wong, M.S. Versatile fluorescent probes for near-infrared imaging of amyloid-beta species in Alzheimer’s disease mouse model. J. Mater. Chem. B 2022, 7, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Mu, X.Y.; Yang, J.; Liang, Y.Y.; Zhang, X.D.; Ming, D. Brain imaging with near-infrared fluorophores. Coord. Chem. Rev. 2019, 380, 550–571. [Google Scholar] [CrossRef]
- Pretorius, E.; Mbotwe, S.; Bester, J.; Robinson, C.J.; Kell, D.B. Acute induction of anomalous and amyloidogenic blood clotting by molecular amplification of highly substoichiometric levels of bacterial lipopolysaccharide. J. R. Soc. Interface 2016, 123, 20160539. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Page, M.J.; Hendricks, L.; Nkosi, N.B.; Benson, S.R.; Kell, D.B. Both lipopolysaccharide and lipoteichoic acids potently induce anomalous fibrin amyloid formation: Assessment with novel Amytracker™ stains. J. R. Soc. Interface 2018, 15, 20170941. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Page, M.J.; Engelbrecht, L.; Ellis, G.C.; Kell, D.B. Substantial fibrin amyloidogenesis in type 2 diabetes assessed using amyloid-selective fluorescent stains. Cardiovasc. Diabetol. 2017, 16, 141. [Google Scholar] [CrossRef]
- Pretorius, E.; Venter, C.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B. Prevalence of readily detected amyloid blood clots in ‘unclotted’ Type 2 Diabetes Mellitus and COVID-19 plasma: A preliminary report. Cardiovasc. Diabetol. 2020, 19, 193. [Google Scholar] [CrossRef]
- Kell, D.B.; Laubscher, G.J.; Pretorius, E. A central role for amyloid fibrin microclots in long COVID/PASC: Origins and therapeutic implications. Biochem. J. 2022, 479, 537–559. [Google Scholar] [CrossRef]
- Pretorius, E.; Venter, C.; Laubscher, G.J.; Kotze, M.J.; Oladejo, S.; Watson, L.R.; Rajaratnam, K.; Watson, B.W.; Kell, D.B. Prevalence of symptoms, comorbidities, fibrin amyloid microclots and platelet pathology in individuals with Long COVID/ Post-Acute Sequelae of COVID-19 (PASC). Cardiovasc. Diabetol. 2022, 21, 148. [Google Scholar] [CrossRef]
- Turner, S.; Khan, M.A.; Putrino, D.; Woodcock, A.; Kell, D.B.; Pretorius, E. Long COVID: Pathophysiological factors and abnormal coagulation. Trends Endocrinol. Metab. 2023, 34, 321–344. [Google Scholar] [CrossRef]
- Turner, S.; Laubscher, G.J.; Khan, M.A.; Kell, D.B.; Pretorius, E. Accelerating discovery: A novel flow cytometric method for detecting fibrin(ogen) amyloid microclots using long COVID as a model. Heliyon 2023, 9, e19605. [Google Scholar] [CrossRef]
- Kell, D.B.; Khan, M.A.; Kane, B.; Lip, G.Y.H.; Pretorius, E. Possible role of fibrinaloid microclots in Postural Orthostatic Tachycardia Syndrome (POTS): Focus on Long COVID. J. Pers. Med. 2024, 14, 170. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Kell, D.B. A Perspective on How Fibrinaloid Microclots and Platelet Pathology May be Applied in Clinical Investigations. Semin. Thromb. Hemost. 2024, 50, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Dalton, C.F.; de Oliveira, M.I.R.; Stafford, P.; Peake, N.; Kane, B.; Higham, A.; Singh, D.; Jackson, N.; Davies, H.; Price, D.; et al. Increased fibrinaloid microclot counts in platelet-poor plasma are associated with Long COVID. medRxiv 2024. [Google Scholar] [CrossRef]
- Schofield, J.; Abrams, S.T.; Jenkins, R.; Lane, S.; Wang, G.; Toh, C.H. Amyloid-fibrinogen aggregates (“microclots”) predict risks of Disseminated Intravascular Coagulation and mortality. Blood Adv. 2024, 8, 2499–2508. [Google Scholar] [CrossRef]
- Tang, H.; Fu, Y.; Zhan, S.; Luo, Y. Alpha(E)C, the C-terminal extension of fibrinogen, has chaperone-like activity. Biochemistry 2009, 48, 3967–3976. [Google Scholar] [CrossRef]
- Tang, H.; Fu, Y.; Cui, Y.; He, Y.; Zeng, X.; Ploplis, V.A.; Castellino, F.J.; Luo, Y. Fibrinogen has chaperone-like activity. Biochem. Biophys. Res. Commun. 2009, 378, 662–667. [Google Scholar] [CrossRef]
- Yamamoto, N.; Chatani, E. Multistep growth of amyloid intermediates and its inhibition toward exploring therapeutic way: A case study using insulin B chain and fibrinogen. Biophys. Physicobiol. 2022, 19, e190017. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Lachmann, H.J.; Wechalekar, A.; Hawkins, P.N. Hereditary fibrinogen A alpha-chain amyloidosis: Clinical phenotype and role of liver transplantation. Blood 2010, 115, 4313, author reply 4314–4315. [Google Scholar] [CrossRef]
- Stangou, A.J.; Banner, N.R.; Hendry, B.M.; Rela, M.; Portmann, B.; Wendon, J.; Monaghan, M.; Maccarthy, P.; Buxton-Thomas, M.; Mathias, C.J.; et al. Hereditary fibrinogen A alpha-chain amyloidosis: Phenotypic characterization of a systemic disease and the role of liver transplantation. Blood 2010, 115, 2998–3007. [Google Scholar] [CrossRef]
- Chapman, J.; Dogan, A. Fibrinogen alpha amyloidosis: Insights from proteomics. Expert. Rev. Proteom. 2019, 16, 783–793. [Google Scholar] [CrossRef]
- Overoye-Chan, K.; Koerner, S.; Looby, R.J.; Kolodziej, A.F.; Zech, S.G.; Deng, Q.; Chasse, J.M.; McMurry, T.J.; Caravan, P. EP-2104R: A fibrin-specific gadolinium-Based MRI contrast agent for detection of thrombus. J. Am. Chem. Soc. 2008, 130, 6025–6039. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Bhayana, B.; Thompson, B.; Kessinger, C.W.; Khatri, A.; McCarthy, J.R.; Weissleder, R.; Lin, C.P.; Tearney, G.J.; Jaffer, F.A. Molecular imaging of fibrin deposition in deep vein thrombosis using fibrin-targeted near-infrared fluorescence. JACC Cardiovasc. Imaging 2012, 5, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Schenk, B.; Bachler, M.; Solomon, C.; Fries, D.; Hermann, M. FITC-linked Fibrin-Binding Peptide and real-time live confocal microscopy as a novel tool to visualize fibrin(ogen) in coagulation. J. Clin. Transl. Res. 2017, 3, 276–282. [Google Scholar] [PubMed]
- Rambaran, R.N.; Serpell, L.C. Amyloid fibrils: Abnormal protein assembly. Prion 2008, 2, 112–117. [Google Scholar] [CrossRef]
- Xu, S.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Characterization of tau fibrillization in vitro. Alzheimer’s Dement. 2010, 6, 110–117. [Google Scholar] [CrossRef]
- Amenitsch, H.; Benetti, F.; Ramos, A.; Legname, G.; Requena, J.R. SAXS structural study of PrP(Sc) reveals ~11 nm diameter of basic double intertwined fibers. Prion 2013, 7, 496–500. [Google Scholar] [CrossRef]
- Yang, Y.; Shi, Y.; Schweighauser, M.; Zhang, X.; Kotecha, A.; Murzin, A.G.; Garringer, H.J.; Cullinane, P.W.; Saito, Y.; Foroud, T.; et al. Structures of alpha-synuclein filaments from human brains with Lewy pathology. Nature 2022, 610, 791–795. [Google Scholar] [CrossRef]
- Dhavale, D.D.; Barclay, A.M.; Borcik, C.G.; Basore, K.; Berthold, D.A.; Gordon, I.R.; Liu, J.; Milchberg, M.H.; O’Shea, J.Y.; Rau, M.J.; et al. Structure of alpha-synuclein fibrils derived from human Lewy body dementia tissue. Nat. Commun. 2024, 15, 2750. [Google Scholar] [CrossRef]
- He, S.; He, X.; Liu, L.; Zhang, W.; Yu, L.; Deng, Z.; Feiyi, Z.; Mo, S.; Fan, Y.; Zhao, X.; et al. The Structural Understanding of Transthyretin Misfolding and the Inspired Drug Approaches for the Treatment of Heart Failure Associated with Transthyretin Amyloidosis. Front. Pharmacol. 2021, 12, 628184. [Google Scholar] [CrossRef]
- Yermolenko, I.S.; Lishko, V.K.; Ugarova, T.P.; Magonov, S.N. High-resolution visualization of fibrinogen molecules and fibrin fibers with atomic force microscopy. Biomacromolecules 2011, 12, 370–379. [Google Scholar] [CrossRef]
- Blinc, A.; Magdič, J.; Fric, J.; Muševič. Atomic force microscopy of fibrin networks and plasma clots during fibrinolysis. Fibrinolysis Proteolysis 2000, 14, 288–299. [Google Scholar] [CrossRef]
- Collet, J.P.; Park, D.; Lesty, C.; Soria, J.; Soria, C.; Montalescot, G.; Weisel, J.W. Influence of fibrin network conformation and fibrin fiber diameter on fibrinolysis speed: Dynamic and structural approaches by confocal microscopy. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1354–1361. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sigley, J.; Pieters, M.; Helms, C.C.; Nagaswami, C.; Weisel, J.W.; Guthold, M. Fibrin Fiber Stiffness Is Strongly Affected by Fiber Diameter, but Not by Fibrinogen Glycation. Biophys. J. 2016, 110, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sigley, J.; Baker, S.R.; Helms, C.C.; Kinney, M.T.; Pieters, M.; Brubaker, P.H.; Cubcciotti, R.; Guthold, M. Nonuniform Internal Structure of Fibrin Fibers: Protein Density and Bond Density Strongly Decrease with Increasing Diameter. Biomed. Res. Int. 2017, 2017, 6385628. [Google Scholar] [CrossRef] [PubMed]
- Zhmurov, A.; Protopopova, A.D.; Litvinov, R.I.; Zhukov, P.; Weisel, J.W.; Barsegov, V. Atomic Structural Models of Fibrin Oligomers. Structure 2018, 26, 857–868.e854. [Google Scholar] [CrossRef]
- Zhmurov, A.; Kononova, O.; Litvinov, R.I.; Dima, R.I.; Barsegov, V.; Weisel, J.W. Mechanical transition from alpha-helical coiled coils to beta-sheets in fibrin(ogen). J. Am. Chem. Soc. 2012, 134, 20396–20402. [Google Scholar] [CrossRef]
- Minin, K.A.; Zhmurov, A.; Marx, K.A.; Purohit, P.K.; Barsegov, V. Dynamic Transition from alpha-Helices to beta-Sheets in Polypeptide Coiled-Coil Motifs. J. Am. Chem. Soc. 2017, 139, 16168–16177. [Google Scholar] [CrossRef]
- Risman, R.A.; Belcher, H.A.; Ramanujam, R.K.; Weisel, J.W.; Hudson, N.E.; Tutwiler, V. Comprehensive Analysis of the Role of Fibrinogen and Thrombin in Clot Formation and Structure for Plasma and Purified Fibrinogen. Biomolecules 2024, 14, 230. [Google Scholar] [CrossRef]
- Wolberg, A.S. Thrombin generation and fibrin clot structure. Blood Rev. 2007, 21, 131–142. [Google Scholar] [CrossRef]
- Swanepoel, A.C.; Lindeque, B.G.; Swart, P.J.; Abdool, Z.; Pretorius, E. Estrogen causes ultrastructural changes of fibrin networks during the menstrual cycle: A qualitative investigation. Microsc. Res. Tech. 2014, 77, 594–601. [Google Scholar] [CrossRef]
- Navarro, S.; Díaz-Caballero, M.; Peccati, F.; Roldán-Martin, L.; Sodupe, M.; Ventura, S. Amyloid Fibrils Formed by Short Prion-Inspired Peptides Are Metalloenzymes. ACS Nano 2023, 17, 16968–16979. [Google Scholar] [CrossRef] [PubMed]
- Usuelli, M.; Ruzzi, V.; Buzzaccaro, S.; Nyström, G.; Piazza, R.; Mezzenga, R. Unraveling gelation kinetics, arrested dynamics and relaxation phenomena in filamentous colloids by photon correlation imaging. Soft Matter 2022, 18, 5632–5644. [Google Scholar] [CrossRef] [PubMed]
- Kurniawan, N.A.; van Kempen, T.H.S.; Sonneveld, S.; Rosalina, T.T.; Vos, B.E.; Jansen, K.A.; Peters, G.W.M.; van de Vosse, F.N.; Koenderink, G.H. Buffers Strongly Modulate Fibrin Self-Assembly into Fibrous Networks. Langmuir 2017, 33, 6342–6352. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Bower, K.; Fink, A.L. Synergistic effects of pesticides and metals on the fibrillation of alpha-synuclein: Implications for Parkinson’s disease. Neurotoxicology 2002, 23, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Villaverde, A.; Carrió, M.M. Protein aggregation in recombinant bacteria: Biological role of inclusion bodies. Biotechnol. Lett. 2003, 25, 1385–1395. [Google Scholar] [CrossRef]
- Ventura, S.; Villaverde, A. Protein quality in bacterial inclusion bodies. Trends Biotechnol. 2006, 24, 179–185. [Google Scholar] [CrossRef]
- Hockney, R.C. Recent developments in heterologous protein production in Escherichia coli. Trends Biotechnol. 1994, 12, 456–463. [Google Scholar] [CrossRef]
- Wållberg, F.; Sundström, H.; Ledung, E.; Hewitt, C.J.; Enfors, S.O. Monitoring and quantification of inclusion body formation in Escherichia coli by multi-parameter flow cytometry. Biotechnol. Lett. 2005, 27, 919–926. [Google Scholar] [CrossRef]
- Lee, Y.J.; Jung, K.H. Modulation of the tendency towards inclusion body formation of recombinant protein by the addition of glucose in the araBAD promoter system of Escherichia coli. J. Microbiol. Biotechnol. 2007, 17, 1898–1903. [Google Scholar]
- Kopp, J.; Spadiut, O. Inclusion Bodies: Status Quo and Perspectives. Methods Mol. Biol. 2023, 2617, 1–13. [Google Scholar] [CrossRef]
- Kachhawaha, K.; Singh, S.; Joshi, K.; Nain, P.; Singh, S.K. Bioprocessing of recombinant proteins from Escherichia coli inclusion bodies: Insights from structure-function relationship for novel applications. Prep. Biochem. Biotechnol. 2023, 53, 728–752. [Google Scholar] [CrossRef] [PubMed]
- Housmans, J.A.J.; Wu, G.; Schymkowitz, J.; Rousseau, F. A guide to studying protein aggregation. FEBS J. 2023, 290, 554–583. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Maji, S.K.; Sawaya, M.R.; Eisenberg, D.; Riek, R. Bacterial inclusion bodies contain amyloid-like structure. PLoS Biol. 2008, 6, e195. [Google Scholar] [CrossRef] [PubMed]
- de Groot, N.S.; Sabate, R.; Ventura, S. Amyloids in bacterial inclusion bodies. Trends Biochem. Sci. 2009, 34, 408–416. [Google Scholar] [CrossRef] [PubMed]
- García-Fruitós, E.; Sabate, R.; de Groot, N.S.; Villaverde, A.; Ventura, S. Biological role of bacterial inclusion bodies: A model for amyloid aggregation. FEBS J. 2011, 278, 2419–2427. [Google Scholar] [CrossRef]
- de Marco, A.; Ferrer-Miralles, N.; Garcia-Fruitós, E.; Mitraki, A.; Peternel, S.; Rinas, U.; Trujillo-Roldán, M.A.; Valdez-Cruz, N.A.; Vázquez, E.; Villaverde, A. Bacterial inclusion bodies are industrially exploitable amyloids. FEMS Microbiol. Rev. 2019, 43, 53–72. [Google Scholar] [CrossRef]
- Carratalá, J.V.; Cisneros, A.; Hellman, E.; Villaverde, A.; Ferrer-Miralles, N. Insoluble proteins catch heterologous soluble proteins into inclusion bodies by intermolecular interaction of aggregating peptides. Microb. Cell Fact. 2021, 20, 30. [Google Scholar] [CrossRef]
- Cano-Garrido, O.; Rodríguez-Carmona, E.; Díez-Gil, C.; Vázquez, E.; Elizondo, E.; Cubarsi, R.; Seras-Franzoso, J.; Corchero, J.L.; Rinas, U.; Ratera, I.; et al. Supramolecular organization of protein-releasing functional amyloids solved in bacterial inclusion bodies. Acta Biomater. 2013, 9, 6134–6142. [Google Scholar] [CrossRef]
- Villaverde, A.; Corchero, J.L.; Seras-Franzoso, J.; Garcia-Fruitos, E. Functional protein aggregates: Just the tip of the iceberg. Nanomedicine 2015, 10, 2881–2891. [Google Scholar] [CrossRef]
- Hrabarova, E.; Belkova, M.; Koszagova, R.; Nahalka, J. Pull-Down Into Active Inclusion Bodies and Their Application in the Detection of (Poly)-Phosphates and Metal-Ions. Front. Bioeng. Biotechnol. 2022, 10, 833192. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef]
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Iadanza, M.G.; Jackson, M.P.; Hewitt, E.W.; Ranson, N.A.; Radford, S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Biswas, A.; Radhakrishna, M. Advanced computational approaches to understand protein aggregation. Biophys. Rev. 2024, 5, 021302. [Google Scholar] [CrossRef]
- Hauser, C.A.E.; Maurer-Stroh, S.; Martins, I.C. Amyloid-based nanosensors and nanodevices. Chem. Soc. Rev. 2014, 43, 5326–5345. [Google Scholar] [CrossRef]
- Ke, P.C.; Zhou, R.; Serpell, L.C.; Riek, R.; Knowles, T.P.J.; Lashuel, H.A.; Gazit, E.; Hamley, I.W.; Davis, T.P.; Fandrich, M.; et al. Half a century of amyloids: Past, present and future. Chem. Soc. Rev. 2020, 49, 5473–5509. [Google Scholar] [CrossRef]
- Morris, K.L.; Serpell, L.C. X-ray fibre diffraction studies of amyloid fibrils. Methods Mol. Biol. 2012, 849, 121–135. [Google Scholar] [CrossRef]
- Al-Garawi, Z.S.; Morris, K.L.; Marshall, K.E.; Eichler, J.; Serpell, L.C. The diversity and utility of amyloid fibrils formed by short amyloidogenic peptides. Interface Focus. 2017, 7, 20170027. [Google Scholar] [CrossRef]
- Serpell, L.C.; Sunde, M.; Benson, M.D.; Tennent, G.A.; Pepys, M.B.; Fraser, P.E. The protofilament substructure of amyloid fibrils. J. Mol. Biol. 2000, 300, 1033–1039. [Google Scholar] [CrossRef]
- Makin, O.S.; Atkins, E.; Sikorski, P.; Johansson, J.; Serpell, L.C. Molecular basis for amyloid fibril formation and stability. Proc. Natl. Acad. Sci. USA 2005, 102, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Cremades, N.; Dobson, C.M. The contribution of biophysical and structural studies of protein self-assembly to the design of therapeutic strategies for amyloid diseases. Neurobiol. Dis. 2018, 109, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Sunde, M.; Blake, C.C.F. From the globular to the fibrous state: Protein structure and structural conversion in amyloid formation. Q. Rev. Biophys. 1998, 31, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Leapman, R.D.; Guo, Z.; Yau, W.M.; Mattson, M.P.; Tycko, R. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science 2005, 307, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Meier, B.H.; Böckmann, A. The structure of fibrils from ‘misfolded’ proteins. Curr. Opin. Struct. Biol. 2015, 30, 43–49. [Google Scholar] [CrossRef]
- Lutter, L.; Serpell, C.J.; Tuite, M.F.; Xue, W.F. The molecular lifecycle of amyloid—Mechanism of assembly, mesoscopic organisation, polymorphism, suprastructures, and biological consequences. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 140257. [Google Scholar] [CrossRef]
- Stefani, M. Structural polymorphism of amyloid oligomers and fibrils underlies different fibrillization pathways: Immunogenicity and cytotoxicity. Curr. Protein Pept. Sci. 2010, 11, 343–354. [Google Scholar] [CrossRef]
- Tycko, R. Physical and structural basis for polymorphism in amyloid fibrils. Protein Sci. 2014, 23, 1528–1539. [Google Scholar] [CrossRef]
- Riek, R. The Three-Dimensional Structures of Amyloids. Cold Spring Harb. Perspect. Biol. 2017, 9, a023572. [Google Scholar] [CrossRef]
- Wilkinson, M.; Xu, Y.; Thacker, D.; Taylor, A.I.P.; Fisher, D.G.; Gallardo, R.U.; Radford, S.E.; Ranson, N.A. Structural evolution of fibril polymorphs during amyloid assembly. Cell 2023, 186, 5798–5811.e5726. [Google Scholar] [CrossRef]
- Guenther, E.L.; Ge, P.; Trinh, H.; Sawaya, M.R.; Cascio, D.; Boyer, D.R.; Gonen, T.; Zhou, Z.H.; Eisenberg, D.S. Atomic-level evidence for packing and positional amyloid polymorphism by segment from TDP-43 RRM2. Nat. Struct. Mol. Biol. 2018, 25, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Ostermeier, L.; de Oliveira, G.A.P.; Dzwolak, W.; Silva, J.L.; Winter, R. Exploring the polymorphism, conformational dynamics and function of amyloidogenic peptides and proteins by temperature and pressure modulation. Biophys. Chem. 2021, 268, 106506. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C. Conformational strains of pathogenic amyloid proteins in neurodegenerative diseases. Nat. Rev. Neurosci. 2022, 23, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C. Molecular rules governing the structural polymorphism of amyloid fibrils in neurodegenerative diseases. Structure 2023, 31, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.I.P.; Staniforth, R.A. General Principles Underpinning Amyloid Structure. Front. Neurosci. 2022, 16, 878869. [Google Scholar] [CrossRef]
- Lövestam, S.; Li, D.; Wagstaff, J.L.; Kotecha, A.; Kimanius, D.; McLaughlin, S.H.; Murzin, A.G.; Freund, S.M.V.; Goedert, M.; Scheres, S.H.W. Disease-specific tau filaments assemble via polymorphic intermediates. Nature 2024, 625, 119–125. [Google Scholar] [CrossRef]
- Tycko, R. Amyloid polymorphism: Structural basis and neurobiological relevance. Neuron 2015, 86, 632–645. [Google Scholar] [CrossRef]
- Aguzzi, A.; Calella, A.M. Prions: Protein aggregation and infectious diseases. Physiol. Rev. 2009, 89, 1105–1152. [Google Scholar] [CrossRef]
- Igel-Egalon, A.; Béringue, V.; Rezaei, H.; Sibille, P. Prion Strains and Transmission Barrier Phenomena. Pathogens 2018, 7, 5. [Google Scholar] [CrossRef]
- Arifin, M.I.; Hannaoui, S.; Chang, S.C.; Thapa, S.; Schatzl, H.M.; Gilch, S. Cervid Prion Protein Polymorphisms: Role in Chronic Wasting Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22, 2271. [Google Scholar] [CrossRef]
- Igel, A.; Fornara, B.; Rezaei, H.; Béringue, V. Prion assemblies: Structural heterogeneity, mechanisms of formation, and role in species barrier. Cell Tissue Res. 2023, 392, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Shoup, D.; Priola, S.A. Cell biology of prion strains in vivo and in vitro. Cell Tissue Res. 2022, 392, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Rajendran, L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009, 64, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Ashe, K.H.; Aguzzi, A. Prions, prionoids and pathogenic proteins in Alzheimer disease. Prion 2013, 7, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Gosset, P.; Camu, W.; Raoul, C.; Mezghrani, A. Prionoids in amyotrophic lateral sclerosis. Brain Commun. 2022, 4, fcac145. [Google Scholar] [CrossRef]
- Hafner Bratkovič, I. Prions, prionoid complexes and amyloids: The bad, the good and something in between. Swiss Med. Wkly. 2017, 147, w14424. [Google Scholar] [CrossRef]
- Wells, C.; Brennan, S.E.; Keon, M.; Saksena, N.K. Prionoid Proteins in the Pathogenesis of Neurodegenerative Diseases. Front. Mol. Neurosci. 2019, 12, 271. [Google Scholar] [CrossRef]
- Gasset-Rosa, F.; Coquel, A.S.; Moreno-Del Alamo, M.; Chen, P.; Song, X.; Serrano, A.M.; Fernandez-Tresguerres, M.E.; Moreno-Diaz de la Espina, S.; Lindner, A.B.; Giraldo, R. Direct assessment in bacteria of prionoid propagation and phenotype selection by Hsp70 chaperone. Mol. Microbiol. 2014, 91, 1070–1087. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef]
- Li, D.; Liu, C. Structural Diversity of Amyloid Fibrils and Advances in Their Structure Determination. Biochemistry 2020, 59, 639–646. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.; Saibil, H.R. Cryo-EM of amyloid fibrils and cellular aggregates. Curr. Opin. Struct. Biol. 2019, 58, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.J.; Fitzpatrick, A.W.; Meehan, S.; Mott, H.R.; Vendruscolo, M.; Dobson, C.M.; Welland, M.E. Role of intermolecular forces in defining material properties of protein nanofibrils. Science 2007, 318, 1900–1903. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, P.A.; Rolich, V.I.; Vezo, O.S.; Belousov, M.V.; Bondarev, S.A.; Zhouravleva, G.A.; Kasyanenko, N.A. Amyloid fibril length distribution from dynamic light scattering data. Eur. Biophys. J. 2022, 51, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Modi, P.; Sharma, G.; Deep, S. Kinetics theories to understand the mechanism of aggregation of a protein and to design strategies for its inhibition. Biophys. Chem. 2021, 278, 106665. [Google Scholar] [CrossRef]
- Kumar, E.K.; Haque, N.; Prabhu, N.P. Kinetics of protein fibril formation: Methods and mechanisms. Int. J. Biol. Macromol. 2017, 100, 3–10. [Google Scholar] [CrossRef]
- Horvath, I.; Welte, H.; Schmit, J.D.; Kovermann, M.; Wittung-Stafshede, P. Distinct growth regimes of alpha-synuclein amyloid elongation. Biophys. J. 2023, 122, 2556–2563. [Google Scholar] [CrossRef]
- Knowles, T.P.J.; Waudby, C.A.; Devlin, G.L.; Cohen, S.I.; Aguzzi, A.; Vendruscolo, M.; Terentjev, E.M.; Welland, M.E.; Dobson, C.M. An analytical solution to the kinetics of breakable filament assembly. Science 2009, 326, 1533–1537. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Wu, J.; Cao, C.; Loch, R.A.; Tiiman, A.; Luo, J. Single-molecule studies of amyloid proteins: From biophysical properties to diagnostic perspectives. Q. Rev. Biophys. 2020, 53, e12. [Google Scholar] [CrossRef]
- Michaels, T.C.T.; Saric, A.; Habchi, J.; Chia, S.; Meisl, G.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Chemical Kinetics for Bridging Molecular Mechanisms and Macroscopic Measurements of Amyloid Fibril Formation. Annu. Rev. Phys. Chem. 2018, 69, 273–298. [Google Scholar] [CrossRef]
- Meisl, G.; Michaels, T.C.T.; Linse, S.; Knowles, T.P.J. Kinetic Analysis of Amyloid Formation. Methods Mol. Biol. 2018, 1779, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Dear, A.J.; Michaels, T.C.T.; Meisl, G.; Klenerman, D.; Wu, S.; Perrett, S.; Linse, S.; Dobson, C.M.; Knowles, T.P.J. Kinetic diversity of amyloid oligomers. Proc. Natl. Acad. Sci. USA 2020, 117, 12087–12094. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Knowles, T.P.J.; Linse, S. On the lag phase in amyloid fibril formation. Phys. Chem. Chem. Phys. 2015, 17, 7606–7618. [Google Scholar] [CrossRef] [PubMed]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef]
- Bucciantini, M.; Calloni, G.; Chiti, F.; Formigli, L.; Nosi, D.; Dobson, C.M.; Stefani, M. Prefibrillar amyloid protein aggregates share common features of cytotoxicity. J. Biol. Chem. 2004, 279, 31374–31382. [Google Scholar] [CrossRef] [PubMed]
- Michaels, T.C.T.; Saric, A.; Meisl, G.; Heller, G.T.; Curk, S.; Arosio, P.; Linse, S.; Dobson, C.M.; Vendruscolo, M.; Knowles, T.P.J. Thermodynamic and kinetic design principles for amyloid-aggregation inhibitors. Proc. Natl. Acad. Sci. USA 2020, 117, 24251–24257. [Google Scholar] [CrossRef]
- Sulatskaya, A.I.; Rodina, N.P.; Sulatsky, M.I.; Povarova, O.I.; Antifeeva, I.A.; Kuznetsova, I.M.; Turoverov, K.K. Investigation of alpha-Synuclein Amyloid Fibrils Using the Fluorescent Probe Thioflavin T. Int. J. Mol. Sci. 2018, 19, 2486. [Google Scholar] [CrossRef]
- Pirt, S.J. Principles of microbe and cell cultivation; Wiley: London, UK, 1975. [Google Scholar]
- Pretorius, E.; Vlok, M.; Venter, C.; Bezuidenhout, J.A.; Laubscher, G.J.; Steenkamp, J.; Kell, D.B. Persistent clotting protein pathology in Long COVID/ Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc. Diabetol. 2021, 20, 172. [Google Scholar] [CrossRef]
- Xia, Q.; Liao, L.; Cheng, D.; Duong, D.M.; Gearing, M.; Lah, J.J.; Levey, A.I.; Peng, J. Proteomic identification of novel proteins associated with Lewy bodies. Front. Biosci. 2008, 13, 3850–3856. [Google Scholar] [CrossRef]
- Tsamourgelis, A.; Swann, P.; Chouliaras, L.; O’Brien, J.T. From protein biomarkers to proteomics in dementia with Lewy Bodies. Ageing Res. Rev. 2023, 83, 101771. [Google Scholar] [CrossRef]
- Horvath, I.; Wittung-Stafshede, P. Cross-talk between amyloidogenic proteins in type-2 diabetes and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2016, 113, 12473–12477. [Google Scholar] [CrossRef] [PubMed]
- Werner, T.; Horvath, I.; Wittung-Stafshede, P. Crosstalk between Alpha-Synuclein and Other Human and Non-Human Amyloidogenic Proteins: Consequences for Amyloid Formation in Parkinson’s Disease. J. Park. Dis. 2020, 10, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Wittung-Stafshede, P. Gut power: Modulation of human amyloid formation by amyloidogenic proteins in the gastrointestinal tract. Curr. Opin. Struct. Biol. 2022, 72, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Slamova, I.; Adib, R.; Ellmerich, S.; Golos, M.R.; Gilbertson, J.A.; Botcher, N.; Canetti, D.; Taylor, G.W.; Rendell, N.; Tennent, G.A.; et al. Plasmin activity promotes amyloid deposition in a transgenic model of human transthyretin amyloidosis. Nat. Commun. 2021, 12, 7112. [Google Scholar] [CrossRef] [PubMed]
- Bhasne, K.; Mukhopadhyay, S. Formation of Heterotypic Amyloids: Alpha-Synuclein in Co-Aggregation. Proteomics 2018, 18, e1800059. [Google Scholar] [CrossRef]
- Bhasne, K.; Sebastian, S.; Jain, N.; Mukhopadhyay, S. Synergistic Amyloid Switch Triggered by Early Heterotypic Oligomerization of Intrinsically Disordered alpha-Synuclein and Tau. J. Mol. Biol. 2018, 430, 2508–2520. [Google Scholar] [CrossRef]
- Hu, R.; Zhang, M.; Chen, H.; Jiang, B.; Zheng, J. Cross-Seeding Interaction between beta-Amyloid and Human Islet Amyloid Polypeptide. ACS Chem. Neurosci. 2015, 6, 1759–1768. [Google Scholar] [CrossRef]
- Ivanova, M.I.; Lin, Y.; Lee, Y.H.; Zheng, J.; Ramamoorthy, A. Biophysical processes underlying cross-seeding in amyloid aggregation and implications in amyloid pathology. Biophys. Chem. 2021, 269, 106507. [Google Scholar] [CrossRef]
- Lundmark, K.; Westermark, G.T.; Olsen, A.; Westermark, P. Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism. Proc. Natl. Acad. Sci. USA 2005, 102, 6098–6102. [Google Scholar] [CrossRef]
- Morales, R.; Moreno-Gonzalez, I.; Soto, C. Cross-Seeding of Misfolded Proteins: Implications for Etiology and Pathogenesis of Protein Misfolding Diseases. PLoS Pathog. 2013, 9, e1003537. [Google Scholar] [CrossRef]
- Ono, K.; Takahashi, R.; Ikeda, T.; Yamada, M. Cross-seeding effects of amyloid beta-protein and alpha-synuclein. J. Neurochem. 2012, 122, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, M.E.; Paulsson, J.F.; Schultz, S.W.; Ingelsson, M.; Westermark, P.; Westermark, G.T. In vivo seeding and cross-seeding of localized amyloidosis: A molecular link between type 2 diabetes and Alzheimer disease. Am. J. Pathol. 2015, 185, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Subedi, S.; Sasidharan, S.; Nag, N.; Saudagar, P.; Tripathi, T. Amyloid Cross-Seeding: Mechanism, Implication, and Inhibition. Molecules 2022, 27, 1776. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hu, R.; Chen, H.; Chang, Y.; Ma, J.; Liang, G.; Mi, J.; Wang, Y.; Zheng, J. Polymorphic cross-seeding amyloid assemblies of amyloid-beta and human islet amyloid polypeptide. Phys. Chem. Chem. Phys. 2015, 17, 23245–23256. [Google Scholar] [CrossRef] [PubMed]
- Ge, W.Y.; Deng, X.; Shi, W.P.; Lin, W.J.; Chen, L.L.; Liang, H.; Wang, X.T.; Zhang, T.D.; Zhao, F.Z.; Guo, W.H.; et al. Amyloid Protein Cross-Seeding Provides a New Perspective on Multiple Diseases In Vivo. Biomacromolecules 2023, 24, 1–18. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, M.; Liu, Y.; Zhang, D.; Tang, Y.; Ren, B.; Zheng, J. Dual amyloid cross-seeding reveals steric zipper-facilitated fibrillization and pathological links between protein misfolding diseases. J. Mater. Chem. B 2021, 9, 3300–3316. [Google Scholar] [CrossRef]
- Ren, B.; Zhang, Y.; Zhang, M.; Liu, Y.; Zhang, D.; Gong, X.; Feng, Z.; Tang, J.; Chang, Y.; Zheng, J. Fundamentals of cross-seeding of amyloid proteins: An introduction. J. Mater. Chem. B 2019, 7, 7267–7282. [Google Scholar] [CrossRef]
- Ren, B.; Hu, R.; Zhang, M.; Liu, Y.; Xu, L.; Jiang, B.; Ma, J.; Ma, B.; Nussinov, R.; Zheng, J. Experimental and Computational Protocols for Studies of Cross-Seeding Amyloid Assemblies. Methods Mol. Biol. 2018, 1777, 429–447. [Google Scholar] [CrossRef]
- Hong, J.Y.; Wang, J.Y.; Yue, H.W.; Zhang, X.L.; Zhang, S.X.; Jiang, L.L.; Hu, H.Y. Coaggregation of polyglutamine (polyQ) proteins is mediated by polyQ-tract interactions and impairs cellular proteostasis. Acta Biochim. Biophys. Sin. 2023, 55, 736–748. [Google Scholar] [CrossRef]
- Chaudhuri, P.; Prajapati, K.P.; Anand, B.G.; Dubey, K.; Kar, K. Amyloid cross-seeding raises new dimensions to understanding of amyloidogenesis mechanism. Ageing Res. Rev. 2019, 56, 100937. [Google Scholar] [CrossRef]
- Murakami, K.; Ono, K. Interactions of amyloid coaggregates with biomolecules and its relevance to neurodegeneration. FASEB J. 2022, 36, e22493. [Google Scholar] [CrossRef] [PubMed]
- Westermark, G.T.; Fandrich, M.; Lundmark, K.; Westermark, P. Noncerebral Amyloidoses: Aspects on Seeding, Cross-Seeding, and Transmission. Cold Spring Harb. Perspect. Med. 2018, 8, a024323. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Matsuzaki, K.; Yanagisawa, K. Cross-seeding of wild-type and hereditary variant-type amyloid beta-proteins in the presence of gangliosides. J. Neurochem. 2005, 95, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Peim, A.; Hortschansky, P.; Christopeit, T.; Schroeckh, V.; Richter, W.; Fandrich, M. Mutagenic exploration of the cross-seeding and fibrillation propensity of Alzheimer’s beta-amyloid peptide variants. Protein Sci. 2006, 15, 1801–1805. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A.; Malmström, S.; Westermark, P. Signs of cross-seeding: Aortic medin amyloid as a trigger for protein AA deposition. Amyloid 2011, 18, 229–234. [Google Scholar] [CrossRef]
- Zhou, Y.; Smith, D.; Leong, B.J.; Brannstrom, K.; Almqvist, F.; Chapman, M.R. Promiscuous cross-seeding between bacterial amyloids promotes interspecies biofilms. J. Biol. Chem. 2012, 287, 35092–35103. [Google Scholar] [CrossRef]
- Hartman, K.; Brender, J.R.; Monde, K.; Ono, A.; Evans, M.L.; Popovych, N.; Chapman, M.R.; Ramamoorthy, A. Bacterial curli protein promotes the conversion of PAP248-286 into the amyloid SEVI: Cross-seeding of dissimilar amyloid sequences. PeerJ 2013, 1, e5. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, R.; Liang, G.; Chang, Y.; Sun, Y.; Peng, Z.; Zheng, J. Structural and energetic insight into the cross-seeding amyloid assemblies of human IAPP and rat IAPP. J. Phys. Chem. B 2014, 118, 7026–7036. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, R.; Chen, H.; Gong, X.; Zhou, F.; Zhang, L.; Zheng, J. Polymorphic Associations and Structures of the Cross-Seeding of Abeta1-42 and hIAPP1-37 Polypeptides. J. Chem. Inf. Model. 2015, 55, 1628–1639. [Google Scholar] [CrossRef]
- Alexandrov, A.I.; Serpionov, G.V.; Kushnirov, V.V.; Ter-Avanesyan, M.D. Wild type huntingtin toxicity in yeast: Implications for the role of amyloid cross-seeding in polyQ diseases. Prion 2016, 10, 221–227. [Google Scholar] [CrossRef][Green Version]
- Hu, R.; Ren, B.; Zhang, M.; Chen, H.; Liu, Y.; Liu, L.; Gong, X.; Jiang, B.; Ma, J.; Zheng, J. Seed-Induced Heterogeneous Cross-Seeding Self-Assembly of Human and Rat Islet Polypeptides. ACS Omega 2017, 2, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hu, R.; Ren, B.; Chen, H.; Jiang, B.; Ma, J.; Zheng, J. Molecular Understanding of Abeta-hIAPP Cross-Seeding Assemblies on Lipid Membranes. ACS Chem. Neurosci. 2017, 8, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Anand, B.G.; Prajapati, K.P.; Kar, K. Abeta(1-40) mediated aggregation of proteins and metabolites unveils the relevance of amyloid cross-seeding in amyloidogenesis. Biochem. Biophys. Res. Commun. 2018, 501, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Ho, G.; Takamatsu, Y.; Wada, R.; Sugama, S.; Takenouchi, T.; Waragai, M.; Masliah, E. Possible Role of Amyloid Cross-Seeding in Evolvability and Neurodegenerative Disease. J. Park. Dis. 2019, 9, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Bardin, T.; Daskalov, A.; Barrouilhet, S.; Granger-Farbos, A.; Salin, B.; Blancard, C.; Kauffmann, B.; Saupe, S.J.; Coustou, V. Partial Prion Cross-Seeding between Fungal and Mammalian Amyloid Signaling Motifs. mBio 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Daskalov, A.; Martinez, D.; Coustou, V.; El Mammeri, N.; Berbon, M.; Andreas, L.B.; Bardiaux, B.; Stanek, J.; Noubhani, A.; Kauffmann, B.; et al. Structural and molecular basis of cross-seeding barriers in amyloids. Proc. Natl. Acad. Sci. USA 2021, 118, e2014085118. [Google Scholar] [CrossRef]
- Koloteva-Levine, N.; Aubrey, L.D.; Marchante, R.; Purton, T.J.; Hiscock, J.R.; Tuite, M.F.; Xue, W.F. Amyloid particles facilitate surface-catalyzed cross-seeding by acting as promiscuous nanoparticles. Proc. Natl. Acad. Sci. USA 2021, 118, e2104148118. [Google Scholar] [CrossRef]
- Nirwal, S.; Bharathi, V.; Patel, B.K. Amyloid-like aggregation of bovine serum albumin at physiological temperature induced by cross-seeding effect of HEWL amyloid aggregates. Biophys. Chem. 2021, 278, 106678. [Google Scholar] [CrossRef]
- Vaneyck, J.; Segers-Nolten, I.; Broersen, K.; Claessens, M.M.A.E. Cross-seeding of alpha-synuclein aggregation by amyloid fibrils of food proteins. J. Biol. Chem. 2021, 296, 100358. [Google Scholar] [CrossRef]
- Yuzu, K.; Yamamoto, N.; Noji, M.; So, M.; Goto, Y.; Iwasaki, T.; Tsubaki, M.; Chatani, E. Multistep Changes in Amyloid Structure Induced by Cross-Seeding on a Rugged Energy Landscape. Biophys. J. 2021, 120, 284–295. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, D.; Liu, Y.; Zhang, Y.; Zhou, Y.; Chang, Y.; Zheng, B.; Xu, A.; Zheng, J. A new strategy to reconcile amyloid cross-seeding and amyloid prevention in a binary system of alpha-synuclein fragmental peptide and hIAPP. Protein Sci. 2022, 31, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhang, D.; Chang, Y.; Zheng, J. Atrial Natriuretic Peptide Associated with Cardiovascular Diseases Inhibits Amyloid-beta Aggregation via Cross-Seeding. ACS Chem. Neurosci. 2023, 14, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhang, D.; Zheng, J. Repurposing Antimicrobial Protegrin-1 as a Dual-Function Amyloid Inhibitor via Cross-seeding. ACS Chem. Neurosci. 2023, 14, 3143–3155. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Zhang, X.; Yan, J.; Xu, H.; Zhao, W.; Ding, F.; Huang, F.; Sun, Y. Computational Investigation of Coaggregation and Cross-Seeding between Abeta and hIAPP Underpinning the Cross-Talk in Alzheimer’s Disease and Type 2 Diabetes. J. Chem. Inf. Model. 2024, 64, 5303–5316. [Google Scholar] [CrossRef]
- Wang, Y.; Bergstrom, J.; Ingelsson, M.; Westermark, G.T. Studies on alpha-synuclein and islet amyloid polypeptide interaction. Front. Mol. Biosci. 2023, 10, 1080112. [Google Scholar] [CrossRef]
- Wang, Y.; Westermark, G.T. The Amyloid Forming Peptides Islet Amyloid Polypeptide and Amyloid beta Interact at the Molecular Level. Int. J. Mol. Sci. 2021, 22, 11153. [Google Scholar] [CrossRef]
- Chen, S.; Berthelier, V.; Yang, W.; Wetzel, R. Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J. Mol. Biol. 2001, 311, 173–182. [Google Scholar] [CrossRef]
- Chen, S.; Berthelier, V.; Hamilton, J.B.; O’Nuallain, B.; Wetzel, R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry 2002, 41, 7391–7399. [Google Scholar] [CrossRef]
- Schneider, R.; Schumacher, M.C.; Mueller, H.; Nand, D.; Klaukien, V.; Heise, H.; Riedel, D.; Wolf, G.; Behrmann, E.; Raunser, S.; et al. Structural characterization of polyglutamine fibrils by solid-state NMR spectroscopy. J. Mol. Biol. 2011, 412, 121–136. [Google Scholar] [CrossRef]
- Punihaole, D.; Workman, R.J.; Hong, Z.; Madura, J.D.; Asher, S.A. Polyglutamine Fibrils: New Insights into Antiparallel beta-Sheet Conformational Preference and Side Chain Structure. J. Phys. Chem. B 2016, 120, 3012–3026. [Google Scholar] [CrossRef]
- Huang, R.K.; Baxa, U.; Aldrian, G.; Ahmed, A.B.; Wall, J.S.; Mizuno, N.; Antzutkin, O.; Steven, A.C.; Kajava, A.V. Conformational switching in PolyGln amyloid fibrils resulting from a single amino acid insertion. Biophys. J. 2014, 106, 2134–2142. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B. Towards a unifying, systems biology understanding of large-scale cellular death and destruction caused by poorly liganded iron: Parkinson’s, Huntington’s, Alzheimer’s, prions, bactericides, chemical toxicology and others as examples. Arch. Toxicol. 2010, 577, 825–889. [Google Scholar]
- Bondarev, S.A.; Antonets, K.S.; Kajava, A.V.; Nizhnikov, A.A.; Zhouravleva, G.A. Protein Co-Aggregation Related to Amyloids: Methods of Investigation, Diversity, and Classification. Int. J. Mol. Sci. 2018, 19, 2292. [Google Scholar] [CrossRef] [PubMed]
- Kajava, A.V.; Baxa, U.; Steven, A.C. Beta arcades: Recurring motifs in naturally occurring and disease-related amyloid fibrils. FASEB J. 2010, 24, 1311–1319. [Google Scholar] [CrossRef]
- Luckgei, N.; Schutz, A.K.; Bousset, L.; Habenstein, B.; Sourigues, Y.; Gardiennet, C.; Meier, B.H.; Melki, R.; Bockmann, A. The conformation of the prion domain of Sup35p in isolation and in the full-length protein. Angew. Chem. Int. Ed. Engl. 2013, 52, 12741–12744. [Google Scholar] [CrossRef]
- Lührs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Dobeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer’s amyloid-beta(1-42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef]
- Sanchez de Groot, N.; Torrent, M.; Villar-Piqué, A.; Lang, B.; Ventura, S.; Gsponer, J.; Babu, M.M. Evolutionary selection for protein aggregation. Biochem. Soc. Trans. 2012, 40, 1032–1037. [Google Scholar] [CrossRef]
- Weirich, F.; Gremer, L.; Mirecka, E.A.; Schiefer, S.; Hoyer, W.; Heise, H. Structural Characterization of Fibrils from Recombinant Human Islet Amyloid Polypeptide by Solid-State NMR: The Central FGAILS Segment Is Part of the beta-Sheet Core. PLoS ONE 2016, 11, e0161243. [Google Scholar] [CrossRef]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef]
- Kajava, A.V.; Klopffleisch, K.; Chen, S.; Hofmann, K. Evolutionary link between metazoan RHIM motif and prion-forming domain of fungal heterokaryon incompatibility factor HET-s/HET-s. Sci. Rep. 2014, 4, 7436. [Google Scholar] [CrossRef]
- Azizyan, R.A.; Wang, W.; Anikeenko, A.; Radkova, Z.; Bakulina, A.; Garro, A.; Charlier, L.; Dumas, C.; Ventura, S.; Kajava, A.V. Amyloidogenicity as a driving force for the formation of functional oligomers. J. Struct. Biol. 2020, 212, 107604. [Google Scholar] [CrossRef]
- Ahn, H.J.; Zamolodchikov, D.; Cortes-Canteli, M.; Norris, E.H.; Glickman, J.F.; Strickland, S. Alzheimer’s disease peptide beta-amyloid interacts with fibrinogen and induces its oligomerization. Proc. Natl. Acad. Sci. USA 2010, 107, 21812–21817. [Google Scholar] [CrossRef] [PubMed]
- Biza, K.V.; Nastou, K.C.; Tsiolaki, P.L.; Mastrokalou, C.V.; Hamodrakas, S.J.; Iconomidou, V.A. The amyloid interactome: Exploring protein aggregation. PLoS ONE 2017, 12, e0173163. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B.; Pretorius, E. Proteomic evidence for amyloidogenic cross-seeding in fibrinaloid microclots. bioRxiv 2024. [Google Scholar] [CrossRef]
- Kruger, A.; Vlok, M.; Turner, S.; Venter, C.; Laubscher, G.J.; Kell, D.B.; Pretorius, E. Proteomics of fibrin amyloid microclots in Long COVID/ Post-Acute Sequelae of COVID-19 (PASC) shows many entrapped pro-inflammatory molecules that may also contribute to a failed fibrinolytic system. Cardiovasc. Diabetol. 2022, 21, 190. [Google Scholar] [CrossRef] [PubMed]
- Hortin, G.L.; Sviridov, D.; Anderson, N.L. High-abundance polypeptides of the human plasma proteome comprising the top 4 logs of polypeptide abundance. Clin. Chem. 2008, 54, 1608–1616. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell. Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef]
- Khan, S.A.; Joyce, J.; Tsuda, T. Quantification of active and total transforming growth factor-beta levels in serum and solid organ tissues by bioassay. BMC Res. Notes 2012, 5, 636. [Google Scholar] [CrossRef]
- Wang, F.; Yang, C.; Song, Y.; Jiang, Y.; Ding, Z. Periostin gene polymorphisms, protein levels and risk of incident coronary artery disease. Mol. Biol. Rep. 2012, 39, 359–367. [Google Scholar] [CrossRef]
- Morgan, B.P.; Campbell, A.K.; Luzio, J.P.; Siddle, K. Immunoradiometric assay for human complement component C9 utilising monoclonal antibodies. Clin. Chim. Acta 1983, 134, 85–94. [Google Scholar] [CrossRef]
- Weeks, I.; Morgan, B.P.; Campbell, A.K.; Woodhead, J.S. Measurement of C9 concentrations using an immunochemiluminometric assay. J. Immunol. Methods 1985, 80, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Kopp, A.; Hebecker, M.; Svobodova, E.; Jozsi, M. Factor H: A complement regulator in health and disease, and a mediator of cellular interactions. Biomolecules 2012, 2, 46–75. [Google Scholar] [CrossRef] [PubMed]
- Refetoff, S. Thyroid Hormone Serum Transport Proteins. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Kanai, M.; Raz, A.; Goodman, D.S. Retinol-binding protein: The transport protein for vitamin A in human plasma. J. Clin. Investig. 1968, 47, 2025–2044. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, L.; Tian, Y.; Zhang, T.; Duan, G.; Liu, Y.; Yin, Y.; Hua, D.; Qi, X.; Mao, Y. Serum Chemokine CXCL7 as a Diagnostic Biomarker for Colorectal Cancer. Front. Oncol. 2019, 9, 921. [Google Scholar] [CrossRef]
- Visser, M.; Heitmeier, S.; Ten Cate, H.; Spronk, H.M.H. Role of Factor XIa and Plasma Kallikrein in Arterial and Venous Thrombosis. Thromb. Haemost. 2020, 120, 883–993. [Google Scholar] [CrossRef]
- Nielsen, C.T.; Lood, C.; Østergaard, O.; Iversen, L.V.; Voss, A.; Bengtsson, A.; Jacobsen, S.; Heegaard, N.H.H. Plasma levels of galectin-3-binding protein reflect type I interferon activity and are increased in patients with systemic lupus erythematosus. Lupus Sci. Med. 2014, 1, e000026. [Google Scholar] [CrossRef]
- Buda, V.; Andor, M.; Cristescu, C.; Tomescu, M.C.; Muntean, D.M.; Baibata, D.E.; Bordejevic, D.A.; Danciu, C.; Dalleur, O.; Coricovac, D.; et al. Thrombospondin-1 Serum Levels In Hypertensive Patients with Endothelial Dysfunction after One Year Of Treatment with Perindopril. Drug Des. Dev. Ther. 2019, 13, 3515–3526. [Google Scholar] [CrossRef]
- Kalaidopoulou Nteak, S.; Völlmy, F.; Lukassen, M.V.; van den Toorn, H.; den Boer, M.A.; Bondt, A.; van der Lans, S.P.A.; Haas, P.J.; van Zuilen, A.D.; Rooijakkers, S.H.M.; et al. Longitudinal Fluctuations in Protein Concentrations and Higher-Order Structures in the Plasma Proteome of Kidney Failure Patients Subjected to a Kidney Transplant. J. Proteome Res. 2024, 23, 2124–2136. [Google Scholar] [CrossRef]
- Park, J.C.; Han, S.H.; Lee, H.; Jeong, H.; Byun, M.S.; Bae, J.; Kim, H.; Lee, D.Y.; Yi, D.; Shin, S.A.; et al. Prognostic plasma protein panel for Abeta deposition in the brain in Alzheimer’s disease. Prog. Neurobiol. 2019, 183, 101690. [Google Scholar] [CrossRef]
- Son, S.M.; Nam, D.W.; Cha, M.Y.; Kim, K.H.; Byun, J.; Ryu, H.; Mook-Jung, I. Thrombospondin-1 prevents amyloid beta-mediated synaptic pathology in Alzheimer’s disease. Neurobiol. Aging 2015, 36, 3214–3227. [Google Scholar] [CrossRef]
- Drolle, E.; Hane, F.; Lee, B.; Leonenko, Z. Atomic force microscopy to study molecular mechanisms of amyloid fibril formation and toxicity in Alzheimer’s disease. Drug Metab. Rev. 2014, 46, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.J.; Park, J.W. Nanoaggregates Derived from Amyloid-beta and Alpha-synuclein Characterized by Sequential Quadruple Force Mapping. Nano Lett. 2021, 21, 3789–3797. [Google Scholar] [CrossRef] [PubMed]
- Vadukul, D.M.; Papp, M.; Thrush, R.J.; Wang, J.; Jin, Y.; Arosio, P.; Aprile, F.A. alpha-Synuclein Aggregation Is Triggered by Oligomeric Amyloid-beta 42 via Heterogeneous Primary Nucleation. J. Am. Chem. Soc. 2023, 145, 18276–18285. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Swasthi, H.M.; Bhasne, K.; Mahapatra, S.; Mukhopadhyay, S. Human Fibrinogen Inhibits Amyloid Assembly of Biofilm-Forming CsgA. Biochemistry 2018, 57, 6270–6273. [Google Scholar] [CrossRef]
- Najarzadeh, Z.; Nielsen, J.; Farzadfard, A.; Sereikaite, V.; Stromgaard, K.; Meyer, R.L.; Otzen, D.E. Human Fibrinogen Inhibits Amyloid Assembly of Most Phenol-Soluble Modulins from Staphylococcus aureus. ACS Omega 2021, 6, 21960–21970. [Google Scholar] [CrossRef]
- Niemietz, C.; Bezerra, F.; Almeida, M.R.; Guo, S.; Monia, B.P.; Saraiva, M.J.; Schutz, P.; Schmidt, H.H.; Zibert, A. SERPINA1 modulates expression of amyloidogenic transthyretin. Exp. Cell Res. 2020, 395, 112217. [Google Scholar] [CrossRef]
- Bezerra, F.; Niemietz, C.; Schmidt, H.H.J.; Zibert, A.; Guo, S.; Monia, B.P.; Goncalves, P.; Saraiva, M.J.; Almeida, M.R. In Vitro and In Vivo Effects of SerpinA1 on the Modulation of Transthyretin Proteolysis. Int. J. Mol. Sci. 2021, 22, 9488. [Google Scholar] [CrossRef]
- Niemietz, C.; Fleischhauer, L.; Sandfort, V.; Guttmann, S.; Zibert, A.; Schmidt, H.H. Hepatocyte-like cells reveal novel role of SERPINA1 in transthyretin amyloidosis. J. Cell Sci. 2018, 131, jcs.219824. [Google Scholar] [CrossRef]
- Berndsen, Z.T.; Cassidy, C.K. The Structure of ApoB100 from Human Low-density Lipoprotein. bioRxiv 2024. [Google Scholar] [CrossRef]
- Namba, Y.; Tsuchiya, H.; Ikeda, K. Apolipoprotein B immunoreactivity in senile plaque and vascular amyloids and neurofibrillary tangles in the brains of patients with Alzheimer’s disease. Neurosci. Lett. 1992, 134, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Grobler, C.; Maphumulo, S.C.; Grobbelaar, L.M.; Bredenkamp`, J.; Laubscher, J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B.; Pretorius, E. COVID-19: The Rollercoaster of Fibrin(ogen), D-dimer, von Willebrand Factor, P-selectin and Their Interactions with Endothelial Cells, Platelets and Erythrocytes. Int. J. Mol. Sci. 2020, 21, 5168. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Byun, M.S.; Yi, D.; Kim, M.J.; Jung, J.H.; Kong, N.; Jung, G.; Ahn, H.; Lee, J.Y.; Kang, K.M.; et al. Serum Adiponectin and In Vivo Brain Amyloid Deposition in Cognitively Normal Older Adults: A Cohort Study. Aging Dis. 2023, 14, 904–918. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.H.; Lam, K.S.; Cheng, O.Y.; Kwan, J.S.; Ho, P.W.; Cheng, K.K.; Chung, S.K.; Ho, J.W.; Guo, V.Y.; Xu, A. Adiponectin is protective against oxidative stress induced cytotoxicity in amyloid-beta neurotoxicity. PLoS ONE 2012, 7, e52354. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Byun, J.; Son, S.M.; Mook-Jung, I. Thrombospondin-1 protects against Abeta-induced mitochondrial fragmentation and dysfunction in hippocampal cells. Cell Death Discov. 2018, 4, 31. [Google Scholar] [CrossRef]
- Kim, D.H.; Lim, H.; Lee, D.; Choi, S.J.; Oh, W.; Yang, Y.S.; Oh, J.S.; Hwang, H.H.; Jeon, H.B. Thrombospondin-1 secreted by human umbilical cord blood-derived mesenchymal stem cells rescues neurons from synaptic dysfunction in Alzheimer’s disease model. Sci. Rep. 2018, 8, 354. [Google Scholar] [CrossRef]
- Singh, S.; Saleem, S.; Reed, G.L. Alpha2-Antiplasmin: The Devil You Don’t Know in Cerebrovascular and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 608899. [Google Scholar] [CrossRef]
- Huber, R.; Carrell, R.W. Implications of the three-dimensional structure of alpha 1-antitrypsin for structure and function of serpins. Biochemistry 1989, 28, 8951–8966. [Google Scholar] [CrossRef]
- Gettins, P.G.W. Serpin structure, mechanism, and function. Chem. Rev. 2002, 102, 4751–4804. [Google Scholar] [CrossRef]
- Huntington, J.A. Serpin structure, function and dysfunction. J. Thromb. Haemost. 2011, 9 (Suppl. S1), 26–34. [Google Scholar] [CrossRef]
- de Serres, F.; Blanco, I. Role of alpha-1 antitrypsin in human health and disease. J. Intern. Med. 2014, 276, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zheng, Y.; Zhang, F.; Wei, Z.; Wang, Y.; Carrell, R.W.; Read, R.J.; Chen, G.Q.; Zhou, A. Molecular Mechanism of Z alpha1-Antitrypsin Deficiency. J. Biol. Chem. 2016, 291, 15674–15686. [Google Scholar] [CrossRef] [PubMed]
- Dobó, J.; Gettins, P.G.W. alpha1-Proteinase inhibitor forms initial non-covalent and final covalent complexes with elastase analogously to other serpin-proteinase pairs, suggesting a common mechanism of inhibition. J. Biol. Chem. 2004, 279, 9264–9269. [Google Scholar] [CrossRef] [PubMed]
- Devlin, G.L.; Bottomley, S.P. A protein family under ‘stress’—Serpin stability, folding and misfolding. Front. Biosci. 2005, 10, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Dolmer, K.; Gettins, P.G.W. How the serpin alpha1-proteinase inhibitor folds. J. Biol. Chem. 2012, 287, 12425–12432. [Google Scholar] [CrossRef] [PubMed]
- Pintanel-Raymundo, M.; Menao-Guillén, S.; Perales-Afán, J.J.; García-Gutiérrez, A.; Moreno-Gázquez, I.; Julián-Ansón, M.; Ramos-Álvarez, M.; Olivera-González, S.; Gutiérrez-Cia, I.; Torralba-Cabeza, M.A. Analysis of the expression of the Serpina1 gene in SARS-CoV-2 infection: Study of a new biomarker. Rev. Clínica Española 2024, 224, 253–258. [Google Scholar] [CrossRef]
- Buhimschi, I.A.; Nayeri, U.A.; Zhao, G.; Shook, L.L.; Pensalfini, A.; Funai, E.F.; Bernstein, I.M.; Glabe, C.G.; Buhimschi, C.S. Protein misfolding, congophilia, oligomerization, and defective amyloid processing in preeclampsia. Sci. Transl. Med. 2014, 6, 245ra292. [Google Scholar] [CrossRef]
- Kouza, M.; Banerji, A.; Kolinski, A.; Buhimschi, I.A.; Kloczkowski, A. Oligomerization of FVFLM peptides and their ability to inhibit beta amyloid peptides aggregation: Consideration as a possible model. Phys. Chem. Chem. Phys. 2017, 19, 2990–2999. [Google Scholar] [CrossRef]
- Tajiri, M.; Okamoto, M.; Fujimoto, K.; Johkoh, T.; Ono, J.; Tominaga, M.; Azuma, K.; Kawayama, T.; Ohta, S.; Izuhara, K.; et al. Serum level of periostin can predict long-term outcome of idiopathic pulmonary fibrosis. Respir. Investig. 2015, 53, 73–81. [Google Scholar] [CrossRef]
- Izuhara, K.; Nunomura, S.; Nanri, Y.; Ono, J.; Takai, M.; Kawaguchi, A. Periostin: An emerging biomarker for allergic diseases. Allergy 2019, 74, 2116–2128. [Google Scholar] [CrossRef]
- Yoshihara, T.; Morimoto, T.; Hirata, H.; Murayama, M.; Nonaka, T.; Tsukamoto, M.; Toda, Y.; Kobayashi, T.; Izuhara, K.; Mawatari, M. Mechanisms of tissue degeneration mediated by periostin in spinal degenerative diseases and their implications for pathology and diagnosis: A review. Front. Med. 2023, 10, 1276900. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Roberts, J.; Lykins, D.; Fujii, Y.; Auclair, D.; Chen, L.B. Novel chemiluminescence assay for serum periostin levels in women with preeclampsia and in normotensive pregnant women. Am. J. Obstet. Gynecol. 2002, 186, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Park, J.C.; Jung, K.S.; Kim, J.; Jang, J.S.; Kwon, S.; Byun, M.S.; Yi, D.; Byeon, G.; Jung, G.; et al. Application of QPLEX(TM) biomarkers in cognitively normal individuals across a broad age range and diverse regions with cerebral amyloid deposition. Exp. Mol. Med. 2022, 54, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.K.; Bozyk, P.D.; Bentley, J.K.; Popova, A.P.; Birch, C.M.; Wilke, C.A.; Fry, C.D.; White, E.S.; Sisson, T.H.; Tayob, N.; et al. Periostin promotes fibrosis and predicts progression in patients with idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L1046–L1056. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, D.N.; Moore, B.B. The role of periostin in lung fibrosis and airway remodeling. Cell. Mol. Life Sci. 2017, 74, 4305–4314. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Duan, Z.; Yang, B.; Xiao, C. The Effective Regulation of Pro- and Anti-inflammatory Cytokines Induced by Combination of PA-MSHA and BPIFB1 in Initiation of Innate Immune Responses. Open Med. 2017, 12, 299–307. [Google Scholar] [CrossRef]
- Li, J.; Xu, P.; Wang, L.; Feng, M.; Chen, D.; Yu, X.; Lu, Y. Molecular biology of BPIFB1 and its advances in disease. Ann. Transl. Med. 2020, 8, 651. [Google Scholar] [CrossRef]
- Pretorius, E.; Page, M.J.; Mbotwe, S.; Kell, D.B. Lipopolysaccharide-binding protein (LBP) can reverse the amyloid state of fibrin seen or induced in Parkinson’s disease. PLoS ONE 2018, 13, e0192121. [Google Scholar] [CrossRef]
- Xu, Y.; Tao, Z.; Jiang, Y.; Liu, T.; Xiang, Y. Overexpression of BPIFB1 promotes apoptosis and inhibits proliferation via the MEK/ERK signal pathway in nasopharyngeal carcinoma. Int. J. Clin. Exp. Pathol. 2019, 12, 356–364. [Google Scholar]
- Cai, J.; Xiao, L.; Liu, J.; Wang, D.; Zhou, Y.; Liao, Z.; Chen, G. BPIFB1, Serving as a Downstream Effector of EBV-miR-BART4, Blocks Immune Escape of Nasopharyngeal Carcinoma via Inhibiting PD-L1 Expression. Biochem. Genet. 2024. [Google Scholar] [CrossRef]
- Richardo, T.; Prattapong, P.; Ngernsombat, C.; Wisetyaningsih, N.; Iizasa, H.; Yoshiyama, H.; Janvilisri, T. Epstein-Barr Virus Mediated Signaling in Nasopharyngeal Carcinoma Carcinogenesis. Cancers 2020, 12, 2441. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Bohmwald, K.; Diethelm-Varela, B.; Rodríguez-Guilarte, L.; Rivera, T.; Riedel, C.A.; González, P.A.; Kalergis, A.M. Pathophysiological, immunological, and inflammatory features of long COVID. Front. Immunol. 2024, 15, 1341600. [Google Scholar] [CrossRef] [PubMed]
- Cervia-Hasler, C.; Bruningk, S.C.; Hoch, T.; Fan, B.; Muzio, G.; Thompson, R.C.; Ceglarek, L.; Meledin, R.; Westermann, P.; Emmenegger, M.; et al. Persistent complement dysregulation with signs of thromboinflammation in active Long Covid. Science 2024, 383, eadg7942. [Google Scholar] [CrossRef]
- Ząbczyk, M.; Stachowicz, A.; Natorska, J.; Olszanecki, R.; Wiśniewski, J.R.; Undas, A. Plasma fibrin clot proteomics in healthy subjects: Relation to clot permeability and lysis time. J. Proteom. 2019, 208, 103487. [Google Scholar] [CrossRef]
- Stathakis, N.E.; Fountas, A.; Tsianos, E. Plasma fibronectin in normal subjects and in various disease states. J. Clin. Pathol. 1981, 34, 504–508. [Google Scholar] [CrossRef]
- Mosher, D.F. Plasma fibronectin concentration: A risk factor for arterial thrombosis? Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1193–1195. [Google Scholar] [CrossRef]
- Fucikova, A.; Lenco, J.; Tambor, V.; Rehulkova, H.; Pudil, R.; Stulik, J. Plasma concentration of fibronectin is decreased in patients with hypertrophic cardiomyopathy. Clin. Chim. Acta 2016, 463, 62–66. [Google Scholar] [CrossRef]
- Patel, S.; Chaffotte, A.F.; Goubard, F.; Pauthe, E. Urea-induced sequential unfolding of fibronectin: A fluorescence spectroscopy and circular dichroism study. Biochemistry 2004, 43, 1724–1735. [Google Scholar] [CrossRef]
- Dalton, C.J.; Lemmon, C.A. Fibronectin: Molecular Structure, Fibrillar Structure and Mechanochemical Signaling. Cells 2021, 10, 2443. [Google Scholar] [CrossRef]
- To, W.S.; Midwood, K.S. Plasma and cellular fibronectin: Distinct and independent functions during tissue repair. Fibrogenesis Tissue Repair. 2011, 4, 21. [Google Scholar] [CrossRef]
- Ingham, K.C.; Brew, S.A.; Broekelmann, T.J.; McDonald, J.A. Thermal stability of human plasma fibronectin and its constituent domains. J. Biol. Chem. 1984, 259, 11901–11907. [Google Scholar] [CrossRef] [PubMed]
- Porebski, B.T.; Nickson, A.A.; Hoke, D.E.; Hunter, M.R.; Zhu, L.; McGowan, S.; Webb, G.I.; Buckle, A.M. Structural and dynamic properties that govern the stability of an engineered fibronectin type III domain. Protein Eng. Des. Sel. 2015, 28, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Muszbek, L.; Bereczky, Z.; Bagoly, Z.; Komaromi, I.; Katona, E. Factor XIII: A coagulation factor with multiple plasmatic and cellular functions. Physiol. Rev. 2011, 91, 931–972. [Google Scholar] [CrossRef] [PubMed]
- Bagoly, Z.; Koncz, Z.; Hársfalvi, J.; Muszbek, L. Factor XIII, clot structure, thrombosis. Thromb. Res. 2012, 129, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.G.; Kopec, A.K.; Groeneveld, D.J.; Pant, A.; Baker, K.S.; Cline-Fedewa, H.M.; Flick, M.J.; Luyendyk, J.P. Factor XIII cross-links fibrin(ogen) independent of fibrin polymerization in experimental acute liver injury. Blood 2021, 137, 2520–2531. [Google Scholar] [CrossRef]
- Standeven, K.F.; Carter, A.M.; Grant, P.J.; Weisel, J.W.; Chernysh, I.; Masova, L.; Lord, S.T.; Ariëns, R.A.S. Functional analysis of fibrin {gamma}-chain cross-linking by activated factor XIII: Determination of a cross-linking pattern that maximizes clot stiffness. Blood 2007, 110, 902–907. [Google Scholar] [CrossRef]
- Murphy, R.M.; Pallitto, M.M. Probing the kinetics of beta-amyloid self-association. J. Struct. Biol. 2000, 130, 109–122. [Google Scholar] [CrossRef]
- Kelly, J.W. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr. Opin. Struct. Biol. 1998, 8, 101–106. [Google Scholar] [CrossRef]
- Rayan, B.; Barnea, E.; Khokhlov, A.; Upcher, A.; Landau, M. Differential fibril morphologies and thermostability determine functional roles of Staphylococcus aureus PSMalpha1 and PSMalpha3. Front. Mol. Biosci. 2023, 10, 1184785. [Google Scholar] [CrossRef]
- Idicula-Thomas, S.; Balaji, P.V. Understanding the relationship between the primary structure of proteins and their amyloidogenic propensity: Clues from inclusion body formation. Protein Eng. Des. Sel. 2005, 18, 175–180. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Klimtchuk, E.S.; Gursky, O.; Patel, R.S.; Laporte, K.L.; Connors, L.H.; Skinner, M.; Seldin, D.C. The critical role of the constant region in thermal stability and aggregation of amyloidogenic immunoglobulin light chain. Biochemistry 2010, 49, 9848–9857. [Google Scholar] [CrossRef] [PubMed]
- Poshusta, T.L.; Katoh, N.; Gertz, M.A.; Dispenzieri, A.; Ramirez-Alvarado, M. Thermal stability threshold for amyloid formation in light chain amyloidosis. Int. J. Mol. Sci. 2013, 14, 22604–22617. [Google Scholar] [CrossRef]
- Rubin, J.; Khosravi, H.; Bruce, K.L.; Lydon, M.E.; Behrens, S.H.; Chernoff, Y.O.; Bommarius, A.S. Ion-specific effects on prion nucleation and strain formation. J. Biol. Chem. 2013, 288, 30300–30308. [Google Scholar] [CrossRef] [PubMed]
- Proal, A.D.; VanElzakker, M.B. Long COVID or Post-acute Sequelae of COVID-19 (PASC): An Overview of Biological Factors That May Contribute to Persistent Symptoms. Front. Microbiol. 2021, 12, 698169. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.M.; Kruger, A.; Proal, A.; Kell, D.B.; Pretorius, E. The Occurrence of Hyperactivated Platelets and Fibrinaloid Microclots in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Pharmaceuticals 2022, 15, 931. [Google Scholar] [CrossRef]
- Nunes, J.M.; Kell, D.B.; Pretorius, E. Cardiovascular and haematological pathology in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): A role for Viruses. Blood Rev. 2023, 60, 101075. [Google Scholar] [CrossRef]
- Ryabkova, V.A.; Gavrilova, N.Y.; Fedotkina, T.V.; Churilov, L.P.; Shoenfeld, Y. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome and Post-COVID Syndrome: A Common Neuroimmune Ground. Diagnostics 2023, 13, 66. [Google Scholar] [CrossRef]
- Annesley, S.J.; Missailidis, D.; Heng, B.; Josev, E.K.; Armstrong, C.W. Unravelling shared mechanisms: Insights from recent ME/CFS research to illuminate long COVID pathologies. Trends Mol. Med. 2024, 30, 443–458. [Google Scholar] [CrossRef]
- Nunes, J.M.; Vlok, M.; Proal, A.; Kell, D.B.; Pretorius, E. Data-independent LC-MS/MS analysis of ME/CFS plasma reveals a dysregulated coagulation system, endothelial dysfunction, downregulation of complement machinery. Cardiovasc. Diabetol. 2024, 23, 254. [Google Scholar] [CrossRef]
- Komaroff, A.L.; Lipkin, W.I. ME/CFS and Long COVID share similar symptoms and biological abnormalities: Road map to the literature. Front. Med. 2023, 10, 1187163. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B.; Lip, G.Y.H.; Pretorius, E. Fibrinaloid Microclots and Atrial Fibrillation. Biomedicines 2024, 12, 891. [Google Scholar] [CrossRef] [PubMed]
- Rinauro, D.J.; Chiti, F.; Vendruscolo, M.; Limbocker, R. Misfolded protein oligomers: Mechanisms of formation, cytotoxic effects, and pharmacological approaches against protein misfolding diseases. Mol. Neurodegener. 2024, 19, 20. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, M.K.; Majid, N.; Malik, S.; Alam, P.; Khan, R.H. Amyloid Oligomers, Protofibrils and Fibrils. Subcell. Biochem. 2019, 93, 471–503. [Google Scholar] [CrossRef] [PubMed]
- Makin, O.S.; Serpell, L.C. Examining the structure of the mature amyloid fibril. Biochem. Soc. Trans. 2002, 30, 521–525. [Google Scholar] [CrossRef]
- Stromer, T.; Serpell, L.C. Structure and morphology of the Alzheimer’s amyloid fibril. Microsc. Res. Tech. 2005, 67, 210–217. [Google Scholar] [CrossRef]
- Aubrey, L.D.; Blakeman, B.J.F.; Lutter, L.; Serpell, C.J.; Tuite, M.F.; Serpell, L.C.; Xue, W.F. Quantification of amyloid fibril polymorphism by nano-morphometry reveals the individuality of filament assembly. Commun. Chem. 2020, 3, 125. [Google Scholar] [CrossRef]
- Vrana, J.A.; Theis, J.D.; Dasari, S.; Mereuta, O.M.; Dispenzieri, A.; Zeldenrust, S.R.; Gertz, M.A.; Kurtin, P.J.; Grogg, K.L.; Dogan, A. Clinical diagnosis and typing of systemic amyloidosis in subcutaneous fat aspirates by mass spectrometry-based proteomics. Haematologica 2014, 99, 1239–1247. [Google Scholar] [CrossRef]
- Buxbaum, J.N.; Dispenzieri, A.; Eisenberg, D.S.; Fandrich, M.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Westermark, P. Amyloid nomenclature 2022: Update, novel proteins, and recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid 2022, 29, 213–219. [Google Scholar] [CrossRef]
- Theis, J.D.; Dasari, S.; Vrana, J.A.; Kurtin, P.J.; Dogan, A. Shotgun-proteomics-based clinical testing for diagnosis and classification of amyloidosis. J. Mass. Spectrom. 2013, 48, 1067–1077. [Google Scholar] [CrossRef]
- Gallo, G.; Wisniewski, T.; Choi-Miura, N.H.; Ghiso, J.; Frangione, B. Potential role of apolipoprotein-E in fibrillogenesis. Am. J. Pathol. 1994, 145, 526–530. [Google Scholar]
- Winter, M.; Tholey, A.; Krüger, S.; Schmidt, H.; Röcken, C. MALDI-mass spectrometry imaging identifies vitronectin as a common constituent of amyloid deposits. J. Histochem. Cytochem. 2015, 63, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Misumi, Y.; Tabata, Y.; Tasaki, M.; Obayashi, K.; Yamakawa, S.; Nomura, T.; Ueda, M. Binding of serum-derived amyloid-associated proteins to amyloid fibrils. Amyloid 2023, 30, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B.; Pretorius, E. The simultaneous occurrence of both hypercoagulability and hypofibrinolysis in blood and serum during systemic inflammation, and the roles of iron and fibrin(ogen). Integr. Biol. 2015, 7, 24–52. [Google Scholar] [CrossRef] [PubMed]
- Grobbelaar, L.M.; Venter, C.; Vlok, M.; Ngoepe, M.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B.; Pretorius, E. SARS-CoV-2 spike protein S1 induces fibrin(ogen) resistant to fibrinolysis: Implications for microclot formation in COVID-19. Biosci. Rep. 2021, 41, BSR20210611. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Mohan, M.L.; Luo, X.; Kundu, B.; Kong, Q.; Singh, N. Modulation of proteinase K-resistant prion protein in cells and infectious brain homogenate by redox iron: Implications for prion replication and disease pathogenesis. Mol. Biol. Cell 2007, 18, 3302–3312. [Google Scholar] [CrossRef][Green Version]
- Candelise, N.; Schmitz, M.; Da Silva Correia, S.M.; Arora, A.S.; Villar-Pique, A.; Zafar, S.; Llorens, F.; Cramm, M.; Zerr, I. Applications of the real-time quaking-induced conversion assay in diagnosis, prion strain-typing, drug pre-screening and other amyloidopathies. Expert. Rev. Mol. Diagn. 2017, 17, 897–904. [Google Scholar] [CrossRef]
- Wang, F.; Wang, X.; Abskharon, R.; Ma, J. Prion infectivity is encoded exclusively within the structure of proteinase K-resistant fragments of synthetically generated recombinant PrP(Sc). Acta Neuropathol. Commun. 2018, 6, 30. [Google Scholar] [CrossRef]
- Hailemariam, D.; Goldansaz, S.A.; Daude, N.; Wishart, D.S.; Ametaj, B.N. Mice Treated Subcutaneously with Mouse LPS-Converted PrP(res) or LPS Alone Showed Brain Gene Expression Profiles Characteristic of Prion Disease. Vet. Sci. 2021, 8, 200. [Google Scholar] [CrossRef]
- Saverioni, D.; Notari, S.; Capellari, S.; Poggiolini, I.; Giese, A.; Kretzschmar, H.A.; Parchi, P. Analyses of protease resistance and aggregation state of abnormal prion protein across the spectrum of human prions. J. Biol. Chem. 2013, 288, 27972–27985. [Google Scholar] [CrossRef]
- Bester, J.; Soma, P.; Kell, D.B.; Pretorius, E. Viscoelastic and ultrastructural characteristics of whole blood and plasma in Alzheimer-type dementia, and the possible role of bacterial lipopolysaccharides (LPS). Oncotarget Gerontol. 2015, 6, 35284–35303. [Google Scholar] [CrossRef] [PubMed]
- de Waal, G.M.; Engelbrecht, L.; Davis, T.; de Villiers, W.J.S.; Kell, D.B.; Pretorius, E. Correlative Light-Electron Microscopy detects lipopolysaccharide and its association with fibrin fibres in Parkinson’s Disease, Alzheimer’s Disease and Type 2 Diabetes Mellitus. Sci. Rep. 2018, 8, 16798. [Google Scholar] [CrossRef]
- Kell, D.B.; Pretorius, E. On the translocation of bacteria and their lipopolysaccharides between blood and peripheral locations in chronic, inflammatory diseases: The central roles of LPS and LPS-induced cell death. Integr. Biol. 2015, 7, 1339–1377. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Bester, J.; Page, M.J.; Kell, D.B. The potential of LPS-binding protein to reverse amyloid formation in plasma fibrin of individuals with Alzheimer-type dementia. Front. Aging Neurosci. 2018, 10, 257. [Google Scholar] [CrossRef] [PubMed]
- Kruger, A.; Joffe, D.; Lloyd-Jones, G.; Khan, M.A.; Šalamon, Š.; Laubscher, G.J.; Putrino, D.; Kell, D.B.; Pretorius, E. Vascular pathogenesis in acute and long covid: Current insights and therapeutic outlook. Semin. Throm Hemost. 2024; online ahead of print. [Google Scholar] [CrossRef]
- Hailemariam, D.; Lam, T.H.; Dervishi, E.; Zwierzchowski, G.; Wishart, D.S.; Ametaj, B.N. Combination of mouse prion protein with detoxified lipopolysaccharide triggers colon genes related to inflammatory, antibacterial, and apoptotic responses. Res. Vet. Sci. 2022, 144, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Saleem, F.; Bjorndahl, T.C.; Ladner, C.L.; Perez-Pineiro, R.; Ametaj, B.N.; Wishart, D.S. Lipopolysaccharide induced conversion of recombinant prion protein. Prion 2014, 8, 221–232. [Google Scholar] [CrossRef]
- Zwierzchowski, G.; Hailemariam, D.; Dunn, S.; Wishart, D.; Ametaj, B. Subcutaneously administered LPS-converted recombinant mouse prion protein alone or in combination with LPS modulates the content of prion-related proteins in the brain of FVB/N mice. Prion 2015, 9, S24–S25. [Google Scholar]
- Okuducu, Y.K.; Boribong, B.; Ellett, F.; Hajizadeh, S.; VanElzakker, M.; Haas, W.; Pillai, S.; Fasano, A.; Irimia, D.; Yonker, L. Evidence Circulating Microclots and Activated Platelets Contribute to Hyperinflammation within Pediatric Post Acute Sequala of COVID. Am. J. Respir. Crit. Care Med. 2024, 209, A2247. [Google Scholar]
- Protopopova, A.D.; Barinov, N.A.; Zavyalova, E.G.; Kopylov, A.M.; Sergienko, V.I.; Klinov, D.V. Visualization of fibrinogen alphaC regions and their arrangement during fibrin network formation by high-resolution AFM. J. Thromb. Haemost. 2015, 13, 570–579. [Google Scholar] [CrossRef]
- Biyani, R.; Hirata, K.; Oqmhula, K.; Yurtsever, A.; Hongo, K.; Maezono, R.; Takagi, M.; Fukuma, T.; Biyani, M. Biophysical Properties of the Fibril Structure of the Toxic Conformer of Amyloid-beta42: Characterization by Atomic Force Microscopy in Liquid and Molecular Docking. ACS Appl. Mater. Interfaces 2023, 15, 27789–27800. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).