
Mining of Oil Content Genes in Recombinant Maize Inbred Lines with Introgression from Temperate and Tropical Germplasm
 and
and
Abstract
:1. Introduction
2. Results
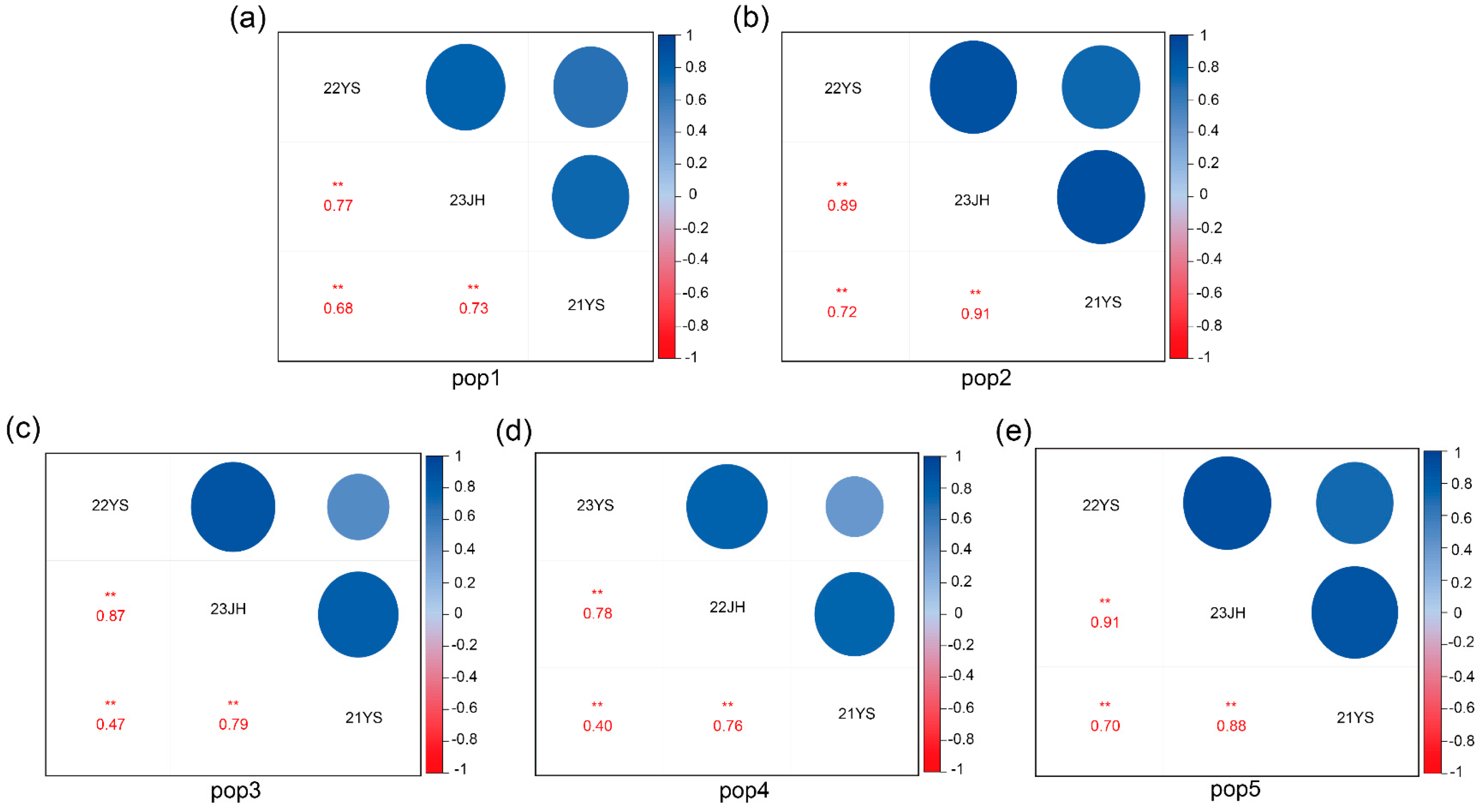
2.1. Phenotypic Analysis of TOC in Five RIL Subpopulations
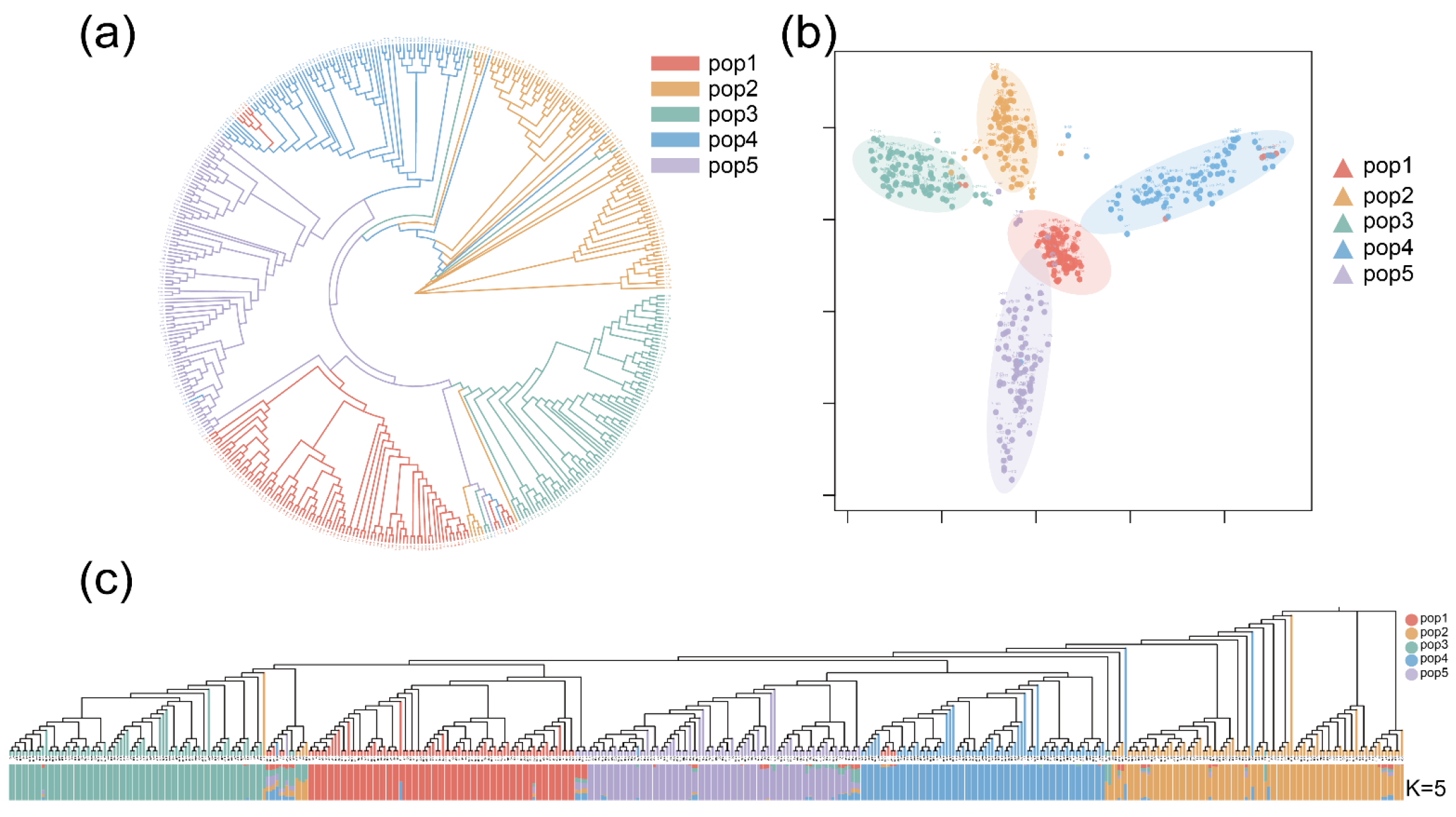
2.2. Phylogenetic Tree, PCA, and Population Structure Analysis
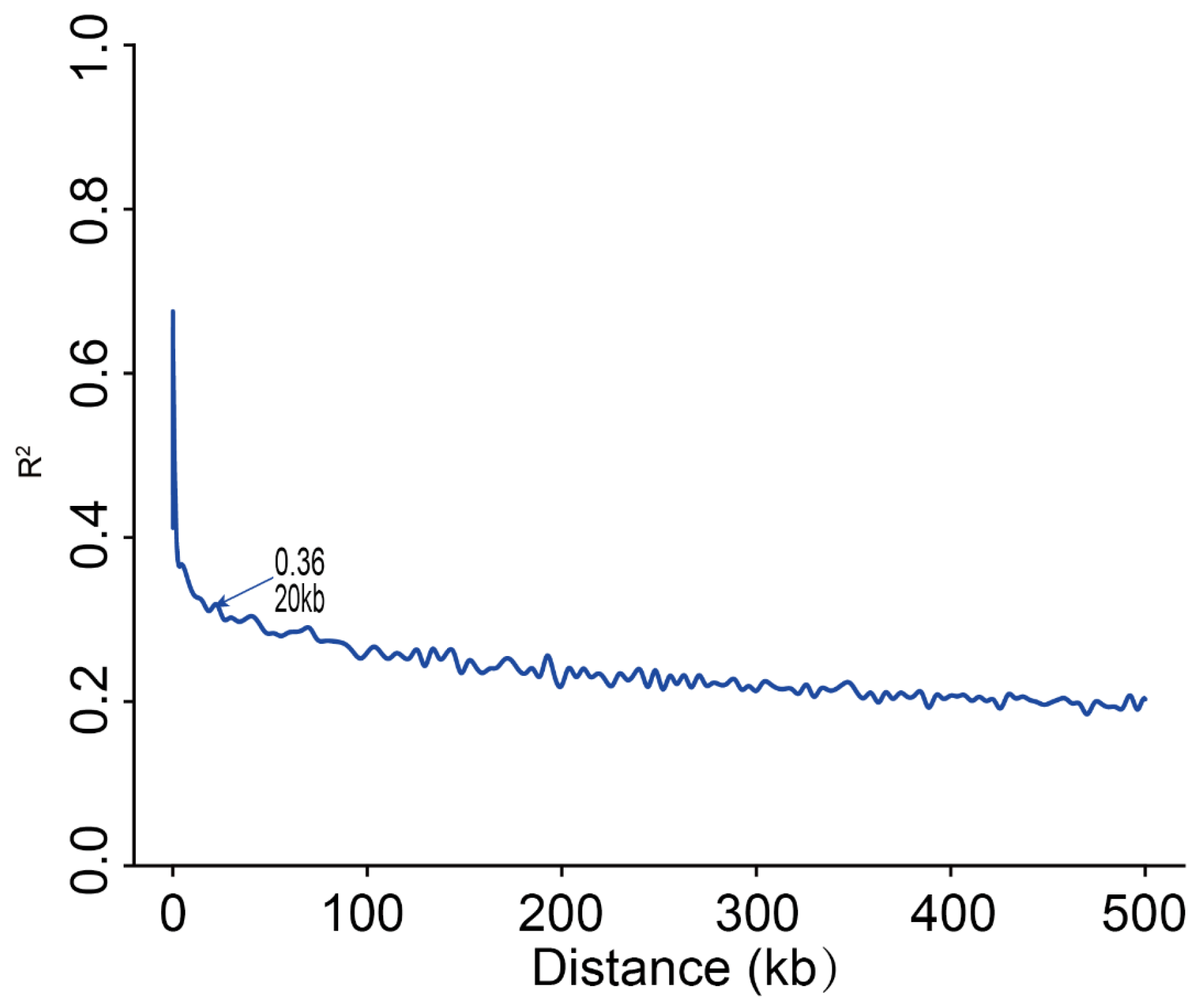
2.3. LD Decay Analysis
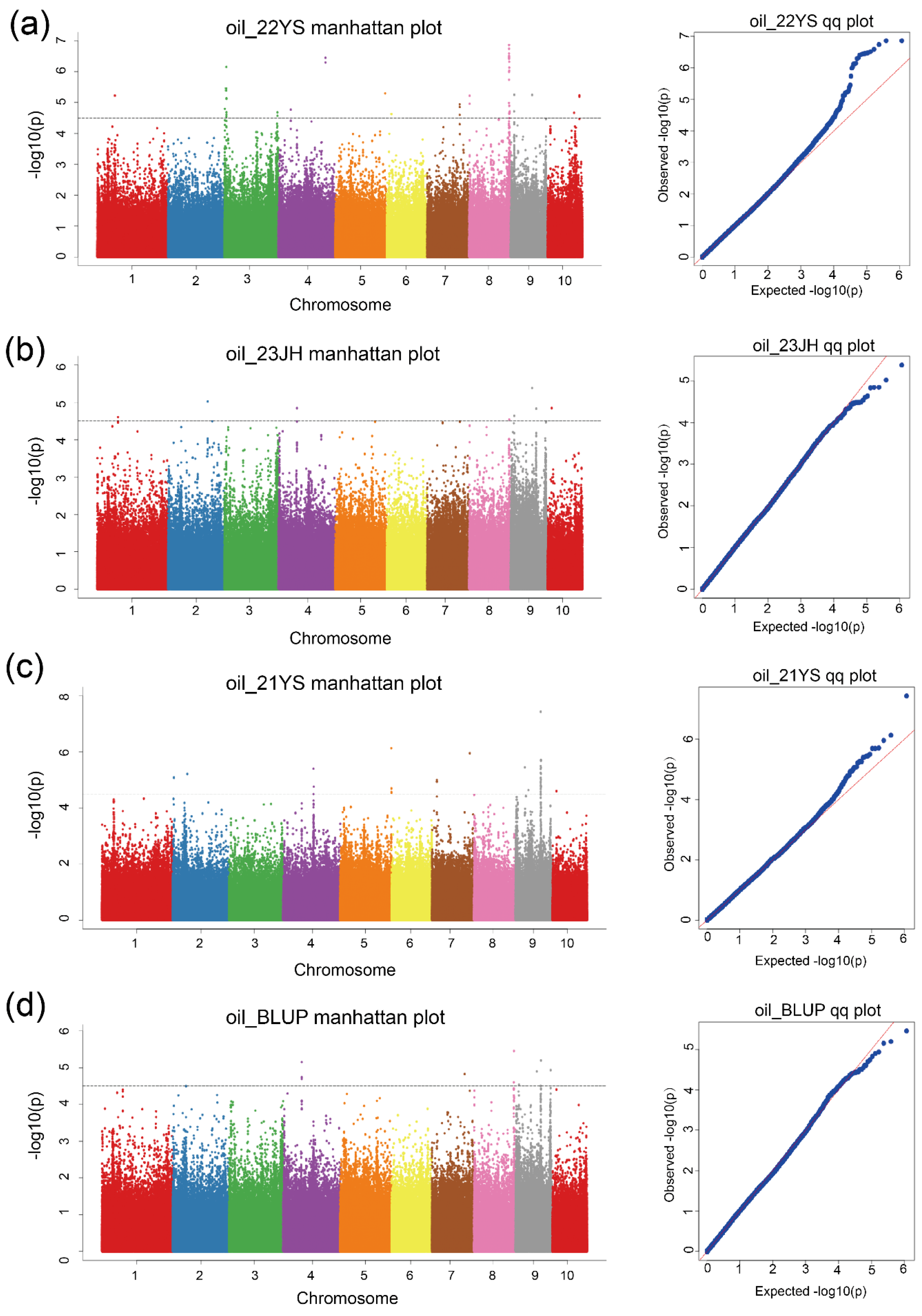
2.4. Genome-Wide Association Analysis for TOC in Maize
2.5. Genetic Map Construction and QTL Mapping of TOC in the Five RIL Subpopulations
2.6. Identification of Candidate Genes Related to TOC and Haplotype Analysis
3. Discussion
3.1. Comparison of Loci Significantly Associated with TOC with Previously Reported QTLs
3.2. Functional Annotation of Candidate Genes
3.3. Mechanism of Synthesis of TOC in Maize
3.4. Genetic Effects of Oil Content in Tropical Maize
4. Materials and Methods
4.1. Plant Materials and Population Development
4.2. Experimental Design and Oil Content Estimation
4.3. Statistical Analysis of TOC and Estimation of Heritability
4.4. DNA Extraction and Genotyping-by-Sequencing (GBS)
4.5. Phylogenetic Tree, PCA, and Linkage Disequilibrium Analysis
4.6. Genome-Wide Association Analysis
4.7. Construction of Genetic Map and QTL Mapping
4.8. Identification and Functional Annotation of Candidate Genes
4.9. Haplotype Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fareghi, S.; Mirlohi, A.F.; Saeidi, G.; Khamisabadi, H. Evaluation of SRAP marker efficiency in identifying the relationship between genetic diversities of corn inbred lines with seed quantity and quality in derived hybrids. Cell. Mol. Biol. 2019, 65, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zeng, B.; Zhang, M.; Xie, S.; Wang, G.; Hauck, A.; Lai, J. Dynamic Transcriptome Landscape of Maize Embryo and Endosperm Development. Plant Physiol. 2014, 166, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, J. High-Oil Maize Genomics. In The Maize Genome; Springer: Berlin/Heidelberg, Germany, 2018; pp. 305–317. [Google Scholar] [CrossRef]
- Lambert, R.J.; Alexander, D.E.; Mejaya, I.J. Single Kernel Selection for Increased Grain Oil in Maize Synthetics and High-Oil Hybrid Development. In Plant Breeding Reviews; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003; pp. 153–175. [Google Scholar] [CrossRef]
- Li, H.; Peng, Z.; Yang, X.; Wang, W.; Fu, J.; Wang, J.; Han, Y.; Chai, Y.; Guo, T.; Yang, N.; et al. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 2013, 45, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Ndlovu, N.; Kachapur, R.M.; Beyene, Y.; Das, B.; Ogugo, V.; Makumbi, D.; Spillane, C.; McKeown, P.C.; Prasanna, B.M.; Gowda, M. Linkage mapping and genomic prediction of grain quality traits in tropical maize (Zea mays L.). Front. Genet. 2024, 15, 1353289. [Google Scholar] [CrossRef]
- Wei, M.; Fu, J.; Li, X.; Wang, Y.; Li, Y. Influence of dent corn genetic backgrounds on QTL detection for plant-height traits and their relationships in high-oil maize. J. Appl. Genet. 2009, 50, 225–234. [Google Scholar] [CrossRef]
- Fang, H.; Fu, X.; Ge, H.; Zhang, A.; Shan, T.; Wang, Y.; Li, P.; Wang, B. Genetic basis of maize kernel oil-related traits revealed by high-density SNP markers in a recombinant inbred line population. BMC Plant Biol. 2021, 21, 344. [Google Scholar] [CrossRef]
- Song, T.M.; Chen, S.J. Long term selection for oil concentration in five maize populations. Maydica 2004, 49, 9–14. [Google Scholar]
- Dudley, J.W.; Lambert, R.J. 100 Generations of Selection for Oil and Protein in Corn. Plant Breed. Rev. 2004, 24, 79–110. [Google Scholar]
- Yang, X.; Guo, Y.; Yan, J.; Zhang, J.; Song, T.; Rocheford, T.; Li, J.S. Major and minor QTL and epistasis contribute to fatty acid compositions and oil concentration in high-oil maize. Theor. Appl. Genet. 2010, 120, 665–678. [Google Scholar] [CrossRef]
- Jing, H.; Hongwu, W.; Shaojiang, C. QTL Mapping of Kernel Oil Content of Chromosome 6 in a High Oil Maize Mutant (Zea mays L.). Genes Genom. 2008, 30, 373–382. [Google Scholar] [CrossRef]
- Song, X.F.; Song, T.M.; Dai, J.R.; Rocheford, T.; Li, J.S. QTL mapping of kernel oil concentration with high-oil maize by SSR markers. Maydica 2004, 49, 41–48. [Google Scholar]
- Alrefai, R.; Berke, T.G.; Rocheford, T.R. Quantitative trait locus analysis of fatty acid concentrations in maize. Genome 1995, 38, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, M.; Guan, H.; Wen, H.; Zhang, C.; Dai, C.; Wang, J.; Pan, B.; Li, J.; Liao, H. Genetic dissection of QTLs for oil content in four maize DH populations. Front. Plant Sci. 2023, 14, 1174985. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Du, Y.; Huo, D.; Wang, M.; Shen, X.; Yue, B.; Qiu, F.; Zheng, Y.; Yan, J.; Zhang, Z. Genetic architecture of maize kernel row number and whole genome prediction. Theor. Appl. Genet. 2015, 128, 2243–2254. [Google Scholar] [CrossRef]
- Xue, S.; Bradbury, P.J.; Casstevens, T.; Holland, J.B. Genetic Architecture of Domestication-Related Traits in Maize. Genetics 2016, 204, 99–113. [Google Scholar] [CrossRef]
- Xiao, Y.; Tong, H.; Yang, X.; Xu, S.; Pan, Q.; Qiao, F.; Raihan, M.S.; Luo, Y.; Liu, H.; Zhang, X.; et al. Genome-wide dissection of the maize ear genetic architecture using multiple populations. New Phytol. 2016, 210, 1095–1106. [Google Scholar] [CrossRef]
- Dell’Acqua, M.; Gatti, D.M.; Pea, G.; Cattonaro, F.; Coppens, F.; Magris, G.; Hlaing, A.L.; Aung, H.H.; Nelissen, H.; Baute, J.; et al. Genetic properties of the MAGIC maize population: A new platform for high definition QTL mapping in Zea mays. Genome Biol. 2015, 16, 167. [Google Scholar] [CrossRef]
- Beló, A.; Zheng, P.; Luck, S.; Shen, B.; Meyer, D.J.; Li, B.; Tingey, S.; Rafalski, A. Whole genome scan detects an allelic variant of fad2 associated with increased oleic acid levels in maize. Mol. Genet. Genom. 2008, 279, 1–10. [Google Scholar] [CrossRef]
- Yi, G.; Shen, M.; Yuan, J.; Sun, C.; Duan, Z.; Qu, L.; Dou, T.; Ma, M.; Lu, J.; Guo, J.; et al. Genome-wide association study dissects genetic architecture underlying longitudinal egg weights in chickens. BMC Genom. 2015, 16, 746. [Google Scholar] [CrossRef]
- Shi, X.; Zhou, Z.; Li, W.; Qin, M.; Yang, P.; Hou, J.; Huang, F.; Lei, Z.; Wu, Z.; Wang, J. Genome-wide association study reveals the genetic architecture for calcium accumulation in grains of hexaploid wheat (Triticum aestivum L.). BMC Plant Biol. 2022, 22, 229. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, M.; Yu, S.; Li, X.; Zhang, A.; Cui, Z.; Dong, X.; Fan, J.; Zhang, L.; Li, C.; et al. A Genome-Wide Association Study Dissects the Genetic Architecture of the Metaxylem Vessel Number in Maize Brace Roots. Front. Plant Sci. 2022, 13, 847234. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ma, H.; Zhang, P.; Yan, J.; Guo, Y.; Song, T.; Li, J. Characterization of QTL for oil content in maize kernel. Theor. Appl. Genet. 2012, 125, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Allen, W.B.; Nagasawa, N.; Ching, A.S.; Heppard, E.P.; Li, H.; Hao, X.; Li, X.; Yang, X.; Yan, J.; et al. A transposable element insertion within ZmGE2 gene is associated with increase in embryo to endosperm ratio in maize. Theor. Appl. Genet. 2012, 125, 1463–1471. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Wang, C.; Hu, Y.; Dai, W.; Liang, Z.; Zou, C.; Pan, G.; Lübberstedt, T.; Shen, Y. GWAS and transcriptome analysis reveal MADS26 involved in seed germination ability in maize. Theor. Appl. Genet. 2022, 135, 1717–1730. [Google Scholar] [CrossRef]
- Ma, L.; Qing, C.; Zhang, M.; Zou, C.; Pan, G.; Shen, Y. GWAS with a PCA uncovers candidate genes for accumulations of microelements in maize seedlings. Physiol. Plant. 2021, 172, 2170–2180. [Google Scholar] [CrossRef]
- Shu, G.; Wang, A.; Wang, X.; Chen, R.; Gao, F.; Wang, A.; Li, T.; Wang, Y. Identification of QTNs, QTN-by-environment interactions for plant height and ear height in maize multi-environment GWAS. Front. Plant Sci. 2023, 14, 1284403. [Google Scholar] [CrossRef]
- Zhao, M.; Liu, S.; Pei, Y.; Jiang, X.; Jaqueth, J.S.; Li, B.; Han, J.; Jeffers, D.; Wang, J.; Song, X. Identification of genetic loci associated with rough dwarf disease resistance in maize by integrating GWAS and linkage mapping. Plant Sci. 2022, 315, 111100. [Google Scholar] [CrossRef]
- Katagiri, T.; Mizoguchi, T.; Shinozaki, K. Molecular cloning of a cDNA encoding diacylglycerol kinase (DGK) in Arabidopsis thaliana. Plant Mol. Biol. 1996, 30, 647–653. [Google Scholar] [CrossRef]
- Gómez-Merino, F.C.; Arana-Ceballos, F.A.; Trejo-Téllez, L.I.; Skirycz, A.; Brearley, C.A.; Dörmann, P.; Mueller-Roeber, B. Arabidopsis AtDGK7, the smallest member of plant diacylglycerol kinases (DGKs), displays unique biochemical features and saturates at low substrate concentration: The DGK inhibitor R59022 differentially affects AtDGK2 and AtDGK7 activity in vitro and alters plant growth and development. J. Biol. Chem. 2005, 280, 34888–34899. [Google Scholar] [CrossRef]
- Arana-Ceballos, F. Biochemical and Physiological Studies of Arabidopsis thaliana Diacylglycerol Kinase 7 (AtDGK7). Ph.D. Thesis, Universität Potsdam, Potsdam, Germany, 2007. [Google Scholar]
- Yunus, I.S.; Cazenave-Gassiot, A.; Liu, Y.C.; Lin, Y.C.; Wenk, M.R.; Nakamura, Y. Phosphatidic acid is a major phospholipid class in reproductive organs of Arabidopsis thaliana. Plant Signal. Behav. 2015, 10, e1049790. [Google Scholar] [CrossRef] [PubMed]
- Kue Foka, I.C.; Ketehouli, T.; Zhou, Y.; Li, X.; Wang, F.-W.; Li, H. The Emerging Roles of Diacylglycerol Kinase (DGK) in Plant Stress Tolerance, Growth, and Development. Agronomy 2020, 10, 1375. [Google Scholar] [CrossRef]
- Schwarz, Z.; Jolly, S.O.; Steinmetz, A.A.; Bogorad, L. Overlapping divergent genes in the maize chloroplast chromosome and in vitro transcription of the gene for tRNA. Proc. Natl. Acad. Sci. USA 1981, 78, 3423–3427. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, S.-X.; Malhi, S. Effects of fertilization and other agronomic measures on nutritional quality of crops. J. Sci. Food Agric. 2008, 88, 7–23. [Google Scholar] [CrossRef]
- Luo, M.; Lu, B.; Shi, Y.; Zhao, Y.; Liu, J.; Zhang, C.; Wang, Y.; Liu, H.; Shi, Y.; Fan, Y.; et al. Genetic basis of the oil biosynthesis in ultra-high-oil maize grains with an oil content exceeding 20. Front. Plant Sci. 2023, 14, 1168216. [Google Scholar] [CrossRef]
- Ndlovu, N.; Spillane, C.; McKeown, P.C.; Cairns, J.E.; Das, B.; Gowda, M. Genome-wide association studies of grain yield and quality traits under optimum and low-nitrogen stress in tropical maize (Zea mays L.). Theor. Appl. Genet. 2022, 135, 4351–4370. [Google Scholar] [CrossRef]
- Cocuron, J.C.; Koubaa, M.; Kimmelfield, R.; Ross, Z.; Alonso, A.P. A Combined Metabolomics and Fluxomics Analysis Identifies Steps Limiting Oil Synthesis in Maize Embryos. Plant Physiol. 2019, 181, 961–975. [Google Scholar] [CrossRef]
- Laurie, C.C.; Chasalow, S.D.; LeDeaux, J.R.; McCarroll, R.; Bush, D.; Hauge, B.; Lai, C.; Clark, D.; Rocheford, T.R.; Dudley, J.W. The genetic architecture of response to long-term artificial selection for oil concentration in the maize kernel. Genetics 2004, 168, 2141–2155. [Google Scholar] [CrossRef]
- Dudley, J. Epistatic Interactions in Crosses of Illinois High Oil × Illinois Low Oil and of Illinois High Protein × Illinois Low Protein Corn Strains. Crop Sci. 2008, 48, 59–68. [Google Scholar] [CrossRef]
- Gürbüz, B.; Aras, E.; Güz, A.; Kahrıman, F. Prediction performance of NIR calibration models developed with different chemometric techniques to predict oil content in a single kernel of maize. Vib. Spectrosc. 2023, 126, 103528. [Google Scholar] [CrossRef]
- Knapp, S.J. Confidence intervals for heritability for two-factor mating design single environment linear models. Theor. Appl. Genet. 1986, 72, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Saghai Maroof, M.A.; Biyashev, R.M.; Yang, G.P.; Zhang, Q.; Allard, R.W. Extraordinarily polymorphic microsatellite DNA in barley: Species diversity, chromosomal locations, and population dynamics. Proc. Natl. Acad. Sci. USA 1994, 91, 5466–5470. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, Y.; Bian, R.; Fritz, A.; Dong, Y.; Zhao, L.; Xu, Y.; Ghori, N.; Bernardo, A.; Amand, P.; et al. Genetic architecture of quantitative trait loci (QTL) for FHB resistance and agronomic traits in a hard winter wheat population. Crop J. 2023, 11, 1836–1845. [Google Scholar] [CrossRef]
- Li, C.; Guan, H.; Jing, X.; Li, Y.; Wang, B.; Li, Y.; Liu, X.; Zhang, D.; Liu, C.; Xie, X.; et al. Genomic insights into historical improvement of heterotic groups during modern hybrid maize breeding. Nat. Plants 2022, 8, 750–763. [Google Scholar] [CrossRef]
- Pei, S.; Liu, T.; Ren, X.; Li, W.; Chen, C.; Xie, Z. Benchmarking variant callers in next-generation and third-generation sequencing analysis. Brief Bioinform. 2021, 22, bbaa148. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Strable, J.; Wallace, J.G.; Unger-Wallace, E.; Briggs, S.; Bradbury, P.J.; Buckler, E.S.; Vollbrecht, E. Maize YABBY Genes drooping leaf1 and drooping leaf2 Regulate Plant Architecture. Plant Cell. 2017, 29, 1622–1641. [Google Scholar] [CrossRef] [PubMed]
- Ooijen, J.W.V.; Ooijen, J.W.v.; Verlaat, J.V.T.; Ooijen, J.W.V.; Tol, J.; Dalén, J.; Buren, J.B.V.; Meer, J.W.M.V.D.; Krieken, J.H.V.; Ooijen, J.W.V.; et al. JoinMap® 4, Software for the calculation of genetic linkage maps in experimental populations.
- Zeng, Z.B. Precision mapping of quantitative trait loci. Genetics 1994, 136, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Ribaut, J.M.; Hoisington, D.A.; Deutsch, J.A.; Jiang, C.; Gonzalez-de-Leon, D. Identification of quantitative trait loci under drought conditions in tropical maize. 1. Flowering parameters and the anthesis-silking interval. Theor. Appl. Genet. 1996, 92, 905–914. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Environment | Mean | Standard Deviation | Skewness | Kurtosis | Coefficient of Variation (%) | Heritability (h2) (%) | Correlation Coefficient |
|---|---|---|---|---|---|---|---|---|
| (r) | ||||||||
| pop1 | 22YS | 5.273 | 0.662 | −0.158 | −0.481 | 12.60% | 88.1 | 22YS/23JH = 0.77 ** |
| 23JH | 5.189 | 0.623 | 0.375 | −0.226 | 12% | 23JH/21YS = 0.73 ** | ||
| 21YS | 5.199 | 0.711 | 0.070 | −0.080 | 13.70% | 21YS/22YS = 0.68 ** | ||
| total | 5.220 | 0.665 | 0.079 | −0.286 | 12.74% | |||
| pop2 | 22YS | 5.164 | 0.590 | 0.146 | −0.010 | 11.40% | 93.1 | 22YS/23JH = 0.89 ** |
| 23JH | 5.050 | 0.550 | 0.314 | −0.334 | 10.90% | 23JH/21YS = 0.91 ** | ||
| 21YS | 4.910 | 0.610 | 0.203 | −0.217 | 12.40% | 21YS/22YS = 0.72 ** | ||
| total | 5.041 | 0.591 | 0.185 | −0.192 | 11.72% | |||
| pop3 | 22YS | 5.501 | 0.809 | −0.243 | −0.071 | 14.70% | 83.9 | 22YS/23JH = 0.87 ** |
| 23JH | 5.320 | 0.623 | 0.052 | −0.441 | 11.70% | 23JH/21YS = 0.79 ** | ||
| 21YS | 5.120 | 0.620 | 0.019 | −0.387 | 12.10% | 21YS/22YS = 0.47 ** | ||
| total | 5.313 | 0.704 | 0.041 | −0.160 | 13.25% | |||
| pop4 | 22YS | 4.884 | 0.432 | 0.533 | −0.045 | 8.80% | 79.4 | 22YS/23JH = 0.78 ** |
| 23JH | 4.888 | 0.371 | 0.163 | −0.925 | 7.60% | 23JH/21YS = 0.76 ** | ||
| 21YS | 4.912 | 0.439 | 0.353 | −0.087 | 8.90% | 21YS/22YS = 0.40 ** | ||
| total | 4.895 | 0.413 | 0.378 | −0.233 | 8.44% | |||
| pop5 | 22YS | 5.276 | 0.592 | 0.241 | −0.402 | 11.20% | 91.6 | 22YS/23JH = 0.91 ** |
| 23JH | 5.214 | 0.542 | 0.242 | −0.644 | 10.40% | 23JH/21YS = 0.88 ** | ||
| 21YS | 5.167 | 0.504 | 0.222 | −0.274 | 9.80% | 21YS/22YS = 0.70 ** | ||
| total | 5.216 | 0.547 | 0.263 | −0.412 | 10.49% |
| Env. | Chr. | SNP | ref | alt | −log(P) | Additive Effect | Dominance Effect | PVE |
|---|---|---|---|---|---|---|---|---|
| 22YS | 1 | 75,791,466 | G | T | 5.23 | −0.26 | 0.35 | 0.035 |
| 22YS | 3 | 2,108,126 | G | A | 4.80 | 0.20 | 0.20 | 0.075 |
| 22YS | 3 | 7,453,745 | G | A | 5.46 | 0.30 | 0.04 | 0.053 |
| 22YS | 3 | 8,646,933 | C | A | 6.15 | 0.33 | −0.11 | 0.046 |
| 22YS | 3 | 9,226,566 | G | T | 4.54 | −0.28 | −0.21 | 0.036 |
| 22YS | 3 | 9,371,935 | C | T | 5.14 | −0.29 | −0.20 | 0.038 |
| 22YS | 3 | 230,340,051 | C | T | 4.68 | −0.24 | 0.14 | 0.062 |
| 22YS | 3 | 230,499,437 | G | A | 4.57 | 0.25 | 0.04 | 0.028 |
| 22YS | 4 | 52,876,804 | A | G | 4.77 | −0.19 | −0.48 | 0.035 |
| 22YS | 4 | 203,717,068 | A | G | 6.45 | −0.41 | −0.01 | 0.073 |
| 22YS | 5 | 216,419,106 | A | T | 5.30 | −0.26 | −0.05 | 0.049 |
| 22YS | 6 | 19,088,018 | C | T | 4.63 | NaN | NaN | 0.029 |
| 22YS | 7 | 140,826,856 | C | T | 4.94 | 0.31 | 0.06 | 0.042 |
| 22YS | 8 | 1,952,449 | C | T | 5.22 | 0.15 | −0.13 | 0.038 |
| 22YS | 8 | 172,972,407 | T | C | 4.99 | −0.27 | 0.13 | 0.055 |
| 22YS, BLUP | 8 | 173,247,098 | A | T | 6.86 | −0.37 | −0.18 | 0.070 |
| 22YS, 23JH, BLUP | 8 | 174,055,891 | T | C | 6.32 | −0.27 | −0.48 | 0.098 |
| 22YS | 8 | 177,414,430 | C | T | 4.71 | −0.22 | −0.01 | 0.077 |
| 22YS | 9 | 13,835,261 | C | T | 4.72 | −0.25 | 0.00 | 0.092 |
| 22YS, 23JH, BLUP | 9 | 14,820,336 | C | A | 5.25 | 0.24 | −0.14 | 0.055 |
| 22YS, 23JH, BLUP | 9 | 92,493,718 | T | C | 5.24 | −0.57 | −0.16 | 0.053 |
| 22YS | 10 | 115,482,753 | G | A | 4.67 | 0.23 | 0.27 | 0.110 |
| 22YS | 10 | 138,012,512 | T | G | 5.23 | 0.35 | −0.61 | 0.117 |
| 23JH | 1 | 89,810,991 | G | T | 4.61 | −0.36 | −0.11 | 0.053 |
| 23JH | 2 | 172,364,269 | T | C | 5.02 | NaN | NaN | 0.083 |
| 23JH, BLUP | 4 | 80,064,051 | C | A | 4.84 | 0.26 | 0.12 | 0.034 |
| 23JH, BLUP | 9 | 110,672,521 | T | C | 4.83 | −0.25 | 0.21 | 0.071 |
| 23JH, 21YS | 10 | 17,500,491 | C | G | 4.85 | NaN | NaN | 0.057 |
| 21YS | 2 | 5,102,776 | G | T | 5.08 | −0.27 | −0.58 | 0.064 |
| 21YS | 2 | 62,659,851 | G | A | 5.21 | NaN | NaN | 0.088 |
| 21YS | 4 | 131,018,543 | C | A | 5.40 | NaN | NaN | 0.072 |
| 21YS | 4 | 132,312,280 | C | T | 4.76 | NaN | NaN | 0.055 |
| 21YS | 5 | 221,373,665 | G | A | 6.13 | 0.22 | −0.01 | 0.059 |
| 21YS | 5 | 222,162,464 | T | C | 4.70 | 0.17 | −0.01 | 0.044 |
| 21YS | 7 | 21,816,794 | C | G | 5.00 | 0.24 | −0.13 | 0.073 |
| 21YS | 7 | 165,218,537 | A | G | 5.96 | 0.26 | −0.17 | 0.084 |
| 21YS | 9 | 40,627,693 | G | A | 5.45 | NaN | NaN | 0.075 |
| 21YS | 9 | 54,914,157 | T | C | 4.64 | NaN | NaN | 0.057 |
| 21YS | 9 | 108,901,209 | A | G | 4.89 | 0.20 | 0.34 | 0.053 |
| 21YS, BLUP | 9 | 108,933,426 | A | G | 7.44 | 0.29 | −0.09 | 0.108 |
| 21YS | 9 | 109,017,561 | A | G | 5.50 | 0.21 | −0.15 | 0.075 |
| 21YS | 9 | 109,122,650 | G | A | 4.58 | −0.20 | 0.14 | 0.080 |
| 21YS | 9 | 109,283,271 | G | A | 4.94 | −0.16 | −0.34 | 0.086 |
| 21YS | 9 | 109,407,646 | C | T | 5.07 | 0.22 | 0.16 | 0.063 |
| 21YS | 9 | 109,451,171 | G | C | 4.94 | −0.20 | 0.09 | 0.057 |
| 21YS | 9 | 110,611,574 | G | T | 5.70 | 0.29 | −0.32 | 0.132 |
| 21YS | 9 | 110,672,521 | T | C | 5.71 | −0.25 | −0.19 | 0.110 |
| 21YS | 10 | 17,500,491 | C | G | 4.60 | NaN | NaN | 0.074 |
| BLUP | 7 | 142,297,954 | A | G | 4.83 | 0.19 | −0.09 | 0.076 |
| BLUP | 9 | 108,933,426 | A | G | 4.51 | 0.21 | −0.03 | 0.081 |
| BLUP | 9 | 152,304,134 | T | A | 4.94 | 0.17 | −0.11 | 0.056 |
| Mapping Population | QTL | Chromosome | Position (cM) | Mapping Interval (bp) | LOD | Additive Effect | R2 |
|---|---|---|---|---|---|---|---|
| pop1 | qTOC2-1 | 2 | 115 | 29,801,036–37,206,712 | 2.90 | 0.163 | 0.090 |
| qTOC2-2 | 2 | 106.98 | 14,177,571–47,384,437 | 4.63 | 0.130 | 0.060 | |
| qTOC2-3 | 2 | 117.03 | 14,177,571–37,206,712 | 3.75 | 0.130 | 0.050 | |
| qTOC3-1 | 3 | 33.25 | 193,353,260–227,027,669 | 3.82 | −0.098 | 0.030 | |
| qTOC9-1 | 9 | 75.92 | 110,367,967–125,469,868 | 3.39 | −0.106 | 0.030 | |
| pop3 | qTOC1-1 | 1 | 27.11 | 273,307,800–273,997,185 | 3.81 | 0.400 | 0.131 |
| qTOC1-2 | 1 | 198.72 | 61,742,795–76,153,784 | 3.82 | 0.250 | 0.014 | |
| qTOC4-1 | 4 | 232.73 | 161,621,304–164,227,248 | 5.65 | 0.650 | 0.199 | |
| qTOC4-2 | 4 | 599.88 | 172,418,126–175,502,163 | 3.46 | 0.320 | 0.143 | |
| qTOC7-1 | 7 | 199.1 | 50,416,681–50,735,457 | 4.33 | −0.560 | 0.151 | |
| pop4 | qTOC5-1 | 5 | 34.22 | 79,346,283–81,012,543 | 3.31 | 0.038 | 0.005 |
| qTOC7-1 | 7 | 29.17 | 132,794,775–153,544,218 | 3.01 | −0.146 | 0.110 | |
| qTOC8-1 | 8 | 89.73 | 10,663,910–30,775,820 | 4.61 | −0.166 | 0.124 | |
| pop5 | qTOC2-1 | 2 | 1.57 | 222,088,040–232,416,169 | 3.75 | −0.290 | 0.144 |
| qTOC2-2 | 2 | 15.67 | 159,679,997–163,407,700 | 5.73 | 0.360 | 0.231 | |
| qTOC3-1 | 3 | 33.53 | 114,248,818–166,417,378 | 4.27 | −0.240 | 0.177 | |
| qTOC5-1 | 5 | 22.99 | 135,409,128–153,772,976 | 3.92 | 0.280 | 0.146 | |
| qTOC5-2 | 5 | 55.99 | 31,369,431–38,866,833 | 2.80 | −0.230 | 0.103 |
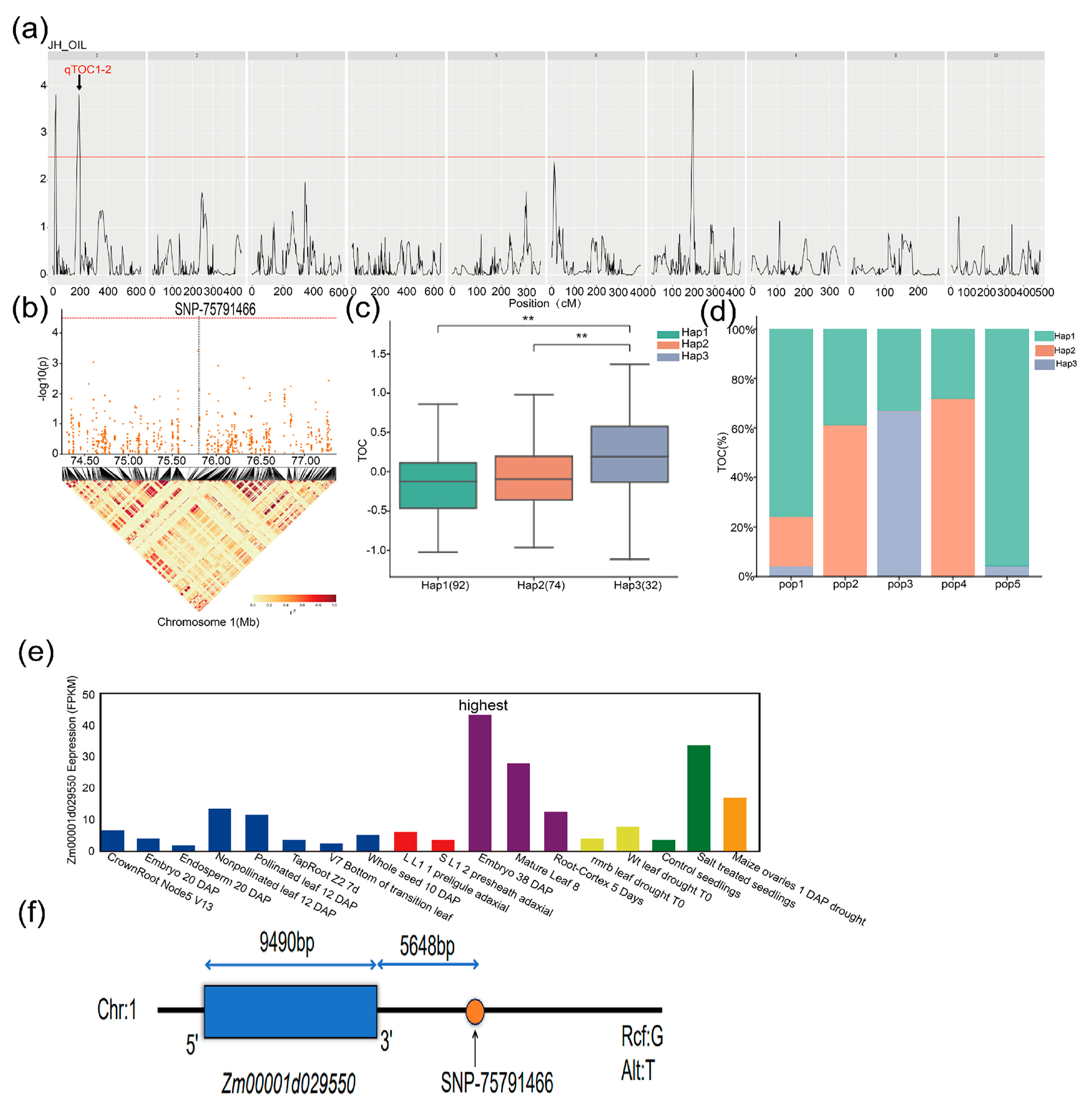
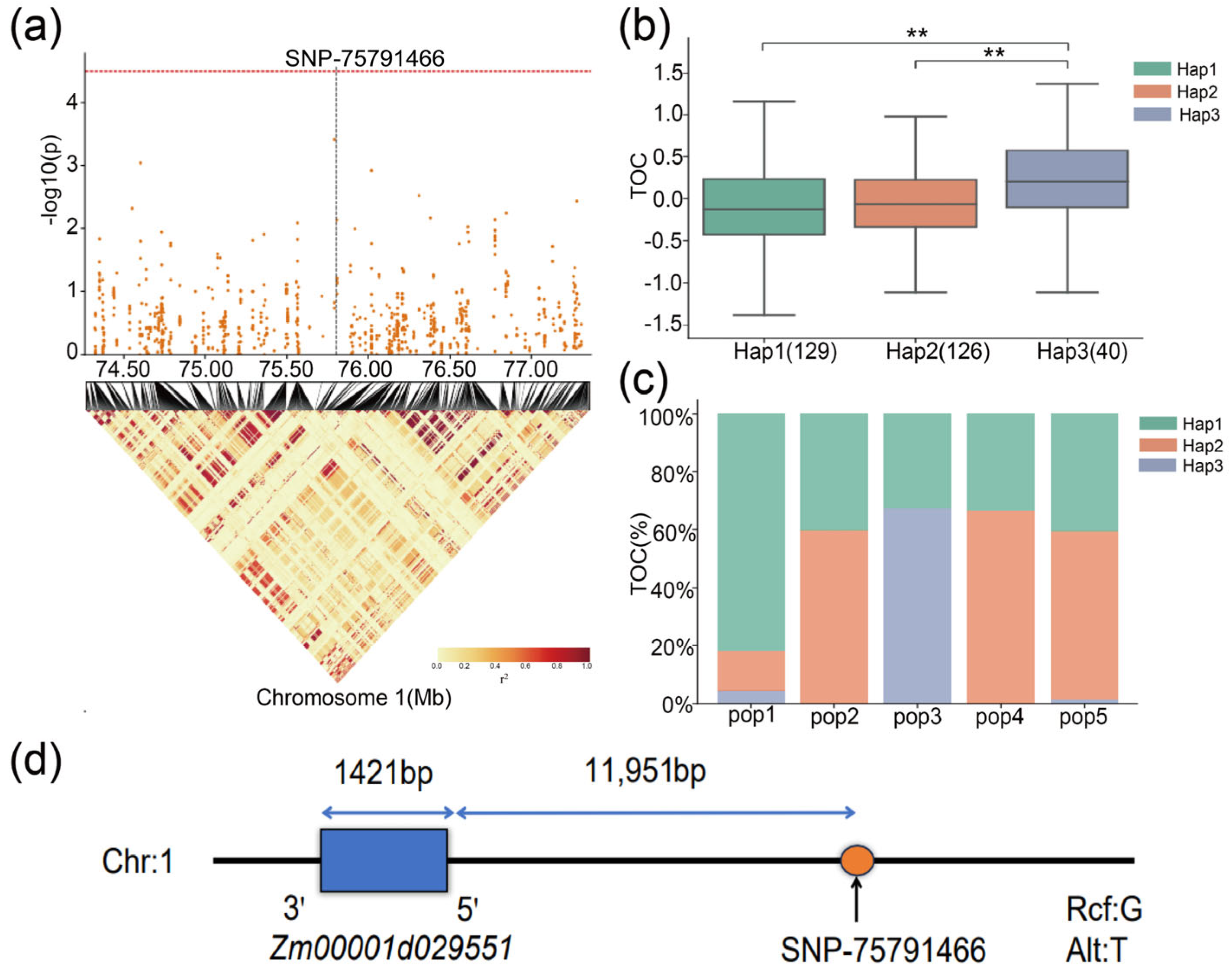
| SNP/QTL | Chromosome | Position | Mapping Interval (bp) | Candidate Gene | Candidate Gene Range (bp) | Gene Annotation |
|---|---|---|---|---|---|---|
| SNP-75791466 | 1 | 75,791,466 bp | [75,771,466–75,811,466] | Zm00001d029550 | 75,797,114–75,806,603 | Diacylglycerol Kinase 1 |
| qTOC1-2 | 1 | 198.72 cM | 61,742,795–76,153,784 | |||
| SNP-75791466 | 1 | 75,791,466 bp | [75,771,466–75,811,466] | Zm00001d029551 | 75,803,417–75,804,837 | NaN |
| qTOC1-2 | 1 | 198.72 cM | 61,742,795–76,153,784 |
| Gene ID | SNP Position | Haplotype | Hap_Sample_Num 1 |
|---|---|---|---|
| Zm00001d029550 | Chr1: 75,791,466 bp | GTTCTACG(Hap1) | 192 |
| ATCTGGTA(Hap2) | 74 | ||
| ACCTGGTA(Hap3) | 32 | ||
| Zm00001d029551 | Chr1: 75,791,466 bp | GTTCT(Hap1) | 129 |
| ATCTG(Hap2) | 126 | ||
| ACCTG(Hap3) | 40 |
| Materials | Population Type | Trait | QTL | Marker/Physical Interval | LOD | PVE (%) | Reference |
|---|---|---|---|---|---|---|---|
| B73, By804 | RIL | KO | qKO1-1 | umc1598–umc1884 | - | 14.3 | [25] |
| Ku13, Sc55 | RIL | OIL | qOLE1-1 | 66.4–71.0 Mb | 4.84 | 11.06 | [8] |
| B73, By804 | RIL | OIL | OIL1-1 | umc2217–bnlg2086 | - | - | [11] |
| B73, By804 | RIL | EER | qEEWR1-1 | 73,374,836–73,376,998 bp | - | - | [26] |
| Parent | Pedigree | Heterotic Group | Ecological Type | Total Oil Content (%) |
|---|---|---|---|---|
| Ye107 | Derived from US hybrid DeKalb XL80 | Reid | Temperate | 3.51 |
| CML312 | S89500-F2-2-2-1-1-B*5-2-1-6-1(DH) | nonReid | Subtropical | 6.8 |
| CML384 | P502c1#-771-2-2-1-3-B-1-1-3-1(DH) | Reid | Subtropical | 7.03 |
| CML395 | 90323B-1-B-1-B*4-1-1-2-1(DH) | nonReid | Tropical | 7.1 |
| YML46 | SW1-1-1-2-1-2-1 | Suwan | Tropical | 5.9 |
| YML32 | Suwan 1(S)C9-S8-346-2 (Kei 8902)-3-4-4-6 | Suwan | Tropical | 7.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, M.; Sun, J.; Jiang, F.; Shaw, R.K.; Ijaz, B.; Fan, X. Mining of Oil Content Genes in Recombinant Maize Inbred Lines with Introgression from Temperate and Tropical Germplasm. Int. J. Mol. Sci. 2024, 25, 10813. https://doi.org/10.3390/ijms251910813
Shi M, Sun J, Jiang F, Shaw RK, Ijaz B, Fan X. Mining of Oil Content Genes in Recombinant Maize Inbred Lines with Introgression from Temperate and Tropical Germplasm. International Journal of Molecular Sciences. 2024; 25(19):10813. https://doi.org/10.3390/ijms251910813
Chicago/Turabian StyleShi, Mengfei, Jiachen Sun, Fuyan Jiang, Ranjan K. Shaw, Babar Ijaz, and Xingming Fan. 2024. "Mining of Oil Content Genes in Recombinant Maize Inbred Lines with Introgression from Temperate and Tropical Germplasm" International Journal of Molecular Sciences 25, no. 19: 10813. https://doi.org/10.3390/ijms251910813