
Leveraging Therapeutic Proteins and Peptides from Lumbricus Earthworms: Targeting SOCS2 E3 Ligase for Cardiovascular Therapy through Molecular Dynamics Simulations




Abstract
1. Introduction
2. Results
2.1. Three-Dimensional Structure Modeling
2.2. Protein–Protein Docking Simulation
2.3. Molecular Dynamics (MD) Simulation
2.4. Molecular Mechanics/Poisson–Boltzmann Surface Area (MM/PBSA) Calculations
3. Discussion
4. Limitations, Clinical Implications, and Future Works
5. Materials and Methods
5.1. Materials
5.2. Computing Power
5.3. Three-Dimensional Structure Modeling
5.4. Protein–Protein Docking Simulation
5.5. Molecular Dynamics (MD) Simulation
5.6. Molecular Mechanics/Poisson–Boltzmann Surface Area (MM/PBSA) Calculations
5.7. Statistical Analysis
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Cardiovascular Diseases (CVDs): World Health Organization. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 1 April 2024).
- Roth Gregory, A.; Mensah George, A.; Johnson Catherine, O.; Addolorato, G.; Ammirati, E.; Baddour Larry, M.; Barengo Noël, C.; Beaton Andrea, Z.; Benjamin Emelia, J.; Benziger Catherine, P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Del Buono, M.G.; Montone, R.A.; Camilli, M.; Carbone, S.; Narula, J.; Lavie, C.J.; Niccoli, G.; Crea, F. Coronary Microvascular Dysfunction Across the Spectrum of Cardiovascular Diseases: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 1352–1371. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Marc, T. Cardiovascular Disease: An Introduction. Vasculopathies 2018, 8, 1–90. [Google Scholar]
- Ullah, A.; Kumar, M.; Sayyar, M.; Sapna, F.; John, C.; Memon, S.; Qureshi, K.; Agbo, E.C.; Ariri, H.I.; Chukwu, E.J.; et al. Revolutionizing Cardiac Care: A Comprehensive Narrative Review of Cardiac Rehabilitation and the Evolution of Cardiovascular Medicine. Cureus 2023, 15, e46469. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jansen-Chaparro, S.; López-Carmona, M.D.; Cobos-Palacios, L.; Sanz-Cánovas, J.; Bernal-López, M.R.; Gómez-Huelgas, R. Statins and Peripheral Arterial Disease: A Narrative Review. Front. Cardiovasc. Med. 2021, 8, 1–18. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rousan, T.A.; Thadani, U. Stable Angina Medical Therapy Management Guidelines: A Critical Review of Guidelines from the European Society of Cardiology and National Institute for Health and Care Excellence. Eur. Cardiol. 2019, 14, 18–22. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kario, K.; Hoshide, S.; Narita, K.; Okawara, Y.; Kanegae, H.; Aoki, K.; Kihara, H.; Koga, T.; Nakata, T.; Oku, K.; et al. Cardiovascular Prognosis in Drug-Resistant Hypertension Stratified by 24-Hour Ambulatory Blood Pressure: The JAMP Study. Hypertension 2021, 78, 1781–1790. [Google Scholar] [CrossRef]
- Wijkman, M.O.; Malachias, M.V.B.; Claggett, B.L.; Cheng, S.; Matsushita, K.; Shah, A.M.; Jhund, P.S.; Coresh, J.; Solomon, S.D.; Vardeny, O. Resistance to antihypertensive treatment and long-term risk: The Atherosclerosis Risk in Communities study. J. Clin. Hypertens. 2021, 23, 1887–1896. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liang, C.; Zhang, L.; Lian, X.; Zhu, T.; Zhang, Y.; Gu, N. Circulating Exosomal SOCS2-AS1 Acts as a Novel Biomarker in Predicting the Diagnosis of Coronary Artery Disease. BioMed Res. Int. 2020, 2020, 9182091. [Google Scholar] [CrossRef]
- Zhang, H.; Dhalla, N.S. The Role of Pro-Inflammatory Cytokines in the Pathogenesis of Cardiovascular Disease. Int. J. Mol. Sci. 2024, 25, 1082. [Google Scholar] [CrossRef]
- Shook, P.L.; Singh, M.; Singh, K. Macrophages in the Inflammatory Phase following Myocardial Infarction: Role of Exogenous Ubiquitin. Biology 2023, 12, 1258. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Klaeske, K.; Dix, M.; Adams, V.; Jawad, K.; Eifert, S.; Etz, C.; Saeed, D.; Borger, M.A.; Dieterlen, M.T. Differential Regulation of Myocardial E3 Ligases and Deubiquitinases in Ischemic Heart Failure. Life 2021, 11, 1430. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rico-Bautista, E.; Flores-Morales, A.; Fernández-Pérez, L. Suppressor of cytokine signaling (SOCS) 2, a protein with multiple functions. Cytokine Growth Factor Rev. 2006, 17, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Durham, G.; Williams, J.; Nasim, T.; Palmer, T. Targeting SOCS Proteins to Control JAK-STAT Signalling in Disease. Trends Pharmacol. Sci. 2019, 40, 298–308. [Google Scholar] [CrossRef]
- Mustafa, R.; Saiqa, A.; Domínguez, J.; Jamil, M.; Manzoor, S.; Wazir, S.; Shaheen, B.; Parveen, A.; Khan, R.; Ali, S. Therapeutic Values of Earthworm Species Extract from Azad Kashmir as Anticoagulant, Antibacterial, and Antioxidant Agents. Can. J. Infect. Dis. Med. Microbiol. 2022, 2022, 1–20. [Google Scholar] [CrossRef]
- Cooper, E.L.; Balamurugan, M.; Huang, C.Y.; Tsao, C.R.; Heredia, J.; Tommaseo-Ponzetta, M.; Paoletti, M.G. Earthworms dilong: Ancient, inexpensive, noncontroversial models may help clarify approaches to integrated medicine emphasizing neuroimmune systems. Evid. Based Complement. Altern. Med. 2012, 2012, 164152. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Woo, Y.M.; Lee, Y.H.; Ahn, M.Y.; Lee, D.G.; Lee, S.H.; Ha, J.M.; Park, C.I.; Kim, A. Data on the potent fibrinolytic effects of the Lumbricus rubellus earthworm and the Perinereis linea lugworm. Data Brief 2019, 26, 104484. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.H.; Han, C.K.; Shibu, M.A.; Pai, P.Y.; Ho, T.J.; Day, C.H.; Tsai, F.J.; Tsai, C.H.; Yao, C.H.; Huang, C.Y. Lumbrokinase from earthworm extract ameliorates second-hand smoke-induced cardiac fibrosis. Environ. Toxicol. 2015, 30, 1216–1225. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, L.; Zheng, C.; Ma, A.; Hu, K.; Xiang, A.; Sun, Z.; Xie, B.; Xiong, G.; Shi, L.; et al. Novel ACE inhibitory peptides derived from bighead carp (Aristichthys nobilis) hydrolysates: Screening, inhibition mechanisms and the bioconjugation effect with graphene oxide. Food Biosci. 2023, 52, 102399. [Google Scholar] [CrossRef]
- Prakash, M.; Balamurugan, M.; Parthasarathi, K.; Gunasekaran, G.; Cooper, E.; Ranganathan, L. Anti-ulceral and anti-oxidative properties of “earthworm paste” of Lampito mauritii (Kinberg) on Rattus Norvegicus. Eur. Rev. Med. Pharmacol. Sci. 2006, 11, 9–15. [Google Scholar]
- Wang, Y.-H.; Chen, K.-M.; Chiu, P.-S.; Lai, S.-C.; Su, H.-H.; Jan, M.-S.; Lin, C.-W.; Lu, D.-Y.; Fu, Y.-T.; Liao, J.-M.; et al. Lumbrokinase attenuates myocardial ischemia-reperfusion injury by inhibiting TLR4 signaling. J. Mol. Cell. Cardiol. 2016, 99, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Li, P.; Yang, Q. Improving the absorption of earthworm fibrinolytic enzymes with mucosal enhancers. Pharm. Biol. 2010, 48, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Dermawan, D.; Prabowo, B.A.; Rakhmadina, C.A. In silico study of medicinal plants with cyclodextrin inclusion complex as the potential inhibitors against SARS-CoV-2 main protease (Mpro) and spike (S) receptor. Inform. Med. Unlocked 2021, 25, 100645. [Google Scholar] [CrossRef] [PubMed]
- Challapa-Mamani, M.R.; Tomás-Alvarado, E.; Espinoza-Baigorria, A.; León-Figueroa, D.A.; Sah, R.; Rodriguez-Morales, A.J.; Barboza, J.J. Molecular Docking and Molecular Dynamics Simulations in Related to Leishmania donovani: An Update and Literature Review. Trop. Med. Infect. Dis. 2023, 8, 457. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sánchez-Gloria, J.L.; Arellano-Buendía, A.S.; Juárez-Rojas, J.G.; García-Arroyo, F.E.; Argüello-García, R.; Sánchez-Muñoz, F.; Sánchez-Lozada, L.G.; Osorio-Alonso, H. Cellular Mechanisms Underlying the Cardioprotective Role of Allicin on Cardiovascular Diseases. Int. J. Mol. Sci. 2022, 23, 9082. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Choudhury, T.Z.; Garg, V. Molecular genetic mechanisms of congenital heart disease. Curr. Opin. Genet. Dev. 2022, 75, 101949. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins 2009, 77 (Suppl. S9), 100–113. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, S.; Narimani, Z.; Ashouri, M.; Firouzi, R.; Karimi-Jafari, M.H. Ensemble learning from ensemble docking: Revisiting the optimum ensemble size problem. Sci. Rep. 2022, 12, 410. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shyamal, M.; Mandal, T.K.; Panja, A.; Saha, A. Influence of anionic co-ligands on the structural diversity and catecholase activity of copper(II) complexes with 2-methoxy-6-(8-iminoquinolinylmethyl)phenol. RSC Adv. 2014, 4, 53520–53530. [Google Scholar] [CrossRef]
- Kufareva, I.; Abagyan, R. Methods of Protein Structure Comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar] [CrossRef]
- Russell, R.; Alber, F.; Aloy, P.; Davis, F.; Korkin, D.; Pichaud, M.; Topf, M.; Sali, A. A structural perspective on protein-protein interactions. Curr. Opin. Struct. Biol. 2004, 14, 313–324. [Google Scholar] [CrossRef]
- Dagliyan, O.; Proctor, E.A.; D’Auria, K.M.; Ding, F.; Dokholyan, N.V. Structural and dynamic determinants of protein-peptide recognition. Structure 2011, 19, 1837–1845. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guedes, I.A.; Pereira, F.S.S.; Dardenne, L.E. Empirical Scoring Functions for Structure-Based Virtual Screening: Applications, Critical Aspects, and Challenges. Front. Pharmacol. 2018, 9, 1089. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Desantis, F.; Miotto, M.; Di Rienzo, L.; Milanetti, E.; Ruocco, G. Spatial organization of hydrophobic and charged residues affects protein thermal stability and binding affinity. Sci. Rep. 2022, 12, 12087. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bitencourt-Ferreira, G.; Veit-Acosta, M.; De Azevedo, W., Jr. Van der Waals Potential in Protein Complexes. Methods Mol. Biol. 2019, 2053, 79–91. [Google Scholar]
- Cramer, J.; Krimmer, S.; Heine, A.; Klebe, G. Paying the Price of Desolvation in Solvent-Exposed Protein Pockets: Impact of Distal Solubilizing Groups on Affinity and Binding Thermodynamics in a Series of Thermolysin Inhibitors. J. Med. Chem. 2017, 60, 5791–5799. [Google Scholar] [CrossRef]
- Zhou, H.-X.; Pang, X. Electrostatic Interactions in Protein Structure, Folding, Binding, and Condensation. Chem. Rev. 2018, 118, 1691–1741. [Google Scholar] [CrossRef] [PubMed]
- Kastritis, P.L.; Rodrigues, J.P.G.L.M.; Bonvin, A.M.J.J. HADDOCK2P2I: A Biophysical Model for Predicting the Binding Affinity of Protein–Protein Interaction Inhibitors. J. Chem. Inf. Model. 2014, 54, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Kastritis, P.L.; Bonvin, A.M. On the binding affinity of macromolecular interactions: Daring to ask why proteins interact. J. R. Soc. Interface 2013, 10, 20120835. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dermawan, D.; Sumirtanurdin, R.; Dewantisari, D. Simulasi dinamika molekular reseptor estrogen alfa dengan andrografolid sebagai anti kanker payudara. Indones J. Pharm. Sci. Technol. 2019, 6, 65–76. [Google Scholar] [CrossRef]
- Craveur, P.; Joseph, A.P.; Esque, J.; Narwani, T.J.; Noël, F.; Shinada, N.; Goguet, M.; Leonard, S.; Poulain, P.; Bertrand, O.; et al. Protein flexibility in the light of structural alphabets. Front. Mol. Biosci. 2015, 2, 20. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Maspero, E.; Mari, S.; Valentini, E.; Musacchio, A.; Fish, A.; Pasqualato, S.; Polo, S. Structure of the HECT:ubiquitin complex and its role in ubiquitin chain elongation. EMBO Rep. 2011, 12, 342–349. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sanusi, Z.K.; Lobb, K.A. Insights into the Dynamics and Binding of Two Polyprotein Substrate Cleavage Points in the Context of the SARS-CoV-2 Main and Papain-like Proteases. Molecules 2022, 27, 8251. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical assessment of methods of protein structure prediction (CASP)-Round XIII. Proteins 2019, 87, 1011–1020. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lenselink, E.B.; Louvel, J.; Forti, A.F.; van Veldhoven, J.P.D.; de Vries, H.; Mulder-Krieger, T.; McRobb, F.M.; Negri, A.; Goose, J.; Abel, R.; et al. Predicting Binding Affinities for GPCR Ligands Using Free-Energy Perturbation. ACS Omega 2016, 1, 293–304. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Salsbury, F.R., Jr. Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr. Opin. Pharmacol. 2010, 10, 738–744. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lear, T.B.; McKelvey, A.C.; Evankovich, J.W.; Rajbhandari, S.; Coon, T.A.; Dunn, S.R.; Londino, J.D.; McVerry, B.J.; Zhang, Y.; Valenzi, E.; et al. KIAA0317 regulates pulmonary inflammation through SOCS2 degradation. JCI Insight 2019, 4, e129110. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bulatov, E.; Ciulli, A. Targeting Cullin–RING E3 ubiquitin ligases for drug discovery: Structure, assembly and small-molecule modulation. Biochem. J. 2015, 467, 365–386. [Google Scholar] [CrossRef] [PubMed]
- Piessevaux, J.; Lavens, D.; Montoye, T.; Wauman, J.; Catteeuw, D.; Vandekerckhove, J.; Belsham, D.; Peelman, F.; Tavernier, J. Functional Cross-modulation between SOCS Proteins Can Stimulate Cytokine Signaling. J. Biol. Chem. 2006, 281, 32953–32966. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L.; Brown, W.M. Complete sequence of the mitochondrial DNA of the annelid worm Lumbricus terrestris. Genetics 1995, 141, 305–319. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Parkos, C.A.; Dinauer, M.C.; Walker, L.E.; Allen, R.A.; Jesaitis, A.J.; Orkin, S.H. Primary structure and unique expression of the 22-kilodalton light chain of human neutrophil cytochrome b. Proc. Natl. Acad. Sci. USA 1988, 85, 3319–3323. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nguyen, Q.T.T.; Rhee, H.; Kim, M.; Lee, M.Y.; Lee, E.J. Lumbrokinase, a Fibrinolytic Enzyme, Prevents Intra-Abdominal Adhesion by Inhibiting the Migrative and Adhesive Activities of Fibroblast via Attenuation of the AP-1/ICAM-1 Signaling Pathway. Biomed. Res. Int. 2023, 2023, 4050730. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, S.; Fan, G.; Li, J. Improving completeness and accuracy of 3D point clouds by using deep learning for applications of digital twins to civil structures. Adv. Eng. Inform. 2023, 58, 102196. [Google Scholar] [CrossRef]
- Davis, E.M.; Sun, Y.; Liu, Y.; Kolekar, P.; Shao, Y.; Szlachta, K.; Mulder, H.L.; Ren, D.; Rice, S.V.; Wang, Z.; et al. SequencErr: Measuring and suppressing sequencer errors in next-generation sequencing data. Genome Biol. 2021, 22, 37. [Google Scholar] [CrossRef] [PubMed]
- Faits, T.; Odom, A.; Castro-Nallar, E.; Crandall, K.; Johnson, W. Metagenomic profiling pipelines improve taxonomic classification for 16S amplicon sequencing data2022. Sci. Rep. 2023, 13, 13957. [Google Scholar]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Kung, W.-W.; Ramachandran, S.; Makukhin, N.; Bruno, E.; Ciulli, A. Structural insights into substrate recognition by the SOCS2 E3 ubiquitin ligase. Nat. Commun. 2019, 10, 2534. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Johansson, M.U.; Zoete, V.; Michielin, O.; Guex, N. Defining and searching for structural motifs using DeepView/Swiss-PdbViewer. BMC Bioinform. 2012, 13, 173. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gitlin, A.D.; Maltzman, A.; Kanno, Y.; Heger, K.; Reja, R.; Schubert, A.F.; Wierciszewski, L.J.; Pantua, H.; Kapadia, S.B.; Harris, S.F.; et al. N4BP1 coordinates ubiquitin-dependent crosstalk within the IκB kinase family to limit Toll-like receptor signaling and inflammation. Immunity 2024, 57, 973–986. [Google Scholar] [CrossRef]
- Ramachandran, S.; Makukhin, N.; Haubrich, K.; Nagala, M.; Forrester, B.; Lynch, D.M.; Casement, R.; Testa, A.; Bruno, E.; Gitto, R.; et al. Structure-based design of a phosphotyrosine-masked covalent ligand targeting the E3 ligase SOCS2. Nat. Commun. 2023, 14, 6345. [Google Scholar] [CrossRef]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A Protein−Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef]
- van Zundert, G.C.P.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.; et al. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLSAA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yuet, P.; Blankschtein, D. Molecular Dynamics Simulation Study of Water Surfaces: Comparison of Flexible Water Models. J. Phys. Chem. B. 2010, 114, 13786–13795. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Chan, H.C.S.; Hu, Z. Using PyMOL as a platform for computational drug design. WIREs Comput Mol Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Sun, H.; Pan, P.; Li, D.; Zhen, X.; Li, Y.; Hou, T. Assessing an ensemble docking-based virtual screening strategy for kinase targets by considering protein flexibility. J. Chem. Inf. Model. 2014, 54, 2664–2679. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Chen, X.; Fan, S.; Chang, L.; Chu, L.; Zhang, Y.; Wang, J.; Li, S.; Xie, J.; Hu, J.; et al. Binding Free Energy Calculation Based on the Fragment Molecular Orbital Method and Its Application in Designing Novel SHP-2 Allosteric Inhibitors. Int. J. Mol. Sci. 2024, 25, 671. [Google Scholar] [CrossRef]
- Rifai, E.A.; Ferrario, V.; Pleiss, J.; Geerke, D.P. Combined Linear Interaction Energy and Alchemical Solvation Free-Energy Approach for Protein-Binding Affinity Computation. J. Chem. Theory Comput. 2020, 16, 1300–1310. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A. Moreno E gmx_MMPBSA: ANew Tool to Perform End-State Free Energy Calculations with, G.R.O.M.A.C.S. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Miller, B.R., 3rd; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: Effic. Program End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Panday, S.K.; Alexov, E. Protein-Protein Binding Free Energy Predictions with the MM/PBSA Approach Complemented with the Gaussian-Based Method for Entropy Estimation. ACS Omega 2022, 7, 11057–11067. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- IBM. IBM SPSS Statistics for Windows, Version 25.0 ed.; IBM Corp: New York, NY, USA, 2017. [Google Scholar]
- OriginLab. Origin(Pro); 2022 ed.; OriginLab Corporation: Northampton, MA, USA, 2022. [Google Scholar]

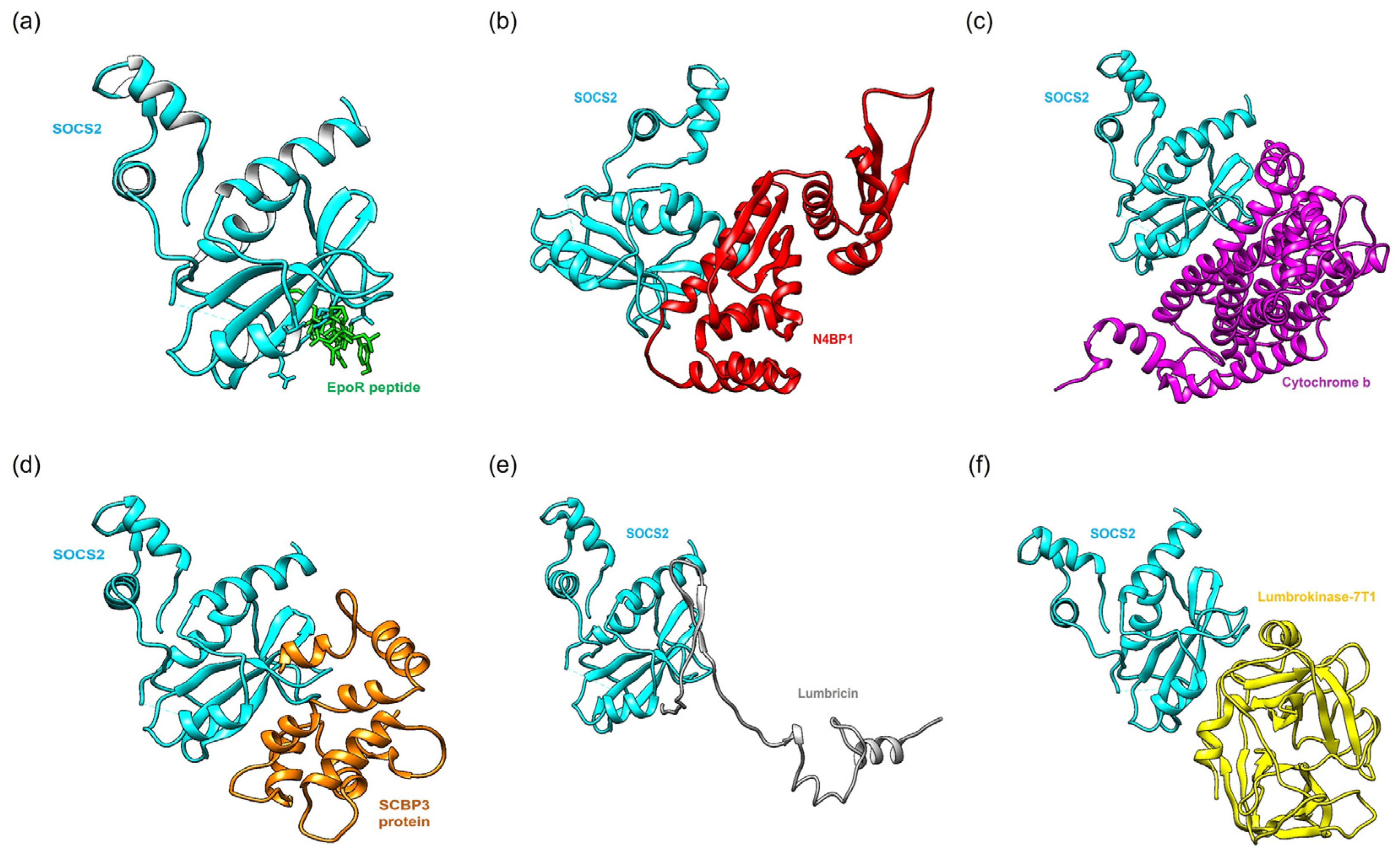
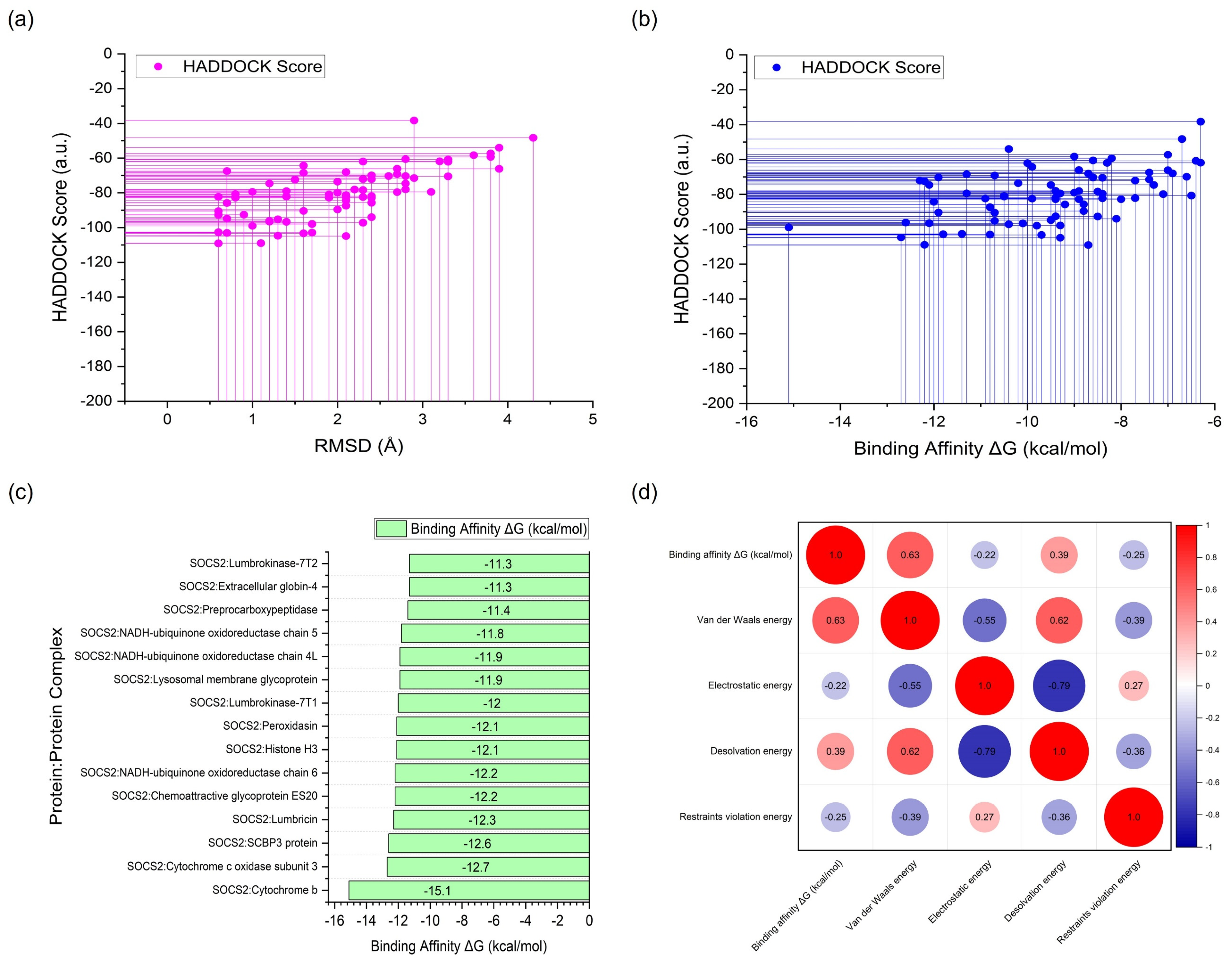

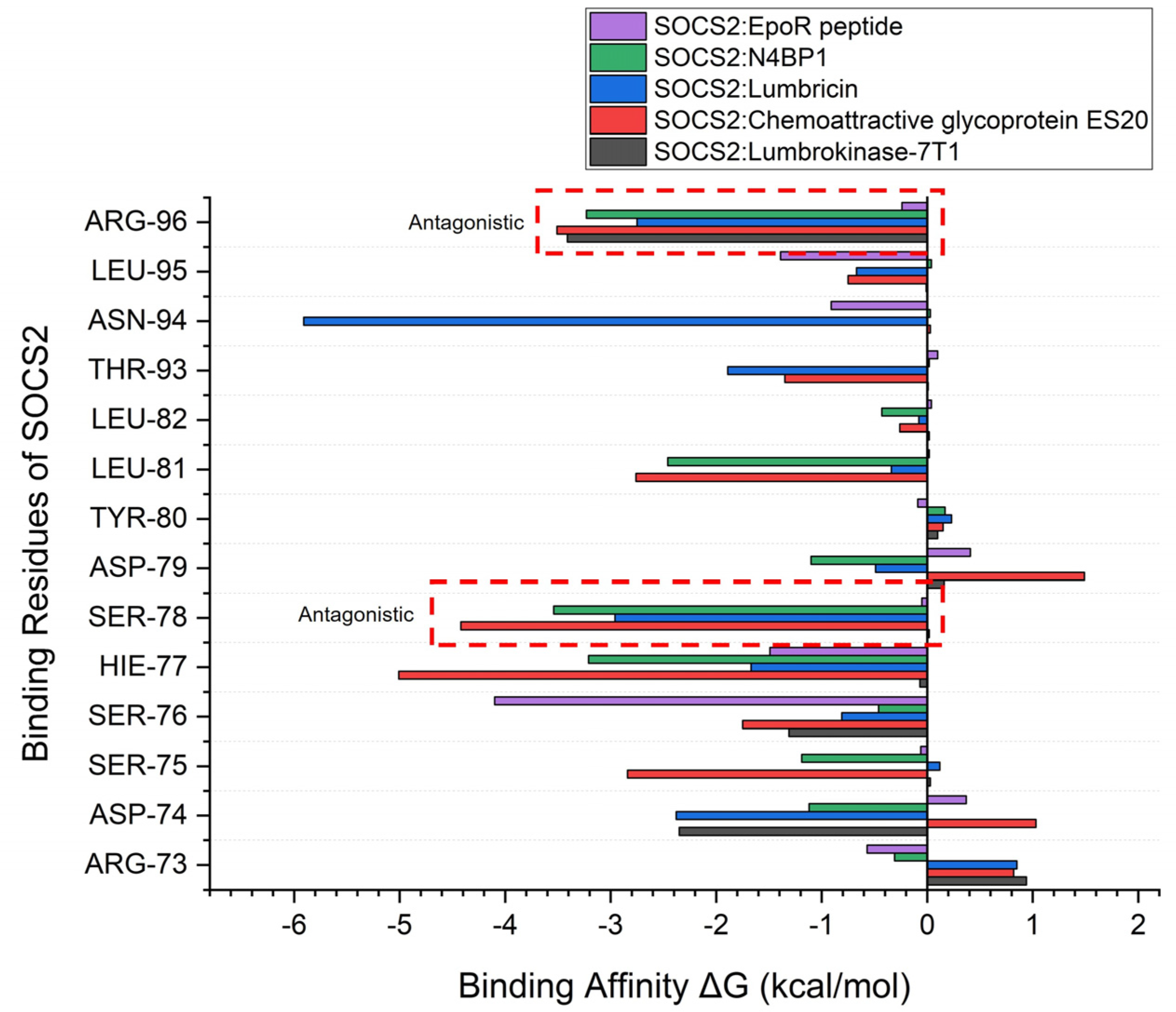

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein/Peptide | UniProt ID | Sequence | Size (kDa) |
|---|---|---|---|
| Actin-1 | P92182 | MCDEEVTALVVDNGSGMCKAGFAGDDAPRAVFPSIVGRPRHQGVMVGMGQKDSYVGDEAQSKRGILTLKYPIEHGIVTNWDDMEKIWHHTFYNELRVAPEEHPVLLTEAPLNPKANREKMTQIMFETFNSPAMYVAIQAVLSLYASGRTTGIVLDSGDGVTHTVPIYEGYALPHAILRLDLAGRDLTDYLMKILTERGYSFTTTAEREIVRDIKEKLCYVALDFDQEMGTAASSSSLEKSYELPDGQVITIGNERFRCPESMFQPAFLGMESAGIHETTFNSIMKCDVDIRKDLYANTVMSGGTTMFPGIADRMQKEITSMAPSTMKIKIIAPPERKYSVWIGGSILASLSTFQQMWISKQEYDESGPSIVHRKCF | 41.85 |
| Chemoattractive glycoprotein ES20 | O44335 | MKTYLLLVFLVGAHALVCPPGFTYLPAGESCYKVIFESHDWHSATERCRQESRGLAAISTPEESIAVKEFIDTEISKDSAGAAVCHPTGQSGIRFWTSGLQTKDTCTKTSFLLKITNTFEVPFDFTNWADGEPTLPRKTEKFSALIVGSSERTPSGTTMTATSSCVHSANISNDTLKRISVLPHLYVGFCDEIWLYLNFCLISIQILI | 22.96 |
| Cytochrome b | Q34945 | MFKPIRTTHPAIKIINSTLIDLPAPNNISIWWNYGSLLGLCLVIQVLTGLFLSMHYVPNIEMAFSSVALISRDVNYGWLLRSIHANGASMFFLFIYLHAGRGLYYGSYNLSETWNIGVILFLLTMATAFMGYVLPWGQMSFWGATVITNLFSAIPYIGKTLVEWIWGGFAVDNATLNRFFAFHFILPFAIMGATILHIMFLHESGSNNPIGLNADSDRIPFHPYYSIKDTLGYTLAISALSLMVLFEPNLFTDPENFLMANPLVTPIHIKPEWYFLWMYAILRSIPNKLGGVMALFAAIVILFIPPLTSVMNKRSLSFYPLNKTMFWGLVASWAILTWIGGRPVEDPFIIIGQVFTSLYFIYFISSPTISKLWDDSIII | 42.88 |
| Fibrinolytic enzyme | P83298 | VIGGTNASPGEFPWQLSQQRQSGSWSHSCGASLLSSTSALSASHCVDGVLPNNIRVIAGLWQQSDTSGTQTANVDSYTMHENYGAGTASYSNDIAILHLATSISLGGNIQAAVLPANNNNDYAGTTCVISGWGRTDGTNNLPDILQKSSIPVITTAQCTAAMVGVGGANIWDNHICVQDPAGNTGACNGDSGGPLNCPDGGTRVVGVTSWVVSSGLGTCLPDYPSVYTRVSAYLGWIGDNSR | 24.84 |
| Histone H3 | A0A1C9UP21 | GGKAPRKQLATKAARKSAPATGGVKKPHRYRPGTVALREIRRYQKSTELLIRKLPFQRLVREIAQDFKTDLRFQSSAVMALQEASEAYLVGLFEDTNLCAIH | 11.47 |
| Lumbricin | O96447 | MSLCISDYLYLTLTFSKYERQKDKRPYSERKNQYTGPQFLYPPERIPPQKVIKWNEEGLPIYEIPGEGGHAEPAAA | 8.85 |
| Lumbrokinase-7T1 | B8ZZ01 | MRSFVAFLAALSLCQARPQKFLDGARPSFRMGGEQYIIGGSNASPGEFPWQLSQTRGGSHSCGASLLNALNGLSAFHCVDGAAPGTITVIAGLHDRSGTPGSQEVDITGYTMHENYNQGTNTYANDIAILHFASAINIGGNGQAALLPANNDNDYSGLTCVISGWGRKGSSNVLPDTLQKASIQVIGTTQCQSLMGSIGHIWDNHICLYNNTNNVGSCNGDSGGPLNCPDGGTRVAGVTSWGVSSGAGNCLQTYPTVYTRTSAYLSWIANNS | 28.35 |
| Ribosomal protein S27 | Q9U5N5 | MPLTRDLLHPTLKDEKRKCKLKRLVQSPNSFFMDVKCPGCYKITTVFSHAQTVVLCVGCNTVLCQPTGGKARLTEGCSFRRKQH | 9.50 |
| SCBP3 protein | Q7YWL4 | VWEQYLKGVVSDGTRLTQAVFVEAVKKQLGDPNFKKVLAGPLPLFFSAVDGNGDGLIQKDEFQLFFKLLGIPESAEKSFEAIDTNKDGDISKEEFVIAGTDFFTSTDESSPSKYFWGPLV | 13.25 |
| Ubiquitin | P84589 | TITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLR | 7.20 |
| Complex | HADDOCK Score (a.u.) | Binding Affinity ΔG (kcal/mol) | Kd (nM) | Cluster Size | RMSD (Å) |
|---|---|---|---|---|---|
| Standard | |||||
| SOCS2: EpoR peptide (standard agonist) | −81.0 ± 3.5 | −8.8 | 650 | 22 | 1.7 ± 0.1 |
| SOCS2: N4BP1 (standard antagonist) | −94.3 ± 11.4 | −8.3 | 1500 | 17 | 1.2 ± 0.3 |
| Protein and peptide derived from earthworm (Lumbricus genus) | |||||
| SOCS2: Cytochrome b | −99.0 ± 8.2 | −15.1 | 0.02 | 27 | 1.0 ± 0.6 |
| SOCS2: Cytochrome c oxidase subunit 3 | −104.7 ± 6.3 | −12.7 | 1.09 | 15 | 1.3 ± 0.5 |
| SOCS2: SCBP3 protein | −96.2 ± 6.2 | −12.6 | 1.30 | 41 | 1.2 ± 0.9 |
| SOCS2: Lumbricin | −72.1 ± 3.7 | −12.3 | 2.10 | 43 | 2.3 ± 0.3 |
| SOCS2: Chemoattractive glycoprotein ES20 | −72.4 ± 5.3 | −12.2 | 2.30 | 20 | 1.5 ± 1.0 |
| SOCS2: NADH-ubiquinone oxidoreductase chain 6 | −109.0 ± 7.1 | −12.2 | 2.30 | 14 | 1.1 ± 0.7 |
| SOCS2: Histone H3 | −96.6 ± 4.0 | −12.1 | 3.09 | 8 | 1.2 ± 0.2 |
| SOCS2: Peroxidasin | −74.6 ± 7.6 | −12.1 | 2.70 | 8 | 2.8 ± 0.0 |
| SOCS2: Lumbrokinase-7T1 | −84.3 ± 2.8 | −12.0 | 3.69 | 43 | 2.1 ± 0.2 |
| SOCS2: Lysosomal membrane glycoprotein | −70.3 ± 2.3 | −11.9 | 4.29 | 5 | 2.6 ± 0.9 |
| SOCS2: NADH-ubiquinone oxidoreductase chain 4L | −90.4 ± 5.4 | −11.9 | 4.10 | 12 | 1.6 ± 1.0 |
| SOCS2: NADH-ubiquinone oxidoreductase chain 5 | −102.9 ± 11.6 | −11.8 | 4.69 | 7 | 1.7 ± 0.3 |
| SOCS2: Preprocarboxypeptidase | −102.7 ± 9.1 | −11.4 | 9.70 | 19 | 0.6 ± 0.4 |
| SOCS2: Extracellular globin-4 | −79.4 ± 10.8 | −11.3 | 12.00 | 6 | 1.0 ± 0.6 |
| SOCS2: Lumbrokinase-7T2 | −68.5 ± 2.5 | −11.3 | 10.99 | 35 | 1.6 ± 0.5 |
| Complex | ICs Charged-Charged | ICs Charged-Polar | ICs Charged-Apolar | ICs Polar-Polar | ICs Polar-Apolar | ICs Apolar-Apolar | NIS Charged | NIS Apolar |
|---|---|---|---|---|---|---|---|---|
| Standard | ||||||||
| SOCS2: EpoR peptide (standard agonist) | 3 | 8 | 12 | 2 | 11 | 12 | 25.55 | 38.69 |
| SOCS2: N4BP1 (standard antagonist) | 5 | 11 | 20 | 6 | 11 | 8 | 29.71 | 39.49 |
| Protein and peptide derived from earthworm (Lumbricus genus) | ||||||||
| SOCS2: Cytochrome b | 0 | 0 | 25 | 0 | 38 | 33 | 15.13 | 52.85 |
| SOCS2: Cytochrome c oxidase subunit 3 | 2 | 7 | 22 | 6 | 30 | 26 | 14.71 | 49.41 |
| SOCS2: SCBP3 protein | 2 | 3 | 24 | 2 | 27 | 16 | 27.83 | 41.74 |
| SOCS2: Lumbricin | 7 | 9 | 20 | 0 | 22 | 12 | 26.34 | 40.98 |
| SOCS2: Chemoattractive glycoprotein ES20 | 8 | 15 | 24 | 5 | 23 | 16 | 22.98 | 42.39 |
| SOCS2: NADH-ubiquinone oxidoreductase chain 6 | 0 | 3 | 21 | 7 | 32 | 29 | 14.29 | 52.01 |
| SOCS2: Histone H3 | 7 | 6 | 27 | 0 | 20 | 14 | 27.56 | 42.67 |
| SOCS2: Peroxidasin | 13 | 20 | 24 | 5 | 22 | 6 | 26.23 | 41.86 |
| SOCS2: Lumbrokinase-7T1 | 3 | 15 | 12 | 4 | 24 | 10 | 17.72 | 41.14 |
| SOCS2: Lysosomal membrane glycoprotein | 12 | 17 | 18 | 5 | 18 | 11 | 20.49 | 39.02 |
| SOCS2: NADH-ubiquinone oxidoreductase chain 4L | 0 | 8 | 21 | 2 | 24 | 14 | 17.73 | 46.82 |
| SOCS2: NADH-ubiquinone oxidoreductase chain 5 | 0 | 3 | 17 | 1 | 25 | 29 | 14.12 | 50.10 |
| SOCS2: Preprocarboxypeptidase | 7 | 17 | 23 | 5 | 20 | 28 | 26.18 | 40.05 |
| SOCS2: Extracellular globin-4 | 15 | 18 | 28 | 2 | 12 | 6 | 31.28 | 37.04 |
| SOCS2: Lumbrokinase-7T2 | 5 | 10 | 16 | 9 | 24 | 11 | 19.03 | 41.69 |
| Complex | Residue (Receptor) | Protein Atom (Receptor) | Residue (Interacting Protein/Peptide) | Protein Atom (Interacting Protein/Peptide) | Interaction Distance (Å) |
|---|---|---|---|---|---|
| SOCS2: EpoR peptide (standard agonist) | Val55 | N | Asp8 | OD1 | 2.76 |
| Ser76 | OG | Asp8 | OD1 | 3.09 | |
| Arg96 | NH1 | Glu3 | O | 2.69 | |
| Arg96 | NH2 | Glu3 | O | 3.33 | |
| Lys113 | NZ | Ser1 | OG | 2.73 | |
| SOCS2: N4BP1 (standard antagonist) | Gln32 | NE2 | Glu178 | OE1 | 2.77 |
| Arg41 | NH1 | Glu118 | OE1 | 2.65 | |
| Arg41 | NH2 | Glu118 | OE2 | 2.57 | |
| Tyr49 | OH | Lys132 | NZ | 2.78 | |
| Asp74 | OD2 | Lys132 | NZ | 2.55 | |
| Ser75 | O | Asn136 | ND2 | 3.04 | |
| Ser78 | O | Ser174 | OG | 2.63 | |
| Asp79 | OD2 | Lys145 | NZ | 2.62 | |
| Arg96 | NH2 | Glu138 | OE1 | 2.67 | |
| SOCS2: Cytochrome b | His77 | NE2 | Leu122 | O | 2.74 |
| Arg96 | NH2 | Ile119 | O | 2.72 | |
| SOCS2: SCBP3 protein | Lys59 | NZ | Asp50 | O | 2.58 |
| Arg96 | NH1 | Leu68 | O | 2.66 | |
| SOCS2: Lumbricin | Arg41 | NH1 | Glu56 | O | 3.08 |
| Arg41 | NH1 | Glu56 | OE1 | 2.60 | |
| Arg41 | NH2 | Glu56 | O | 2.98 | |
| Tyr49 | OH | Lys53 | NZ | 2.83 | |
| Ser52 | OG | Glu63 | OE1 | 2.59 | |
| Asp74 | OD2 | Lys53 | NZ | 2.65 | |
| Ser78 | OG | Ile46 | O | 2.67 | |
| Asp79 | OD1 | Arg45 | NH1 | 2.65 | |
| Thr93 | OG1 | Glu72 | OE2 | 2.70 | |
| Asn94 | N | Glu72 | OE2 | 2.73 | |
| Asn94 | ND2 | Glu72 | OE1 | 2.64 | |
| Arg96 | NE | Glu72 | O | 2.74 | |
| Arg96 | NH2 | Glu72 | O | 2.87 | |
| SOCS2: Lumbrokinase-7T1 | Glu57 | OE2 | Lys20 | NZ | 2.55 |
| Asp74 | OD2 | Arg17 | NH1 | 2.69 | |
| Asp74 | OD2 | Arg17 | NH2 | 2.72 | |
| Ser76 | O | Gln19 | NE2 | 3.20 | |
| Asp101 | OD1 | Asn249 | ND2 | 2.66 | |
| Asp101 | OD2 | Ser170 | OG | 2.66 | |
| Cys111 | O | Lys168 | NZ | 2.91 | |
| Lys113 | NZ | Gly32 | O | 2.87 | |
| Lys113 | NZ | Gln35 | OE1 | 2.66 | |
| Leu116 | O | Asn214 | ND2 | 2.98 |
| Complex | Average RMSD (Å) | Average RMSF (Å) | Average RoG (Å) | Number of Hydrogen Bonds between the Two Proteins | Potential Energy (kcal/mol) |
|---|---|---|---|---|---|
| Standard | |||||
| SOCS2 (apo-protein) | 2.417 | 1.089 | 1.674 | N/A | −158,603.87 |
| SOCS2: EpoR peptide (standard agonist) | 2.423 | 1.397 | 2.101 | 11 | −159,708.72 |
| SOCS2: N4BP1 (standard antagonist) | 2.496 | 1.179 | 2.162 | 24 | −459,214.66 |
| Protein and peptide derived from earthworm (Lumbricus genus) | |||||
| SOCS2: Cytochrome b | 2.467 | 0.876 | 2.182 | 39 | −471,304.36 |
| SOCS2: Cytochrome c oxidase subunit 3 | 2.504 | 1.122 | 2.176 | 30 | −450,869.63 |
| SOCS2: SCBP3 protein | 2.587 | 1.301 | 2.198 | 22 | −316,369.57 |
| SOCS2: Lumbricin | 2.495 | 1.031 | 2.148 | 18 | −694,628.89 |
| SOCS2: Chemoattractive glycoprotein ES20 | 2.512 | 0.941 | 2.176 | 22 | −621,068.14 |
| SOCS2: NADH-ubiquinone oxidoreductase chain 6 | 2.487 | 1.001 | 2.178 | 20 | −466,577.25 |
| SOCS2: Histone H3 | 2.413 | 0.917 | 2.287 | 16 | −629,402.13 |
| SOCS2: Peroxidasin | 2.599 | 1.106 | 2.678 | 35 | −2,208,424.10 |
| SOCS2: Lumbrokinase-7T1 | 2.523 | 1.123 | 2.213 | 27 | −1,065,029.39 |
| SOCS2: Lysosomal membrane glycoprotein | 2.511 | 1.115 | 2.298 | 31 | −1,645,741.31 |
| SOCS2: NADH-ubiquinone oxidoreductase chain 4L | 2.498 | 1.178 | 2.199 | 26 | −406,049.87 |
| SOCS2: NADH-ubiquinone oxidoreductase chain 5 | 2.487 | 1.101 | 2.190 | 21 | −1,187,770.16 |
| SOCS2: Preprocarboxypeptidase | 2.596 | 1.259 | 2.188 | 30 | −560,689.83 |
| SOCS2: Extracellular globin-4 | 2.543 | 1.028 | 2.287 | 23 | −473,808.36 |
| SOCS2: Lumbrokinase-7T2 | 2.524 | 1.060 | 2.214 | 24 | −764,436.84 |
| Complex | MM/PBSA Calculation Results ΔGbinding (kcal/mol) | Average (kcal/mol) | ||
|---|---|---|---|---|
| I | II | III | ||
| Standard | ||||
| SOCS2: EpoR peptide (standard agonist) | −42.84 | −43.48 | −41.48 | −42.60 |
| SOCS2: N4BP1 (standard antagonist) | −42.34 | −42.34 | −42.85 | −42.51 |
| Protein and peptide derived from earthworm (Lumbricus genus) | ||||
| SOCS2: Cytochrome b | −50.57 | −49.93 | −49.82 | −50.11 |
| SOCS2: Cytochrome c oxidase subunit 3 | −44.93 | −44.93 | −44.87 | −44.91 |
| SOCS2: SCBP3 protein | −29.33 | −29.45 | −29.45 | −29.41 |
| SOCS2: Lumbricin | −59.22 | −59.26 | −59.26 | −59.25 |
| SOCS2: Chemoattractive glycoprotein ES20 | −53.69 | −57.76 | −53.60 | −55.02 |
| SOCS2: NADH-ubiquinone oxidoreductase chain 6 | −52.81 | −51.48 | −53.15 | −52.48 |
| SOCS2: Histone H3 | −44.05 | −44.84 | −45.82 | −44.90 |
| SOCS2: Peroxidasin | −42.91 | −42.91 | −42.91 | −42.91 |
| SOCS2: Lumbrokinase-7T1 | −69.22 | −69.35 | −69.27 | −69.28 |
| SOCS2: Lysosomal membrane glycoprotein | −34.42 | −34.89 | −34.75 | −34.69 |
| SOCS2: NADH-ubiquinone oxidoreductase chain 4L | −48.14 | −48.35 | −48.35 | −48.28 |
| SOCS2: NADH-ubiquinone oxidoreductase chain 5 | −37.74 | −36.11 | −36.11 | −36.65 |
| SOCS2: Preprocarboxypeptidase | −39.55 | −38.03 | −39.58 | −39.05 |
| SOCS2: Extracellular globin-4 | −50.09 | −49.68 | −50.30 | −50.02 |
| SOCS2: Lumbrokinase-7T2 | −46.18 | −45.80 | −47.11 | −46.36 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alotaiq, N.; Dermawan, D.; Elwali, N.E. Leveraging Therapeutic Proteins and Peptides from Lumbricus Earthworms: Targeting SOCS2 E3 Ligase for Cardiovascular Therapy through Molecular Dynamics Simulations. Int. J. Mol. Sci. 2024, 25, 10818. https://doi.org/10.3390/ijms251910818
Alotaiq N, Dermawan D, Elwali NE. Leveraging Therapeutic Proteins and Peptides from Lumbricus Earthworms: Targeting SOCS2 E3 Ligase for Cardiovascular Therapy through Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2024; 25(19):10818. https://doi.org/10.3390/ijms251910818
Chicago/Turabian StyleAlotaiq, Nasser, Doni Dermawan, and Nasr Eldin Elwali. 2024. "Leveraging Therapeutic Proteins and Peptides from Lumbricus Earthworms: Targeting SOCS2 E3 Ligase for Cardiovascular Therapy through Molecular Dynamics Simulations" International Journal of Molecular Sciences 25, no. 19: 10818. https://doi.org/10.3390/ijms251910818
APA StyleAlotaiq, N., Dermawan, D., & Elwali, N. E. (2024). Leveraging Therapeutic Proteins and Peptides from Lumbricus Earthworms: Targeting SOCS2 E3 Ligase for Cardiovascular Therapy through Molecular Dynamics Simulations. International Journal of Molecular Sciences, 25(19), 10818. https://doi.org/10.3390/ijms251910818