
Mitochondrial Dysfunction in Periodontitis and Associated Systemic Diseases: Implications for Pathomechanisms and Therapeutic Strategies

 ,
,
Abstract
1. Introduction
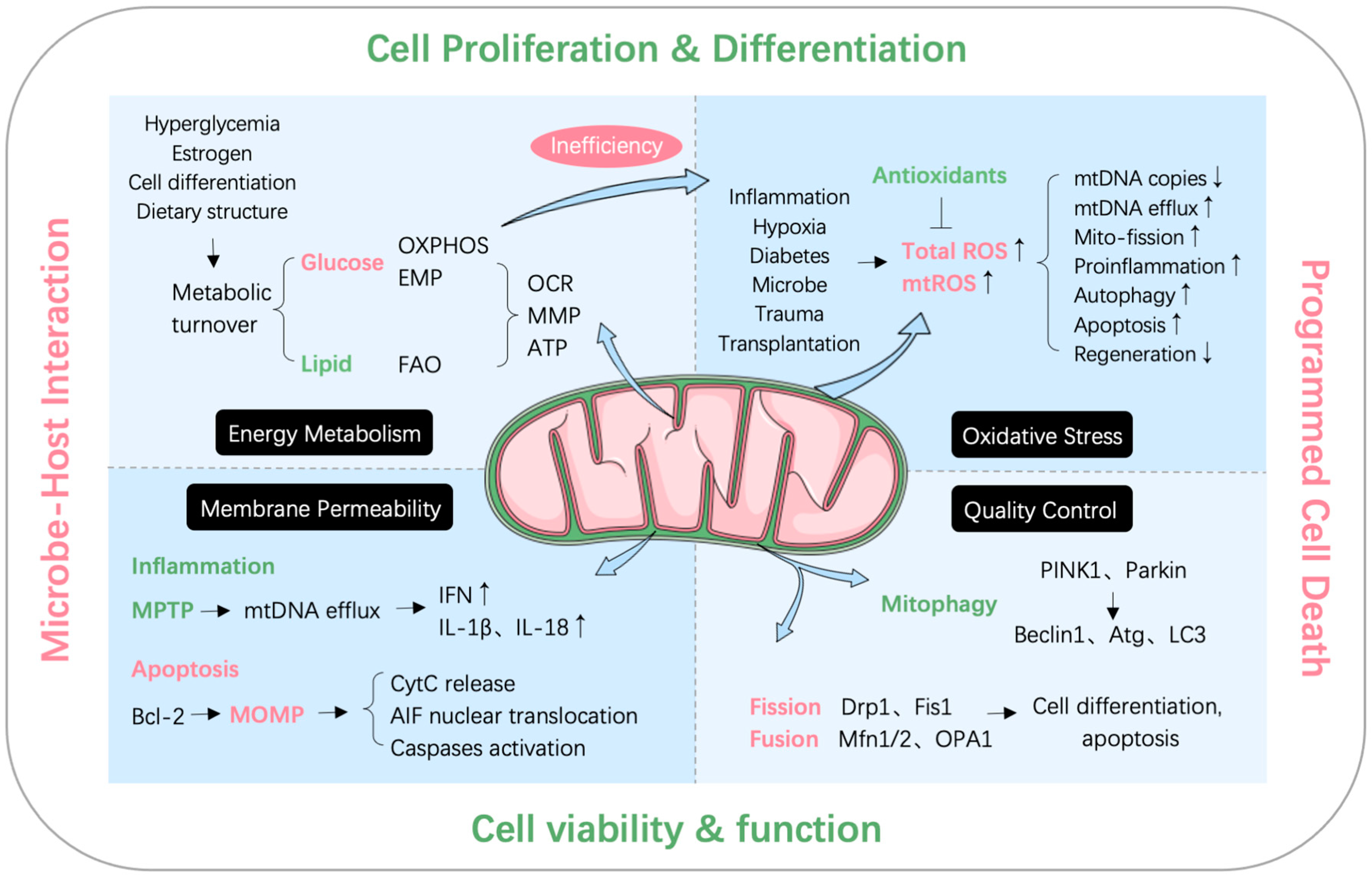
2. Mitochondrial Quality Control and Dysfunction
2.1. Mitochondrial Quality Control System
2.1.1. Mitochondrial Dynamics
2.1.2. Mitophagy
2.1.3. Mitochondrial Biogenesis
2.2. Mitochondrial Dysfunction
2.2.1. MtDNA Efflux
2.2.2. Oxidative Stress
2.2.3. Mitochondria-Mediated Apoptosis
3. Effects of Periodontal Pathogens and Their Virulence Factors on Mitochondria
4. The Relationship between Mitochondria and Periodontal Tissue Cells
4.1. Gingiva
4.1.1. Gingival Epithelial Cells
4.1.2. Gingival Fibroblasts
4.2. Periodontal Ligament
4.2.1. Periodontal Ligament Cells
4.2.2. Periodontal Ligament Stem Cells
4.3. Alveolar Bone
4.3.1. Osteoclasts
4.3.2. Osteoblasts
4.4. Cementum
4.5. Others
4.5.1. Macrophages
4.5.2. Neutrophils
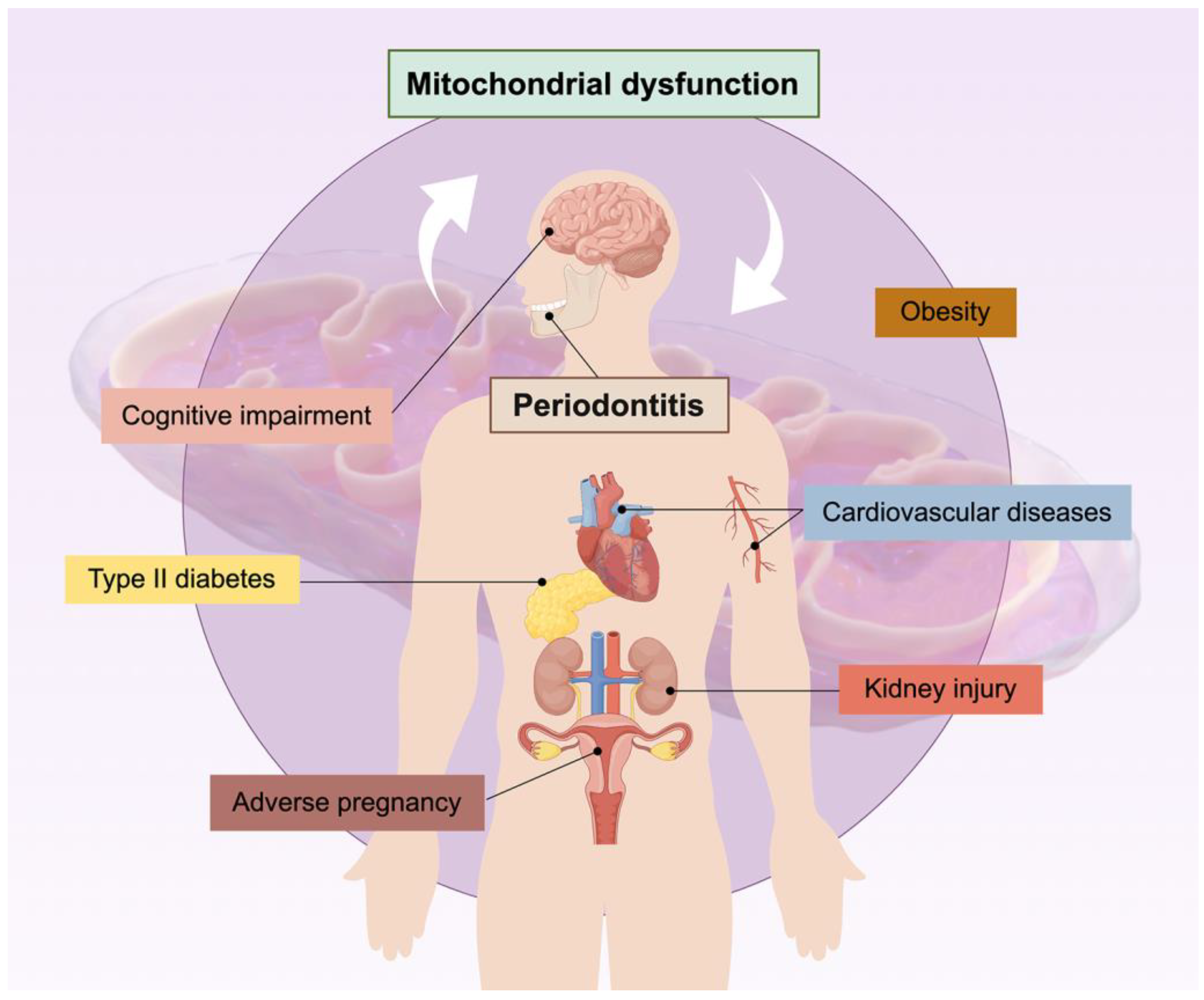
5. The Relationship between Mitochondria and Periodontitis-Related Systemic Diseases
5.1. Type II Diabetes Mellitus
5.2. Cognitive Impairment
5.3. Obesity
5.4. Cardiovascular Diseases
5.5. Other Systemic Diseases
5.5.1. Adverse Pregnancy
5.5.2. Kidney Injury
6. Mitochondria-Targeted Therapy

{kind=link}
{kind=link}
{kind=link}
| Type | Therapeutic Regulator | Cell Type | Function | Mechanism | Ref. |
|---|---|---|---|---|---|
| Antioxidant | CoQ10 | Fibroblast | Improve mitochondria biogenesis | Activate PGC-1α and TFAM | [26] |
| OC | Inhibit RANKL-induced osteoclastogenesis | Regulate mitochondrial apoptosis and oxidative stress | [91] | ||
| Resveratrol | MØ | Anti-inflammation | Induce PINK1-mediated mitophagy | [39] | |
| OB | Induce OB differentiation | Stimulate mitochondrial respiration and biogenesis via PGC-1α upregulation | [24] | ||
| Renal tissue cell | Protect renal tissue from periodontitis-induced damage | Prevent mitochondrial dysfunction | [136] | ||
| Hydroxytyrosol | Pre-OB; OC | Protect pre-OB from OS damage, inhibit OC differentiation, attenuate bone loss in periodontitis mice | Modulate mitochondria function, ERK and JNK signaling pathways | [90] | |
| OB | Prevent OS-induced OB apoptosis | Decrease OPA1 cleavage and increase AKT and GSK3β phosphorylation to inhibit OS-induced mitochondrial dysfunction | [104] | ||
| EGCG | OC | Inhibit OC differentiation | Modulate mitophagy through AKT and p38 signaling pathways; block RANK-RANKL binding | [92] | |
| OB | Induce OB differentiation | Stimulate mitochondrial respiration and biogenesis via PGC-1α upregulation | [24] | ||
| Polyphenolic Compounds (tannic acid, gallic acid, and ellagic acid) | OC | Inhibit OC differentiation | Inactivate Akt to suppress autophagy; downregulate ROS, cellular Ca2+ and MMP | [93] | |
| Isovitexin | OB | Promote OB differentiation | Upregulate PGC-1α via AdipoRs to induce OxPhos and ATP production | [25] | |
| Proanthocyanidins | OB | Ameliorate OB oxidative stress and mitochondrial dysfunction | Activate Nrf2 signaling | [105] | |
| Mito-TEMPO; MnSOD | hGF | Prevent LPS-induced pro-inflammatory response in HGF | Inhibit mtROS to downregulate IL-1β, IL-6 and TNF-α, inhibit p38, c-Jun N-terminal kinase and inhibitor of nuclear factor-κB kinase | [68] | |
| Madecassic acid | hPDLC | Protect HPDLC from oxidative stress and apoptosis | Reduce ROS and maintain MMP; downregulate Bax, upregulate Bcl-2 and Bcl-xL | [79] | |
| Moringin | hPDLSC | Inhibit OS-induced mitochondria dysfunction | Downregulate PINK1 to prevent mitophagy, downregulate Bax and caspases, upregulate Bal2L12 and MCL1 to inactivate apoptosis | [86] | |
| Curcumin | hPDLSC | Reduce apoptosis and promote osteogenesis in HPDLSC | Reduce ROS, modulate Erk1/2 signaling pathway | [81] | |
| Baicalein | hGEC | Prevent HGEC oxidative stress and reduce bone loss in CPDM mice | Modulate Nrf2 signaling pathway | [125] | |
| Hormone, antioxidant | Melatonin | OB | Induce OB mitochondrial apoptosis | Upregulate STIM1/cytosolic calcium elevation/ERK pathway | [107] |
| hGF | Attenuate ER stress, fibrosis and mitochondrial apoptosis markers | Decrease VEGF; significantly decrease CHOP, GRP78, and XBP1s; significantly increase cytochrome c, significantly decrease cyclophilin D | [72] | ||
| hPDLC | Promote HPDLC osteogenesis (pharmacological concentration) | Upregulate TOM20 to enhance mitochondrial function | [76] | ||
| hPDLSC | Suppress HPDLSC osteogenic differentiation and alter mitochondrial function (physiological concentration) | Downregulate OPN and OCN; decrease ATP, increase ROS and NAD+/NADH ratio | [77] | ||
| Hormone | Estrogen | OC | Increase OC precursor mitochondria-induced apoptosis, decrease OC number | Attenuate mitochondria oxidative phosphorylation and ATP production, promote OC precursor apoptosis through Bak/Bax | [88] |
| Mitochondrial fission inhibitor | Mdivi-1 | Endothelial cell | Inhibit Pg-induced mitochondrial fission and dysfunction | Inhibit assembly and activity of Drp1 | [11] |
| Mitochondrial fusion promoter | M1 | OB | Impair osteogenesis | Upregulate OPA1 expression | [103] |
| Ferroptosis inhibitor | Fer-1 | hGF | Attenuate Pg-LPS-induced inflammation | - | [47] |
| ER stress inhibitor | 4PBA | hGF | Attenuate ER stress, fibrosis and mitochondrial apoptosis markers | Decrease CTGF, VEGF, TGFβ, and α-SMA; significantly decrease ER stress markers, CHOP, GRP78, XBP1, XBP1s; significantly increase cytochrome c, decrease both Bcl-2 and cyclophilin D | [72] |
| Hypoxia mimetic | CoCl2 | hPDLSC | Induce HPDLSC apoptosis | Increase ROS and Drp1 to promote mitochondrial fission | [84] |
| CB | Inhibits CB mineralization and mitochondrial biogenesis | Downregulate PGC-1α via p38 and Erk1/2 signaling pathways | [22] | ||
| Cardiovascular drug | Simvastatin | OB | Inhibit mitophagy-related OB apoptosis and alleviate bone resorption | Alleviated hypoxia-induced mitochondrial dysfunction, mitophagy and apoptosis | [51] |
| hGF | Attenuate ER stress, fibrosis and mitochondrial apoptosis markers | Decrease VEGF, CTGF, TGFβ; significantly decrease CHOP, GRP78, and XBP1s; significantly increase cytochrome c, decrease cyclophilin D | [72] | ||
| Nitric oxide (Sodium nitroprusside) | hGF | Induce HGF apoptosis | Induce the loss of MMP, increase Bax/Bcl-2 ratio and activate JNK and caspases | [71] | |
| Chemotherapy drug | Doxorubicin | OC | Induce bone loss by increasing OC autophagy | Increase mitochondrial ROS/TRPML1/TFEB axis in OC | [94] |
| Ginsenoside Rg1 | hPDLC | Alleviate LPS-induced HPDLC pyroptosis | Upregulate AMPK-dependent Drp-1 phosphorylation to inhibit mitochondrial fission; reduce NLRP3, ASC, caspase-1 and GSDMD-NT | [80] | |
| Immunosuppressant | Cyclosporine A | hGEC | Enhance mitochondrial apoptosis in gingival keratinocytes | Upregulate Bax, AIF, caspase-3/9 expression | [59] |
| Inhibit mitochondrial apoptosis in spinous and basal layer cells | Upregulate Bcl-2, downregulate caspase-3 | [60] | |||
| hGF | Induce ER stress | Interference with mitochondrial pro- and anti-apoptotic factors | [72] | ||
| Cell component | Myo-EV | OC | Suppress OC formation and mitochondrial energy metabolism | Suppress oxygen consumption and mitochondria biogenesis | [139] |
| Mitochondria and MDV | OB | Promote osteogenesis | Elevate BMP signaling in osteoprogenitor cell | [103] | |
| Peptide | 7R-ETGE | OC | Attenuate RANKL-dependent osteoclastogenesis and bone destruction, decrease ROS level | Prevent Keap1 from binding to Nrf2, induce Nrf2 nuclear translocation and Nrf2-dependent cytoprotective enzyme expression | [140] |
| Dental material | PRF/BCP | OC | Impair OC differentiation, promote mitochondria-induced apoptosis | Inhibit NF-kappa B and Bcl-2/xL, promote caspase-3 and Bax levels | [89] |
| TEGDMA | hGF | Induce oxidative stress and mitochondrial dysfunction in HGF | Increase ROS, aberrant mitochondrial morphology and functions | [69] | |
| Molybdenum | MØ | Promote periodontal wound healing | Effect mitochondrial structure, MMP and promotes metabolic shift from glycolysis toward mitochondrial OXPHOS | [118] | |
| Physical treatment | Magnetic stimulation | hPDLSC | Induce proliferation and osteogenesis in HPDLSC | Promote Erk phosphorylation; Alter mitochondrial respiration | [83] |
| Methylene blue-mediated photodynamic therapy | MØ | Alleviate bone loss in periodontitis | Induce mitochondrial-mediated MØ apoptosis | [119] |
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Graves, D. Cytokines That Promote Periodontal Tissue Destruction. J. Periodontol. 2008, 79, 1585–1591. [Google Scholar] [CrossRef]
- Bascones, A.; Noronha, S.; Gómez, M.; Mota, P.; Gónzalez Moles, M.A.; Villarroel Dorrego, M. Tissue Destruction in Periodontitis: Bacteria or Cytokines Fault? Quintessence Int. 2005, 36, 299–306. [Google Scholar]
- Dahiya, P.; Kamal, R.; Gupta, R.; Bhardwaj, R.; Chaudhary, K.; Kaur, S. Reactive Oxygen Species in Periodontitis. J. Indian Soc. Periodontol. 2013, 17, 411–416. [Google Scholar] [CrossRef]
- Mottis, A.; Herzig, S.; Auwerx, J. Mitocellular Communication: Shaping Health and Disease. Science 2019, 366, 827–832. [Google Scholar] [CrossRef]
- Jiang, W.; Wang, Y.; Cao, Z.; Chen, Y.; Si, C.; Sun, X.; Huang, S. The Role of Mitochondrial Dysfunction in Periodontitis: From Mechanisms to Therapeutic Strategy. J. Periodontal Res. 2023, 58, 853–863. [Google Scholar] [CrossRef]
- Govindaraj, P.; Khan, N.A.; Gopalakrishna, P.; Chandra, R.V.; Vanniarajan, A.; Reddy, A.A.; Singh, S.; Kumaresan, R.; Srinivas, G.; Singh, L.; et al. Mitochondrial Dysfunction and Genetic Heterogeneity in Chronic Periodontitis. Mitochondrion 2011, 11, 504–512. [Google Scholar] [CrossRef]
- Bullon, P.; Newman, H.N.; Battino, M. Obesity, Diabetes Mellitus, Atherosclerosis and Chronic Periodontitis: A Shared Pathology via Oxidative Stress and Mitochondrial Dysfunction? Periodontol. 2000 2014, 64, 139–153. [Google Scholar] [CrossRef]
- Zheng, M.; Wang, C.; Ali, A.; Shih, Y.A.; Xie, Q.; Guo, C. Prevalence of Periodontitis in People Clinically Diagnosed with Diabetes Mellitus: A Meta-Analysis of Epidemiologic Studies. Acta Diabetol. 2021, 58, 1307–1327. [Google Scholar] [CrossRef]
- Guo, H.; Chang, S.; Pi, X.; Hua, F.; Jiang, H.; Liu, C.; Du, M. The Effect of Periodontitis on Dementia and Cognitive Impairment: A Meta-Analysis. Int. J. Environ. Res. Public Health 2021, 18, 6823. [Google Scholar] [CrossRef]
- Sanz, M.; Del Castillo, A.M.; Jepsen, S.; Gonzalez-Juanatey, J.R.; D’Aiuto, F.; Bouchard, P.; Chapple, I.; Dietrich, T.; Gotsman, I.; Graziani, F.; et al. Periodontitis and Cardiovascular Diseases. Consensus Report. Glob. Heart 2020, 15, 1. [Google Scholar] [CrossRef]
- Xu, T.; Dong, Q.; Luo, Y.; Liu, Y.; Gao, L.; Pan, Y.; Zhang, D. Porphyromonas gingivalis Infection Promotes Mitochondrial Dysfunction through Drp1-Dependent Mitochondrial Fission in Endothelial Cells. Int. J. Oral Sci. 2021, 13, 28. [Google Scholar] [CrossRef]
- Ferreira, I.L.; Costa, S.; Moraes, B.J.; Costa, A.; Fokt, O.; Marinho, D.; Alves, V.; Baptista, I.P.; Rego, A.C. Mitochondrial and Redox Changes in Periodontitis and Type 2 Diabetes Human Blood Mononuclear Cells. Antioxidants 2023, 12, 226. [Google Scholar] [CrossRef]
- Li, A.; Du, M.; Chen, Y.; Marks, L.A.M.; Visser, A.; Xu, S.; Tjakkes, G.-H.E. Periodontitis and Cognitive Impairment in Older Adults: The Mediating Role of Mitochondrial Dysfunction. J. Periodontol. 2022, 93, 1302–1313. [Google Scholar] [CrossRef]
- Zhao, P.; Xu, A.; Leung, W.K. Obesity, Bone Loss, and Periodontitis: The Interlink. Biomolecules 2022, 12, 865. [Google Scholar] [CrossRef]
- Atabay, V.E.; Lutfioğlu, M.; Avci, B.; Sakallioglu, E.E.; Aydoğdu, A. Obesity and Oxidative Stress in Patients with Different Periodontal Status: A Case-Control Study. J. Periodontal Res. 2017, 52, 51–60. [Google Scholar] [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The Cell Biology of Mitochondrial Membrane Dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial Quality Control as a Therapeutic Target. Pharmacol. Rev. 2016, 68, 20–48. [Google Scholar] [CrossRef]
- Roca-Portoles, A.; Tait, S.W.G. Mitochondrial Quality Control: From Molecule to Organelle. Cell. Mol. Life Sci. 2021, 78, 3853–3866. [Google Scholar] [CrossRef]
- Fu, W.; Liu, Y.; Yin, H. Mitochondrial Dynamics: Biogenesis, Fission, Fusion, and Mitophagy in the Regulation of Stem Cell Behaviors. Stem Cells Int. 2019, 2019, 9757201. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of Mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Ma, L.; Huang, X.; Peng, Y.; Huang, H.; Gao, X.; Chen, Y.; Cao, Z. PGC-1 Alpha Regulates Mitochondrial Biogenesis to Ameliorate Hypoxia-Inhibited Cementoblast Mineralization. Ann. N. Y. Acad. Sci. 2022, 1516, 300–311. [Google Scholar] [CrossRef]
- Singh, S.P.; Huck, O.; Abraham, N.G.; Amar, S. Kavain Reduces Porphyromonas gingivalis–Induced Adipocyte Inflammation: Role of PGC-1α Signaling. J. Immunol. 2018, 201, 1491–1499. [Google Scholar] [CrossRef]
- Pal, S.; Porwal, K.; Rajak, S.; Sinha, R.A.; Chattopadhyay, N. Selective Dietary Polyphenols Induce Differentiation of Human Osteoblasts by Adiponectin Receptor 1-Mediated Reprogramming of Mitochondrial Energy Metabolism. Biomed. Pharmacother. 2020, 127, 110207. [Google Scholar] [CrossRef]
- Pal, S.; Singh, M.; Porwal, K.; Rajak, S.; Das, N.; Rajput, S.; Trivedi, A.K.; Maurya, R.; Sinha, R.A.; Siddiqi, M.I.; et al. Adiponectin Receptors by Increasing Mitochondrial Biogenesis and Respiration Promote Osteoblast Differentiation: Discovery of Isovitexin as a New Class of Small Molecule Adiponectin Receptor Modulator with Potential Osteoanabolic Function. Eur. J. Pharmacol. 2021, 913, 174634. [Google Scholar] [CrossRef]
- Bullón, P.; Román-Malo, L.; Marín-Aguilar, F.; Alvarez-Suarez, J.M.; Giampieri, F.; Battino, M.; Cordero, M.D. Lipophilic Antioxidants Prevent Lipopolysaccharide-Induced Mitochondrial Dysfunction through Mitochondrial Biogenesis Improvement. Pharmacol. Res. 2015, 91, 1–8. [Google Scholar] [CrossRef]
- Zhong, F.; Liang, S.; Zhong, Z. Emerging Role of Mitochondrial DNA as a Major Driver of Inflammation and Disease Progression. Trends Immunol. 2019, 40, 1120–1133. [Google Scholar] [CrossRef]
- Collins, L.V.; Hajizadeh, S.; Holme, E.; Jonsson, I.-M.; Tarkowski, A. Endogenously Oxidized Mitochondrial DNA Induces In Vivo and In Vitro Inflammatory Responses. J. Leukoc. Biol. 2004, 75, 995–1000. [Google Scholar] [CrossRef]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in Inflammation and Immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Hooftman, A.; Peace, C.G.; Ryan, D.G.; Day, E.A.; Yang, M.; McGettrick, A.F.; Yin, M.; Montano, E.N.; Huo, L.; Toller-Kawahisa, J.E.; et al. Macrophage Fumarate Hydratase Restrains mtRNA-Mediated Interferon Production. Nature 2023, 615, 490–498. [Google Scholar] [CrossRef]
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining Roles of Specific Reactive Oxygen Species (ROS) in Cell Biology and Physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W.G. Mitochondria as Multifaceted Regulators of Cell Death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef]
- Darveau, R.P. Periodontitis: A Polymicrobial Disruption of Host Homeostasis. Nat. Rev. Microbiol. 2010, 8, 481–490. [Google Scholar] [CrossRef]
- Fleetwood, A.J.; Lee, M.K.S.; Singleton, W.; Achuthan, A.; Lee, M.-C.; O’Brien-Simpson, N.M.; Cook, A.D.; Murphy, A.J.; Dashper, S.G.; Reynolds, E.C.; et al. Metabolic Remodeling, Inflammasome Activation, and Pyroptosis in Macrophages Stimulated by Porphyromonas gingivalis and Its Outer Membrane Vesicles. Front. Cell. Infect. Microbiol. 2017, 7, 351. [Google Scholar] [CrossRef]
- Tomi, N.; Fukuyo, Y.; Arakawa, S.; Nakajima, T. Pro-Inflammatory Cytokine Production from Normal Human Fibroblasts Is Induced by Tannerella forsythia Detaching Factor. J. Periodontal Res. 2008, 43, 136–142. [Google Scholar] [CrossRef]
- Lin, L.-T.; Shi, Y.-C.; Choong, C.-Y.; Tai, C.-J. The Fruits of Paris Polyphylla Inhibit Colorectal Cancer Cell Migration Induced by Fusobacterium Nucleatum-Derived Extracellular Vesicles. Molecules 2021, 26, 4081. [Google Scholar] [CrossRef]
- Jiang, K.; Li, J.; Jiang, L.; Li, H.; Lei, L. PINK1-Mediated Mitophagy Reduced Inflammatory Responses to Porphyromonas gingivalis in Macrophages. Oral Dis. 2023, 29, 3665–3676. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; Shi, Q.; Wang, X.; Zou, P.; Zheng, M.; Luan, Q. Mitochondrial DNA Efflux Maintained in Gingival Fibroblasts of Patients with Periodontitis through ROS/mPTP Pathway. Oxidative Med. Cell. Longev. 2022, 2022, 1000213. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Azhar, G.; Zhang, X.; Patyal, P.; Kc, G.; Sharma, S.; Che, Y.; Wei, J.Y. P. gingivalis-LPS Induces Mitochondrial Dysfunction Mediated by Neuroinflammation through Oxidative Stress. Int. J. Mol. Sci. 2023, 24, 950. [Google Scholar] [CrossRef] [PubMed]
- Bullon, P.; Cordero, M.D.; Quiles, J.L.; Morillo, J.M.; del Carmen Ramirez-Tortosa, M.; Battino, M. Mitochondrial Dysfunction Promoted by Porphyromonas gingivalis Lipopolysaccharide as a Possible Link between Cardiovascular Disease and Periodontitis. Free Radic. Biol. Med. 2011, 50, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, H.; Duncan, M.J. Translocation of Porphyromonas gingivalis Gingipain Adhesin Peptide A44 to Host Mitochondria Prevents Apoptosis. Infect. Immun. 2010, 78, 3616–3624. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, J.; Lin, L.; Zhao, H.; Miao, L.; Pan, Y. Porphyromonas gingivalis Degrades Integrin Β1 and Induces AIF-Mediated Apoptosis of Epithelial Cells. Infect. Dis. Lond. Engl. 2019, 51, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.-T.; Zeng, J.-J.; Lu, J.-Y.; Zhang, X.-Y.; Xu, P.-P.; Su, Y. Porphyromonas gingivalis Lipopolysaccharide (Pg-LPS) Influences Adipocytes Injuries through Triggering XBP1 and Activating Mitochondria-Mediated Apoptosis. Adipocyte 2021, 10, 28–37. [Google Scholar] [CrossRef]
- Li, Y.; Shibata, Y.; Zhang, L.; Kuboyama, N.; Abiko, Y. Periodontal Pathogen Aggregatibacter actinomycetemcomitans LPS Induces Mitochondria-Dependent-Apoptosis in Human Placental Trophoblasts. Placenta 2011, 32, 11–19. [Google Scholar] [CrossRef]
- Qiao, S.; Li, B.; Cai, Q.; Li, Z.; Yin, Z.; He, J.; Li, Y.; Meng, W. Involvement of Ferroptosis in Porphyromonas gingivalis Lipopolysaccharide-Stimulated Periodontitis In Vitro and In Vivo. Oral Dis. 2023, 29, 3571–3582. [Google Scholar] [CrossRef]
- Hung, S.-C.; Huang, P.-R.; Almeida-da-Silva, C.L.C.; Atanasova, K.R.; Yilmaz, O.; Ojcius, D.M. NLRX1 Modulates Differentially NLRP3 Inflammasome Activation and NF-κB Signaling during Fusobacterium nucleatum Infection. Microbes Infect. 2018, 20, 615–625. [Google Scholar] [CrossRef]
- Mei, Y.-M.; Li, L.; Wang, X.-Q.; Zhang, M.; Zhu, L.-F.; Fu, Y.-W.; Xu, Y. AGEs Induces Apoptosis and Autophagy via Reactive Oxygen Species in Human Periodontal Ligament Cells. J. Cell. Biochem. 2020, 121, 3764–3779. [Google Scholar] [CrossRef]
- Medeiros, C.; Wallace, J.M. High Glucose-Induced Inhibition of Osteoblast like MC3T3-E1 Differentiation Promotes Mitochondrial Perturbations. PLoS ONE 2022, 17, e0270001. [Google Scholar] [CrossRef]
- Yang, C.-N.; Kok, S.-H.; Wang, H.-W.; Chang, J.Z.-C.; Lai, E.H.-H.; Shun, C.-T.; Yang, H.; Chen, M.-H.; Hong, C.-Y.; Lin, S.-K. Simvastatin Alleviates Bone Resorption in Apical Periodontitis Possibly by Inhibition of Mitophagy-Related Osteoblast Apoptosis. Int. Endod. J. 2019, 52, 676–688. [Google Scholar] [CrossRef]
- Fujita, T.; Yoshimoto, T.; Kajiya, M.; Ouhara, K.; Matsuda, S.; Takemura, T.; Akutagawa, K.; Takeda, K.; Mizuno, N.; Kurihara, H. Regulation of Defensive Function on Gingival Epithelial Cells Can Prevent Periodontal Disease. Jpn. Dent. Sci. Rev. 2018, 54, 66–75. [Google Scholar] [CrossRef]
- Roberts, J.S.; Atanasova, K.R.; Lee, J.; Diamond, G.; Deguzman, J.; Hee Choi, C.; Yilmaz, Ö. Opportunistic Pathogen Porphyromonas gingivalis Modulates Danger Signal ATP-Mediated Antibacterial NOX2 Pathways in Primary Epithelial Cells. Front. Cell. Infect. Microbiol. 2017, 7, 291. [Google Scholar] [CrossRef] [PubMed]
- Wielento, A.; Lagosz-Cwik, K.B.; Potempa, J.; Grabiec, A.M. The Role of Gingival Fibroblasts in the Pathogenesis of Periodontitis. J. Dent. Res. 2023, 102, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tian, B.-M.; Deng, D.-K.; Liu, F.; Zhou, H.; Kong, D.-Q.; Qu, H.-L.; Sun, L.-J.; He, X.-T.; Chen, F.-M. LncRNA GACAT2 Binds with Protein PKM1/2 to Regulate Cell Mitochondrial Function and Cementogenesis in an Inflammatory Environment. Bone Res. 2022, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Usui, M.; Onizuka, S.; Sato, T.; Kokabu, S.; Ariyoshi, W.; Nakashima, K. Mechanism of Alveolar Bone Destruction in Periodontitis—Periodontal Bacteria and Inflammation. Jpn. Dent. Sci. Rev. 2021, 57, 201–208. [Google Scholar] [CrossRef]
- Calenic, B.; Yaegaki, K.; Murata, T.; Imai, T.; Aoyama, I.; Sato, T.; Ii, H. Oral Malodorous Compound Triggers Mitochondrial-Dependent Apoptosis and Causes Genomic DNA Damage in Human Gingival Epithelial Cells. J. Periodontal Res. 2010, 45, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.; Baeshen, H.A. Aqueous Extract of Tobacco Induces Mitochondrial Potential Dependent Cell Death and Epithelial-Mesenchymal Transition in Gingival Epithelial Cells. Saudi J. Biol. Sci. 2021, 28, 4613–4618. [Google Scholar] [CrossRef]
- Tu, H.-P.; Chen, Y.-T.; Chiu, H.-C.; Chin, Y.-T.; Huang, S.-M.; Cheng, L.-C.; Fu, E.; Chiang, C.-Y. Cyclosporine A Enhances Apoptosis in Gingival Keratinocytes of Rats and in OECM1 Cells via the Mitochondrial Pathway. J. Periodontal Res. 2009, 44, 767–775. [Google Scholar] [CrossRef]
- Ma, S.; Liu, P.; Li, Y.; Hou, L.; Chen, L.; Qin, C. Cyclosporine a Inhibits Apoptosis of Rat Gingival Epithelium. J. Periodontol. 2014, 85, 1126–1134. [Google Scholar] [CrossRef]
- Tu, H.-P.; Chen, Y.-T.; Shieh, Y.-S.; Chin, Y.-T.; Huang, R.-Y.; Yang, S.-F.; Gau, C.-H.; Fu, E. Cyclosporin-Induced Downregulation of the Expression of E-Cadherin during Proliferation of Edentulous Gingival Epithelium in Rats. J. Periodontol. 2006, 77, 832–839. [Google Scholar] [CrossRef]
- Yoshimoto, T.; Fujita, T.; Kajiya, M.; Matsuda, S.; Ouhara, K.; Shiba, H.; Kurihara, H. Involvement of Smad2 and Erk/Akt Cascade in TGF-Β1-Induced Apoptosis in Human Gingival Epithelial Cells. Cytokine 2015, 75, 165–173. [Google Scholar] [CrossRef]
- Zhu, C.; Zhao, Y.; Pei, D.; Liu, Z.; Liu, J.; Li, Y.; Yu, S.; Ma, L.; Sun, J.; Li, A. PINK1 Mediated Mitophagy Attenuates Early Apoptosis of Gingival Epithelial Cells Induced by High Glucose. BMC Oral Health 2022, 22, 144. [Google Scholar] [CrossRef]
- Mao, S.; Park, Y.; Hasegawa, Y.; Tribble, G.D.; James, C.E.; Handfield, M.; Stavropoulos, M.F.; Yilmaz, O.; Lamont, R.J. Intrinsic Apoptotic Pathways of Gingival Epithelial Cells Modulated by Porphyromonas gingivalis. Cell. Microbiol. 2007, 9, 1997–2007. [Google Scholar] [CrossRef]
- Yao, L.; Jermanus, C.; Barbetta, B.; Choi, C.; Verbeke, P.; Ojcius, D.M.; Yilmaz, O. Porphyromonas gingivalis Infection Sequesters Pro-Apoptotic Bad through Akt in Primary Gingival Epithelial Cells. Mol. Oral Microbiol. 2010, 25, 89–101. [Google Scholar] [CrossRef]
- Choi, C.H.; Spooner, R.; DeGuzman, J.; Koutouzis, T.; Ojcius, D.M.; Yilmaz, Ö. Porphyromonas gingivalis-Nucleoside-Diphosphate-Kinase Inhibits ATP-Induced Reactive-Oxygen-Species via P2X7 Receptor/NADPH-Oxidase Signalling and Contributes to Persistence. Cell. Microbiol. 2013, 15, 961–976. [Google Scholar] [CrossRef]
- Liu, J.; Wang, X.; Xue, F.; Zheng, M.; Luan, Q. Abnormal Mitochondrial Structure and Function Are Retained in Gingival Tissues and Human Gingival Fibroblasts from Patients with Chronic Periodontitis. J. Periodontal Res. 2022, 57, 94–103. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Zheng, M.; Luan, Q.X. Mitochondrial Reactive Oxygen Species Mediate the Lipopolysaccharide-Induced pro-Inflammatory Response in Human Gingival Fibroblasts. Exp. Cell Res. 2016, 347, 212–221. [Google Scholar] [CrossRef]
- Liu, B.; Gan, X.; Zhao, Y.; Chen, J.; Yu, H.; Gao, J.; Yu, H. TEGDMA Releasing in Resin Composites with Different Filler Contents and Its Correlation with Mitochondrial Mediated Cytotoxicity in Human Gingival Fibroblasts. J. Biomed. Mater. Res. A 2019, 107, 1132–1142. [Google Scholar] [CrossRef]
- Yoshida, A.; Yoshino, F.; Makita, T.; Maehata, Y.; Higashi, K.; Miyamoto, C.; Wada-Takahashi, S.; Takahashi, S.; Takahashi, O.; Lee, M.C. Reactive Oxygen Species Production in Mitochondria of Human Gingival Fibroblast Induced by Blue Light Irradiation. J. Photochem. Photobiol. B 2013, 129, 1–5. [Google Scholar] [CrossRef]
- Baek, M.-W.; Seong, K.-J.; Jeong, Y.-J.; Kim, G.-M.; Park, H.-J.; Kim, S.-H.; Chung, H.-J.; Kim, W.-J.; Jung, J.-Y. Nitric Oxide Induces Apoptosis in Human Gingival Fibroblast through Mitochondria-Dependent Pathway and JNK Activation. Int. Endod. J. 2015, 48, 287–297. [Google Scholar] [CrossRef]
- Ranga Rao, S.; Subbarayan, R.; Ajitkumar, S.; Murugan Girija, D. 4PBA Strongly Attenuates Endoplasmic Reticulum Stress, Fibrosis, and Mitochondrial Apoptosis Markers in Cyclosporine Treated Human Gingival Fibroblasts. J. Cell. Physiol. 2018, 233, 60–66. [Google Scholar] [CrossRef]
- Aral, K.; Milward, M.R.; Cooper, P.R. Gene Expression Profiles of Mitochondria-Endoplasmic Reticulum Tethering in Human Gingival Fibroblasts in Response to Periodontal Pathogens. Arch. Oral Biol. 2021, 128, 105173. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, Autophagy, and Mitochondria Crosstalk Underlies the ER Stress Response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Nilsson, B.-O. Mechanisms Involved in Regulation of Periodontal Ligament Cell Production of Pro-Inflammatory Cytokines: Implications in Periodontitis. J. Periodontal Res. 2021, 56, 249–255. [Google Scholar] [CrossRef]
- Sun, H.; Zheng, M.; Liu, J.; Fan, W.; He, H.; Huang, F. Melatonin Promoted Osteogenesis of Human Periodontal Ligament Cells by Regulating Mitochondrial Functions through the Translocase of the Outer Mitochondrial Membrane 20. J. Periodontal Res. 2023, 58, 53–69. [Google Scholar] [CrossRef]
- Zheng, M.; Zhang, F.; Fan, W.; Jiang, L.; Li, J.; Xie, S.; Huang, F.; He, H. Suppression of Osteogenic Differentiation and Mitochondrial Function Change in Human Periodontal Ligament Stem Cells by Melatonin at Physiological Levels. PeerJ 2020, 8, e8663. [Google Scholar] [CrossRef]
- Chen, Y.; Ji, Y.; Jin, X.; Sun, X.; Zhang, X.; Chen, Y.; Shi, L.; Cheng, H.; Mao, Y.; Li, X.; et al. Mitochondrial Abnormalities Are Involved in Periodontal Ligament Fibroblast Apoptosis Induced by Oxidative Stress. Biochem. Biophys. Res. Commun. 2019, 509, 483–490. [Google Scholar] [CrossRef]
- Jin, Y.; Li, J.; Ding, L.; Zhao, Q.; Song, Y.; Li, G.; Ji, J.; Ni, Y.; Hu, Q. Madecassic Acid Protects Human Periodontal Ligament Fibroblasts against Hydrogen Peroxide-Induced Cell Damage by Maintaining Mitochondrial Membrane Potential. Mol. Cell. Toxicol. 2022, 18, 81–90. [Google Scholar] [CrossRef]
- Chu, K.; Zhang, Z.; Chu, Y.; Xu, Y.; Yang, W.; Guo, L. Ginsenoside Rg1 Alleviates Lipopolysaccharide-Induced Pyroptosis in Human Periodontal Ligament Cells via Inhibiting Drp1-Mediated Mitochondrial Fission. Arch. Oral Biol. 2023, 147, 105632. [Google Scholar] [CrossRef]
- Tan, L.; Cao, Z.; Chen, H.; Xie, Y.; Yu, L.; Fu, C.; Zhao, W.; Wang, Y. Curcumin Reduces Apoptosis and Promotes Osteogenesis of Human Periodontal Ligament Stem Cells under Oxidative Stress In Vitro and In Vivo. Life Sci. 2021, 270, 119125. [Google Scholar] [CrossRef]
- Chen, H.; Huang, X.; Fu, C.; Wu, X.; Peng, Y.; Lin, X.; Wang, Y. Recombinant Klotho Protects Human Periodontal Ligament Stem Cells by Regulating Mitochondrial Function and the Antioxidant System during H2O2-Induced Oxidative Stress. Oxidative Med. Cell. Longev. 2019, 2019, 9261565. [Google Scholar] [CrossRef]
- Peluso, V.; Rinaldi, L.; Russo, T.; Oliviero, O.; Di Vito, A.; Garbi, C.; Giudice, A.; De Santis, R.; Gloria, A.; D’Antò, V. Impact of Magnetic Stimulation on Periodontal Ligament Stem Cells. Int. J. Mol. Sci. 2021, 23, 188. [Google Scholar] [CrossRef]
- He, Y.; Gan, X.; Zhang, L.; Liu, B.; Zhu, Z.; Li, T.; Zhu, J.; Chen, J.; Yu, H. CoCl2 Induces Apoptosis via a ROS-Dependent Pathway and Drp1-Mediated Mitochondria Fission in Periodontal Ligament Stem Cells. Am. J. Physiol. Cell Physiol. 2018, 315, C389–C397. [Google Scholar] [CrossRef]
- Fang, H.; Yang, K.; Tang, P.; Zhao, N.; Ma, R.; Luo, X.; Liu, Q. Glycosylation End Products Mediate Damage and Apoptosis of Periodontal Ligament Stem Cells Induced by the JNK-Mitochondrial Pathway. Aging 2020, 12, 12850–12868. [Google Scholar] [CrossRef]
- Chiricosta, L.; Gugliandolo, A.; Diomede, F.; Pizzicannella, J.; Trubiani, O.; Iori, R.; Tardiolo, G.; Guarnieri, S.; Bramanti, P.; Mazzon, E. Moringin Pretreatment Inhibits the Expression of Genes Involved in Mitophagy in the Stem Cell of the Human Periodontal Ligament. Molecules 2019, 24, 3217. [Google Scholar] [CrossRef]
- Elson, A.; Anuj, A.; Barnea-Zohar, M.; Reuven, N. The Origins and Formation of Bone-Resorbing Osteoclasts. Bone 2022, 164, 116538. [Google Scholar] [CrossRef]
- Kim, H.-N.; Ponte, F.; Nookaew, I.; Ucer Ozgurel, S.; Marques-Carvalho, A.; Iyer, S.; Warren, A.; Aykin-Burns, N.; Krager, K.; Sardao, V.A.; et al. Estrogens Decrease Osteoclast Number by Attenuating Mitochondria Oxidative Phosphorylation and ATP Production in Early Osteoclast Precursors. Sci. Rep. 2020, 10, 11933. [Google Scholar] [CrossRef]
- Kumar, A.; Mahendra, J.; Samuel, S.; Govindraj, J.; Loganathan, T.; Vashum, Y.; Mahendra, L.; Krishnamoorthy, T. Platelet-Rich Fibrin/Biphasic Calcium Phosphate Impairs Osteoclast Differentiation and Promotes Apoptosis by the Intrinsic Mitochondrial Pathway in Chronic Periodontitis. J. Periodontol. 2019, 90, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jiang, Y.; Mao, J.; Ren, X.; Ji, Y.; Mao, Y.; Chen, Y.; Sun, X.; Pan, Y.; Ma, J.; et al. Hydroxytyrosol Prevents Periodontitis-Induced Bone Loss by Regulating Mitochondrial Function and Mitogen-Activated Protein Kinase Signaling of Bone Cells. Free Radic. Biol. Med. 2021, 176, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Cui, C.; Shao, C.; Wang, Y.; Ye, C.; Lv, G. Coenzyme Q10 Inhibits RANKL-Induced Osteoclastogenesis by Regulation of Mitochondrial Apoptosis and Oxidative Stress in RAW264.7 Cells. J. Biochem. Mol. Toxicol. 2021, 35, e22778. [Google Scholar] [CrossRef]
- Sarkar, J.; Das, M.; Howlader, M.S.I.; Prateeksha, P.; Barthels, D.; Das, H. Epigallocatechin-3-Gallate Inhibits Osteoclastic Differentiation by Modulating Mitophagy and Mitochondrial Functions. Cell Death Dis. 2022, 13, 908. [Google Scholar] [CrossRef] [PubMed]
- Laha, D.; Sarkar, J.; Maity, J.; Pramanik, A.; Howlader, M.S.I.; Barthels, D.; Das, H. Polyphenolic Compounds Inhibit Osteoclast Differentiation While Reducing Autophagy through Limiting ROS and the Mitochondrial Membrane Potential. Biomolecules 2022, 12, 1220. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-J.; Yoon, S.-Y.; Park, J.-N.; Suh, J.-H.; Choi, H.-S. Doxorubicin Induces Bone Loss by Increasing Autophagy through a Mitochondrial ROS/TRPML1/TFEB Axis in Osteoclasts. Antioxidants 2022, 11, 1476. [Google Scholar] [CrossRef]
- Jung, S.; Kwon, J.-O.; Kim, M.K.; Song, M.-K.; Kim, B.; Lee, Z.H.; Kim, H.-H. Mitofusin 2, a Mitochondria-ER Tethering Protein, Facilitates Osteoclastogenesis by Regulating the Calcium-Calcineurin-NFATc1 Axis. Biochem. Biophys. Res. Commun. 2019, 516, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Lee, W.-C.; Song, C.; Ye, L.; Abel, E.D.; Long, F. Both Aerobic Glycolysis and Mitochondrial Respiration Are Required for Osteoclast Differentiation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 11058–11067. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, P.; Alekos, N.S.; Kim, S.P.; Li, Z.; Wolfgang, M.J.; Riddle, R.C. Mitochondrial Fatty Acid β-Oxidation Is Important for Normal Osteoclast Formation in Growing Female Mice. Front. Physiol. 2022, 13, 997358. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, T.; Ono, N. The Diverse Origin of Bone-Forming Osteoblasts. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2021, 36, 1432–1447. [Google Scholar] [CrossRef]
- Das, B.K.; Wang, L.; Fujiwara, T.; Zhou, J.; Aykin-Burns, N.; Krager, K.J.; Lan, R.; Mackintosh, S.G.; Edmondson, R.; Jennings, M.L.; et al. Transferrin Receptor 1-Mediated Iron Uptake Regulates Bone Mass in Mice via Osteoclast Mitochondria and Cytoskeleton. eLife 2022, 11, e73539. [Google Scholar] [CrossRef]
- Lee, W.-C.; Ji, X.; Nissim, I.; Long, F. Malic Enzyme Couples Mitochondria with Aerobic Glycolysis in Osteoblasts. Cell Rep. 2020, 32, 108108. [Google Scholar] [CrossRef]
- Pahwa, H.; Khan, M.T.; Sharan, K. Hyperglycemia Impairs Osteoblast Cell Migration and Chemotaxis Due to a Decrease in Mitochondrial Biogenesis. Mol. Cell. Biochem. 2020, 469, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; An, H.-J.; Kim, J.M.; Sung, M.-J.; Kim, D.K.; Kim, H.K.; Oh, J.; Jeong, H.Y.; Lee, Y.H.; Yang, T.; et al. PINK1 Deficiency Impairs Osteoblast Differentiation through Aberrant Mitochondrial Homeostasis. Stem Cell Res. Ther. 2021, 12, 589. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.; Kim, N.-K.; Shim, W.; Lee, S.-H.; Kim, H.-J.; Moon, E.; Sesaki, H.; Jang, J.H.; Kim, J.-E.; Lee, Y.-S. Mitochondrial Fragmentation and Donut Formation Enhance Mitochondrial Secretion to Promote Osteogenesis. Cell Metab. 2023, 35, 345–360.e7. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.J.; Chen, Y.; Shi, L.X.; Cheng, H.R.; Banda, I.; Ji, Y.H.; Wang, Y.T.; Li, X.M.; Mao, Y.X.; Zhang, D.F.; et al. AKT-GSK3β Signaling Pathway Regulates Mitochondrial Dysfunction-Associated OPA1 Cleavage Contributing to Osteoblast Apoptosis: Preventative Effects of Hydroxytyrosol. Oxidative Med. Cell. Longev. 2019, 2019, 4101738. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hu, S.-L.; Xie, J.; Yan, D.-Y.; Weng, S.-J.; Tang, J.-H.; Wang, B.-Z.; Xie, Z.-J.; Wu, Z.-Y.; Yang, L. Proanthocyanidins-Mediated Nrf2 Activation Ameliorates Glucocorticoid-Induced Oxidative Stress and Mitochondrial Dysfunction in Osteoblasts. Oxidative Med. Cell. Longev. 2020, 2020, 9102012. [Google Scholar] [CrossRef]
- Gan, X.; Huang, S.; Yu, Q.; Yu, H.; Yan, S.S. Blockade of Drp1 Rescues Oxidative Stress-Induced Osteoblast Dysfunction. Biochem. Biophys. Res. Commun. 2015, 468, 719–725. [Google Scholar] [CrossRef]
- Qiu, S.; Tao, Z.B.; Tao, L.; Zhu, Y. Melatonin Induces Mitochondrial Apoptosis in Osteoblasts by Regulating the STIM1/Cytosolic Calcium Elevation/ERK Pathway. Life Sci. 2020, 248, 117455. [Google Scholar] [CrossRef]
- Cao, Z.; Zhang, H.; Zhou, X.; Han, X.; Ren, Y.; Gao, T.; Xiao, Y.; De Crombrugghe, B.; Somerman, M.J.; Feng, J.Q. Genetic Evidence for the Vital Function of Osterix in Cementogenesis. J. Bone Miner. Res. 2012, 27, 1080–1092. [Google Scholar] [CrossRef]
- Cao, Z.; Liu, R.; Zhang, H.; Liao, H.; Zhang, Y.; Hinton, R.J.; Feng, J.Q. Osterix Controls Cementoblast Differentiation through Downregulation of Wnt-Signaling via Enhancing DKK1 Expression. Int. J. Biol. Sci. 2015, 11, 335–344. [Google Scholar] [CrossRef]
- Ma, L.; Wang, X.; Liu, H.; Jiang, C.; Liao, H.; Xu, S.; Guo, Y.; Cao, Z. CXXC5 Mediates P. gingivalis-Suppressed Cementoblast Functions Partially via MAPK Signaling Network. Int. J. Biol. Sci. 2019, 15, 1685–1695. [Google Scholar] [CrossRef]
- Ma, L.; Liu, H.; Wang, X.; Jiang, C.; Yao, S.; Guo, Y.; Wang, H.; Cao, Z. CXXC5 Orchestrates Stat3/Erk/Akt Signaling Networks to Modulate P. gingivalis-Elicited Autophagy in Cementoblasts. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2021, 1868, 118923. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, H.; Liu, H.; Ma, L.; Jiang, C.; Liao, H.; Xu, S.; Xiang, J.; Cao, Z. MicroRNA-181b-5p Modulates Tumor Necrosis Factor-α-induced Inflammatory Responses by Targeting Interleukin-6 in Cementoblasts. J. Cell. Physiol. 2019, 234, 22719–22730. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Ma, L.; Wang, X.; Wang, H.; Peng, Y.; Gao, X.; Huang, H.; Chen, Y.; Zhang, Y.; Cao, Z. Ckip-1 Mediates P. gingivalis-Suppressed Cementoblast Mineralization. J. Dent. Res. 2022, 101, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, H.; Liao, H.; Wang, C.; Jiang, C.; Zhang, Y.; Cao, Z. MicroRNA-155-3p Mediates TNF-α-Inhibited Cementoblast Differentiation. J. Dent. Res. 2017, 96, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wang, X.; Ma, L.; Wang, H.; Peng, Y.; Liu, H.; Xiao, J.; Cao, Z. M2 Macrophages with Inflammation Tropism Facilitate Cementoblast Mineralization. J. Periodontol. 2022, 94, 290–300. [Google Scholar] [CrossRef]
- Huang, X.; Deng, Y.; Xiao, J.; Wang, H.; Yang, Q.; Cao, Z. Genetically Engineered M2-like Macrophage-Derived Exosomes for P. gingivalis-Suppressed Cementum Regeneration: From Mechanism to Therapy. Bioact. Mater. 2024, 32, 473–487. [Google Scholar] [CrossRef]
- He, P.; Liu, F.; Li, M.; Ren, M.; Wang, X.; Deng, Y.; Wu, X.; Li, Y.; Yang, S.; Song, J. Mitochondrial Calcium Ion Nanogluttons Alleviate Periodontitis via Controlling mPTPs. Adv. Healthc. Mater. 2023, 12, e2203106. [Google Scholar] [CrossRef]
- He, X.-T.; Li, X.; Zhang, M.; Tian, B.-M.; Sun, L.-J.; Bi, C.-S.; Deng, D.-K.; Zhou, H.; Qu, H.-L.; Wu, C.; et al. Role of Molybdenum in Material Immunomodulation and Periodontal Wound Healing: Targeting Immunometabolism and Mitochondrial Function for Macrophage Modulation. Biomaterials 2022, 283, 121439. [Google Scholar] [CrossRef]
- Jiang, C.; Yang, W.; Wang, C.; Qin, W.; Ming, J.; Zhang, M.; Qian, H.; Jiao, T. Methylene Blue-Mediated Photodynamic Therapy Induces Macrophage Apoptosis via ROS and Reduces Bone Resorption in Periodontitis. Oxidative Med. Cell. Longev. 2019, 2019, 1529520. [Google Scholar] [CrossRef]
- Chiu, A.V.; Saigh, M.A.; McCulloch, C.A.; Glogauer, M. The Role of NrF2 in the Regulation of Periodontal Health and Disease. J. Dent. Res. 2017, 96, 975–983. [Google Scholar] [CrossRef]
- Sima, C.; Aboodi, G.M.; Lakschevitz, F.S.; Sun, C.; Goldberg, M.B.; Glogauer, M. Nuclear Factor Erythroid 2-Related Factor 2 Down-Regulation in Oral Neutrophils Is Associated with Periodontal Oxidative Damage and Severe Chronic Periodontitis. Am. J. Pathol. 2016, 186, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Preshaw, P.M.; Alba, A.L.; Herrera, D.; Jepsen, S.; Konstantinidis, A.; Makrilakis, K.; Taylor, R. Periodontitis and Diabetes: A Two-Way Relationship. Diabetologia 2012, 55, 21–31. [Google Scholar] [CrossRef]
- Li, X.; Sun, X.; Zhang, X.; Mao, Y.; Ji, Y.; Shi, L.; Cai, W.; Wang, P.; Wu, G.; Gan, X.; et al. Enhanced Oxidative Damage and Nrf2 Downregulation Contribute to the Aggravation of Periodontitis by Diabetes Mellitus. Oxidative Med. Cell. Longev. 2018, 2018, 9421019. [Google Scholar] [CrossRef]
- Sun, X.; Mao, Y.; Dai, P.; Li, X.; Gu, W.; Wang, H.; Wu, G.; Ma, J.; Huang, S. Mitochondrial Dysfunction Is Involved in the Aggravation of Periodontitis by Diabetes. J. Clin. Periodontol. 2017, 44, 463–471. [Google Scholar] [CrossRef]
- Zhu, C.; Zhao, Y.; Wu, X.; Qiang, C.; Liu, J.; Shi, J.; Gou, J.; Pei, D.; Li, A. The Therapeutic Role of Baicalein in Combating Experimental Periodontitis with Diabetes via Nrf2 Antioxidant Signaling Pathway. J. Periodontal Res. 2020, 55, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxidative Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef] [PubMed]
- Dziedzic, A. Is Periodontitis Associated with Age-Related Cognitive Impairment? The Systematic Review, Confounders Assessment and Meta-Analysis of Clinical Studies. Int. J. Mol. Sci. 2022, 23, 15320. [Google Scholar] [CrossRef]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s Disease Brains: Evidence for Disease Causation and Treatment with Small-Molecule Inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef]
- Kim, C.M.; Lee, S.; Hwang, W.; Son, E.; Kim, T.W.; Kim, K.; Kim, Y.H. Obesity and Periodontitis: A Systematic Review and Updated Meta-Analysis. Front. Endocrinol. 2022, 13, 999455. [Google Scholar] [CrossRef]
- Martínez-Herrera, M.; Abad-Jiménez, Z.; Silvestre, F.J.; López-Domènech, S.; Márquez-Arrico, C.F.; Silvestre-Rangil, J.; Víctor, V.M.; Rocha, M. Effect of Non-Surgical Periodontal Treatment on Oxidative Stress Markers in Leukocytes and Their Interaction with the Endothelium in Obese Subjects with Periodontitis: A Pilot Study. J. Clin. Med. 2020, 9, 2117. [Google Scholar] [CrossRef]
- Quiles, J.M.; Gustafsson, Å.B. The Role of Mitochondrial Fission in Cardiovascular Health and Disease. Nat. Rev. Cardiol. 2022, 19, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.-S.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial Quality Control Mechanisms as Molecular Targets in Cardiac Ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Zahlten, J.; Riep, B.; Nichols, F.C.; Walter, C.; Schmeck, B.; Bernimoulin, J.-P.; Hippenstiel, S. Porphyromonas gingivalis Dihydroceramides Induce Apoptosis in Endothelial Cells. J. Dent. Res. 2007, 86, 635–640. [Google Scholar] [CrossRef]
- Martinez-Herrera, M.; López-Domènech, S.; Silvestre, F.J.; Silvestre-Rangil, J.; Bañuls, C.; Victor, V.M.; Rocha, M. Chronic Periodontitis Impairs Polymorphonuclear Leucocyte-Endothelium Cell Interactions and Oxidative Stress in Humans. J. Clin. Periodontol. 2018, 45, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Jajoo, N.S.; Shelke, A.U.; Bajaj, R.S.; Patil, P.P.; Patil, M.A. Association of Periodontitis with Pre Term Low Birth Weight—A Review. Placenta 2020, 95, 62–68. [Google Scholar] [CrossRef]
- Li, X.; Liu, X.-C.; Ding, X.; Liu, X.-M.; Cao, N.-B.; Deng, Y.; Hou, Y.-B.; Yu, W.-X. Resveratrol Protects Renal Damages Induced by Periodontitis via Preventing Mitochondrial Dysfunction in Rats. Oral Dis. 2023, 29, 1812–1825. [Google Scholar] [CrossRef]
- Scribante, A.; Gallo, S.; Pascadopoli, M.; Frani, M.; Butera, A. Ozonized Gels vs Chlorhexidine in Non-Surgical Periodontal Treatment: A Randomized Clinical Trial. Oral Dis. 2023. [Google Scholar] [CrossRef]
- Elbay, M.; Elbay, Ü.Ş.; Kaya, E.; Kalkan, Ö.P. Effects of Photobiomodulation with Different Application Parameters on Injection Pain in Children: A Randomized Clinical Trial. Available online: https://www.jocpd.com/articles/10.22514/jocpd.2023.035 (accessed on 2 January 2024).
- Takafuji, Y.; Tatsumi, K.; Ishida, M.; Kawao, N.; Okada, K.; Kaji, H. Extracellular Vesicles Secreted from Mouse Muscle Cells Suppress Osteoclast Formation: Roles of Mitochondrial Energy Metabolism. Bone 2020, 134, 115298. [Google Scholar] [CrossRef]
- Kanzaki, H.; Shinohara, F.; Kajiya, M.; Fukaya, S.; Miyamoto, Y.; Nakamura, Y. Nuclear Nrf2 Induction by Protein Transduction Attenuates Osteoclastogenesis. Free Radic. Biol. Med. 2014, 77, 239–248. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, Y.; Xiao, J.; Ma, L.; Wang, C.; Wang, X.; Huang, X.; Cao, Z. Mitochondrial Dysfunction in Periodontitis and Associated Systemic Diseases: Implications for Pathomechanisms and Therapeutic Strategies. Int. J. Mol. Sci. 2024, 25, 1024. https://doi.org/10.3390/ijms25021024
Deng Y, Xiao J, Ma L, Wang C, Wang X, Huang X, Cao Z. Mitochondrial Dysfunction in Periodontitis and Associated Systemic Diseases: Implications for Pathomechanisms and Therapeutic Strategies. International Journal of Molecular Sciences. 2024; 25(2):1024. https://doi.org/10.3390/ijms25021024
Chicago/Turabian StyleDeng, Yifei, Junhong Xiao, Li Ma, Chuan Wang, Xiaoxuan Wang, Xin Huang, and Zhengguo Cao. 2024. "Mitochondrial Dysfunction in Periodontitis and Associated Systemic Diseases: Implications for Pathomechanisms and Therapeutic Strategies" International Journal of Molecular Sciences 25, no. 2: 1024. https://doi.org/10.3390/ijms25021024
APA StyleDeng, Y., Xiao, J., Ma, L., Wang, C., Wang, X., Huang, X., & Cao, Z. (2024). Mitochondrial Dysfunction in Periodontitis and Associated Systemic Diseases: Implications for Pathomechanisms and Therapeutic Strategies. International Journal of Molecular Sciences, 25(2), 1024. https://doi.org/10.3390/ijms25021024