
Mismatch Repair Deficiency as a Predictive and Prognostic Biomarker in Endometrial Cancer: A Review on Immunohistochemistry Staining Patterns and Clinical Implications


 ,
,  ,
,  , ,
, ,
Abstract
:1. Introduction
2. Molecular Landscape of MSI/MMRd EC
- -
- -
- Lynch syndrome (LS), an autosomal dominant disorder characterized by the occurrence of multiple cancers, resulting from constitutional germline mutations, affecting the DNA MMR genes MLH1, MSH2, MSH6 and PMS2; constitutional MLH1 hypermethylation; or deletion of the stop codon (3′ end truncating) of the EPCAM gene causing the epigenetic silencing of the neighboring MSH2.
- -
- Homozygous MLH1 promoter hypermethylation is predominantly associated with sporadic cases;
- -
- Heterozygous signature of the MLH1 promoter hypermethylation, as a second-hit event results in the loss of expression of the wild-type allele in LS tumors;
- -
- The MLH1 pathogenic variant can be associated with MLH1 promoter hypermethylation.
3. The Histo-Molecular Approach
3.1. MMR Deficiency as a Predictive and Prognostic Biomarker in Endometrial Cancer: The Relationship with Molecular and Histological Subtypes
3.2. EC Histological Subtypes and Genomic Alterations: The Relationship with Molecular Classification and MMR Deficiency
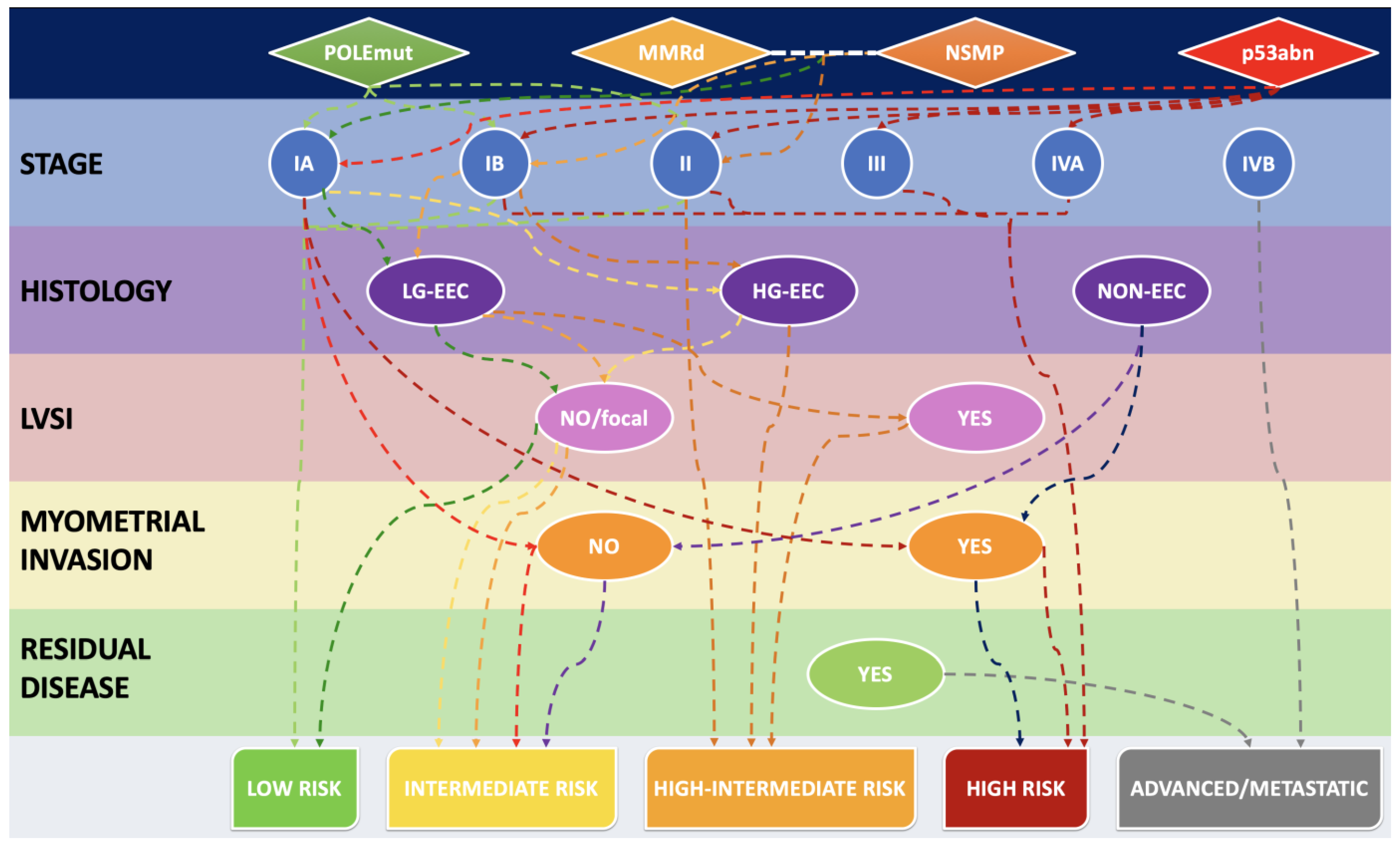
3.3. MMR Deficiency in Light of 2021 ESGO-ESTRO-ESP Guidelines and 2023 FIGO Staging System: A Combined Histo-Molecular Approach for Risk Stratification
4. Immunotherapy for MSI/dMMR Gynecological Cancers
5. MMRd/MSI ECs: Testing Method
- -
- Whenever IHC shows indetermined/ambiguous/equivocal results;
- -
- False negative IHC results due to pre-analytical tissue poor fixation;
- -
- Whenever IHC shows aberrant patterns (e.g., cytoplasmic, dot-like and perinuclear staining);
- -
- Whenever IHC shows the loss of only one heterodimer subunit (i.e., only MLH1 or PMS2 and not both).
6. Practical Issues in IHC MMR Proteins Interpretation: When–Where
- (a)
- Isolated loss of PMS2 or MSH6 regardless of the microsatellite status;
- (b)
- Classical loss of MLH1/PMS2 or MSH2/MSH6 without MSI (MSS tumors) or with MSI low (false positive cases: MSS/MSI-L-MMRd, due to MLH1 promoter hypermethylation or somatic MMR variants);
- (c)
- Four MMR proteins retained expression (non-functional protein with retained antigenicity) with MSI (false negative cases: MSI-MMRp in the case of POLE variants or missense mutation of MMR proteins, in particular MLH1);
- (d)
- Complex loss of MMR proteins regardless of the microsatellite status.
7. Problems of IHC Interpretation
7.1. Poor Fixation, Cauterization Artefacts, Neoadjuvant Chemotherapy and Freezing of Tissues
Solutions
7.2. Weak and or Focal Expression
7.3. Numerous Lymphocytes and Stromal Cells
7.4. Internal Control
- -
- An internal control tissue staining being diffuse and at least faint and adjacent tumor cells with similar or stronger staining intensity throughout the tumors.
- -
- There is a good and strong internal control but tumoral tissue stains less than 5%;
- -
- More than 10% of tumoral tissue presents a sharply demarcated lower staining intensity or absence of the staining compared with the evident internal control.
- -
- Tumoral area with weaker staining than internal control tissue;
- -
- Very faint staining in both internal control and tumor tissue;
- -
- Any other ambiguous pattern after excluding fixation issues.
7.5. Heterogeneous Loss (Subclonal Pattern)
7.6. Unusual Stainings
7.7. Unusual Combinations
7.8. Interlaboratory Reproducibility
7.9. Interobserver Reproducibility
7.10. Primary Vs. Metastasis
8. MMRd and Artificial Intelligence: Futuristic Approaches for MSI Detection
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Allemani, C.; Matsuda, T.; Carlo, V.D.; Harewood, R.; Matz, M.; Nikšić, M.; Bonaventure, A.; Valkov, M.; Johnson, C.J.; Estève, J.; et al. Global Surveillance of Trends in Cancer Survival 2000–14 (CONCORD-3): Analysis of Individual Records for 37 513 025 Patients Diagnosed with One of 18 Cancers from 322 Population-Based Registries in 71 Countries. Lancet 2018, 391, 1023–1075. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer Incidence and Mortality Patterns in Europe: Estimates for 40 Countries and 25 Major Cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef] [PubMed]
- Bokhman, J.V. Two Pathogenetic Types of Endometrial Carcinoma. Gynecol. Oncol. 1983, 15, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.A.; The Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Endometrial Carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Santoro, A.; Angelico, G.; Travaglino, A.; Inzani, F.; Arciuolo, D.; Valente, M.; D’Alessandris, N.; Scaglione, G.; Fiorentino, V.; Raffone, A.; et al. New Pathological and Clinical Insights in Endometrial Cancer in View of the Updated ESGO/ESTRO/ESP Guidelines. Cancers 2021, 13, 2623. [Google Scholar] [CrossRef]
- de Biase, D.; Maloberti, T.; Corradini, A.G.; Rosini, F.; Grillini, M.; Ruscelli, M.; Coluccelli, S.; Altimari, A.; Gruppioni, E.; Sanza, V.; et al. Integrated Clinicopathologic and Molecular Analysis of Endometrial Carcinoma: Prognostic Impact of the New ESGO-ESTRO-ESP Endometrial Cancer Risk Classification and Proposal of Histopathologic Algorithm for Its Implementation in Clinical Practice. Front. Med. 2023, 10, 1146499. [Google Scholar] [CrossRef]
- Santoro, A.; Angelico, G.; Travaglino, A.; Raffone, A.; Arciuolo, D.; D’Alessandris, N.; Inzani, F.; Zannoni, G.F. Clinico-Pathological Significance of TCGA Classification and SWI/SNF Proteins Expression in Undifferentiated/Dedifferentiated Endometrial Carcinoma: A Possible Prognostic Risk Stratification. Gynecol. Oncol. 2021, 161, 629–635. [Google Scholar] [CrossRef]
- Concin, N.; Matias-Guiu, X.; Vergote, I.; Cibula, D.; Mirza, M.R.; Marnitz, S.; Ledermann, J.; Bosse, T.; Chargari, C.; Fagotti, A.; et al. ESGO/ESTRO/ESP Guidelines for the Management of Patients with Endometrial Carcinoma. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2021, 31, 12–39. [Google Scholar] [CrossRef]
- Kommoss, S.; McConechy, M.K.; Kommoss, F.; Leung, S.; Bunz, A.; Magrill, J.; Britton, H.; Kommoss, F.; Grevenkamp, F.; Karnezis, A.; et al. Final Validation of the ProMisE Molecular Classifier for Endometrial Carcinoma in a Large Population-Based Case Series. Ann. Oncol. 2018, 29, 1180–1188. [Google Scholar] [CrossRef]
- Stelloo, E.; Nout, R.A.; Osse, E.M.; Jürgenliemk-Schulz, I.J.; Jobsen, J.J.; Lutgens, L.C.; van der Steen-Banasik, E.M.; Nijman, H.W.; Putter, H.; Bosse, T.; et al. Improved Risk Assessment by Integrating Molecular and Clinicopathological Factors in Early-Stage Endometrial Cancer-Combined Analysis of the PORTEC Cohorts. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4215–4224. [Google Scholar] [CrossRef]
- León-Castillo, A.; de Boer, S.M.; Powell, M.E.; Mileshkin, L.R.; Mackay, H.J.; Leary, A.; Nijman, H.W.; Singh, N.; Pollock, P.M.; Bessette, P.; et al. Molecular Classification of the PORTEC-3 Trial for High-Risk Endometrial Cancer: Impact on Prognosis and Benefit From Adjuvant Therapy. J. Clin. Oncol. 2020, 38, 3388–3397. [Google Scholar] [CrossRef]
- van den Heerik, A.S.V.M.; Horeweg, N.; Nout, R.A.; Lutgens, L.C.H.W.; van der Steen-Banasik, E.M.; Westerveld, G.H.; van den Berg, H.A.; Slot, A.; Koppe, F.L.A.; Kommoss, S.; et al. PORTEC-4a: International Randomized Trial of Molecular Profile-Based Adjuvant Treatment for Women with High-Intermediate Risk Endometrial Cancer. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2020, 30, 2002–2007. [Google Scholar] [CrossRef]
- Jessica McAlpine Tailored Adjuvant Therapy in POLE-Mutated and P53-Wildtype Early Stage Endometrial Cancer-Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04705649 (accessed on 11 July 2023).
- RAINBO Research Consortium Refining Adjuvant Treatment in Endometrial Cancer Based on Molecular Features: The RAINBO Clinical Trial Program. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2022, 33, 109–117. [CrossRef]
- Santoro, A.; Travaglino, A.; Inzani, F.; Arciuolo, D.; Angelico, G.; D’Alessandris, N.; Scaglione, G.; Valente, M.; Martini, M.; Raffone, A.; et al. Clear Cell Endometrial Carcinoma Precursors: Presentation of Two Cases and Diagnostic Issues. Diagn. Pathol. 2021, 16, 95. [Google Scholar] [CrossRef]
- Travaglino, A.; Raffone, A.; Santoro, A.; Raimondo, D.; Angelico, G.; Valente, M.; Arciuolo, D.; Scaglione, G.; D’alessandris, N.; Casadio, P.; et al. Clear Cell Endometrial Carcinomas with Mismatch Repair Deficiency Have a Favorable Prognosis: A Systematic Review and Meta-Analysis. Gynecol. Oncol. 2021, 162, 804–808. [Google Scholar] [CrossRef]
- O’Malley, D.M.; Bariani, G.M.; Cassier, P.A.; Marabelle, A.; Hansen, A.R.; De Jesus Acosta, A.; Miller, W.H.; Safra, T.; Italiano, A.; Mileshkin, L.; et al. Pembrolizumab in Patients With Microsatellite Instability–High Advanced Endometrial Cancer: Results From the KEYNOTE-158 Study. J. Clin. Oncol. 2022, 40, 752–761. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Green, A.K.; Feinberg, J.; Makker, V. A Review of Immune Checkpoint Blockade Therapy in Endometrial Cancer. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, J.M.; Panda, A.; Zhong, H.; Hirshfield, K.; Damare, S.; Lane, K.; Sokol, L.; Stein, M.N.; Rodriguez-Rodriquez, L.; Kaufman, H.L.; et al. Immune Activation and Response to Pembrolizumab in POLE-Mutant Endometrial Cancer. J. Clin. Investig. 2016, 126, 2334–2340. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The Therapeutic Significance of Mutational Signatures from DNA Repair Deficiency in Cancer. Nat. Commun. 2018, 9, 3292. [Google Scholar] [CrossRef]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndrome) and Microsatellite Instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, R.B.; Pyatt, R.E.; Niemann, T.H.; Richards, S.K.; Johnson, C.K.; Stevens, M.W.; Meek, J.E.; Hampel, H.; Prior, T.W.; de la Chapelle, A. Hereditary and Somatic DNA Mismatch Repair Gene Mutations in Sporadic Endometrial Carcinoma. J. Med. Genet. 2001, 38, 461–466. [Google Scholar] [CrossRef]
- Galuppini, F.; Opocher, E.; Tabori, U.; Mammi, I.; Edwards, M.; Campbell, B.; Kelly, J.; Viel, A.; Quaia, M.; Rivieri, F.; et al. Concomitant IDH Wild-Type Glioblastoma and IDH1-Mutant Anaplastic Astrocytoma in a Patient with Constitutional Mismatch Repair Deficiency Syndrome. Neuropathol. Appl. Neurobiol. 2018, 44, 233–239. [Google Scholar] [CrossRef]
- Abedalthagafi, M. Constitutional Mismatch Repair-Deficiency: Current Problems and Emerging Therapeutic Strategies. Oncotarget 2018, 9, 35458–35469. [Google Scholar] [CrossRef]
- Bonadona, V.; Bonaïti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.-J.; Caron, O.; et al. Cancer Risks Associated with Germline Mutations in MLH1, MSH2, and MSH6 Genes in Lynch Syndrome. JAMA 2011, 305, 2304–2310. [Google Scholar] [CrossRef]
- Spinosa, D.; Acosta, T.; Wong, J.; Kurtovic, K.; Mewshaw, J.; Collins, S.; Kauff, N.; Havrilesky, L.J.; Strickland, K.C.; Previs, R.A. Universal Screening for Lynch Syndrome in Uterine Cancer Patients: A Quality Improvement Initiative. Gynecol. Oncol. 2021, 160, 169–174. [Google Scholar] [CrossRef]
- Lancaster, J.M.; Powell, C.B.; Chen, L.-M.; Richardson, D.L.; SGO Clinical Practice Committee. Society of Gynecologic Oncology Statement on Risk Assessment for Inherited Gynecologic Cancer Predispositions. Gynecol. Oncol. 2015, 136, 3–7. [Google Scholar] [CrossRef]
- Esteller, M.; Levine, R.; Baylin, S.B.; Ellenson, L.H.; Herman, J.G. MLH1 Promoter Hypermethylation Is Associated with the Microsatellite Instability Phenotype in Sporadic Endometrial Carcinomas. Oncogene 1998, 17, 2413–2417. [Google Scholar] [CrossRef]
- Simpkins, S.B.; Bocker, T.; Swisher, E.M.; Mutch, D.G.; Gersell, D.J.; Kovatich, A.J.; Palazzo, J.P.; Fishel, R.; Goodfellow, P.J. MLH1 Promoter Methylation and Gene Silencing Is the Primary Cause of Microsatellite Instability in Sporadic Endometrial Cancers. Hum. Mol. Genet. 1999, 8, 661–666. [Google Scholar] [CrossRef]
- Leenen, C.H.M.; van Lier, M.G.F.; van Doorn, H.C.; van Leerdam, M.E.; Kooi, S.G.; de Waard, J.; Hoedemaeker, R.F.; van den Ouweland, A.M.W.; Hulspas, S.M.; Dubbink, H.J.; et al. Prospective Evaluation of Molecular Screening for Lynch Syndrome in Patients with Endometrial Cancer ≤ 70 Years. Gynecol. Oncol. 2012, 125, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, E.; Sato, N.; Sugawara, T.; Noto, A.; Takahashi, K.; Makino, K.; Terada, Y. MLH1 Promoter Hypermethylation Predicts Poorer Prognosis in Mismatch Repair Deficiency Endometrial Carcinomas. J. Gynecol. Oncol. 2021, 32, e79. [Google Scholar] [CrossRef]
- Loukovaara, M.; Pasanen, A.; Bützow, R. Mismatch Repair Protein and MLH1 Methylation Status as Predictors of Response to Adjuvant Therapy in Endometrial Cancer. Cancer Med. 2021, 10, 1034–1042. [Google Scholar] [CrossRef]
- Chow, R.D.; Michaels, T.; Bellone, S.; Hartwich, T.M.P.; Bonazzoli, E.; Iwasaki, A.; Song, E.; Santin, A.D. Distinct Mechanisms of Mismatch-Repair Deficiency Delineate Two Modes of Response to Anti-PD-1 Immunotherapy in Endometrial Carcinoma. Cancer Discov. 2023, 13, 312–331. [Google Scholar] [CrossRef]
- WHO Classification of Tumours Editorial Board. Female Genital Tumours. In WHO Classification of Tumours Series, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2020; Volume 4, Available online: https://tumourclassification.iarc.who.int/chaptercontent/34/536 (accessed on 15 December 2023).
- Murali, R.; Delair, D.F.; Bean, S.M.; Abu-Rustum, N.R.; Soslow, R.A. Evolving Roles of Histologic Evaluation and Molecular/Genomic Profiling in the Management of Endometrial Cancer. J. Natl. Compr. Cancer Netw. 2018, 16, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Talhouk, A.; McConechy, M.K.; Leung, S.; Li-Chang, H.H.; Kwon, J.S.; Melnyk, N.; Yang, W.; Senz, J.; Boyd, N.; Karnezis, A.N.; et al. A Clinically Applicable Molecular-Based Classification for Endometrial Cancers. Br. J. Cancer 2015, 113, 299–310. [Google Scholar] [CrossRef]
- Talhouk, A.; McConechy, M.K.; Leung, S.; Yang, W.; Lum, A.; Senz, J.; Boyd, N.; Pike, J.; Anglesio, M.; Kwon, J.S.; et al. Confirmation of ProMisE: A Simple, Genomics-Based Clinical Classifier for Endometrial Cancer. Cancer 2017, 123, 802–813. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, N.D.; Salvesen, H.B.; Ryan, A.; Iversen, O.E.; Akslen, L.A.; Jacobs, I.J. Frequency and Prognostic Impact of Microsatellite Instability in a Large Population-Based Study of Endometrial Carcinomas. Cancer Res. 2000, 60, 1750–1752. [Google Scholar]
- Travaglino, A.; Raffone, A.; Mascolo, M.; Guida, M.; Insabato, L.; Zannoni, G.F.; Zullo, F. Clear Cell Endometrial Carcinoma and the TCGA Classification. Histopathology 2020, 76, 336–338. [Google Scholar] [CrossRef]
- Travaglino, A.; Raffone, A.; Gencarelli, A.; Mollo, A.; Guida, M.; Insabato, L.; Santoro, A.; Zannoni, G.F.; Zullo, F. TCGA Classification of Endometrial Cancer: The Place of Carcinosarcoma. Pathol. Oncol. Res. POR 2020, 26, 2067–2073. [Google Scholar] [CrossRef]
- Leon-Castillo, A.; Horeweg, N.; Peters, E.E.M.; Rutten, T.; ter Haar, N.; Smit, V.T.H.B.M.; Kroon, C.D.; Boennelycke, M.; Hogdall, E.; Hogdall, C.; et al. Prognostic Relevance of the Molecular Classification in High-Grade Endometrial Cancer for Patients Staged by Lymphadenectomy and without Adjuvant Treatment. Gynecol. Oncol. 2022, 164, 577–586. [Google Scholar] [CrossRef]
- Santoro, A.; Angelico, G.; Inzani, F.; Spadola, S.; Arciuolo, D.; Valente, M.; Musarra, T.; Capelli, G.; Fanfani, F.; Gallotta, V.; et al. Pathological Features, Immunoprofile and Mismatch Repair Protein Expression Status in Uterine Endometrioid Carcinoma: Focus on MELF Pattern of Myoinvasion. Eur. J. Surg. Oncol. 2021, 47, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Segura, S.E.; Pedra Nobre, S.; Hussein, Y.R.; Abu-Rustum, N.R.; Weigelt, B.; Soslow, R.A.; DeLair, D.F. DNA Mismatch Repair–Deficient Endometrial Carcinosarcomas Portend Distinct Clinical, Morphologic, and Molecular Features Compared With Traditional Carcinosarcomas. Am. J. Surg. Pathol. 2020, 44, 1573. [Google Scholar] [CrossRef]
- Howitt, B.E.; Dong, F.; Vivero, M.; Shah, V.; Lindeman, N.; Schoolmeester, J.K.; Baltay, M.; MacConaill, L.; Sholl, L.M.; Nucci, M.R.; et al. Molecular Characterization of Neuroendocrine Carcinomas of the Endometrium: Representation in All 4 TCGA Groups. Am. J. Surg. Pathol. 2020, 44, 1541. [Google Scholar] [CrossRef]
- Horn, L.-C.; Höhn, A.K.; Krücken, I.; Stiller, M.; Obeck, U.; Brambs, C.E. Mesonephric-like Adenocarcinomas of the Uterine Corpus: Report of a Case Series and Review of the Literature Indicating Poor Prognosis for This Subtype of Endometrial Adenocarcinoma. J. Cancer Res. Clin. Oncol. 2020, 146, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Ardighieri, L.; Palicelli, A.; Ferrari, F.; Bugatti, M.; Drera, E.; Sartori, E.; Odicino, F. Endometrial Carcinomas with Intestinal-Type Metaplasia/Differentiation: Does Mismatch Repair System Defects Matter? Case Report and Systematic Review of the Literature. J. Clin. Med. 2020, 9, 2552. [Google Scholar] [CrossRef] [PubMed]
- Moroney, M.R.; Davies, K.D.; Wilberger, A.C.; Sheeder, J.; Post, M.D.; Berning, A.A.; Fisher, C.; Lefkowits, C.; Guntupalli, S.R.; Behbakht, K.; et al. Molecular Markers in Recurrent Stage I, Grade 1 Endometrioid Endometrial Cancers. Gynecol. Oncol. 2019, 153, 517–520. [Google Scholar] [CrossRef]
- Bosse, T.; Nout, R.A.; McAlpine, J.N.; McConechy, M.K.; Britton, H.; Hussein, Y.R.; Gonzalez, C.; Ganesan, R.; Steele, J.C.; Harrison, B.T.; et al. Molecular Classification of Grade 3 Endometrioid Endometrial Cancers Identifies Distinct Prognostic Subgroups. Am. J. Surg. Pathol. 2018, 42, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Joehlin-Price, A.; Van Ziffle, J.; Hills, N.K.; Ladwig, N.; Rabban, J.T.; Garg, K. Molecularly Classified Uterine FIGO Grade 3 Endometrioid Carcinomas Show Distinctive Clinical Outcomes But Overlapping Morphologic Features. Am. J. Surg. Pathol. 2021, 45, 421. [Google Scholar] [CrossRef]
- Kim, S.R.; Cloutier, B.T.; Leung, S.; Cochrane, D.; Britton, H.; Pina, A.; Storness-Bliss, C.; Farnell, D.; Huang, L.; Shum, K.; et al. Molecular Subtypes of Clear Cell Carcinoma of the Endometrium: Opportunities for Prognostic and Predictive Stratification. Gynecol. Oncol. 2020, 158, 3–11. [Google Scholar] [CrossRef]
- DeLair, D.F.; Burke, K.A.; Selenica, P.; Lim, R.S.; Scott, S.N.; Middha, S.; Mohanty, A.S.; Cheng, D.T.; Berger, M.F.; Soslow, R.A.; et al. The Genetic Landscape of Endometrial Clear Cell Carcinomas. J. Pathol. 2017, 243, 230–241. [Google Scholar] [CrossRef]
- McConechy, M.K.; Hoang, L.N.; Chui, M.H.; Senz, J.; Yang, W.; Rozenberg, N.; Mackenzie, R.; McAlpine, J.N.; Huntsman, D.G.; Clarke, B.A.; et al. In-Depth Molecular Profiling of the Biphasic Components of Uterine Carcinosarcomas. J. Pathol. Clin. Res. 2015, 1, 173–185. [Google Scholar] [CrossRef]
- Cherniack, A.D.; Shen, H.; Walter, V.; Stewart, C.; Murray, B.A.; Bowlby, R.; Hu, X.; Ling, S.; Soslow, R.A.; Broaddus, R.R.; et al. Integrated Molecular Characterization of Uterine Carcinosarcoma. Cancer Cell 2017, 31, 411–423. [Google Scholar] [CrossRef]
- Travaglino, A.; Raffone, A.; Gencarelli, A.; Saracinelli, S.; Riccardi, C.; Mollo, A.; Zullo, F.; Insabato, L. Clinico-Pathological Features Associated with Mismatch Repair Deficiency in Endometrial Undifferentiated/Dedifferentiated Carcinoma: A Systematic Review and Meta-Analysis. Gynecol. Oncol. 2021, 160, 579–585. [Google Scholar] [CrossRef]
- Bell, D.W.; Ellenson, L.H. Molecular Genetics of Endometrial Carcinoma. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 339–367. [Google Scholar] [CrossRef]
- Wiegand, K.C.; Lee, A.F.; Al-Agha, O.M.; Chow, C.; Kalloger, S.E.; Scott, D.W.; Steidl, C.; Wiseman, S.M.; Gascoyne, R.D.; Gilks, B.; et al. Loss of BAF250a (ARID1A) Is Frequent in High-Grade Endometrial Carcinomas. J. Pathol. 2011, 224, 328–333. [Google Scholar] [CrossRef]
- Da Cruz Paula, A.; DeLair, D.F.; Ferrando, L.; Fix, D.J.; Soslow, R.A.; Park, K.J.; Chiang, S.; Reis-Filho, J.S.; Zehir, A.; Donoghue, M.T.A.; et al. Genetic and Molecular Subtype Heterogeneity in Newly Diagnosed Early- and Advanced-Stage Endometrial Cancer. Gynecol. Oncol. 2021, 161, 535–544. [Google Scholar] [CrossRef]
- Zighelboim, I.; Schmidt, A.P.; Gao, F.; Thaker, P.H.; Powell, M.A.; Rader, J.S.; Gibb, R.K.; Mutch, D.G.; Goodfellow, P.J. ATR Mutation in Endometrioid Endometrial Cancer Is Associated With Poor Clinical Outcomes. J. Clin. Oncol. 2009, 27, 3091–3096. [Google Scholar] [CrossRef]
- Zighelboim, I.; Mutch, D.G.; Knapp, A.; Ding, L.; Xie, M.; Cohn, D.E.; Goodfellow, P.J. High Frequency Strand Slippage Mutations in CTCF in MSI-Positive Endometrial Cancers. Hum. Mutat. 2014, 35, 63–65. [Google Scholar] [CrossRef]
- Novetsky, A.P.; Zighelboim, I.; Thompson, D.M.; Powell, M.A.; Mutch, D.G.; Goodfellow, P.J. Frequent Mutations in the RPL22 Gene and Its Clinical and Functional Implications. Gynecol. Oncol. 2013, 128, 470–474. [Google Scholar] [CrossRef]
- Kim, T.-M.; Laird, P.W.; Park, P.J. The Landscape of Microsatellite Instability in Colorectal and Endometrial Cancer Genomes. Cell 2013, 155, 858–868. [Google Scholar] [CrossRef]
- Giannakis, M.; Hodis, E.; Jasmine Mu, X.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 Is Frequently Mutated in Colorectal and Endometrial Cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, H.; Isacson, C.; Levine, R.; Kurman, R.J.; Cho, K.R.; Hedrick, L. P53 Gene Mutations Are Common in Uterine Serous Carcinoma and Occur Early in Their Pathogenesis. Am. J. Pathol. 1997, 150, 177–185. [Google Scholar] [PubMed]
- Sherman, M.E.; Bur, M.E.; Kurman, R.J. P53 in Endometrial Cancer and Its Putative Precursors: Evidence for Diverse Pathways of Tumorigenesis. Hum. Pathol. 1995, 26, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Vermij, L.; Léon-Castillo, A.; Singh, N.; Powell, M.E.; Edmondson, R.J.; Genestie, C.; Khaw, P.; Pyman, J.; McLachlin, C.M.; Ghatage, P.; et al. p53 immunohistochemistry in endometrial cancer: Clinical and molecular correlates in the PORTEC-3 trial. Mod Pathol. 2022, 35, 1475–1483. [Google Scholar] [CrossRef]
- Timmers, P.J.; Zwinderman, A.H.; Teodorovic, I.; Vergote, I.; Trimbos, J.B. Clear Cell Carcinoma Compared to Serous Carcinoma in Early Ovarian Cancer: Same Prognosis in a Large Randomized Trial. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2009, 19, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Le Gallo, M.; Rudd, M.L.; Urick, M.E.; Hansen, N.F.; Zhang, S.; Lozy, F.; Sgroi, D.C.; Bel, A.V.; Matias-Guiu, X.; Broaddus, R.R.; et al. Somatic Mutation Profiles of Clear Cell Endometrial Tumors Revealed by Whole Exome and Targeted Gene Sequencing. Cancer 2017, 123, 3261–3268. [Google Scholar] [CrossRef] [PubMed]
- An, H.-J.; Logani, S.; Isacson, C.; Ellenson, L.H. Molecular Characterization of Uterine Clear Cell Carcinoma. Mod. Pathol. 2004, 17, 530–537. [Google Scholar] [CrossRef]
- Hoang, L.N.; McConechy, M.K.; Meng, B.; McIntyre, J.B.; Ewanowich, C.; Gilks, C.B.; Huntsman, D.G.; Köbel, M.; Lee, C.-H. Targeted Mutation Analysis of Endometrial Clear Cell Carcinoma. Histopathology 2015, 66, 664–674. [Google Scholar] [CrossRef]
- Stelloo, E.; Bosse, T.; Nout, R.A.; MacKay, H.J.; Church, D.N.; Nijman, H.W.; Leary, A.; Edmondson, R.J.; Powell, M.E.; Crosbie, E.J.; et al. Refining Prognosis and Identifying Targetable Pathways for High-Risk Endometrial Cancer; a TransPORTEC Initiative. Mod. Pathol. 2015, 28, 836–844. [Google Scholar] [CrossRef]
- Zhao, S.; Bellone, S.; Lopez, S.; Thakral, D.; Schwab, C.; English, D.P.; Black, J.; Cocco, E.; Choi, J.; Zammataro, L.; et al. Mutational Landscape of Uterine and Ovarian Carcinosarcomas Implicates Histone Genes in Epithelial-Mesenchymal Transition. Proc. Natl. Acad. Sci. USA 2016, 113, 12238–12243. [Google Scholar] [CrossRef]
- McConechy, M.K.; Ding, J.; Cheang, M.C.; Wiegand, K.; Senz, J.; Tone, A.; Yang, W.; Prentice, L.; Tse, K.; Zeng, T.; et al. Use of Mutation Profiles to Refine the Classification of Endometrial Carcinomas. J. Pathol. 2012, 228, 20–30. [Google Scholar] [CrossRef]
- Raffone, A.; Travaglino, A.; Raimondo, D.; Maletta, M.; De Vivo, V.; Visiello, U.; Casadio, P.; Seracchioli, R.; Zullo, F.; Insabato, L.; et al. Uterine Carcinosarcoma vs Endometrial Serous and Clear Cell Carcinoma: A Systematic Review and Meta-analysis of Survival. Int. J. Gynecol. Obstet. 2022, 158, 520–527. [Google Scholar] [CrossRef]
- Jones, S.; Stransky, N.; McCord, C.L.; Cerami, E.; Lagowski, J.; Kelly, D.; Angiuoli, S.V.; Sausen, M.; Kann, L.; Shukla, M.; et al. Genomic Analyses of Gynaecologic Carcinosarcomas Reveal Frequent Mutations in Chromatin Remodelling Genes. Nat. Commun. 2014, 5, 5006. [Google Scholar] [CrossRef]
- Le Gallo, M.; Rudd, M.L.; Urick, M.E.; Hansen, N.F.; National Institutes of Health Intramural Sequencing Center Comparative Sequencing Program; Merino, M.J.; Mutch, D.G.; Goodfellow, P.J.; Mullikin, J.C.; Bell, D.W. The FOXA2 Transcription Factor Is Frequently Somatically Mutated in Uterine Carcinosarcomas and Carcinomas. Cancer 2018, 124, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Biscuola, M.; Van de Vijver, K.; Castilla, M.Á.; Romero-Pérez, L.; López-García, M.Á.; Díaz-Martín, J.; Matias-Guiu, X.; Oliva, E.; Palacios Calvo, J. Oncogene Alterations in Endometrial Carcinosarcomas. Hum. Pathol. 2013, 44, 852–859. [Google Scholar] [CrossRef]
- Asami, Y.; Kobayashi Kato, M.; Hiranuma, K.; Matsuda, M.; Shimada, Y.; Ishikawa, M.; Koyama, T.; Komatsu, M.; Hamamoto, R.; Nagashima, M.; et al. Utility of Molecular Subtypes and Genetic Alterations for Evaluating Clinical Outcomes in 1029 Patients with Endometrial Cancer. Br. J. Cancer 2023, 128, 1582–1591. [Google Scholar] [CrossRef] [PubMed]
- Berek, J.S.; Matias-Guiu, X.; Creutzberg, C.; Fotopoulou, C.; Gaffney, D.; Kehoe, S.; Lindemann, K.; Mutch, D.; Concin, N.; Endometrial Cancer Staging Subcommittee; et al. FIGO Staging of Endometrial Cancer: 2023. Int. J. Gynecol. Obstet. 2023, 162, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, A.; Huvila, J.; Chiu, D.; Thompson, E.F.; Scott, S.; Salvador, S.; Vicus, D.; Helpman, L.; Gotlieb, W.; Kean, S.; et al. Grade and Estrogen Receptor Expression Identify a Subset of No Specific Molecular Profile Endometrial Carcinomas at a Very Low Risk of Disease-Specific Death. Mod. Pathol. 2023, 36, 100085. [Google Scholar] [CrossRef]
- Vermij, L.; Jobsen, J.J.; León-Castillo, A.; Brinkhuis, M.; Roothaan, S.; Powell, M.E.; de Boer, S.M.; Khaw, P.; Mileshkin, L.R.; Fyles, A.; et al. Prognostic Refinement of NSMP High-Risk Endometrial Cancers Using Oestrogen Receptor Immunohistochemistry. Br. J. Cancer 2023, 128, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Oaknin, A.; Gilbert, L.; Tinker, A.V.; Brown, J.; Mathews, C.; Press, J.; Sabatier, R.; O’Malley, D.M.; Samouelian, V.; Boni, V.; et al. Safety and Antitumor Activity of Dostarlimab in Patients with Advanced or Recurrent DNA Mismatch Repair Deficient/Microsatellite Instability-High (dMMR/MSI-H) or Proficient/Stable (MMRp/MSS) Endometrial Cancer: Interim Results from GARNET—A Phase I, Single-Arm Study. J. Immunother. Cancer 2022, 10, e003777. [Google Scholar] [CrossRef]
- Raffone, A.; Travaglino, A.; Cerbone, M.; Gencarelli, A.; Mollo, A.; Insabato, L.; Zullo, F. Diagnostic Accuracy of Immunohistochemistry for Mismatch Repair Proteins as Surrogate of Microsatellite Instability Molecular Testing in Endometrial Cancer. Pathol. Oncol. Res. POR 2020, 26, 1417–1427. [Google Scholar] [CrossRef]
- Zannoni, G.F.; Bragantini, E.; Castiglione, F.; Fassan, M.; Troncone, G.; Inzani, F.; Pesci, A.; Santoro, A.; Fraggetta, F. Current Prognostic and Predictive Biomarkers for Endometrial Cancer in Clinical Practice: Recommendations/Proposal from the Italian Study Group. Front. Oncol. 2022, 12, 805613. [Google Scholar] [CrossRef] [PubMed]
- Malapelle, U.; Parente, P.; Pepe, F.; De Luca, C.; Cerino, P.; Covelli, C.; Balestrieri, M.; Russo, G.; Bonfitto, A.; Pisapia, P.; et al. Impact of Pre-Analytical Factors on MSI Test Accuracy in Mucinous Colorectal Adenocarcinoma: A Multi-Assay Concordance Study. Cells 2020, 9, 2019. [Google Scholar] [CrossRef] [PubMed]
- Jaffrelot, M.; Farés, N.; Brunac, A.C.; Laurenty, A.P.; Danjoux, M.; Grand, D.; Icher, S.; Meilleroux, J.; Mery, E.; Buscail, E.; et al. An Unusual Phenotype Occurs in 15% of Mismatch Repair-Deficient Tumors and Is Associated with Non-Colorectal Cancers and Genetic Syndromes. Mod. Pathol. 2022, 35, 427–437. [Google Scholar] [CrossRef]
- Morak, M.; Käsbauer, S.; Kerscher, M.; Laner, A.; Nissen, A.M.; Benet-Pagès, A.; Schackert, H.K.; Keller, G.; Massdorf, T.; Holinski-Feder, E. Loss of MSH2 and MSH6 Due to Heterozygous Germline Defects in MSH3 and MSH6. Fam. Cancer 2017, 16, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Hui, P.; Buza, N. Frequent Loss of Mutation-Specific Mismatch Repair Protein Expression in Nonneoplastic Endometrium of Lynch Syndrome Patients. Mod. Pathol. 2020, 33, 1172–1181. [Google Scholar] [CrossRef]
- Stelloo, E.; Jansen, A.M.L.; Osse, E.M.; Nout, R.A.; Creutzberg, C.L.; Ruano, D.; Church, D.N.; Morreau, H.; Smit, V.T.H.B.M.; van Wezel, T.; et al. Practical Guidance for Mismatch Repair-Deficiency Testing in Endometrial Cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 96–102. [Google Scholar] [CrossRef]
- Catasus, L.; Matias-Guiu, X.; Machin, P.; Zannoni, G.F.; Scambia, G.; Benedetti-Panici, P.; Prat, J. Frameshift Mutations at Coding Mononucleotide Repeat Microsatellites in Endometrial Carcinoma with Microsatellite Instability. Cancer 2000, 88, 2290–2297. [Google Scholar] [CrossRef]
- Graham, R.P.; Kerr, S.E.; Butz, M.L.; Thibodeau, S.N.; Halling, K.C.; Smyrk, T.C.; Dina, M.A.; Waugh, V.M.; Rumilla, K.M. Heterogenous MSH6 Loss Is a Result of Microsatellite Instability within MSH6 and Occurs in Sporadic and Hereditary Colorectal and Endometrial Carcinomas. Am. J. Surg. Pathol. 2015, 39, 1370–1376. [Google Scholar] [CrossRef]
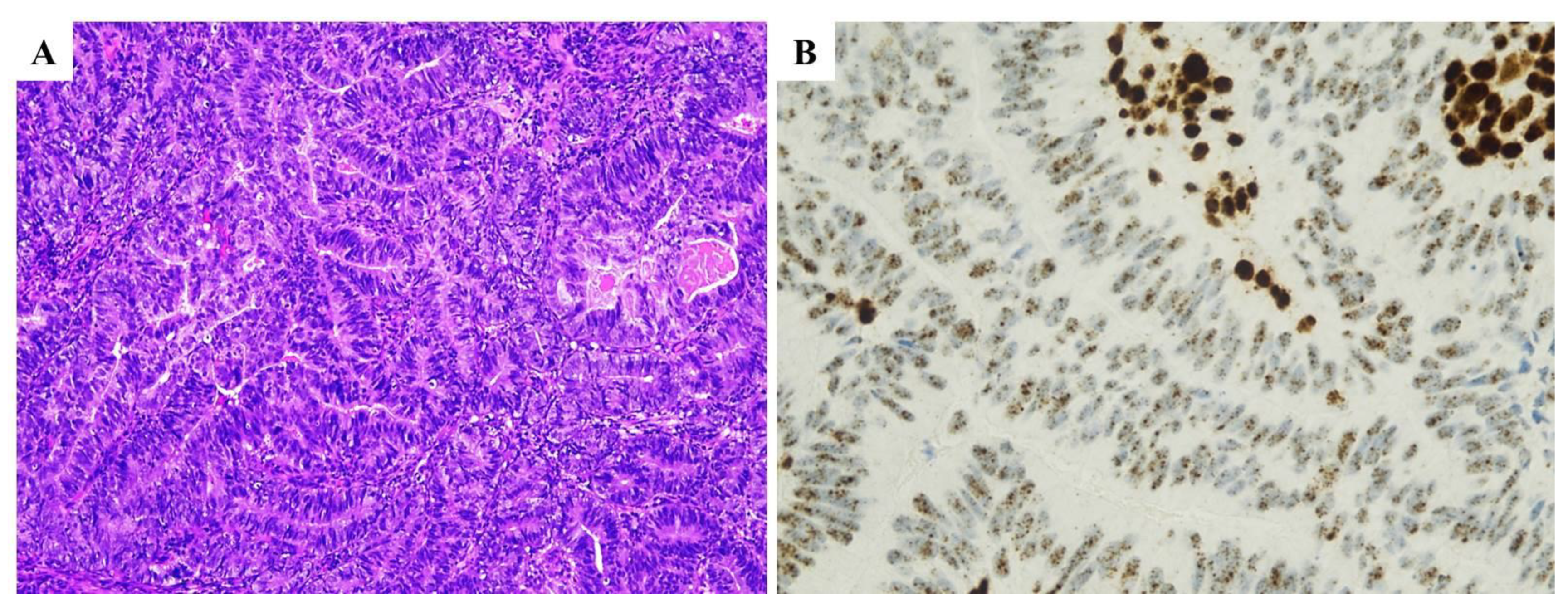
- Dasgupta, S.; Ewing-Graham, P.C.; Groenendijk, F.H.; Stam, O.; Biermann, K.E.; Doukas, M.; Dubbink, H.J.; van Velthuysen, M.F.; Dinjens, W.N.M.; Van Bockstal, M.R. Granular Dot-like Staining with MLH1 Immunohistochemistry Is a Clone-Dependent Artefact. Pathol.-Res. Pract. 2020, 216, 152581. [Google Scholar] [CrossRef]
- Plotkin, A.; Kuzeljevic, B.; De Villa, V.; Thompson, E.F.; Gilks, C.B.; Clarke, B.A.; Köbel, M.; McAlpine, J.N. Interlaboratory Concordance of ProMisE Molecular Classification of Endometrial Carcinoma Based on Endometrial Biopsy Specimens. Int. J. Gynecol. Pathol. Off. J. Int. Soc. Gynecol. Pathol. 2020, 39, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Sari, A.; Pollett, A.; Eiriksson, L.R.; Lumsden-Johanson, B.; Van de Laar, E.; Kazerouni, H.; Salehi, A.; Sur, M.; Lytwyn, A.; Ferguson, S.E. Interobserver Agreement for Mismatch Repair Protein Immunohistochemistry in Endometrial and Nonserous, Nonmucinous Ovarian Carcinomas. Am. J. Surg. Pathol. 2019, 43, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Ta, R.M.; Hecht, J.L.; Lin, D.I. Discordant Loss of Mismatch Repair Proteins in Advanced Endometrial Endometrioid Carcinoma Compared to Paired Primary Uterine Tumors. Gynecol. Oncol. 2018, 151, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Spinosa, D.; Wong, J.; Whitaker, R.; Strickland, K.; Previs, R. To Test or Re-Test, That Is the Question: Comparison of the Mismatch Repair Deficiency between Primary and Recurrent Sites of Uterine Cancers (088). Gynecol. Oncol. 2022, 166, S60. [Google Scholar] [CrossRef]
- Hong, R.; Liu, W.; DeLair, D.; Razavian, N.; Fenyö, D. Predicting Endometrial Cancer Subtypes and Molecular Features from Histopathology Images Using Multi-Resolution Deep Learning Models. Cell Rep. Med. 2021, 2, 100400. [Google Scholar] [CrossRef]
- Alam, M.R.; Abdul-Ghafar, J.; Yim, K.; Thakur, N.; Lee, S.H.; Jang, H.-J.; Jung, C.K.; Chong, Y. Recent Applications of Artificial Intelligence from Histopathologic Image-Based Prediction of Microsatellite Instability in Solid Cancers: A Systematic Review. Cancers 2022, 14, 2590. [Google Scholar] [CrossRef]
- Zhang, R.; Osinski, B.L.; Taxter, T.J.; Perera, J.; Lau, D.J.; Khan, A.A. Adversarial Deep Learning for Microsatellite Instability Prediction from Histopathology Slides. In Proceedings of the 1st Conference on Medical Imaging with Deep Learning (MIDL 2018), Amsterdam, The Netherlands, 4–6 July 2018. [Google Scholar]
- Kather, J.N.; Pearson, A.T.; Halama, N.; Jäger, D.; Krause, J.; Loosen, S.H.; Marx, A.; Boor, P.; Tacke, F.; Neumann, U.P.; et al. Deep Learning Can Predict Microsatellite Instability Directly from Histology in Gastrointestinal Cancer. Nat. Med. 2019, 25, 1054–1056. [Google Scholar] [CrossRef]
- Wang, T.; Lu, W.; Yang, F.; Liu, L.; Dong, Z.; Tang, W.; Chang, J.; Huan, W.; Huang, K.; Yao, J. Microsatellite Instability Prediction of Uterine Corpus Endometrial Carcinoma Based on H&E Histology Whole-Slide Imaging. In Proceedings of the 2020 IEEE 17th International Symposium on Biomedical Imaging (ISBI), Iowa City, IA, USA, 3–7 April 2020; pp. 1289–1292. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Histopathological Features of MMRd/MSI Endometrial Carcinoma |
|---|
| Lower uterine segment (LUS) origin |
| Endometrioid differentiation |
| Severe nuclear atypia with undifferentiated component |
| High mitotic index |
| High tumor-infiltrating lymphocytes (TILs) and/or peri-tumoral lymphocytes (≥40 TIL/10HPFs, with more CD8+, CD45RO+ and PD1+ T cells at the invasive tumoral margin) |
| High morphological heterogeneity |
| Substantial lympho-vascular space invasion (LVSI) |
| Deeper myometrial invasion |
| Synchronous ovarian cancer (clear cell or endometrioid variants) |
| Prevalence of Different Histotypes of MMRd/MSI Endometrial Carcinoma |
|---|
| Undifferentiated/dedifferentiated carcinoma (UEC/DEC): 44% |
| Neuroendocrine carcinoma: 42.9% |
| High-grade endometrioid carcinoma: 39.7% |
| Mixed: 33.3% |
| Low-grade endometrioid carcinoma: 24.7% |
| Clear cell carcinoma: 9.8% |
| Carcinosarcoma: 7.3% |
| Serous carcinoma (sporadic) |
| Mesonephric-like carcinoma (sporadic) |
| 2023 Figo Stage | Defining Criteria |
|---|---|
| IA1 | non-aggressive histological type limited to the endometrium or an endometrial polyp |
| IA2 | non-aggressive histological type involving <50% myometrium, with no/focal LVSI |
| IA3 | low-grade EEC limited to the uterus and ovary |
| IAmPOLEmut | POLEmut EC, confined to the uterine corpus or with cervical extension, regardless of LVSI or histological type |
| IB | non-aggressive histological type involving ≥50% myometrium, and with no/focal LVSI |
| IC | aggressive histological type limited to the endometrium or an endometrial polyp |
| IIA | non-aggressive histological type with invasion of the cervical stroma |
| IIB | non-aggressive histological type with substantial LVSI |
| IIC | aggressive histological type with any myometrial infiltration |
| IICmp53abn | p53abn EC, confined to the uterine corpus with any myometrial infiltration, with or without cervical invasion, and regardless of LVSI or histological type |
| IIIA1 | spread to ovary or fallopian tube (except if it meets the Stage IA3 criteria) |
| IIIA2 | involvement of uterine subserosa/serosa |
| IIIB1 | metastasis or direct spread to the vagina and/or the parametria |
| IIIB2 | metastasis to the pelvic peritoneum |
| IIIC1 | metastasis to the pelvic lymph nodes (micrometastasis = IIIC1i/macrometastasis = IIIC1ii) |
| IIIC2 | metastasis to para-aortic lymph nodes up to the renal vessels, with or without metastasis to the pelvic lymph nodes (micrometastasis = IIIC2i/macrometastasis = IIIC2ii) |
| IVA | invasion of the bladder mucosa and/or the intestinal mucosa |
| IVB | abdominal peritoneal metastasis beyond the pelvis |
| IVC | distant metastasis, including metastasis to any extra- or intra-abdominal lymph nodes above the renal vessels, lungs, liver, brain or bone |
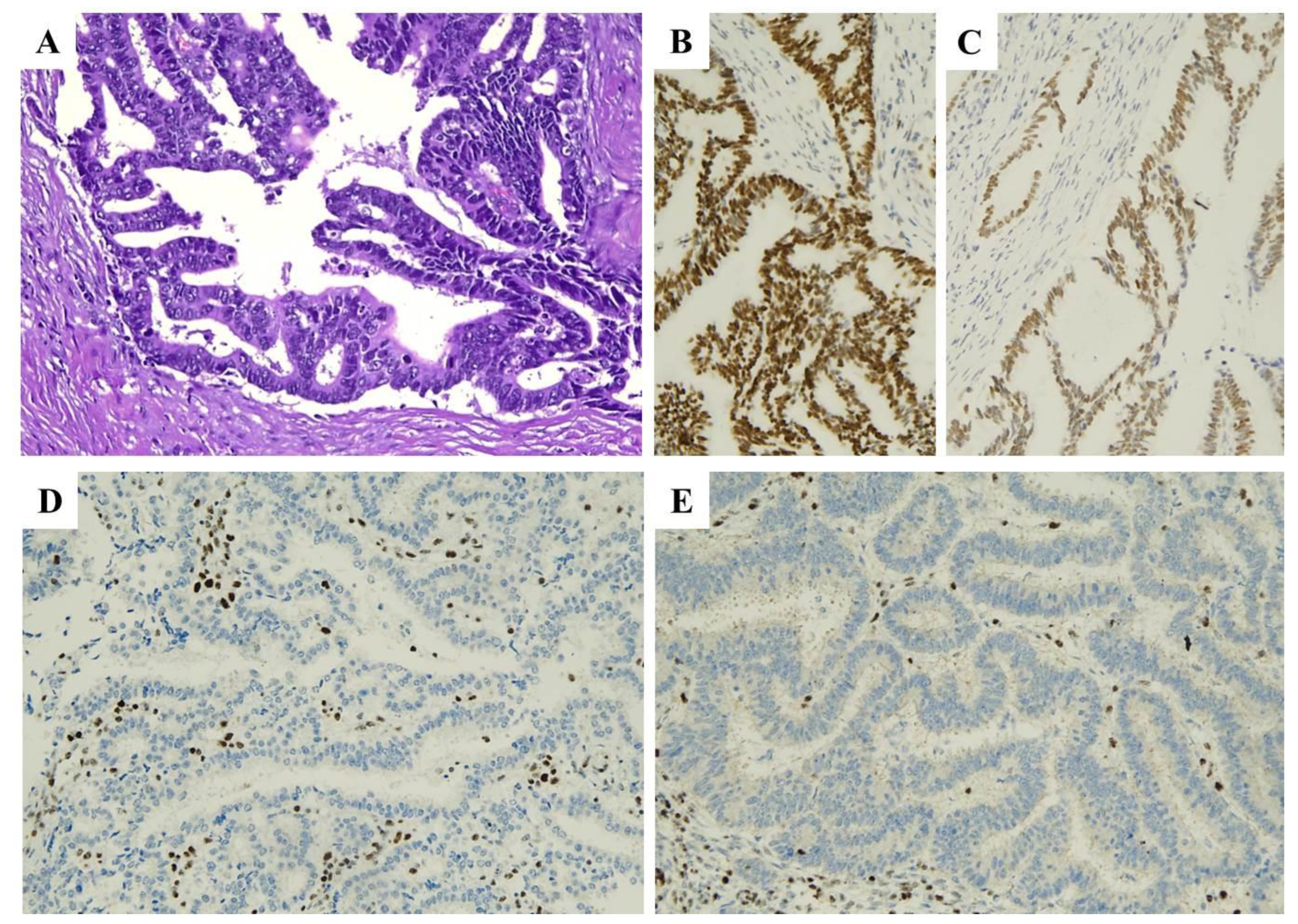
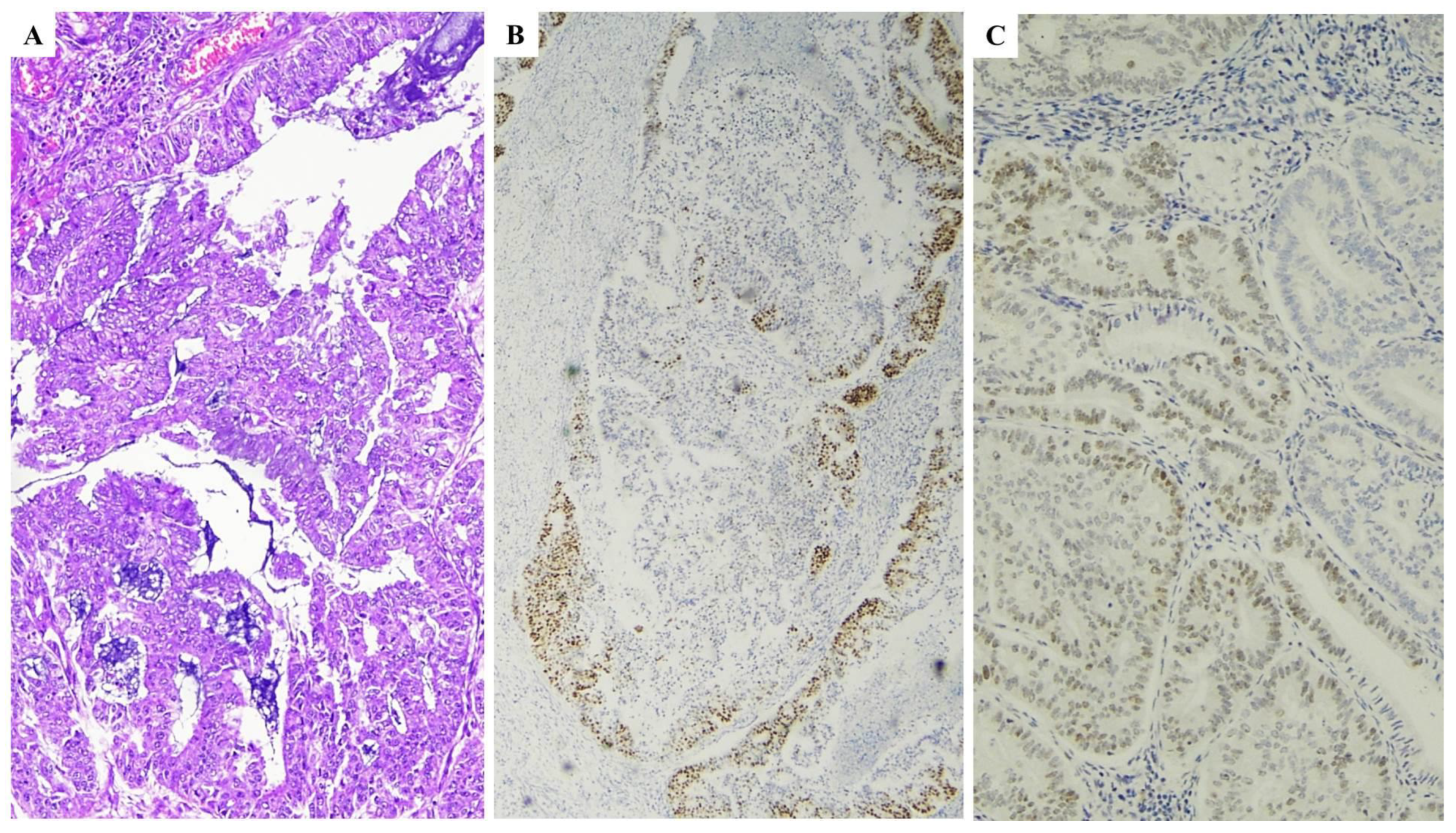
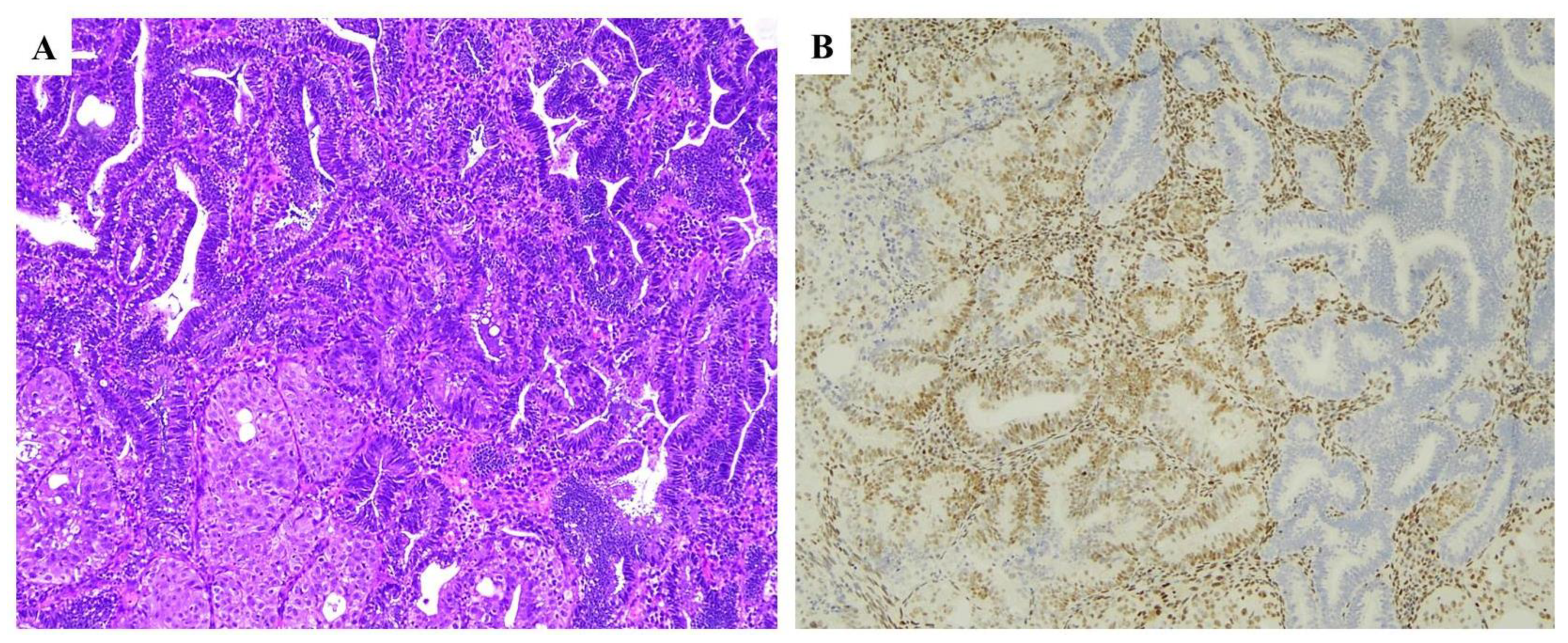
| MMR IHC Pattern | MMR Molecular Defect |
|---|---|
| MLH1+PMS2 loss | MLH1 promoter hypermethylation |
| MLH1+PMS2 loss | MLH1 gene defect (germline or somatic) |
| PMS2 loss | PMS2 gene defect (germline or somatic) |
| MSH2+MSH6 loss | MSH2 gene defect (germline or somatic) |
| MSH6 loss | MSH6 gene defect (germline or somatic) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Addante, F.; d’Amati, A.; Santoro, A.; Angelico, G.; Inzani, F.; Arciuolo, D.; Travaglino, A.; Raffone, A.; D’Alessandris, N.; Scaglione, G.; et al. Mismatch Repair Deficiency as a Predictive and Prognostic Biomarker in Endometrial Cancer: A Review on Immunohistochemistry Staining Patterns and Clinical Implications. Int. J. Mol. Sci. 2024, 25, 1056. https://doi.org/10.3390/ijms25021056
Addante F, d’Amati A, Santoro A, Angelico G, Inzani F, Arciuolo D, Travaglino A, Raffone A, D’Alessandris N, Scaglione G, et al. Mismatch Repair Deficiency as a Predictive and Prognostic Biomarker in Endometrial Cancer: A Review on Immunohistochemistry Staining Patterns and Clinical Implications. International Journal of Molecular Sciences. 2024; 25(2):1056. https://doi.org/10.3390/ijms25021056
Chicago/Turabian StyleAddante, Francesca, Antonio d’Amati, Angela Santoro, Giuseppe Angelico, Frediano Inzani, Damiano Arciuolo, Antonio Travaglino, Antonio Raffone, Nicoletta D’Alessandris, Giulia Scaglione, and et al. 2024. "Mismatch Repair Deficiency as a Predictive and Prognostic Biomarker in Endometrial Cancer: A Review on Immunohistochemistry Staining Patterns and Clinical Implications" International Journal of Molecular Sciences 25, no. 2: 1056. https://doi.org/10.3390/ijms25021056
APA StyleAddante, F., d’Amati, A., Santoro, A., Angelico, G., Inzani, F., Arciuolo, D., Travaglino, A., Raffone, A., D’Alessandris, N., Scaglione, G., Valente, M., Tinnirello, G., Sfregola, S., Padial Urtueta, B., Piermattei, A., Cianfrini, F., Mulè, A., Bragantini, E., & Zannoni, G. F. (2024). Mismatch Repair Deficiency as a Predictive and Prognostic Biomarker in Endometrial Cancer: A Review on Immunohistochemistry Staining Patterns and Clinical Implications. International Journal of Molecular Sciences, 25(2), 1056. https://doi.org/10.3390/ijms25021056