
NAD+ Precursors Reverse Experimental Diabetic Neuropathy in Mice

 ,
,  and
and
Abstract
:1. Introduction
2. Results
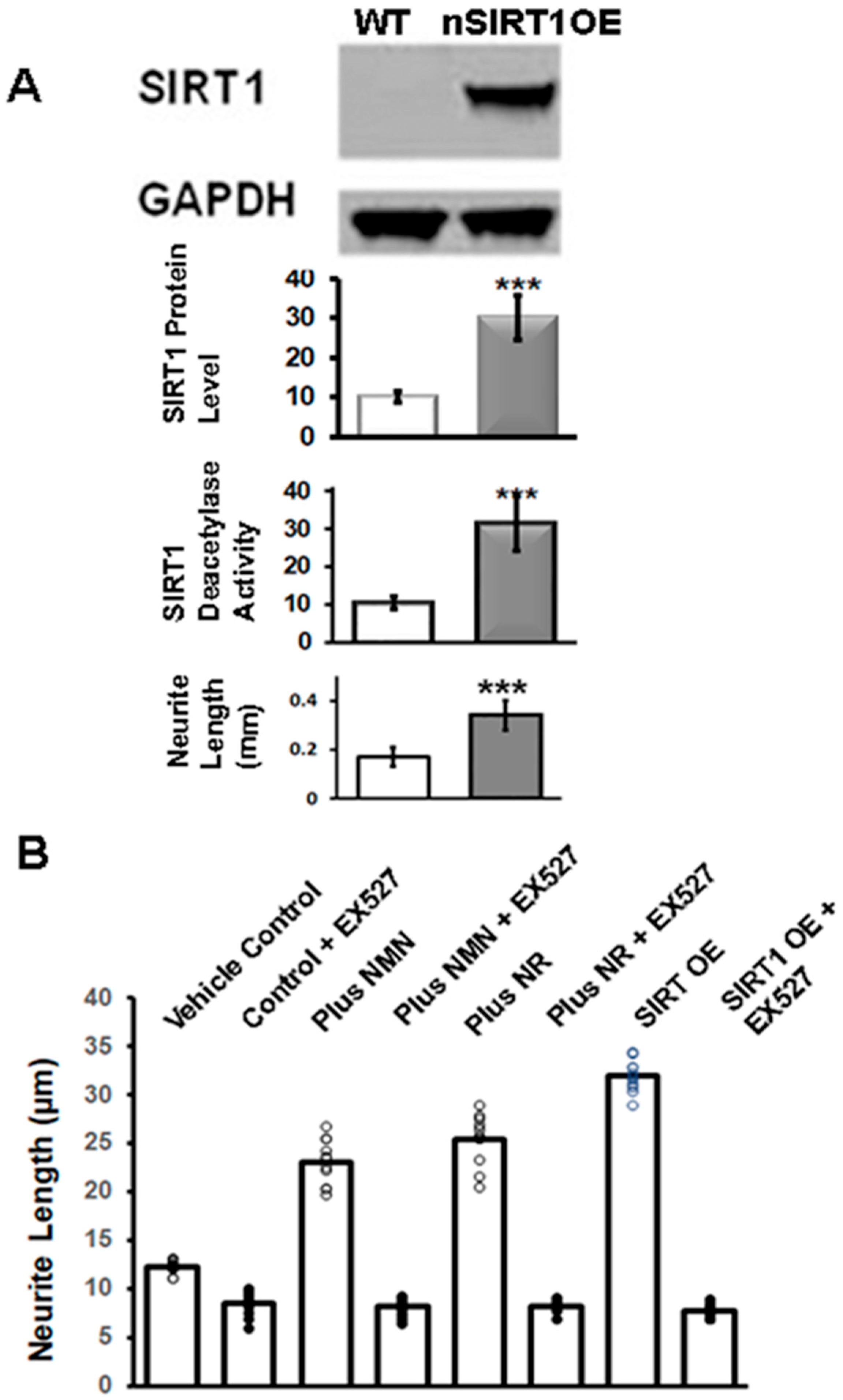
2.1. NR or NMN Increases Neurite Length through a SIRT1-Mediated Mechanism in Immortalized DRG Neuronal Cultures
2.2. Dietary Administration of NR or Subcutaneous Administration of NMN and Peripheral Neuropathy Induced via Streptozotocin (STZ) in Mice
2.3. Dietary Administration of NR or Subcutaneous Administration of NMN Reverses HFD-Induced Peripheral Neuropathy in Mice
2.4. Administration of NMN or NR Corrects Alterations Induced via STZ or HFD in the NAD+ Metabolome
2.5. No Additive Effect of NMN Administration in nSIRT1OE Mice in the Reversal of HFD-Induced Neuropathy in C57BL6 Mice
3. Discussion
3.1. Effect of NAD Precursors on Neurite Growth
3.2. Regulation of Axonal Growth and Mitochondrial Function via SIRT1-Mediated Deacetylation of NEDD4-1
3.3. NMNAT2, SARM, and Axonal Degeneration
3.4. NAD Precursors and Axonal Transport
3.5. NAD Precursors Affect Lipid Regulation but Have No Significant Effect on Glucose Regulation
3.6. There Is No Additive Effect on Neuropathy with SIRT1 Overexpression
4. Materials and Methods
4.1. Neurite Length Measurement
4.2. Diabetes Induction with STZ
4.3. CD and HFD
4.4. Quantification of NAD+ and SIRT1 Activity
4.5. Neuropathy Measurements
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chandrasekaran, K.; Najimi, N.; Sagi, A.R.; Yarlagadda, S.; Salimian, M.; Arvas, M.I.; Hedayat, A.F.; Kevas, Y.; Kadakia, A.; Russell, J.W. NAD(+) Precursors Repair Mitochondrial Function in Diabetes and Prevent Experimental Diabetic Neuropathy. Int. J. Mol. Sci. 2022, 23, 4887. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, K.; Choi, J.; Arvas, M.I.; Salimian, M.; Singh, S.; Xu, S.; Gullapalli, R.P.; Kristian, T.; Russell, J.W. Nicotinamide Mononucleotide Administration Prevents Experimental Diabetes-Induced Cognitive Impairment and Loss of Hippocampal Neurons. Int. J. Mol. Sci. 2020, 21, 3756. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Salimian, M.; Konduru, S.R.; Choi, J.; Kumar, P.; Long, A.; Klimova, N.; Ho, C.-Y.; Kristian, T.; Russell, J.W. Overexpression of Sirtuin 1 protein in neurons prevents and reverses experimental diabetic neuropathy. Brain 2019, 142, 3737–3752. [Google Scholar] [CrossRef] [PubMed]
- Trammell, S.A.; Weidemann, B.J.; Chadda, A.; Yorek, M.S.; Holmes, A.; Coppey, L.J.; Obrosov, A.; Kardon, R.H.; Yorek, M.A.; Brenner, C. Nicotinamide Riboside Opposes Type 2 Diabetes and Neuropathy in Mice. Sci. Rep. 2016, 6, 26933. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Anjaneyulu, M.; Choi, J.; Kumar, P.; Salimian, M.; Ho, C.Y.; Russell, J.W. Role of mitochondria in diabetic peripheral neuropathy: Influencing the NAD(+)-dependent SIRT1-PGC-1alpha-TFAM pathway. Int. Rev. Neurobiol. 2016, 145, 177–209. [Google Scholar] [CrossRef]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. NAD(+) in Aging: Molecular Mechanisms and Translational Implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef]
- Yoshino, J.; Baur, J.A.; Imai, S.-I. NAD(+) Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef]
- Gerdts, J.; Summers, D.W.; Milbrandt, J.; DiAntonio, A. Axon Self-Destruction: New Links among SARM1, MAPKs, and NAD+ Metabolism. Neuron 2016, 89, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.P.; Hoke, A. Programmed axon degeneration: From mouse to mechanism to medicine. Nat. Rev. Neurosci. 2020, 21, 183–196. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Auwerx, J. NAD⁺ metabolism: A therapeutic target for age-related metabolic disease. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 397–408. [Google Scholar] [CrossRef]
- Yang, Y.; Sauve, A.A. NAD(+) metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta 2016, 1864, 1787–1800. [Google Scholar] [CrossRef]
- Osterloh, J.M.; Yang, J.; Rooney, T.M.; Fox, A.N.; Adalbert, R.; Powell, E.H.; Sheehan, A.E.; Avery, M.A.; Hackett, R.; Logan, M.A.; et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 2012, 337, 481–484. [Google Scholar] [CrossRef]
- Geisler, S.; Doan, R.A.; Strickland, A.; Huang, X.; Milbrandt, J.; DiAntonio, A. Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain 2016, 139, 3092–3108. [Google Scholar] [CrossRef] [PubMed]
- Turkiew, E.; Falconer, D.; Reed, N.; Höke, A. Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. J. Peripher. Nerv. Syst. 2017, 22, 162–171. [Google Scholar] [CrossRef]
- Sasaki, Y. Metabolic aspects of neuronal degeneration: From a NAD(+) point of view. Neurosci. Res. 2019, 139, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.T.; Soares, S.M.; Novak, C.M.; Sinclair, D.; Levine, J.A.; Aksoy, P.; Chini, E.N. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007, 21, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Obrosova, I.G.; Xu, W.; Lyzogubov, V.V.; Ilnytska, O.; Mashtalir, N.; Vareniuk, I.; Pavlov, I.A.; Zhang, J.; Slusher, B.; Drel, V.R. PARP inhibition or gene deficiency counteracts intraepidermal nerve fiber loss and neuropathic pain in advanced diabetic neuropathy. Free Radic. Biol. Med. 2008, 44, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, P.; Escande, C.; White, T.A.; Thompson, M.; Soares, S.; Benech, J.C.; Chini, E.N. Regulation of SIRT 1 mediated NAD dependent deacetylation: A novel role for the multifunctional enzyme CD38. Biochem. Biophys. Res. Commun. 2006, 349, 353–359. [Google Scholar] [CrossRef]
- Alexandris, A.S.; Koliatsos, V.E. NAD(+), Axonal Maintenance, and Neurological Disease. Antioxid. Redox Signal. 2023, 39, 1167–1184. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, M.; Loreto, A.; Orsomando, G.; Mori, V.; Zamporlini, F.; Hulse, R.P.; Webster, J.; Donaldson, L.F.; Gering, M.; Raffaelli, N.; et al. NMN Deamidase Delays Wallerian Degeneration and Rescues Axonal Defects Caused by NMNAT2 Deficiency In Vivo. Curr. Biol. 2017, 27, 784–794. [Google Scholar] [CrossRef]
- Araki, T.; Sasaki, Y.; Milbrandt, J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 2004, 305, 1010–1013. [Google Scholar] [CrossRef]
- Tanner, K.G.; Landry, J.; Sternglanz, R.; Denu, J.M. Silent information regulator 2 family of NAD- dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc. Natl. Acad. Sci. USA 2000, 97, 14178–14182. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.D.; Schmidt, M.T.; Oppenheimer, N.J.; Denu, J.M. Mechanism of nicotinamide inhibition and transglycosidation by Sir2 histone/protein deacetylases. J. Biol. Chem. 2003, 278, 50985–50998. [Google Scholar] [CrossRef]
- Chalkiadaki, A.; Guarente, L. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat. Rev. Endocrinol. 2012, 8, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, D.A.; Guarente, L. Small-molecule allosteric activators of sirtuins. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 363–380. [Google Scholar] [CrossRef]
- Zilliox, L.A.; Russell, J.W. Physical activity and dietary interventions in diabetic neuropathy: A systematic review. Clin. Auton. Res. 2019, 4, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.B.; Rinaldi, C.; Lu, Y.; Amorim, J.A.; Sinclair, D.A. Molecular and Cellular Characterization of SIRT1 Allosteric Activators. Methods Mol. Biol. 2019, 1983, 133–149. [Google Scholar] [CrossRef]
- Min, S.W.; Sohn, P.D.; Cho, S.H.; Swanson, R.A.; Gan, L. Sirtuins in neurodegenerative diseases: An update on potential mechanisms. Front. Aging Neurosci. 2013, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Donmez, G.; Outeiro, T.F. SIRT1 and SIRT2: Emerging targets in neurodegeneration. EMBO Mol. Med. 2013, 5, 344–352. [Google Scholar] [CrossRef]
- Tomita, T.; Hamazaki, J.; Hirayama, S.; McBurney, M.W.; Yashiroda, H.; Murata, S. Sirt1-deficiency causes defective protein quality control. Sci. Rep. 2015, 5, 12613. [Google Scholar] [CrossRef]
- Ramadori, G.; Lee, C.E.; Bookout, A.L.; Lee, S.; Williams, K.W.; Anderson, J.; Elmquist, J.K.; Coppari, R. Brain SIRT1: Anatomical distribution and regulation by energy availability. J. Neurosci. 2008, 28, 9989–9996. [Google Scholar] [CrossRef]
- Grozio, A.; Mills, K.F.; Yoshino, J.; Bruzzone, S.; Sociali, G.; Tokizane, K.; Lei, H.C.; Cunningham, R.; Sasaki, Y.; Migaud, M.E.; et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab. 2019, 1, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Yoshino, J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes. Metab. 2013, 15 (Suppl. S3), 26–33. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S.-I. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.N.; Xu, T.Y.; Li, W.L.; Miao, C.Y. Targeting Nicotinamide Phosphoribosyltransferase as a Potential Therapeutic Strategy to Restore Adult Neurogenesis. CNS Neurosci. Ther. 2016, 22, 431–439. [Google Scholar] [CrossRef]
- Di Stefano, M.; Nascimento-Ferreira, I.; Orsomando, G.; Mori, V.; Gilley, J.; Brown, R.; Janeckova, L.; Vargas, M.E.; Worrell, L.A.; Loreto, A.; et al. A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ. 2015, 22, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Gao, M.; Hu, C.; Chen, X.; Zhou, Y. NMNAT2: An important metabolic enzyme affecting the disease progression. Biomed. Pharmacother. 2023, 158, 114143. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, J.Y.; Yang, Z.C.; Liu, M.; Gang, B.Z.; Zhao, Q.J. Nicotinamide mononucleotide adenylyltransferase 1 gene NMNAT1 regulates neuronal dendrite and axon morphogenesis in vitro. Chin. Med. J. 2011, 124, 3373–3377. [Google Scholar]
- Milde, S.; Fox, A.N.; Freeman, M.R.; Coleman, M.P. Deletions within its subcellular targeting domain enhance the axon protective capacity of Nmnat2 in vivo. Sci. Rep. 2023, 3, 2567. [Google Scholar] [CrossRef]
- Liu, D.; Gharavi, R.; Pitta, M.; Gleichmann, M.; Mattson, M.P. Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. Neuromolecular Med. 2009, 11, 28–42. [Google Scholar] [CrossRef]
- DiAntonio, A. Nedd4 branches out. Neuron 2010, 65, 293–294. [Google Scholar] [CrossRef]
- Kawabe, H.; Brose, N. The ubiquitin E3 ligase Nedd4-1 controls neurite development. Cell Cycle 2010, 9, 2477–2478. [Google Scholar] [CrossRef] [PubMed]
- Kawabe, H.; Neeb, A.; Dimova, K.; Young, S.M.; Takeda, M.; Katsurabayashi, S.; Mitkovski, M.; Malakhova, O.A.; Zhang, D.-E.; Umikawa, M.; et al. Regulation of Rap2A by the ubiquitin ligase Nedd4-1 controls neurite development. Neuron 2010, 65, 358–372. [Google Scholar] [CrossRef]
- Hsia, H.-E.; Kumar, R.; Luca, R.; Takeda, M.; Courchet, J.; Nakashima, J.; Wu, S.; Goebbels, S.; An, W.; Eickholt, B.J.; et al. Ubiquitin E3 ligase Nedd4-1 acts as a downstream target of PI3K/PTEN-mTORC1 signaling to promote neurite growth. Proc. Natl. Acad. Sci. USA 2014, 111, 13205–13210. [Google Scholar] [CrossRef]
- Fouladkou, F.; Landry, T.; Kawabe, H.; Neeb, A.; Lu, C.; Brose, N.; Stambolic, V.; Rotin, D. The ubiquitin ligase Nedd4-1 is dispensable for the regulation of PTEN stability and localization. Proc. Natl. Acad. Sci. USA 2008, 105, 8585–8590. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Trotman, L.C.; Koppie, T.; Alimonti, A.; Chen, Z.; Gao, Z.; Wang, J.; Erdjument-Bromage, H.; Tempst, P.; Cordon-Cardo, C.; et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 2007, 128, 129–139. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Pagliei, B.; Ciriolo, M.R. Extranuclear localization of SIRT1 and PGC-1alpha: An insight into possible roles in diseases associated with mitochondrial dysfunction. Curr. Mol. Med. 2013, 13, 140–154. [Google Scholar] [CrossRef]
- Aquilano, K.; Vigilanza, P.; Baldelli, S.; Pagliei, B.; Rotilio, G.; Ciriolo, M.R. Peroxisome proliferator-activated receptor gamma co-activator 1alpha (PGC-1alpha) and sirtuin 1 (SIRT1) reside in mitochondria: Possible direct function in mitochondrial biogenesis. J. Biol. Chem. 2010, 285, 21590–21599. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Oppenheim, R.W.; Sugiura, Y.; Lin, W. Abnormal development of the neuromuscular junction in Nedd4-deficient mice. Dev. Biol. 2009, 330, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Schmeisser, M.J.; Kühl, S.J.; Schoen, M.; Beth, N.H.; Weis, T.M.; Grabrucker, A.M.; Kühl, M.; Boeckers, T.M. The Nedd4-binding protein 3 (N4BP3) is crucial for axonal and dendritic branching in developing neurons. Neural Dev. 2013, 8, 18. [Google Scholar] [CrossRef]
- Drinjakovic, J.; Jung, H.; Campbell, D.S.; Strochlic, L.; Dwivedy, A.; Holt, C.E. E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron 2010, 65, 341–357. [Google Scholar] [CrossRef] [PubMed]
- Christie, K.J.; Martinez, J.A.; Zochodne, D.W. Disruption of E3 ligase NEDD4 in peripheral neurons interrupts axon outgrowth: Linkage to PTEN. Mol. Cell Neurosci. 2012, 50, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Wang, B.; Sastry, N.; Masliah, E.; Nelson, P.T.; Cai, H.; Liao, F.-F. NEDD4-mediated HSF1 degradation underlies alpha-synucleinopathy. Hum. Mol. Genet. 2016, 25, 211–222. [Google Scholar] [CrossRef]
- Huang, X.; Chen, J.; Cao, W.; Yang, L.; Chen, Q.; He, J.; Yi, Q.; Huang, H.; Zhang, E.; Cai, Z. The many substrates and functions of NEDD4-1. Cell Death Dis. 2019, 10, 904. [Google Scholar] [CrossRef]
- Xie, W.; Jin, S.; Wu, Y.; Xian, H.; Tian, S.; Liu, D.-A.; Guo, Z.; Cui, J. Auto-ubiquitination of NEDD4-1 Recruits USP13 to Facilitate Autophagy through Deubiquitinating VPS34. Cell Rep. 2020, 30, 2807–2819.e4. [Google Scholar] [CrossRef] [PubMed]
- Gilley, J.; Ribchester, R.R.; Coleman, M.P. Sarm1 Deletion, but Not Wld(S), Confers Lifelong Rescue in a Mouse Model of Severe Axonopathy. Cell Rep. 2017, 21, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Gilley, J.; Orsomando, G.; Nascimento-Ferreira, I.; Coleman, M.P. Absence of SARM1 rescues development and survival of NMNAT2-deficient axons. Cell Rep. 2015, 10, 1974–1981. [Google Scholar] [CrossRef]
- Marion, C.M.; McDaniel, D.P.; Armstrong, R.C. Sarm1 deletion reduces axon damage, demyelination, and white matter atrophy after experimental traumatic brain injury. Exp. Neurol. 2019, 321, 113040. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, M.; Yong, H.; Zhang, C.; Kang, K.; Liu, Z.; Yang, Y.; Huang, Z.; Wang, S.; Ge, H.; et al. Mitochondrial Localization of SARM1 in Acrylamide Intoxication Induces Mitophagy and Limits Neuropathy. Mol. Neurobiol. 2022, 59, 7337–7353. [Google Scholar] [CrossRef]
- Milde, S.; Gilley, J.; Coleman, M.P. Axonal trafficking of NMNAT2 and its roles in axon growth and survival in vivo. Bioarchitecture 2013, 3, 133–140. [Google Scholar] [CrossRef]
- Yang, S.; Niou, Z.X.; Enriquez, A.; LaMar, J.; Huang, J.Y.; Ling, K.; Jafar-Nejad, P.; Gilley, J.; Coleman, M.P.; Tennessen, J.M.; et al. NMNAT2 supports vesicular glycolysis via NAD homeostasis to fuel fast axonal transport. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Luchniak, A.; Mahamdeh, M.; Howard, J. Nicotinamide adenine dinucleotides and their precursor NMN have no direct effect on microtubule dynamics in purified brain tubulin. PLoS ONE 2019, 14, e0220794. [Google Scholar] [CrossRef]
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic neuropathy. Nat. Rev. Dis. Primers 2019, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Drexel, H. Nicotinic acid in the treatment of hyperlipidaemia. Fundam. Clin. Pharmacol. 2007, 21 (Suppl. S2), 5–6. [Google Scholar] [CrossRef]
- Dollerup, O.L.; Christensen, B.; Svart, M.; Schmidt, M.S.; Sulek, K.; Ringgaard, S.; Stødkilde-Jørgensen, H.; Møller, N.; Brenner, C.; Treebak, J.T.; et al. A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: Safety, insulin-sensitivity, and lipid-mobilizing effects. Am. J. Clin. Nutr. 2018, 108, 343–353. [Google Scholar] [CrossRef]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Imamura, H.; Nagayama, D.; Ishihara, N.; Tanaka, S.; Watanabe, R.; Watanabe, Y.; Sato, Y.; Yamaguchi, T.; Ban, N.; Kawana, H.; et al. Resveratrol attenuates triglyceride accumulation associated with upregulation of Sirt1 and lipoprotein lipase in 3T3-L1 adipocytes. Mol. Genet. Metab. Rep. 2017, 12, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Zhong, O.; Wang, J.; Tan, Y.; Lei, X.; Tang, Z. Effects of NAD+ precursor supplementation on glucose and lipid metabolism in humans: A meta-analysis. Nutr. Metab. 2022, 19, 20. [Google Scholar] [CrossRef]
- Gariani, K.; Menzies, K.J.; Ryu, D.; Wegner, C.J.; Wang, X.; Ropelle, E.R.; Moullan, N.; Zhang, H.; Perino, A.; Lemos, V.; et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 2016, 63, 1190–1204. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Chandrasekaran, K.; Inoue, T.; Muragundla, A.; Russell, J.W. PGC-1α regulation of mitochondrial degeneration in experimental diabetic neuropathy. Neurobiol. Dis. 2014, 64, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, K.; Anjaneyulu, M.; Inoue, T.; Choi, J.; Sagi, A.R.; Chen, C.; Ide, T.; Russell, J.W. Mitochondrial transcription factor A regulation of mitochondrial degeneration in experimental diabetic neuropathy. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E132–E141. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Yang, L.; Qin, A.R.; Ly, J.; Liederer, B.M.; Messick, K.; Ma, S.; Zak, M.; Dragovich, P.S.; Dean, B.J.; et al. Measuring NAD(+) levels in mouse blood and tissue samples via a surrogate matrix approach using LC-MS/MS. Bioanalysis 2014, 6, 1445–1457. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Bril, V.; Calcutt, N.A.; Cameron, N.E.; Cotter, M.A.; Dobrowsky, R.; Feldman, E.L.; Fernyhough, P.; Jakobsen, J.; Malik, R.A.; et al. Phenotyping animal models of diabetic neuropathy: A consensus statement of the diabetic neuropathy study group of the EASD (Neurodiab). J. Peripher. Nerv. Syst. 2014, 19, 77–87. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Muragundla, A.; Demarest, T.G.; Choi, J.; Sagi, A.R.; Najimi, N.; Kumar, P.; Singh, A.; Ho, C.; Fiskum, G.; et al. mGluR2/3 activation of the SIRT1 axis preserves mitochondrial function in diabetic neuropathy. Ann. Clin. Transl. Neurol. 2017, 4, 844–858. [Google Scholar] [CrossRef] [PubMed]
- Lauria, G.; Hsieh, S.T.; Johansson, O.; Kennedy, W.R.; Leger, J.M.; Mellgren, S.I.; Nolano, M.; Merkies, I.S.J.; Polydefkis, M.; Smith, A.G.; et al. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on the use of skin biopsy in the diagnosis of small fiber neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society. Eur. J. Neurol. 2010, 17, 903–912.e44–e49. [Google Scholar] [CrossRef]
- Lauria, G.; Cornblath, D.R.; Johansson, O.; McArthur, J.C.; Mellgren, S.I.; Nolano, M.; Rosenberg, N.; Sommer, C. EFNS guidelines on the use of skin biopsy in the diagnosis of peripheral neuropathy. Eur. J. Neurol. 2005, 12, 747–758. [Google Scholar] [CrossRef]
- Russell, J.W.; Berent-Spillson, A.; Vincent, A.M.; Freimann, C.L.; Sullivan, K.A.; Feldman, E.L. Oxidative injury and neuropathy in diabetes and impaired glucose tolerance. Neurobiol. Dis. 2008, 30, 420–429. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Groups | Non-Diabetic | Diabetic | Diabetic + NMN | Significance p Values | ||
|---|---|---|---|---|---|---|
| Group # | 1 | 2 | 3 | 1 vs. 2 | 2 vs. 3 | 1 vs. 3 |
| SMNCV (m/s) | 48 ± 7 | 29 ± 6 | 44 ± 6.5 | <0.001 | <0.001 | NS |
| TML (m/s) | 1.3 ± 0.2 | 2.2 ± 0.3 | 1.4 ± 0.3 | <0.001 | <0.001 | NS |
| TSNCV (m/s) | 44 ± 3 | 25 ± 5 | 42 ± 6 | <0.001 | <0.001 | NS |
| Von Frey MA | 1.2 ± 0.2 | 0.6 ± 0.1 | 1.2 ± 0.3 | <0.001 | <0.001 | NS |
| IENFD #/mm | 22 ± 3 | 12 ± 2 | 21 ± 3 | <0.001 | <0.001 | NS |

| Groups | CD-Fed | HFD-Fed | HFD-Fed + NMN | Significance p Values | ||
|---|---|---|---|---|---|---|
| Group # | 1 | 2 | 3 | 1 vs. 2 | 2 vs. 3 | 1 vs. 3 |
| SMNCV (m/s) | 49 ± 6 | 31 ± 6 | 45 ± 6.5 | <0.001 | <0.001 | NS |
| TMl (m/s) | 13 ± 0.3 | 2.4 ± 0.3 | 1.4 ± 0.3 | <0.001 | <0.001 | NS |
| TSNCV (m/s) | 41 ± 3 | 31 ± 5 | 42 ± 6 | <0.001 | <0.001 | NS |
| Von Frey MA | 1.2 ± 0.2 | 0.6 ± 0.1 | 1.2 ± 0.3 | <0.001 | <0.001 | NS |
| IENFD #/mm | 27 ± 6 | 12 ± 4 | 24 ± 4 | <0.001 | <0.001 | NS |
| Parameters | nSIRT1OE-OFF | nSIRT1OE-ON | Significance | |||||
|---|---|---|---|---|---|---|---|---|
| CD (n = 6) | HFD (n = 6) | HFD (n = 6) | HFD + NMN (n = 6) | 1 vs. 2 | 2 vs. 4 | 1 vs. 3 | 3 vs. 4 | |
| Group # | 1 | 2 | 3 | 4 | ||||
| SMNCV (m/s) | 45.2 ± 7.7 | 36.6 ± 8.2 | 46.3 ± 4.7 | 47.5 ± 7.1 | <0.001 | <0.001 | NS | NS |
| TML (m Sec) | 1.5 ± 0.12 | 2.2 ± 0.19 | 1.4 ± 0.14 | 1.4 ± 0.12 | <0.001 | <0.001 | NS | NS |
| TSNCV (m/s) | 33.9 ± 2.5 | 27.3 ± 3.6 | 35.5 ± 4.9 | 35.6 ± 1.4 | <0.001 | <0.001 | NS | NS |
| Von Frey (g) | 11 ± 0.2 | 0.4 ± 0.2 | 1.2 ± 0.12 | 1.0 ± 0.3 | <0.001 | <0.001 | NS | NS |
| Hargreaves (sec) | 8.4 ± 1 | 12 ± 1.9 | 8.6 ± 1 | 7.8 ± 1.4 | <0.001 | <0.001 | NS | NS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandrasekaran, K.; Najimi, N.; Sagi, A.R.; Yarlagadda, S.; Salimian, M.; Arvas, M.I.; Hedayat, A.F.; Kevas, Y.; Kadakia, A.; Kristian, T.; et al. NAD+ Precursors Reverse Experimental Diabetic Neuropathy in Mice. Int. J. Mol. Sci. 2024, 25, 1102. https://doi.org/10.3390/ijms25021102
Chandrasekaran K, Najimi N, Sagi AR, Yarlagadda S, Salimian M, Arvas MI, Hedayat AF, Kevas Y, Kadakia A, Kristian T, et al. NAD+ Precursors Reverse Experimental Diabetic Neuropathy in Mice. International Journal of Molecular Sciences. 2024; 25(2):1102. https://doi.org/10.3390/ijms25021102
Chicago/Turabian StyleChandrasekaran, Krish, Neda Najimi, Avinash R. Sagi, Sushuma Yarlagadda, Mohammad Salimian, Muhammed Ikbal Arvas, Ahmad F. Hedayat, Yanni Kevas, Anand Kadakia, Tibor Kristian, and et al. 2024. "NAD+ Precursors Reverse Experimental Diabetic Neuropathy in Mice" International Journal of Molecular Sciences 25, no. 2: 1102. https://doi.org/10.3390/ijms25021102
APA StyleChandrasekaran, K., Najimi, N., Sagi, A. R., Yarlagadda, S., Salimian, M., Arvas, M. I., Hedayat, A. F., Kevas, Y., Kadakia, A., Kristian, T., & Russell, J. W. (2024). NAD+ Precursors Reverse Experimental Diabetic Neuropathy in Mice. International Journal of Molecular Sciences, 25(2), 1102. https://doi.org/10.3390/ijms25021102