

Performance of a Shotgun Prediction Model for Colorectal Cancer When Using 16S rRNA Sequencing Data

 , , , and
, , , and
Abstract
1. Introduction
2. Results
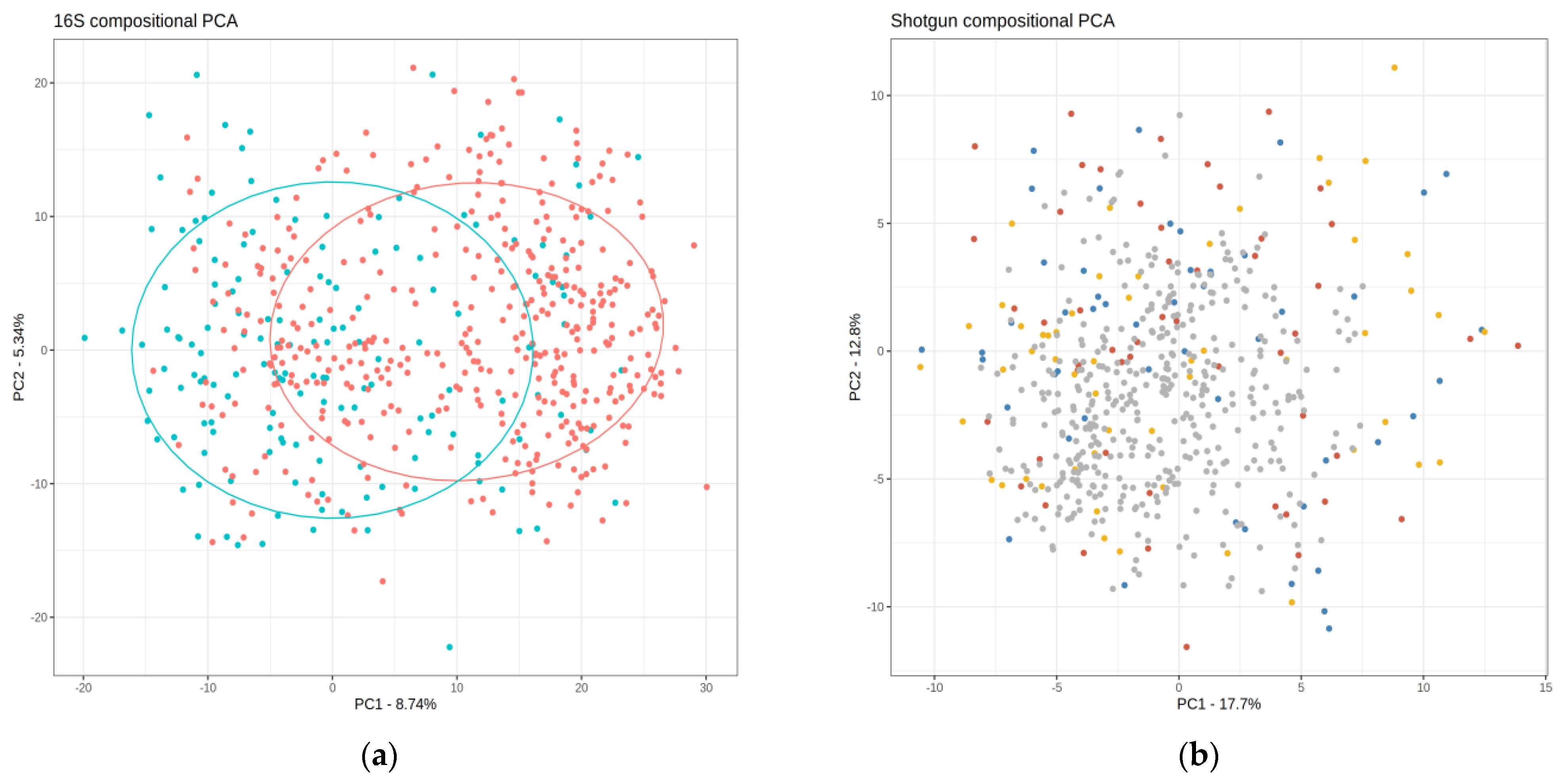
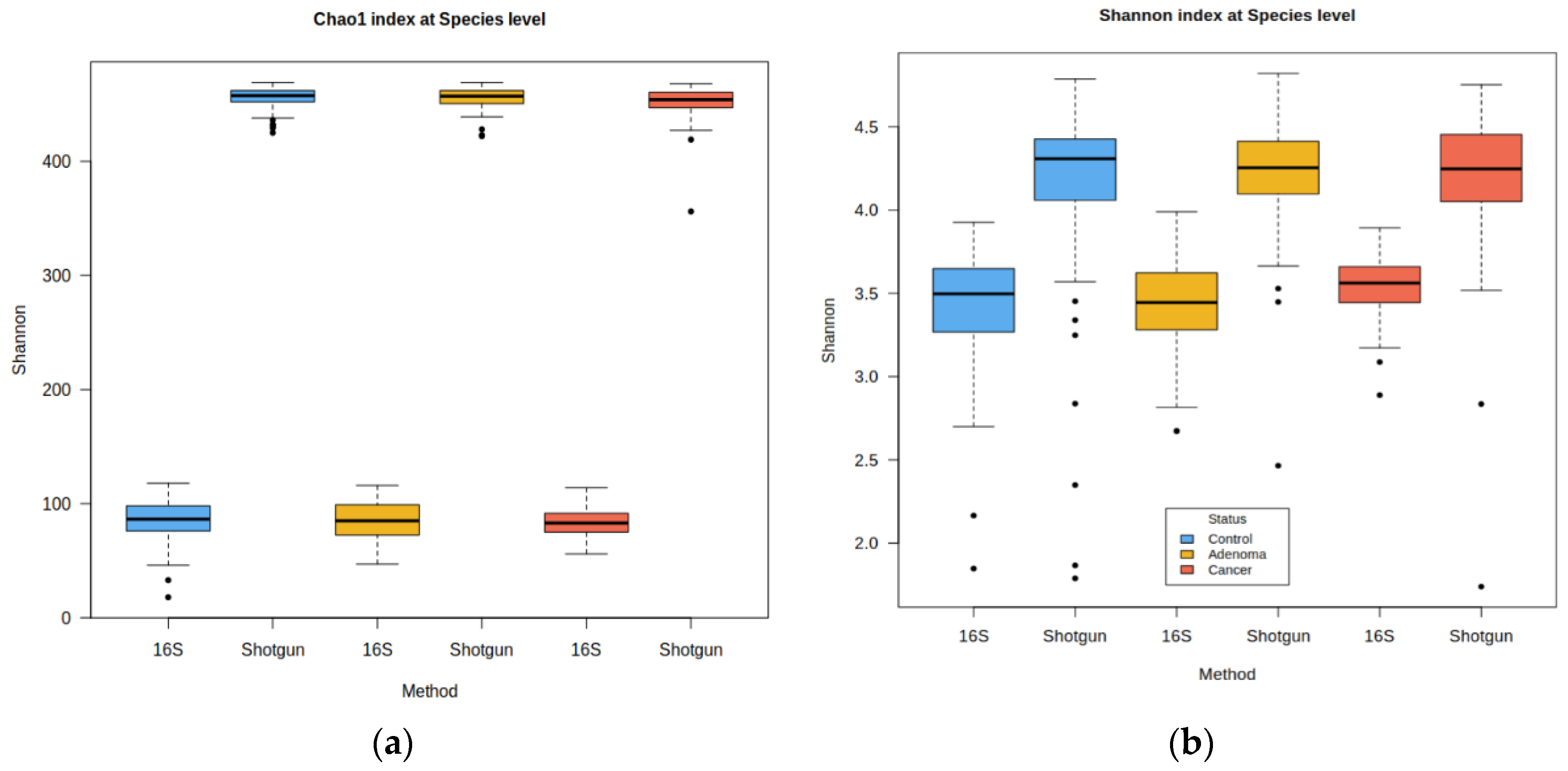
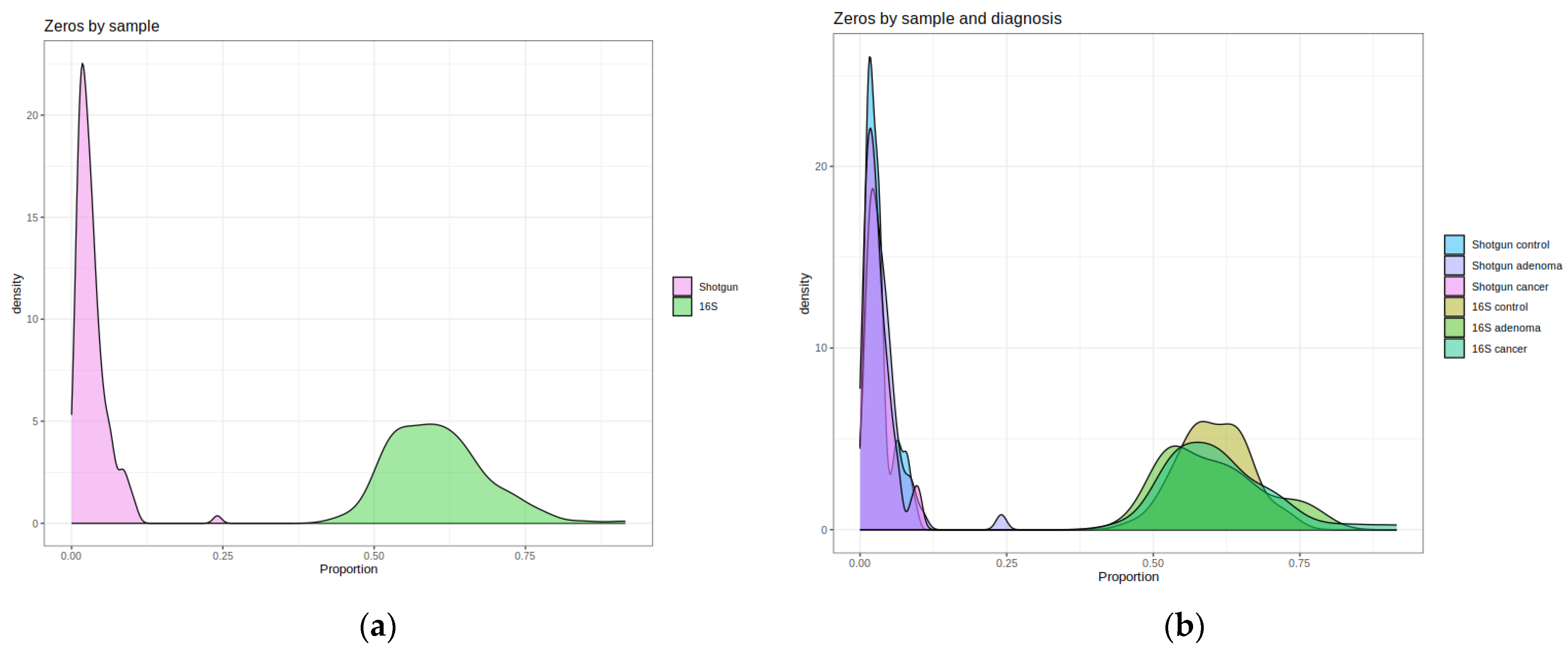
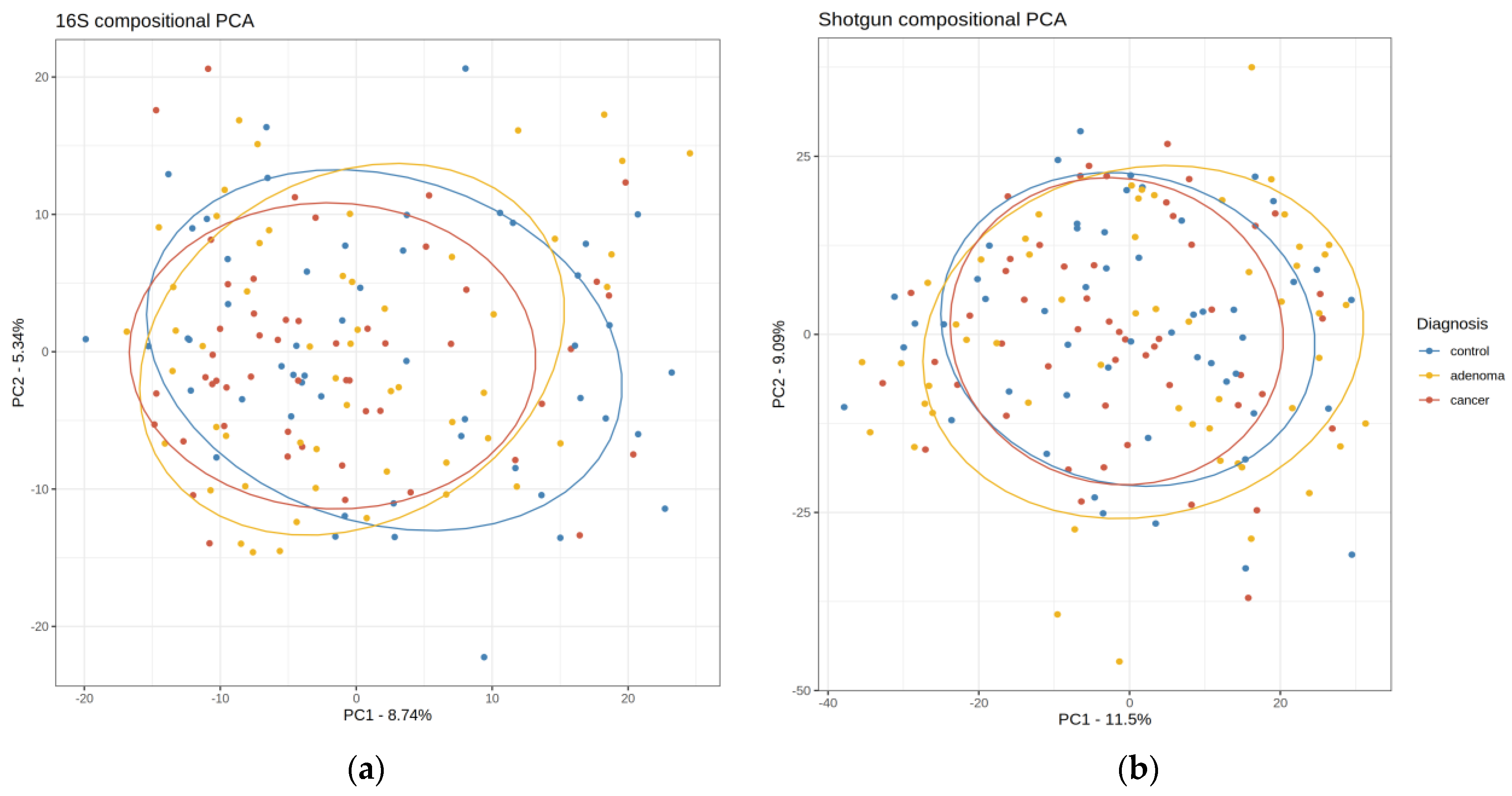
2.1. Description of the Shotgun and 16S Matrices
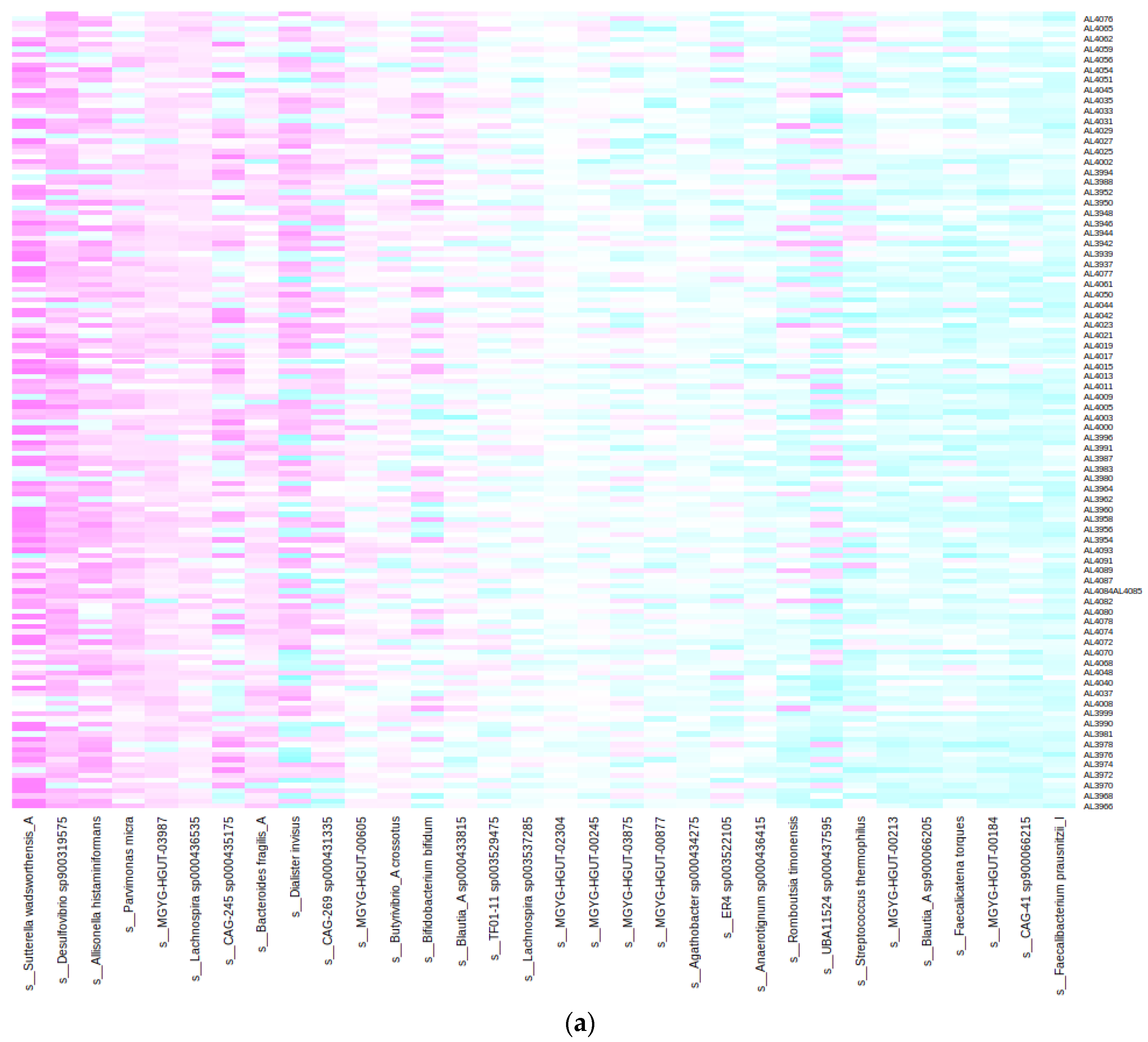
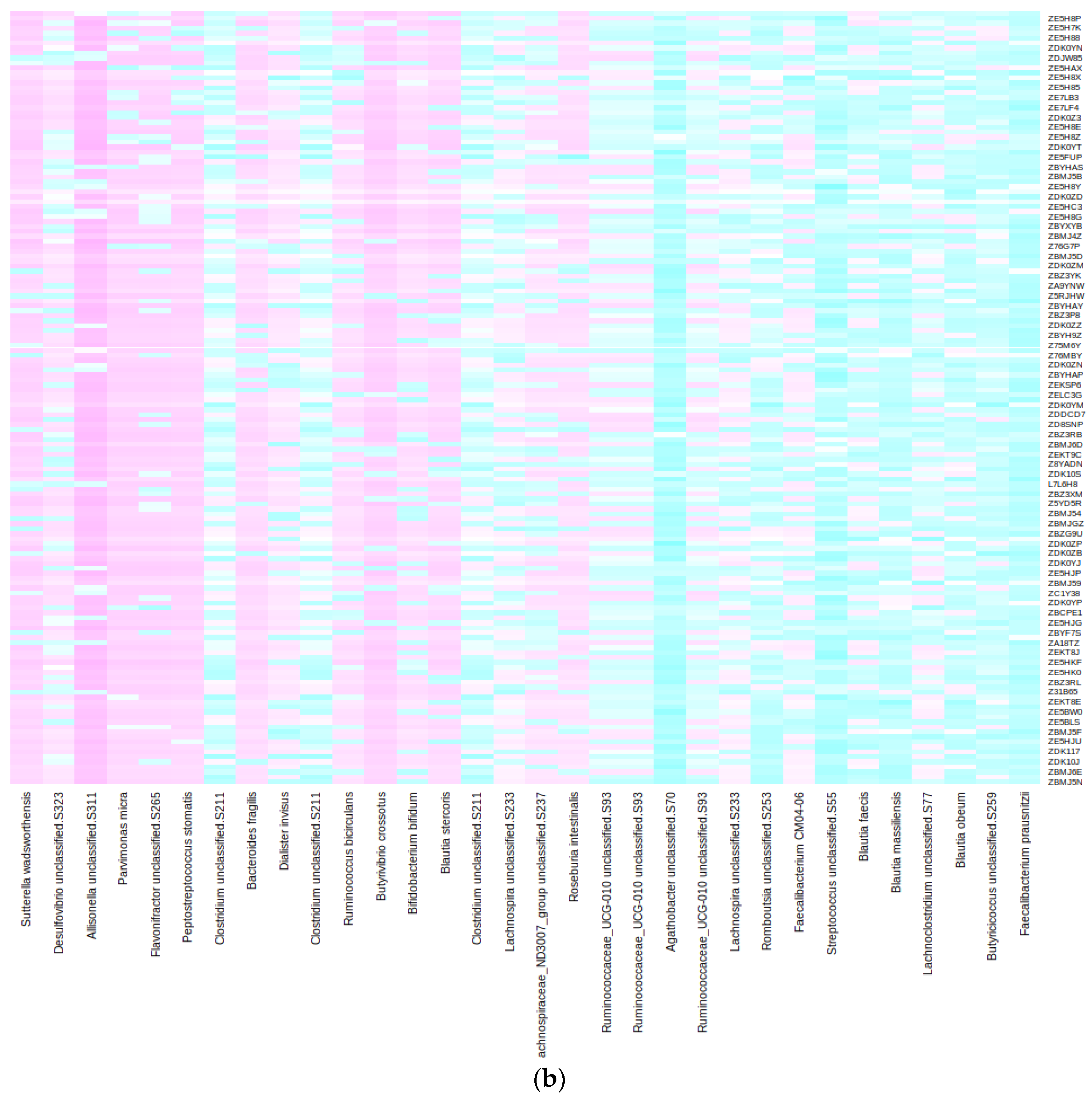
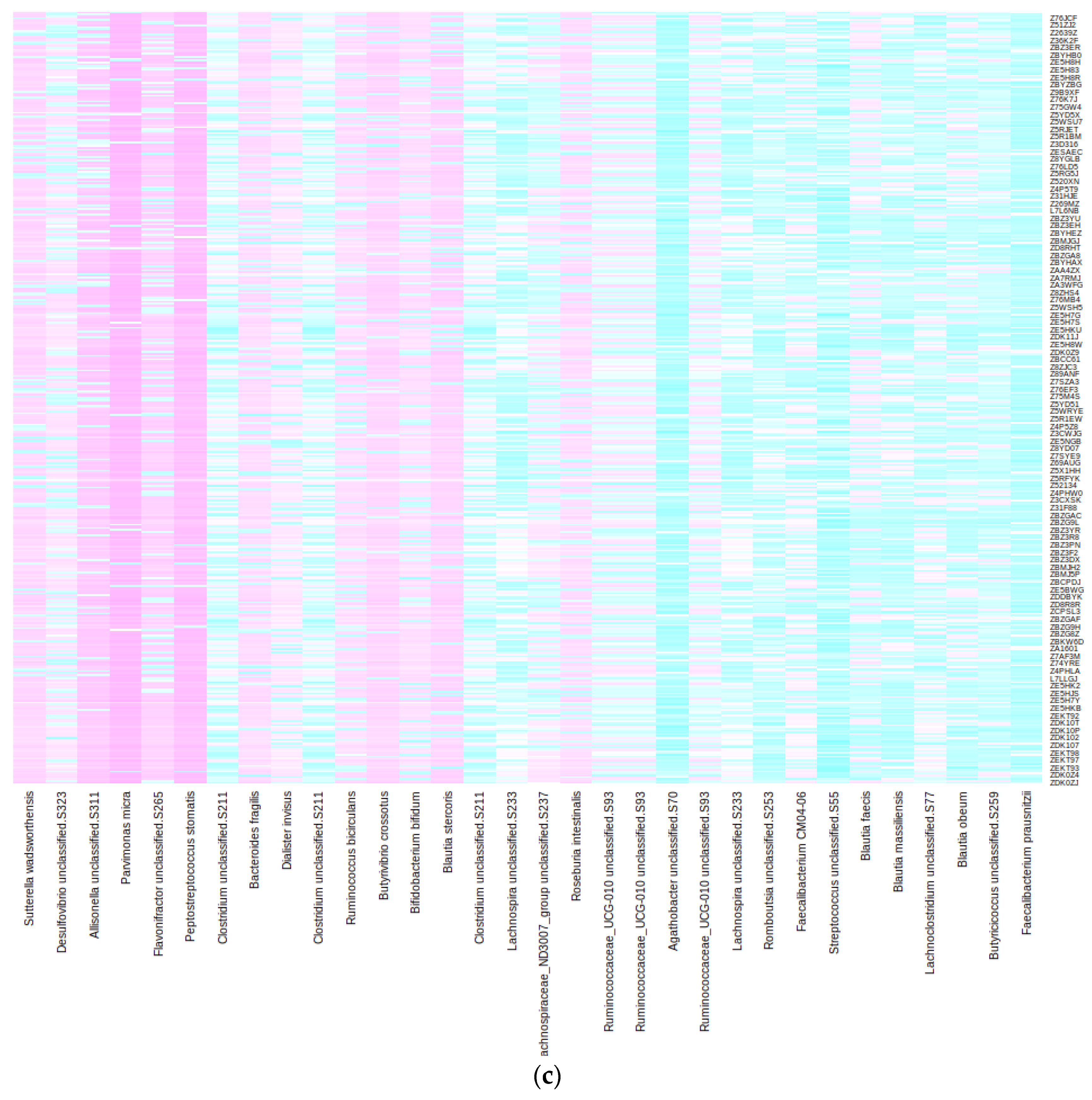
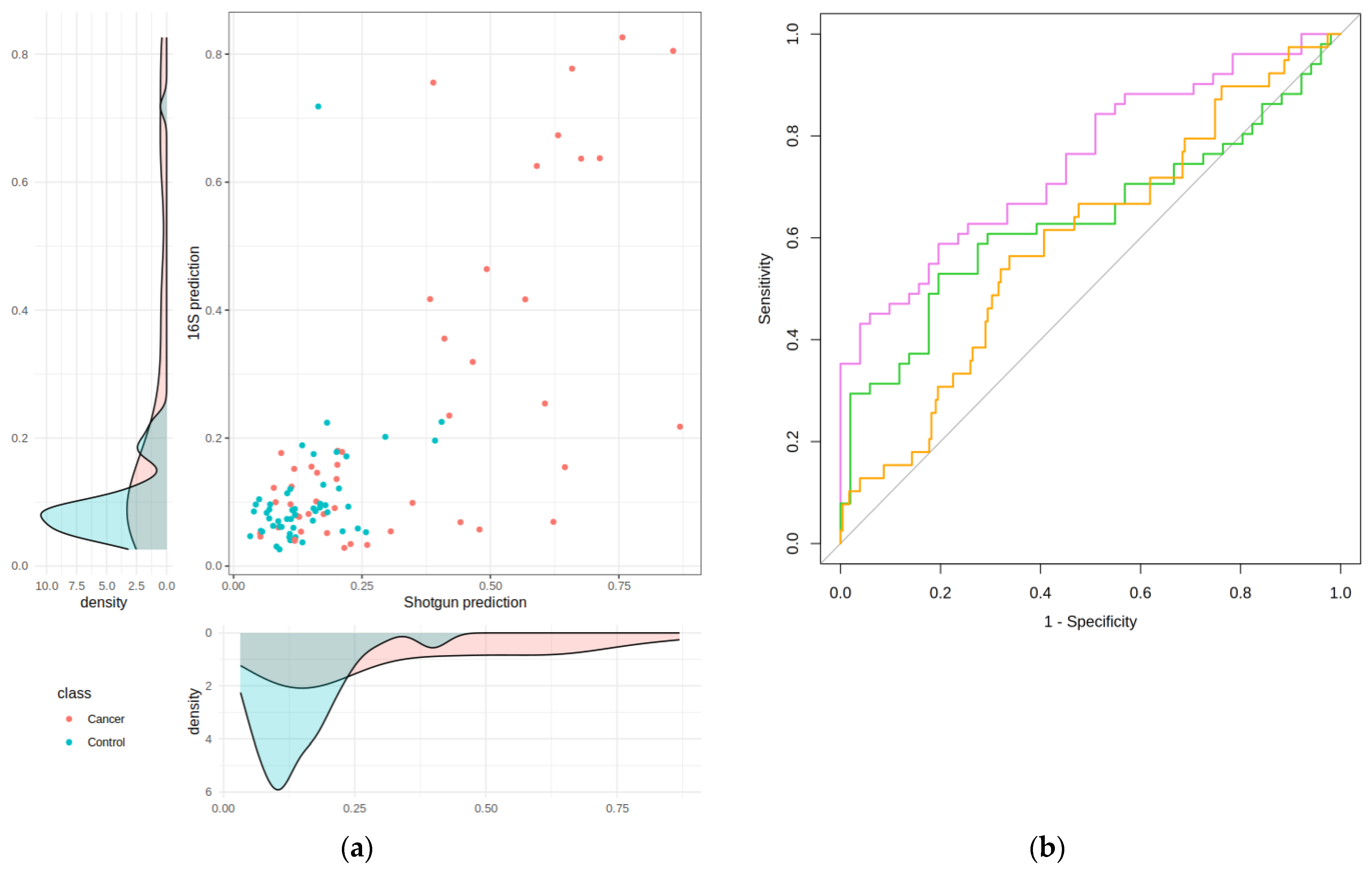
2.2. Taxonomic and Distance-Based Mapping
2.3. Performance of the Mapping in the Validation and Test Sets
3. Discussion
4. Materials and Methods
4.1. Study Population and Design
4.2. DNA Extraction, Sequencing, and Bioinformatics Analysis
4.3. Pre-Processing of the Abundance Matrices
4.4. Description of the Abundance Matrices
4.5. Mapping Shotgun to 16S Abundance Data
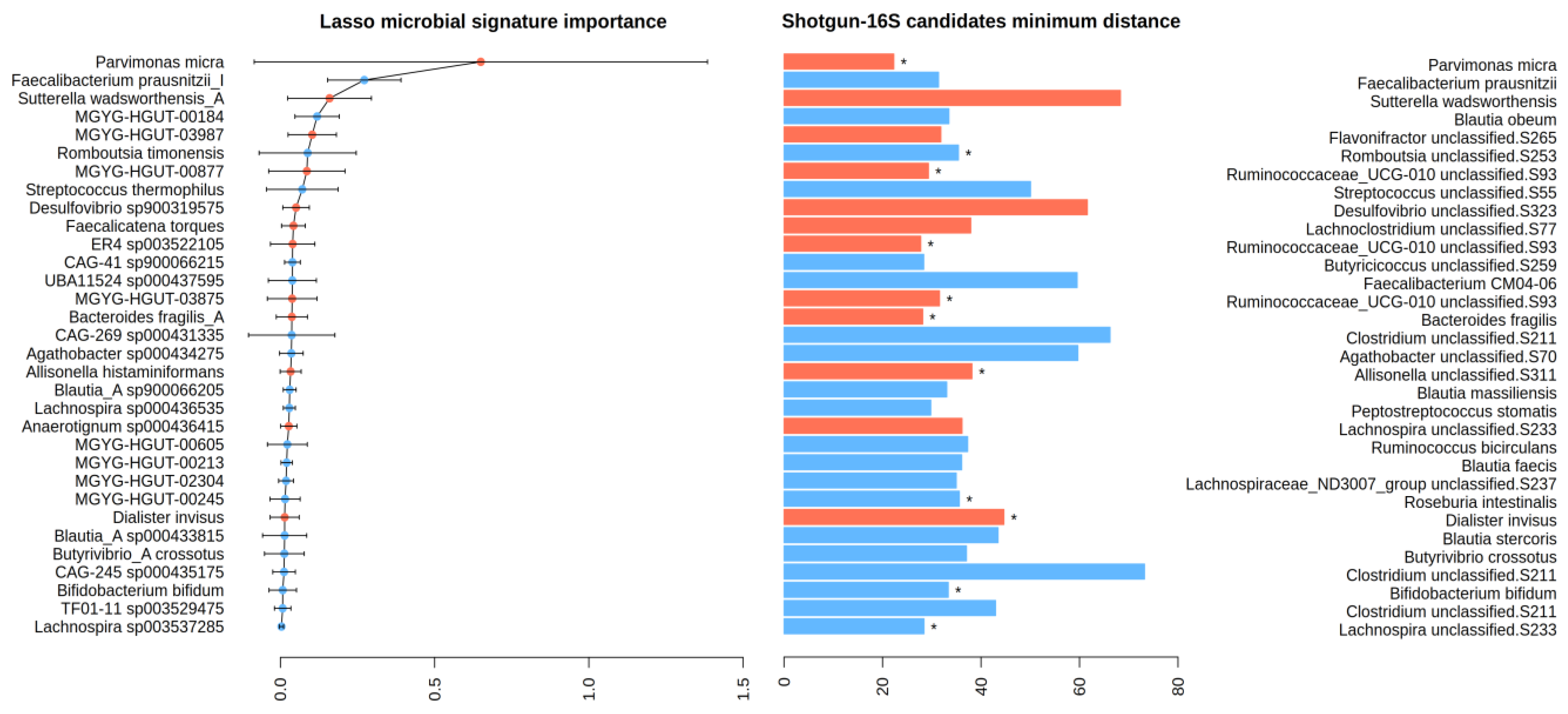
- Taxonomic: Here, we compared the taxonomic trees of shotgun and 16S and searched the latter for the species of interest. In our case, the first step involved checking whether the 32 species in the Lasso model’s signature were among the 16S taxa. If a direct match was not found, the algorithm was extended to higher taxonomic ranks (such as family or order) until a match was identified. This taxonomic strategy is “universal” in that it only requires the availability of taxonomic tables for both 16S and shotgun sequencing, which is generally the case. Notably, the datasets for shotgun and 16S sequencing do not need to be paired; they can originate from different individuals. However, this approach faces several challenges and limitations. Firstly, it requires that taxa lineages be identical in both phylogenetic trees, meaning that the same microorganism should be classified with the same species name and lineage in both datasets. This is often not the case due to rapid updates in microorganism phylogeny and variations in reference databases. As a result, taxa classification must be standardized to the same nomenclature in both shotgun and 16S datasets. Secondly, the lower resolution of 16S sequencing means that many taxa identified at the species level in the shotgun may not be present in the 16S dataset, or only identifiable at the genus level or higher. Consequently, a single taxon identified in shotgun sequencing could correspond to multiple candidate taxa in the 16S dataset. In those cases, we proceeded with the second step.
- Data-driven: For this second step, it is essential to have paired samples, i.e., samples that are sequenced using both shotgun and 16S techniques. Then, some metrics may be devised to select the “closest” 16S taxon to a particular shotgun taxon in a data-driven way. We propose computing the Euclidean distance between relevant shotgun taxa and all 16S taxa using transposed abundance matrices where samples are treated as variables. The chosen 16S taxon is the one with the “closest” clr-transformed abundance profile across all samples to the target shotgun species. The main advantage of this approach is that no information about bacterial phylogeny is needed. The disparate taxonomic resolutions of the shotgun and 16S sequencing techniques are by-passed; in fact, knowing the species or genus name (or even their lineage) is not mandatory. However, if the sample size is limited, this method may face difficulties in obtaining significant separation of 16S taxa and result in wrong mappings due to perceiving noise in the abundance data as meaningful variation. The shotgun model’s performance using these 16S biomarkers may be misleadingly optimistic; thus, using an independent test set (additional samples sequenced with 16S) is advisable to estimate the true performance.
4.6. Performance Evaluation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Colella, M.; Charitos, I.A.; Ballini, A.; Cafiero, C.; Topi, S.; Palmirotta, R.; Santacroce, L. Microbiota Revolution: How Gut Microbes Regulate Our Lives. World J. Gastroenterol. 2023, 29, 4368–4383. [Google Scholar] [CrossRef]
- Wong, C.C.; Yu, J. Gut Microbiota in Colorectal Cancer Development and Therapy. Nat. Rev. Clin. Oncol. 2023, 20, 429–452. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Jovel, J.; Patterson, J.; Wang, W.; Hotte, N.; O’Keefe, S.; Mitchel, T.; Perry, T.; Kao, D.; Mason, A.L.; Madsen, K.L.; et al. Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics. Front. Microbiol. 2016, 7, 459. [Google Scholar] [CrossRef] [PubMed]
- Durazzi, F.; Sala, C.; Castellani, G.; Manfreda, G.; Remondini, D.; De Cesare, A. Comparison between 16S rRNA and Shotgun Sequencing Data for the Taxonomic Characterization of the Gut Microbiota. Sci. Rep. 2021, 11, 3030. [Google Scholar] [CrossRef] [PubMed]
- Mas-Lloret, J.; Obon-Santacana, M.; Ibanez-Sanz, G.; Guino, E.; Pato, M.L.; Rodriguez-Moranta, F.; Mata, A.; Garcia-Rodriguez, A.; Moreno, V.; Pimenoff, V.N. Gut Microbiome Diversity Detected by High-Coverage 16S and Shotgun Sequencing of Paired Stool and Colon Sample. Sci. Data 2020, 7, 92. [Google Scholar] [CrossRef] [PubMed]
- Bars-Cortina, D.; Moratalla-Navarro, F.; García-Serrano, A.; Mach, N.; Riobó-Mayo, L.; Vea-Barbany, J.; Rius-Sansalvador, B.; Murcia, S.; Obón-Santacana, M.; Moreno, V. Improving Species Level-Taxonomic Assignment from 16S rRNA Sequencing Technologies. Curr. Protoc. 2023, 3, e930. [Google Scholar] [CrossRef]
- Marcos-Zambrano, L.J.; Karaduzovic-Hadziabdic, K.; Loncar Turukalo, T.; Przymus, P.; Trajkovik, V.; Aasmets, O.; Berland, M.; Gruca, A.; Hasic, J.; Hron, K.; et al. Applications of Machine Learning in Human Microbiome Studies: A Review on Feature Selection, Biomarker Identification, Disease Prediction and Treatment. Front. Microbiol. 2021, 12, 634511. [Google Scholar] [CrossRef]
- Wirbel, J.; Pyl, P.T.; Kartal, E.; Zych, K.; Kashani, A.; Milanese, A.; Fleck, J.S.; Voigt, A.Y.; Palleja, A.; Ponnudurai, R.; et al. Meta-Analysis of Fecal Metagenomes Reveals Global Microbial Signatures That Are Specific for Colorectal Cancer. Nat. Med. 2019, 25, 679–689. [Google Scholar] [CrossRef]
- Thomas, A.M.; Manghi, P.; Asnicar, F.; Pasolli, E.; Armanini, F.; Zolfo, M.; Beghini, F.; Manara, S.; Karcher, N.; Pozzi, C.; et al. Metagenomic Analysis of Colorectal Cancer Datasets Identifies Cross-Cohort Microbial Diagnostic Signatures and a Link with Choline Degradation. Nat. Med. 2019, 25, 667–678. [Google Scholar] [CrossRef]
- Obón-Santacana, M.; Mas-Lloret, J.; Bars-Cortina, D.; Criado-Mesas, L.; Carreras-Torres, R.; Díez-Villanueva, A.; Moratalla-Navarro, F.; Guinó, E.; Ibáñez-Sanz, G.; Rodríguez-Alonso, L.; et al. Meta-Analysis and Validation of a Colorectal Cancer Risk Prediction Model Using Deep Sequenced Fecal Metagenomes. Cancers 2022, 14, 4214. [Google Scholar] [CrossRef] [PubMed]
- Pepe, M.S.; Cai, T. The Analysis of Placement Values for Evaluating Discriminatory Measures. Biometrics 2004, 60, 528–535. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Jiang, Y.; Balaban, M.; Cantrell, K.; Zhu, Q.; Gonzalez, A.; Morton, J.T.; Nicolaou, G.; Parks, D.H.; Karst, S.M.; et al. Greengenes2 Unifies Microbial Data in a Single Reference Tree. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Böhm, J.; Brunetti, F.; Habermann, N.; et al. Potential of Fecal Microbiota for Early-Stage Detection of Colorectal Cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef]
- Hannigan, G.D.; Duhaime, M.B.; Ruffin, M.T.; Koumpouras, C.C.; Schloss, P.D. Diagnostic Potential and Interactive Dynamics of the Colorectal Cancer Virome. mBio 2018, 9, e02248-18. [Google Scholar] [CrossRef]
- Nagata, N.; Nishijima, S.; Kojima, Y.; Hisada, Y.; Imbe, K.; Miyoshi-Akiyama, T.; Suda, W.; Kimura, M.; Aoki, R.; Sekine, K.; et al. Metagenomic Identification of Microbial Signatures Predicting Pancreatic Cancer From a Multinational Study. Gastroenterology 2022, 163, 222–238. [Google Scholar] [CrossRef]
- Saus, E.; Iraola-Guzmán, S.; Willis, J.R.; Brunet-Vega, A.; Gabaldón, T. Microbiome and Colorectal Cancer: Roles in Carcinogenesis and Clinical Potential. Mol. Asp. Med. 2019, 69, 93–106. [Google Scholar] [CrossRef]
- Castells, A.; Andreu, M.; Binefa, G.; Fité, A.; Font, R.; Espinàs, J.A. Postpolypectomy Surveillance in Patients with Adenomas and Serrated Lesions: A Proposal for Risk Stratification in the Context of Organized Colorectal Cancer-Screening Programs. Endoscopy 2015, 47, 86–87. [Google Scholar] [CrossRef]
- Almeida, A.; Nayfach, S.; Boland, M.; Strozzi, F.; Beracochea, M.; Shi, Z.J.; Pollard, K.S.; Sakharova, E.; Parks, D.H.; Hugenholtz, P.; et al. A Unified Catalog of 204,938 Reference Genomes from the Human Gut Microbiome. Nat. Biotechnol. 2021, 39, 105–114. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Drost, H.-G.; Gabel, A.; Liu, J.; Quint, M.; Grosse, I. myTAI: Evolutionary Transcriptomics with R. Bioinformatics 2018, 34, 1589–1590. [Google Scholar] [CrossRef]
- Palarea-Albaladejo, J.; Martín-Fernández, J.A. zCompositions—R Package for Multivariate Imputation of Left-Censored Data under a Compositional Approach. Chemom. Intell. Lab. Syst. 2015, 143, 85–96. [Google Scholar] [CrossRef]
- Oksanen, J.; Simpson, G.L.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. Vegan: Community Ecology Package. Available online: https://github.com/vegandevs/vegan (accessed on 29 November 2023).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Genus | Family | Order | |
|---|---|---|---|---|
| Bacterial signature (original Lasso model [11]) | Bacteroides fragilis A | Agathobacter sp000434275 | Anaerotignum sp000436415 | Lachnospira sp000436535 |
| Bifidobacterium bifidum | Allisonella histaminiformans | CAG-41 sp900066215 | MGYG-HGUT-03875 | |
| Butyrivibrio A crossotus | Blautia A sp000433815 | ER4 sp003522105 | ||
| Dialister invisus | Blautia A sp900066205 | Faecalicatena torques | ||
| Faecalibacterium prausnitzii I | CAG-245 sp000435175 | MGYG-HGUT-00877 | ||
| Parvimonas micra | CAG-269 sp000431335 | MGYG-HGUT-02304 | ||
| Sutterella wadsworthensis A | Desulfovibrio sp900319575 | MGYG-HGUT-03987 | ||
| Lachnospira sp003537285 | TF01-11 sp003529475 | |||
| MGYG-HGUT-00184 | ||||
| MGYG-HGUT-00213 | ||||
| MGYG-HGUT-00245 | ||||
| MGYG-HGUT-00605 | ||||
| Romboutsia timonensis | ||||
| Streptococcus thermophilus | ||||
| N (% of total) | 7 (21.9%) | 15 (46.9%) | 8 (25%) | 2 (6.2%) |
| Median (Range) number of candidates | 1 (0) | 6 (1–68) | 47 (14–68) | 87.5 (2–173) |
| Diagnosis | N | Woman (%) | Age Median (IQR) | BMI Median (IQR) | |
|---|---|---|---|---|---|
| Validation | Controls | 51 | 47.1% | 57 (7) | 26.2 (4.4) |
| High-risk lesions | 54 | 33.3% | 60 (9.5) | 28.1 (5.6) | |
| CRC cases | 51 | 27.5% | 65 (13.5) | 26.9 (4.3) | |
| Total | 156 | 35.9% | 60 (7.9) | 27.1 (4.2) | |
| Test | Controls | 231 | 64.1% | 60 (10) | 27.1 (6.7) |
| High-risk lesions | 146 | 35.6% | 63 (7) | 27.7 (4.9) | |
| CRC cases | 39 | 25.6% | 66 (6.5) | 27.5 (5.8) | |
| Total | 416 | 50.5% | 62 (6.0) | 27.4 (5.1) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramon, E.; Obón-Santacana, M.; Khannous-Lleiffe, O.; Saus, E.; Gabaldón, T.; Guinó, E.; Bars-Cortina, D.; Ibáñez-Sanz, G.; Rodríguez-Alonso, L.; Mata, A.; et al. Performance of a Shotgun Prediction Model for Colorectal Cancer When Using 16S rRNA Sequencing Data. Int. J. Mol. Sci. 2024, 25, 1181. https://doi.org/10.3390/ijms25021181
Ramon E, Obón-Santacana M, Khannous-Lleiffe O, Saus E, Gabaldón T, Guinó E, Bars-Cortina D, Ibáñez-Sanz G, Rodríguez-Alonso L, Mata A, et al. Performance of a Shotgun Prediction Model for Colorectal Cancer When Using 16S rRNA Sequencing Data. International Journal of Molecular Sciences. 2024; 25(2):1181. https://doi.org/10.3390/ijms25021181
Chicago/Turabian StyleRamon, Elies, Mireia Obón-Santacana, Olfat Khannous-Lleiffe, Ester Saus, Toni Gabaldón, Elisabet Guinó, David Bars-Cortina, Gemma Ibáñez-Sanz, Lorena Rodríguez-Alonso, Alfredo Mata, and et al. 2024. "Performance of a Shotgun Prediction Model for Colorectal Cancer When Using 16S rRNA Sequencing Data" International Journal of Molecular Sciences 25, no. 2: 1181. https://doi.org/10.3390/ijms25021181
APA StyleRamon, E., Obón-Santacana, M., Khannous-Lleiffe, O., Saus, E., Gabaldón, T., Guinó, E., Bars-Cortina, D., Ibáñez-Sanz, G., Rodríguez-Alonso, L., Mata, A., García-Rodríguez, A., & Moreno, V. (2024). Performance of a Shotgun Prediction Model for Colorectal Cancer When Using 16S rRNA Sequencing Data. International Journal of Molecular Sciences, 25(2), 1181. https://doi.org/10.3390/ijms25021181