
Cancer Nano-Immunotherapy: The Novel and Promising Weapon to Fight Cancer

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Immunotherapy: A Strategy to Enhance Anticancer Efficacy
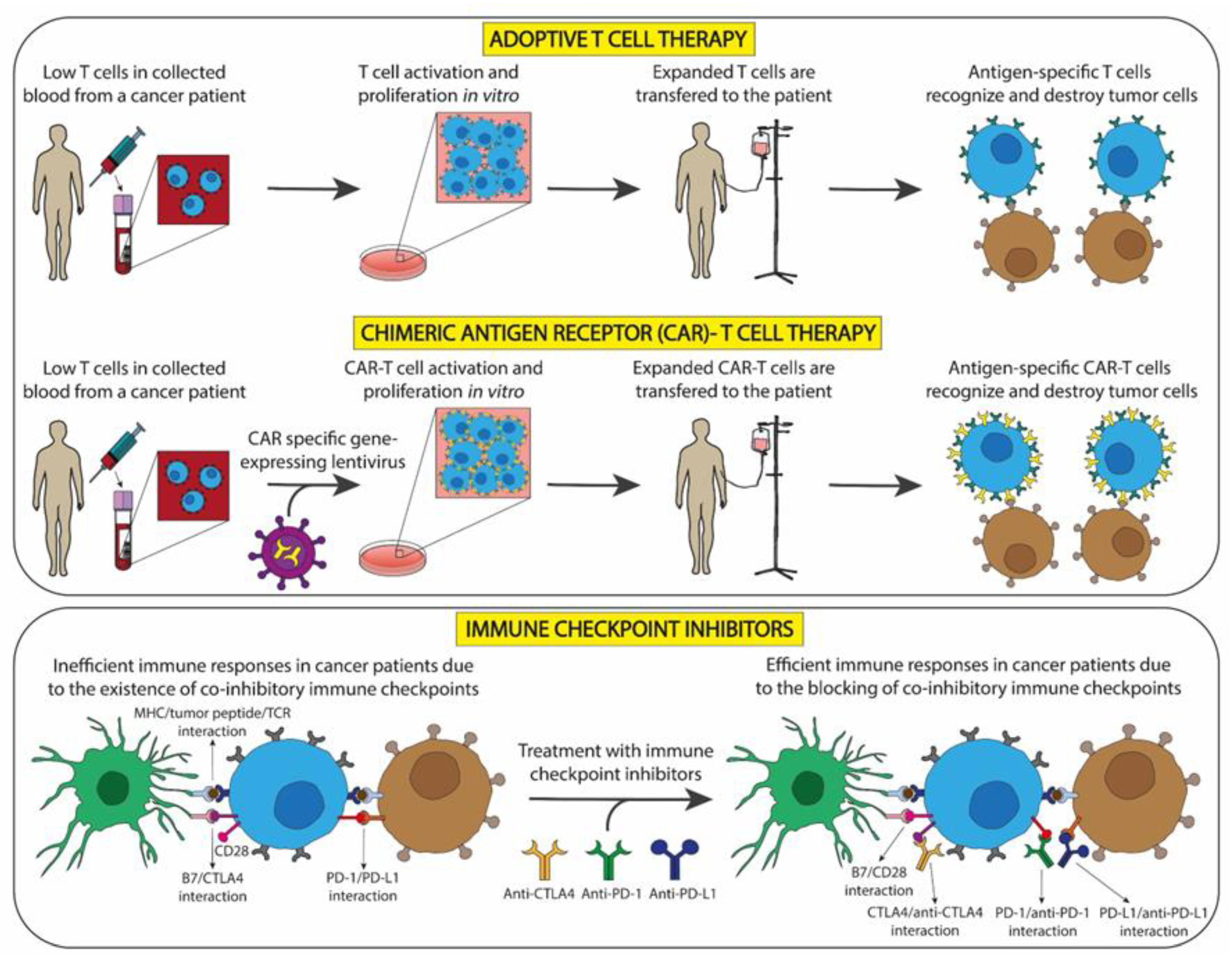
2.1. Cellular Immunotherapy for Tumors
2.1.1. Immunotherapy with Non-Genetically Modified T Cells
2.1.2. Immunotherapy with Genetically Modified T Cells
3. Nanomedicine in Cancer
3.1. Nanocarriers for Cancer Treatment
3.1.1. Inorganic Materials
3.1.2. Organic Materials
3.1.3. Biological Materials
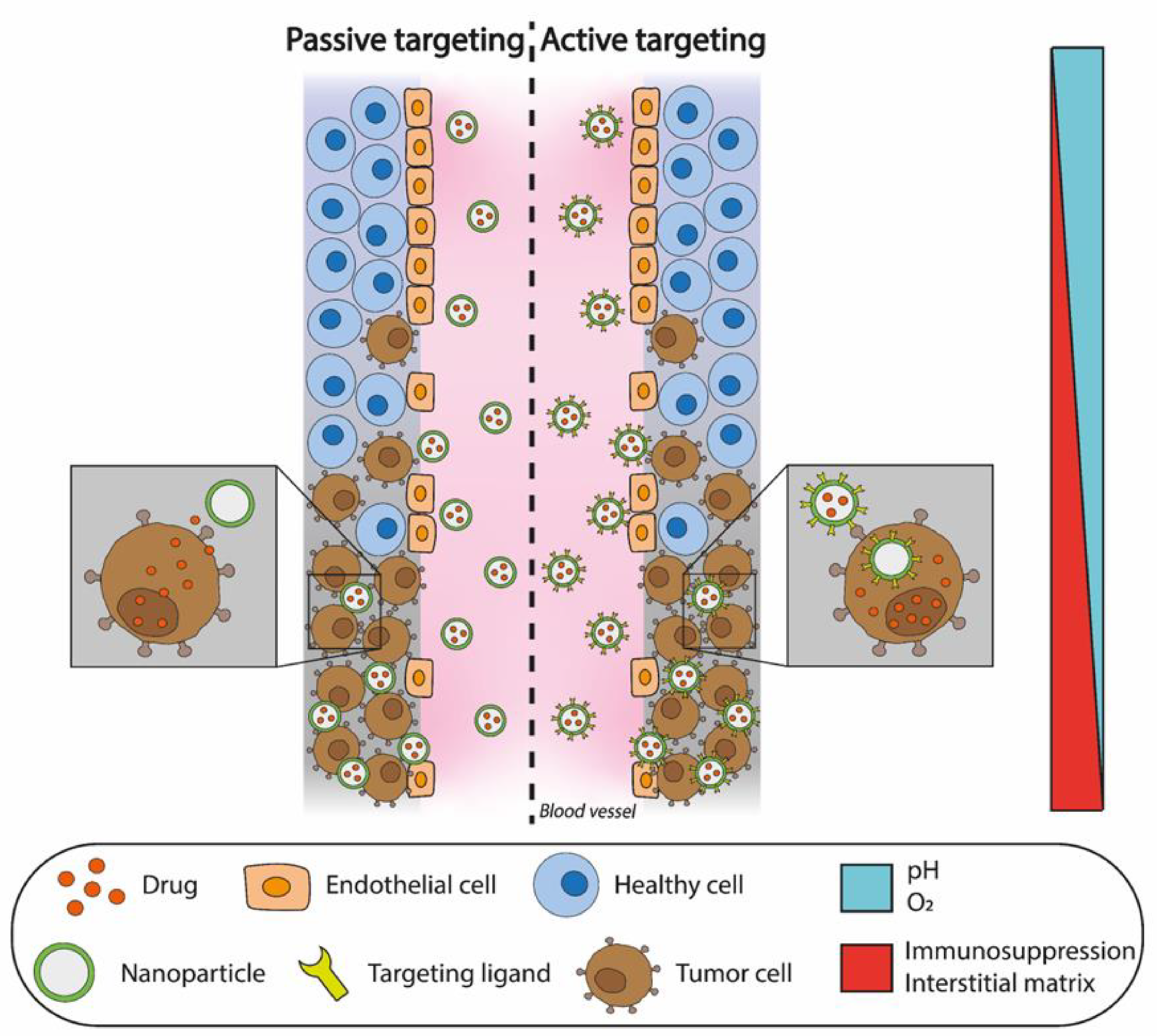
3.2. Targeting Strategies
3.2.1. Passive Targeting for Cancer Treatment
3.2.2. Active Targeting for Cancer Treatment
3.2.3. Smart Nanomedicines
4. Nano-Immunotherapy: A Reality in Cancer Treatment
- (a).
- Nano-immunotherapy against tumor cells: CT drugs (e.g., doxorubicin, oxaliplatin, and paclitaxel), RT, photodynamic and photothermal therapies, as well as other physical stimuli are able to induce immunogenic cell death [127,128,129,130]. Examples of this kind of treatment are:
- -
- Oxaliplatin: This CT drug encapsulated in monomethoxy-poly(ethylene glycol)-poly (d,l-lactide-co-glycolide (PLGA-mPEG) NPs (OXA-NPs) triggered more damage-associated molecular pattern (DAMP) release and induced a stronger DC and T lymphocyte infiltration and activation in pancreatic tumor cells in vitro than oxaliplatin. Moreover, OXA-NPs inhibited tumor growth in immunocompetent mice, exhibiting stronger therapeutic effects than the OXA group [131]. Similarly, magnetic NPs, as a delivery system of oxaliplatin, reinforced immunogenic cell death induction of that CT drug [132]. Additionally, the combination of oxaliplatin and PD-L1 trap fusion protein using liposomal NPs inhibited tumor growth in an orthotopic colorectal mice model and showed T cell activation [133].
- -
- Paclitaxel: 1-NP, a paclitaxel NP, exhibited much lower cytotoxicity to macrophages than did PTX at the same high PTX concentration and maintained the capacity to stimulate macrophages to polarize into M1 and inhibit their M2 differentiation both on phenotypical and functional levels in a dose-dependent in vitro and in melanoma tumor-bearing mouse model [134]. Moreover, paclitaxel and SP-LPS, a Toll-like receptor (TLR) 4 agonist, were encapsulated into a bio-polymer, and their combination increased chemotherapeutic and immunotherapeutic activity both in vitro and in vivo as compared to the paclitaxel-treated group in melanoma [135].
- -
- Doxorubicin: Doxorubicin was integrated into mesoporous silica NPs (DOX@HIMSNs) for a systemic treatment of triple negative BC (TNBC). DOX@HIMSNs enhanced antitumor efficacy and induced DC maturation and antitumor cytokine release as compared with doxorubicin [136]. Gao et al. developed an NP in which doxorubicin was conjugated with anionic polymer hyaluronic acid via a tumor overexpressed matrix metalloproteinase sensitive peptide [137]. This NP combined with anti-PD-1 led to better results in vivo, improving the antitumor efficiency [137]. Moreover, doxorubicin and recombinant human IL-2 were co-delivered in hydrophilic cationic polymer NPs [138]. This combination delayed tumor growth and increased tumor-infiltrated cytotoxic T lymphocytes in a hepatocellular carcinoma model [138].
- -
- Radiotherapy: The combination of localized radiation with NBTXR3 (a radio-enhancing NP) and systemic anti-PD1 treatment in a lung cancer mouse model was explored. This combination significantly delayed tumor growth in anti-PD1-resistant and -sensitive metastatic lung cancer cells, which could open the possibility of its use to treat patients with metastatic lung cancer regardless of their sensitivity (or resistance) to immunotherapies [139]. Additionally, PLGA-R837@Cat NP is an NP based on poly (lactic-co-glycolic) acid (PLGA) loading with hydrophobic imiquimod (R837), a TLR-7 agonist, and water-soluble catalase (Cat) [140]. These NPs have enhanced RT efficacy, inhibited tumor metastases, and induced antitumor immune responses [140].
- -
- Photothermal therapy (PTT): In neuroblastoma, Cano-Mejia et al. developed a combination of nano-immunotherapy and PTT, which resulted in CpG oligodeoxynucleotide-coated Prussian blue (CpG-PB) NPs. PPT triggered tumor cell death and released TAA. Immunogenicity increased in vitro, and a high survival and a complete tumor regression of 70% at 60 days in treated mice was observed with this combination in a syngeneic neuroblastoma mouse model [141]. Subsequently, the same authors used this combination with anti-CTLA-4 in neuroblastoma, obtaining similar results. Additionally, an alteration of the surface levels of co-stimulatory, antigen-presenting, and co-inhibitory molecules on neuroblastoma cell lines resulted with the use of an anti-CTLA-4 therapy [141]. In BC, ovalbumin-coated PEGylated MnFe2O4 NPs loaded with R837 immunoadjuvant (R837-OVA-PEG-MnFe2O4) NPs were used together with PTT. These NPs downregulated M2-associated cytokines, tumor growth was inhibited, and lung metastasis was prevented [142].
- (b).
- Nano-immunotherapy against the TME: NPs can be used to modulate TME by inhibiting immunosuppression or by increasing the immune system activation.
- -
- Inhibition of immunosuppression: The inhibition of immunosuppression in cancer has been evaluated with different nano-formulations, particularly affecting immune cells. For example:
- Regulatory T cells (Treg): PEG-modified single-walled carbon nanotubes (PEG-SWCNTs) against specific receptors of Tregs, which are highly found within the TME, were designed by Sacchetti et al. [143]. In vivo assays showed that Tregs, residing in the melanoma TME, uptake PEG-SWCNTs more efficiently than intratumor non-Tregs or splenic Tregs [143].
- Macrophages: In vitro co-cultures with adenocarcinoma cell lines, macrophages, and ferumoxytol (an iron oxide NP) increased pro-inflammatory Th1-type responses. In vivo, ferumoxytol reduced tumor growth of subcutaneous adenocarcinomas in mice and increased pro-inflammatory M1 macrophages in tumor tissues [144]. Lastly, Glycocalyx-mimicking NPs (GNPs) self-assembled into amphiphilic copolymers to target tumor-associated macrophages (TAMs) in Lewis lung cancer were evaluated [145]. GNPs were internalized by TAMs via lectin receptors, which resulted in a phenotypic change towards M1 macrophages. Additionally, GNPs reduced tumor growth by depleting Tregs, and its combination with anti-PD-L1 increased IL-12 levels, decreased IL-10, arginase, and C-C motif chemokine ligand (CCL) 22 in tumor-induced mice [145].
- MDSCs: MDSCs are predominantly polarized into M2 in BC. However, two cationic polymers (cationic dextran and polyethyleneimine) were able to repolarize M2-MDSCs into the M1-type. In a BC mouse model, intratumoral administration of these NPs reduced both tumor growth and the percentage of tumor-induced MDSCs in blood, spleen, tumors, and bone marrow [146]. Moreover, both cationic polymers promoted the proliferation and activity of CD4+ and CD8+ T cells in vivo, which showed that restoring T lymphocyte function in the tumor environment is critical because it effectively induces massive necrosis of tumor cells [146]. The reduction of the MDSC activity using synthetic high-density lipoprotein-like NPs (HDL NP) specifically binding the scavenger receptor type B-1 (SCARB1) has also been studied. These NPs reduced tumor growth and metastatic tumor burden, as well as increased CD8+ T cells and reduced Tregs in the metastatic TME in a melanoma mouse model [147].
- -
- Activation of immune responses: Anti-immunosuppressive factors can be delivered by NPs to the TME in order to enhance the immune system activation. Thus, TGF-β inhibitor and IL-2 were co-delivered to the TME by nanoscale liposomal polymeric gels (nLGs) [148]. An increase in the number and activity of NK cells and CD8+ T cells was observed together with a reduction in tumor growth in a melanoma mouse model [148]. Similarly, Xu et al. used liposome-protamine-hyaluronic acid (LPH) NPs loaded with TGF-β small interfering (si)RNA promoting TGF-β downregulation in melanoma. This inhibition increased tumor-infiltrating CD8+ T cells, decreased Tregs, and inhibited tumor growth [149]. Inhibitors of metabolic enzymes have also been evaluated due to their participation in the immune modulation. NPs loaded with the Indoleamine 2,3-dioxygenase 1 (IDO) inhibitor 1-MT modified with hyaluronic acid and with an anti-PD-1 antibody showed an increment of T cell immunity and an immunosuppression reduction, which resulted in a beneficial antitumor effect in melanoma-bearing mice [150]. Furthermore, IDO inhibitor and oxaliplatin were conjugated in mesoporous silica NPs. This combination caused tumor reduction or eradication, as well as an increase in the recruitment of CD8+ T cells along with a reduction in Foxp3+ T cells in a pancreatic ductal adenocarcinoma mouse model [151].
4.1. Nano-Immunotherapy Clinical Trials in Cancer
4.1.1. Immune Checkpoint Inhibitors
4.1.2. Other Combinatorial Therapies
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Garcia-Dominguez, D.J.; Hontecillas-Prieto, L.; Palazon-Carrion, N.; Jimenez-Cortegana, C.; Sanchez-Margalet, V.; de la Cruz-Merino, L. Tumor Immune Microenvironment in Lymphoma: Focus on Epigenetics. Cancers 2022, 14, 1469. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, J.; Esteller, M. Cancer epigenomics: Beyond genomics. Curr. Opin. Genet. Dev. 2012, 22, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Arruebo, M.; Vilaboa, N.; Saez-Gutierrez, B.; Lambea, J.; Tres, A.; Valladares, M.; Gonzalez-Fernandez, A. Assessment of the evolution of cancer treatment therapies. Cancers 2011, 3, 3279–3330. [Google Scholar] [CrossRef] [PubMed]
- Beil, D.R.; Wein, L.M. Sequencing surgery, radiotherapy and chemotherapy: Insights from a mathematical analysis. Breast Cancer Res. Treat. 2002, 74, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, X.; Chen, D.; Yu, J. Radiotherapy combined with immunotherapy: The dawn of cancer treatment. Signal Transduct. Target. Ther. 2022, 7, 258. [Google Scholar] [CrossRef] [PubMed]
- Pucci, C.; Martinelli, C.; Ciofani, G. Innovative approaches for cancer treatment: Current perspectives and new challenges. Ecancermedicalscience 2019, 13, 961. [Google Scholar] [CrossRef] [PubMed]
- Urruticoechea, A.; Alemany, R.; Balart, J.; Villanueva, A.; Vinals, F.; Capella, G. Recent advances in cancer therapy: An overview. Curr. Pharm. Des. 2010, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Huang, D.; Peppas, N.A. Advanced engineered nanoparticulate platforms to address key biological barriers for delivering chemotherapeutic agents to target sites. Adv. Drug Deliv. Rev. 2020, 167, 170–188. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Wang, Y.; Wargo, J.A.; Lang, F.F.; Kim, B.Y.S. Considerations for designing preclinical cancer immune nanomedicine studies. Nat. Nanotechnol. 2021, 16, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A. Cytokines in Cancer Immunotherapy. Cold Spring Harb. Perspect. Biol. 2018, 10, a028472. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, Z.; Gu, Z. Bioinspired and Biomimetic Nanomedicines. Acc. Chem. Res. 2019, 52, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Cellular and Molecular Immunology, 10th ed.; Elsevier: Amsterdam, The Netherlands, 2022. [Google Scholar]
- Smyth, M.J.; Dunn, G.P.; Schreiber, R.D. Cancer immunosurveillance and immunoediting: The roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv. Immunol. 2006, 90, 1–50. [Google Scholar] [CrossRef] [PubMed]
- Taefehshokr, N.; Baradaran, B.; Baghbanzadeh, A.; Taefehshokr, S. Promising approaches in cancer immunotherapy. Immunobiology 2020, 225, 151875. [Google Scholar] [CrossRef]
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Forget, M.A.; Chacon, J.; Bernatchez, C.; Haymaker, C.; Chen, J.Q.; Hwu, P.; Radvanyi, L.G. Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: Current status and future outlook. Cancer J. 2012, 18, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.; Donia, M.; Ellebaek, E.; Borch, T.H.; Kongsted, P.; Iversen, T.Z.; Holmich, L.R.; Hendel, H.W.; Met, O.; Andersen, M.H.; et al. Long-Lasting Complete Responses in Patients with Metastatic Melanoma after Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes and an Attenuated IL2 Regimen. Clin. Cancer Res. 2016, 22, 3734–3745. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.U.; Malekzadeh, P.; Shelton, T.; White, D.E.; Butman, J.A.; Yang, J.C.; Kammula, U.S.; Goff, S.L.; Rosenberg, S.A.; Sherry, R.M. Outcomes of Adoptive Cell Transfer with Tumor-infiltrating Lymphocytes for Metastatic Melanoma Patients with and without Brain Metastases. J. Immunother. 2018, 41, 241–247. [Google Scholar] [CrossRef]
- Hamoud, B.H.; Sima, R.M.; Vacaroiu, I.A.; Georgescu, M.T.; Bobirca, A.; Gaube, A.; Bobirca, F.; Georgescu, D.E. The Evolving Landscape of Immunotherapy in Uterine Cancer: A Comprehensive Review. Life 2023, 13, 1502. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Wunderlich, J.R.; Shelton, T.E.; Even, J.; Rosenberg, S.A. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J. Immunother. 2003, 26, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Chacon, J.A.; Wu, R.C.; Sukhumalchandra, P.; Molldrem, J.J.; Sarnaik, A.; Pilon-Thomas, S.; Weber, J.; Hwu, P.; Radvanyi, L. Co-stimulation through 4-1BB/CD137 improves the expansion and function of CD8(+) melanoma tumor-infiltrating lymphocytes for adoptive T-cell therapy. PLoS ONE 2013, 8, e60031. [Google Scholar] [CrossRef]
- Ye, Q.; Song, D.G.; Poussin, M.; Yamamoto, T.; Best, A.; Li, C.; Coukos, G.; Powell, D.J., Jr. CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin. Cancer Res. 2014, 20, 44–55. [Google Scholar] [CrossRef]
- Khammari, A.; Knol, A.C.; Nguyen, J.M.; Bossard, C.; Denis, M.G.; Pandolfino, M.C.; Quereux, G.; Bercegeay, S.; Dreno, B. Adoptive TIL transfer in the adjuvant setting for melanoma: Long-term patient survival. J. Immunol. Res. 2014, 2014, 186212. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.K. The Surprising Story of IL-2: From Experimental Models to Clinical Application. Am. J. Pathol. 2020, 190, 1776–1781. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, M.T.; Trifanescu, O.G.; Serbanescu, G.L.; Mitrica, R.I.; Georgescu, D.E.; Mihaila, R.I.; Neagu, A.; Gaube, A.; Botezatu, C.; Mastalier, B.S.M. Navigating a Complex Intersection: Immunotherapy and Radiotherapy Synergy in Squamous Cell Carcinoma of the Skin—A Comprehensive Literature Review. Cosmetics 2023, 10, 165. [Google Scholar] [CrossRef]
- Deniger, D.C.; Kwong, M.L.; Pasetto, A.; Dudley, M.E.; Wunderlich, J.R.; Langhan, M.M.; Lee, C.R.; Rosenberg, S.A. A Pilot Trial of the Combination of Vemurafenib with Adoptive Cell Therapy in Patients with Metastatic Melanoma. Clin. Cancer Res. 2017, 23, 351–362. [Google Scholar] [CrossRef]
- Tarhini, A.A.; Edington, H.; Butterfield, L.H.; Lin, Y.; Shuai, Y.; Tawbi, H.; Sander, C.; Yin, Y.; Holtzman, M.; Johnson, J.; et al. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PLoS ONE 2014, 9, e87705. [Google Scholar] [CrossRef]
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Vella, L.J.; Pasam, A.; Dimopoulos, N.; Andrews, M.; Knights, A.; Puaux, A.L.; Louahed, J.; Chen, W.; Woods, K.; Cebon, J.S. MEK inhibition, alone or in combination with BRAF inhibition, affects multiple functions of isolated normal human lymphocytes and dendritic cells. Cancer Immunol. Res. 2014, 2, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, J.; Inozume, T.; Sax, N.; Ariyasu, R.; Ishikawa, M.; Yamashita, K.; Kawazu, M.; Ueno, T.; Irie, T.; Tanji, E.; et al. PD-1 blockade therapy promotes infiltration of tumor-attacking exhausted T cell clonotypes. Cell Rep. 2022, 38, 110331. [Google Scholar] [CrossRef]
- Chan, C.Y.; Chiu, D.K.; Yuen, V.W.; Law, C.T.; Wong, B.P.; Thu, K.L.; Cescon, D.W.; Soria-Bretones, I.; Cheu, J.W.; Lee, D.; et al. CFI-402257, a TTK inhibitor, effectively suppresses hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2022, 119, e2119514119. [Google Scholar] [CrossRef]
- Zacharakis, N.; Huq, L.M.; Seitter, S.J.; Kim, S.P.; Gartner, J.J.; Sindiri, S.; Hill, V.K.; Li, Y.F.; Paria, B.C.; Ray, S.; et al. Breast Cancers Are Immunogenic: Immunologic Analyses and a Phase II Pilot Clinical Trial Using Mutation-Reactive Autologous Lymphocytes. J. Clin. Oncol. 2022, 40, 1741–1754. [Google Scholar] [CrossRef] [PubMed]
- Creelan, B.C.; Wang, C.; Teer, J.K.; Toloza, E.M.; Yao, J.; Kim, S.; Landin, A.M.; Mullinax, J.E.; Saller, J.J.; Saltos, A.N.; et al. Tumor-infiltrating lymphocyte treatment for anti-PD-1-resistant metastatic lung cancer: A phase 1 trial. Nat. Med. 2021, 27, 1410–1418. [Google Scholar] [CrossRef]
- Yee, C. The use of endogenous T cells for adoptive transfer. Immunol. Rev. 2014, 257, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Ou, Y.; Wang, T.; Shen, H.; Wu, F.; Zhang, W.; Tao, C.; Yuan, Y.; Bo, H.; Wang, H.; et al. Differences in TCR-Vbeta repertoire and effector phenotype between tumor infiltrating lymphocytes and peripheral blood lymphocytes increase with age. PLoS ONE 2014, 9, e102327. [Google Scholar] [CrossRef] [PubMed]
- Yee, C. Adoptive T-cell therapy of cancer. Hematol. Oncol. Clin. N. Am. 2006, 20, 711–733. [Google Scholar] [CrossRef]
- Chapuis, A.G.; Lee, S.M.; Thompson, J.A.; Roberts, I.M.; Margolin, K.A.; Bhatia, S.; Sloan, H.L.; Lai, I.; Wagener, F.; Shibuya, K.; et al. Combined IL-21-primed polyclonal CTL plus CTLA4 blockade controls refractory metastatic melanoma in a patient. J. Exp. Med. 2016, 213, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Brentjens, R.J. CD19-Targeted CAR T cells as novel cancer immunotherapy for relapsed or refractory B-cell acute lymphoblastic leukemia. Clin. Adv. Hematol. Oncol. 2016, 14, 802–808. [Google Scholar] [PubMed]
- Kwok, A.; Eggimann, G.A.; Reymond, J.L.; Darbre, T.; Hollfelder, F. Peptide dendrimer/lipid hybrid systems are efficient DNA transfection reagents: Structure–activity relationships highlight the role of charge distribution across dendrimer generations. ACS Nano 2013, 7, 4668–4682. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Li, Y.; Bonini, C.; Nair, S.; Gilboa, E.; Greenberg, P.D.; Yee, C. Transfection of RNA encoding tumor antigens following maturation of dendritic cells leads to prolonged presentation of antigen and the generation of high-affinity tumor-reactive cytotoxic T lymphocytes. Mol. Ther. 2004, 9, 757–764. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Yao, X.L.; Tabata, Y.; Nakagawa, S.; Gao, J.Q. Gene carriers and transfection systems used in the recombination of dendritic cells for effective cancer immunotherapy. Clin. Dev. Immunol. 2010, 2010, 565643. [Google Scholar] [CrossRef]
- Butler, M.O.; Lee, J.S.; Ansen, S.; Neuberg, D.; Hodi, F.S.; Murray, A.P.; Drury, L.; Berezovskaya, A.; Mulligan, R.C.; Nadler, L.M.; et al. Long-lived antitumor CD8+ lymphocytes for adoptive therapy generated using an artificial antigen-presenting cell. Clin. Cancer Res. 2007, 13, 1857–1867. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, A.G.; Thompson, J.A.; Margolin, K.A.; Rodmyre, R.; Lai, I.P.; Dowdy, K.; Farrar, E.A.; Bhatia, S.; Sabath, D.E.; Cao, J.; et al. Transferred melanoma-specific CD8+ T cells persist, mediate tumor regression, and acquire central memory phenotype. Proc. Natl. Acad. Sci. USA 2012, 109, 4592–4597. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, A.G.; Desmarais, C.; Emerson, R.; Schmitt, T.M.; Shibuya, K.; Lai, I.; Wagener, F.; Chou, J.; Roberts, I.M.; Coffey, D.G.; et al. Tracking the Fate and Origin of Clinically Relevant Adoptively Transferred CD8(+) T Cells In Vivo. Sci. Immunol. 2017, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bleakley, M.; Yee, C. IL-21 influences the frequency, phenotype, and affinity of the antigen-specific CD8 T cell response. J. Immunol. 2005, 175, 2261–2269. [Google Scholar] [CrossRef]
- Li, Y.; Yee, C. IL-21 mediated Foxp3 suppression leads to enhanced generation of antigen-specific CD8+ cytotoxic T lymphocytes. Blood 2008, 111, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Pollack, S.M.; Jones, R.L.; Farrar, E.A.; Lai, I.P.; Lee, S.M.; Cao, J.; Pillarisetty, V.G.; Hoch, B.L.; Gullett, A.; Bleakley, M.; et al. Tetramer guided, cell sorter assisted production of clinical grade autologous NY-ESO-1 specific CD8(+) T cells. J. Immunother. Cancer 2014, 2, 36. [Google Scholar] [CrossRef]
- Ishihara, M.; Kitano, S.; Kageyama, S.; Miyahara, Y.; Yamamoto, N.; Kato, H.; Mishima, H.; Hattori, H.; Funakoshi, T.; Kojima, T.; et al. NY-ESO-1-specific redirected T cells with endogenous TCR knockdown mediate tumor response and cytokine release syndrome. J. Immunother. Cancer 2022, 10, e003811. [Google Scholar] [CrossRef]
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 2018, 559, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Kebriaei, P.; Singh, H.; Huls, M.H.; Figliola, M.J.; Bassett, R.; Olivares, S.; Jena, B.; Dawson, M.J.; Kumaresan, P.R.; Su, S.; et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J. Clin. Investig. 2016, 126, 3363–3376. [Google Scholar] [CrossRef]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef]
- Amir, A.L.; van der Steen, D.M.; van Loenen, M.M.; Hagedoorn, R.S.; de Boer, R.; Kester, M.D.; de Ru, A.H.; Lugthart, G.J.; van Kooten, C.; Hiemstra, P.S.; et al. PRAME-specific Allo-HLA-restricted T cells with potent antitumor reactivity useful for therapeutic T-cell receptor gene transfer. Clin. Cancer Res. 2011, 17, 5615–5625. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bennett, A.D.; Zheng, Z.; Wang, Q.J.; Robbins, P.F.; Yu, L.Y.; Li, Y.; Molloy, P.E.; Dunn, S.M.; Jakobsen, B.K.; et al. High-affinity TCRs generated by phage display provide CD4+ T cells with the ability to recognize and kill tumor cell lines. J. Immunol. 2007, 179, 5845–5854. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Cameron, B.J.; Gerry, A.B.; Dukes, J.; Harper, J.V.; Kannan, V.; Bianchi, F.C.; Grand, F.; Brewer, J.E.; Gupta, M.; Plesa, G.; et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 2013, 5, 197ra103. [Google Scholar] [CrossRef]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef]
- Chambers, C.A.; Allison, J.P. Co-stimulation in T cell responses. Curr. Opin. Immunol. 1997, 9, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.A.; Heiders, J.; Foppe, M.; Chmielewski, M.; Abken, H. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4(+) T cells. Oncoimmunology 2012, 1, 458–466. [Google Scholar] [CrossRef]
- Jaspers, J.E.; Brentjens, R.J. Development of CAR T cells designed to improve antitumor efficacy and safety. Pharmacol. Ther. 2017, 178, 83–91. [Google Scholar] [CrossRef]
- Thanindratarn, P.; Dean, D.C.; Nelson, S.D.; Hornicek, F.J.; Duan, Z. Chimeric antigen receptor T (CAR-T) cell immunotherapy for sarcomas: From mechanisms to potential clinical applications. Cancer Treat. Rev. 2020, 82, 101934. [Google Scholar] [CrossRef]
- Fathi Maroufi, N.; Aghayi, E.; Garshasbi, H.; Gholampour Matin, M.; Babazadeh Bedoustani, A.; Firouzi Amoudizaj, F.; Hajazimian, S.; Isazadeh, A.; Taefehshokr, S.; Taefehshokr, N.; et al. Association of rs1946518 C/A Polymorphism in Promoter Region of Interleukin 18 Gene and Breast Cancer Risk in Iranian Women: A Case-control Study. Iran. J. Allergy Asthma Immunol. 2019, 18, 671–678. [Google Scholar] [CrossRef]
- Posey, A.D., Jr.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef]
- Vairy, S.; Garcia, J.L.; Teira, P.; Bittencourt, H. CTL019 (tisagenlecleucel): CAR-T therapy for relapsed and refractory B-cell acute lymphoblastic leukemia. Drug Des. Devel Ther. 2018, 12, 3885–3898. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jager, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Geyer, M.B. First CAR to Pass the Road Test: Tisagenlecleucel’s Drive to FDA Approval. Clin. Cancer Res. 2019, 25, 1133–1135. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, S.H.; Murad, J.; Werner, L.; Daley, H.; Trebeden-Negre, H.; Gicobi, J.K.; Schmucker, A.; Reder, J.; Sentman, C.L.; Gilham, D.E.; et al. Phase I Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients with AML/MDS and Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 100–112. [Google Scholar] [CrossRef]
- Sallman, D.A.; Brayer, J.; Sagatys, E.M.; Lonez, C.; Breman, E.; Agaugue, S.; Verma, B.; Gilham, D.E.; Lehmann, F.F.; Davila, M.L. NKG2D-based chimeric antigen receptor therapy induced remission in a relapsed/refractory acute myeloid leukemia patient. Haematologica 2018, 103, e424–e426. [Google Scholar] [CrossRef]
- Zhang, H.; Gan, W.T.; Hao, W.G.; Wang, P.F.; Li, Z.Y.; Chang, L.J. Successful Anti-CLL1 CAR T-Cell Therapy in Secondary Acute Myeloid Leukemia. Front. Oncol. 2020, 10, 685. [Google Scholar] [CrossRef]
- Jetani, H.; Garcia-Cadenas, I.; Nerreter, T.; Thomas, S.; Rydzek, J.; Meijide, J.B.; Bonig, H.; Herr, W.; Sierra, J.; Einsele, H.; et al. CAR T-cells targeting FLT3 have potent activity against FLT3(−)ITD(+) AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia 2018, 32, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, Y.; Li, S.; Liu, J.; Xing, Y.; Xing, H.; Tian, Z.; Tang, K.; Rao, Q.; Wang, M.; et al. Targeting FLT3 in acute myeloid leukemia using ligand-based chimeric antigen receptor-engineered T cells. J. Hematol. Oncol. 2018, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.; Cheng, H.Y.; Nguyen, D.; Dettling, D.; Yeung, Y.A.; Sutton, J.; Hamze, M.; Valton, J.; Smith, J.; Djuretic, I.; et al. Allogeneic FLT3 CAR T Cells with an Off-Switch Exhibit Potent Activity against AML and Can Be Depleted to Expedite Bone Marrow Recovery. Mol. Ther. 2020, 28, 2237–2251. [Google Scholar] [CrossRef]
- Marin, V.; Pizzitola, I.; Agostoni, V.; Attianese, G.M.; Finney, H.; Lawson, A.; Pule, M.; Rousseau, R.; Biondi, A.; Biagi, E. Cytokine-induced killer cells for cell therapy of acute myeloid leukemia: Improvement of their immune activity by expression of CD33-specific chimeric receptors. Haematologica 2010, 95, 2144–2152. [Google Scholar] [CrossRef]
- Dutour, A.; Marin, V.; Pizzitola, I.; Valsesia-Wittmann, S.; Lee, D.; Yvon, E.; Finney, H.; Lawson, A.; Brenner, M.; Biondi, A.; et al. In Vitro and In Vivo Antitumor Effect of Anti-CD33 Chimeric Receptor-Expressing EBV-CTL against CD33 Acute Myeloid Leukemia. Adv. Hematol. 2012, 2012, 683065. [Google Scholar] [CrossRef]
- Economides, M.P.; McCue, D.; Lane, A.A.; Pemmaraju, N. Tagraxofusp, the first CD123-targeted therapy and first targeted treatment for blastic plasmacytoid dendritic cell neoplasm. Expert. Rev. Clin. Pharmacol. 2019, 12, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 2022, 22, 85–96. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Slaney, C.Y.; Kershaw, M.H.; Darcy, P.K. Trafficking of T cells into tumors. Cancer Res. 2014, 74, 7168–7174. [Google Scholar] [CrossRef] [PubMed]
- Craddock, J.A.; Lu, A.; Bear, A.; Pule, M.; Brenner, M.K.; Rooney, C.M.; Foster, A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 2010, 33, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, L.; Sek, K.; Henderson, M.A.; House, I.G.; Lai, J.; Chen, A.X.Y.; Todd, K.L.; Petley, E.V.; Mardiana, S.; Todorovski, I.; et al. IL-15 Preconditioning Augments CAR T Cell Responses to Checkpoint Blockade for Improved Treatment of Solid Tumors. Mol. Ther. 2020, 28, 2379–2393. [Google Scholar] [CrossRef]
- Song, Y.; Gao, Q.; Zhang, H.; Fan, L.; Zhou, J.; Zou, D.; Li, W.; Yang, H.; Liu, T.; Wang, Q.; et al. Treatment of relapsed or refractory classical Hodgkin lymphoma with the anti-PD-1, tislelizumab: Results of a phase 2, single-arm, multicenter study. Leukemia 2020, 34, 533–542. [Google Scholar] [CrossRef]
- Guedan, S.; Madar, A.; Casado-Medrano, V.; Shaw, C.; Wing, A.; Liu, F.; Young, R.M.; June, C.H.; Posey, A.D., Jr. Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J. Clin. Investig. 2020, 130, 3087–3097. [Google Scholar] [CrossRef]
- Ghosn, M.; Cheema, W.; Zhu, A.; Livschitz, J.; Maybody, M.; Boas, F.E.; Santos, E.; Kim, D.; Beattie, J.A.; Offin, M.; et al. Image-guided interventional radiological delivery of chimeric antigen receptor (CAR) T cells for pleural malignancies in a phase I/II clinical trial. Lung Cancer 2022, 165, 1–9. [Google Scholar] [CrossRef]
- Adusumilli, P.S.; Zauderer, M.G.; Riviere, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti-PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef]
- Chen, L.; Chen, F.; Niu, H.; Li, J.; Pu, Y.; Yang, C.; Wang, Y.; Huang, R.; Li, K.; Lei, Y.; et al. Chimeric Antigen Receptor (CAR)-T Cell Immunotherapy against Thoracic Malignancies: Challenges and Opportunities. Front. Immunol. 2022, 13, 871661. [Google Scholar] [CrossRef] [PubMed]
- Tinkle, S.; McNeil, S.E.; Muhlebach, S.; Bawa, R.; Borchard, G.; Barenholz, Y.C.; Tamarkin, L.; Desai, N. Nanomedicines: Addressing the scientific and regulatory gap. Ann. N. Y. Acad. Sci. 2014, 1313, 35–56. [Google Scholar] [CrossRef]
- Cheng, Z.; Li, M.; Dey, R.; Chen, Y. Nanomaterials for cancer therapy: Current progress and perspectives. J. Hematol. Oncol. 2021, 14, 85. [Google Scholar] [CrossRef]
- Heshmati Aghda, N.; Dabbaghianamiri, M.; Tunnell, J.W.; Betancourt, T. Design of smart nanomedicines for effective cancer treatment. Int. J. Pharm. 2022, 621, 121791. [Google Scholar] [CrossRef] [PubMed]
- Sriraman, S.K.; Aryasomayajula, B.; Torchilin, V.P. Barriers to drug delivery in solid tumors. Tissue Barriers 2014, 2, e29528. [Google Scholar] [CrossRef]
- Garnacho, C. Intracellular Drug Delivery: Mechanisms for Cell Entry. Curr. Pharm. Des. 2016, 22, 1210–1226. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Meir, R.; Shamalov, K.; Betzer, O.; Motiei, M.; Horovitz-Fried, M.; Yehuda, R.; Popovtzer, A.; Popovtzer, R.; Cohen, C.J. Nanomedicine for Cancer Immunotherapy: Tracking Cancer-Specific T-Cells in Vivo with Gold Nanoparticles and CT Imaging. ACS Nano 2015, 9, 6363–6372. [Google Scholar] [CrossRef]
- Chenthamara, D.; Subramaniam, S.; Ramakrishnan, S.G.; Krishnaswamy, S.; Essa, M.M.; Lin, F.H.; Qoronfleh, M.W. Therapeutic efficacy of nanoparticles and routes of administration. Biomater. Res. 2019, 23, 20. [Google Scholar] [CrossRef]
- Haist, M.; Mailander, V.; Bros, M. Nanodrugs Targeting T Cells in Tumor Therapy. Front. Immunol. 2022, 13, 912594. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, D.; Trad, M.; Hanke, N.T.; Larmonier, C.B.; Janikashvili, N.; Bonnotte, B.; Katsanis, E.; Larmonier, N. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. 2014, 74, 104–118. [Google Scholar] [CrossRef]
- Din, F.U.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int. J. Nanomed. 2017, 12, 7291–7309. [Google Scholar] [CrossRef]
- Adriano, B.; Cotto, N.M.; Chauhan, N.; Jaggi, M.; Chauhan, S.C.; Yallapu, M.M. Milk exosomes: Nature’s abundant nanoplatform for theranostic applications. Bioact. Mater. 2021, 6, 2479–2490. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Liu, D.; Booth, G.; Gao, W.; Lu, Y. Virus-Like Particle Engineering: From Rational Design to Versatile Applications. Biotechnol. J. 2018, 13, e1700324. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, Y.; Zhang, F.; Zhao, Q.; Zhong, H. Increased anti-tumour activity by exosomes derived from doxorubicin-treated tumour cells via heat stress. Int. J. Hyperth. 2015, 31, 498–506. [Google Scholar] [CrossRef]
- Hadla, M.; Palazzolo, S.; Corona, G.; Caligiuri, I.; Canzonieri, V.; Toffoli, G.; Rizzolio, F. Exosomes increase the therapeutic index of doxorubicin in breast and ovarian cancer mouse models. Nanomedicine 2016, 11, 2431–2441. [Google Scholar] [CrossRef]
- Gehrmann, U.; Hiltbrunner, S.; Georgoudaki, A.M.; Karlsson, M.C.; Naslund, T.I.; Gabrielsson, S. Synergistic induction of adaptive antitumor immunity by codelivery of antigen with alpha-galactosylceramide on exosomes. Cancer Res. 2013, 73, 3865–3876. [Google Scholar] [CrossRef]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release Off. J. Control. Release Soc. 2000, 65, 271–284. [Google Scholar]
- Zi, Y.; Yang, K.; He, J.; Wu, Z.; Liu, J.; Zhang, W. Strategies to enhance drug delivery to solid tumors by harnessing the EPR effects and alternative targeting mechanisms. Adv. Drug Deliv. Rev. 2022, 188, 114449. [Google Scholar] [CrossRef]
- Neri, D.; Bicknell, R. Tumour vascular targeting. Nat. Rev. Cancer 2005, 5, 436–446. [Google Scholar] [CrossRef]
- Liao, J.; Zheng, H.; Fei, Z.; Lu, B.; Li, D.; Xiong, X.; Yi, Y. Tumor-targeting and pH-responsive nanoparticles from hyaluronic acid for the enhanced delivery of doxorubicin. Int. J. Biol. Macromol. 2018, 113, 737–747. [Google Scholar] [CrossRef]
- Liao, J.; Zheng, H.; Hu, R.; Cao, J.; Wei, X.; Li, D.; Yin, Y. Hyaluronan Based Tumor-Targeting and pH-Responsive Shell Cross-Linkable Nanoparticles for the Controlled Release of Doxorubicin. J. Biomed. Nanotechnol. 2018, 14, 496–509. [Google Scholar] [CrossRef]
- Bansal, K.K.; Özliseli, E.; Rosling, A.; Rosenholm, J.M. Synthesis and Evaluation of Novel Functional Polymers Derived from Renewable Jasmine Lactone for StimuliResponsive Drug Delivery. Adv. Funct. Mater. 2021, 31, 2101998. [Google Scholar] [CrossRef]
- Tian, M.; Xin, X.; Wu, R.; Guan, W.; Zhou, W. Advances in intelligent-responsive nanocarriers for cancer therapy. Pharmacol. Res. 2022, 178, 106184. [Google Scholar] [CrossRef]
- Graff, B.A.; Vangberg, L.; Rofstad, E.K. Quantitative assessment of uptake and distribution of iron oxide particles (NC100150) in human melanoma xenografts by contrast-enhanced MRI. Magn. Reson. Med. 2004, 51, 727–735. [Google Scholar] [CrossRef]
- Pun, S.H.; Tack, F.; Bellocq, N.C.; Cheng, J.; Grubbs, B.H.; Jensen, G.S.; Davis, M.E.; Brewster, M.; Janicot, M.; Janssens, B.; et al. Targeted delivery of RNA-cleaving DNA enzyme (DNAzyme) to tumor tissue by transferrin-modified, cyclodextrin-based particles. Cancer Biol. Ther. 2004, 3, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.J.; Finch, S.K.; Hallahan, D.E.; Giorgio, T.D. Proteolytic surface functionalization enhances in vitro magnetic nanoparticle mobility through extracellular matrix. Nano Lett. 2006, 6, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Goodman, T.T.; Olive, P.L.; Pun, S.H. Increased nanoparticle penetration in collagenase-treated multicellular spheroids. Int. J. Nanomed. 2007, 2, 265–274. [Google Scholar]
- Dietz, H.C. TGF-beta in the pathogenesis and prevention of disease: A matter of aneurysmic proportions. J. Clin. Investig. 2010, 120, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Diop-Frimpong, B.; Chauhan, V.P.; Krane, S.; Boucher, Y.; Jain, R.K. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 2909–2914. [Google Scholar] [CrossRef]
- Shan, X.; Gong, X.; Li, J.; Wen, J.; Li, Y.; Zhang, Z. Current approaches of nanomedicines in the market and various stage of clinical translation. Acta Pharm. Sin. B 2022, 12, 3028–3048. [Google Scholar] [CrossRef]
- Moon, J.J.; Huang, B.; Irvine, D.J. Engineering nano- and microparticles to tune immunity. Adv. Mater. 2012, 24, 3724–3746. [Google Scholar] [CrossRef]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef]
- Duan, X.; Chan, C.; Lin, W. Nanoparticle-Mediated Immunogenic Cell Death Enables and Potentiates Cancer Immunotherapy. Angew. Chem. Int. Ed. Engl. 2019, 58, 670–680. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, K.; Zhao, R.; Ji, T.; Wang, X.; Yang, X.; Zhang, Y.; Cheng, K.; Liu, S.; Hao, J.; et al. Inducing enhanced immunogenic cell death with nanocarrier-based drug delivery systems for pancreatic cancer therapy. Biomaterials 2016, 102, 187–197. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, L.; Lu, Y.; Chen, X.; Zhang, Y.; Zhou, W.; Guo, Q.; Li, C.; Zhang, Y.; Zhang, Y.; et al. Tumor Microenvironment-Triggered Aggregated Magnetic Nanoparticles for Reinforced Image-Guided Immunogenic Chemotherapy. Adv. Sci. 2019, 6, 1802134. [Google Scholar] [CrossRef]
- Song, W.; Shen, L.; Wang, Y.; Liu, Q.; Goodwin, T.J.; Li, J.; Dorosheva, O.; Liu, T.; Liu, R.; Huang, L. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat. Commun. 2018, 9, 2237. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Yang, J.; Yuan, Y.; Zhao, Z.; Lian, Z.; Liang, G. Paclitaxel nanoparticle awakens immune system to fight against cancer. Nanoscale 2017, 9, 6529–6536. [Google Scholar] [CrossRef]
- Roy, A.; Chandra, S.; Mamilapally, S.; Upadhyay, P.; Bhaskar, S. Anticancer and immunostimulatory activity by conjugate of paclitaxel and non-toxic derivative of LPS for combined chemo-immunotherapy. Pharm. Res. 2012, 29, 2294–2309. [Google Scholar] [CrossRef]
- Zheng, D.W.; Chen, J.L.; Zhu, J.Y.; Rong, L.; Li, B.; Lei, Q.; Fan, J.X.; Zou, M.Z.; Li, C.; Cheng, S.X.; et al. Highly Integrated Nano-Platform for Breaking the Barrier between Chemotherapy and Immunotherapy. Nano Lett. 2016, 16, 4341–4347. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, C.; Qiu, W.X.; Dong, X.; Zheng, D.W.; Wu, W.; Zhang, X.Z. PD-1 Blockade for Improving the Antitumor Efficiency of Polymer-Doxorubicin Nanoprodrug. Small 2018, 14, e1802403. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Tang, C.; Yin, C. Co-delivery of doxorubicin and interleukin-2 via chitosan based nanoparticles for enhanced antitumor efficacy. Acta Biomater. 2017, 47, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Paris, S.; Barsoumian, H.; Abana, C.O.; He, K.; Wasley, M.; Younes, A.I.; Masrorpour, F.; Chen, D.; Yang, L.; et al. Radiation Therapy Enhanced by NBTXR3 Nanoparticles Overcomes Anti-PD1 Resistance and Evokes Abscopal Effects. Int. J. Radiat. Oncol. Biol. Phys. 2021, 111, 647–657. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, J.; Yang, Z.; Xu, J.; Xu, L.; Liang, C.; Han, X.; Liu, Z. Nanoparticle-Enhanced Radiotherapy to Trigger Robust Cancer Immunotherapy. Adv. Mater. 2019, 31, e1802228. [Google Scholar] [CrossRef]
- Cano-Mejia, J.; Shukla, A.; Ledezma, D.K.; Palmer, E.; Villagra, A.; Fernandes, R. CpG-coated prussian blue nanoparticles-based photothermal therapy combined with anti-CTLA-4 immune checkpoint blockade triggers a robust abscopal effect against neuroblastoma. Transl. Oncol. 2020, 13, 100823. [Google Scholar] [CrossRef]
- Zhou, B.; Wu, Q.; Wang, M.; Hoover, A.; Wang, X.; Zhou, F.; Towner, R.A.; Smith, N.; Saunders, D.; Song, J.; et al. Immunologically modified MnFe2O4 nanoparticles to synergize photothermal therapy and immunotherapy for cancer treatment. Chem. Eng. J. 2020, 396, 125239. [Google Scholar] [CrossRef]
- Sacchetti, C.; Rapini, N.; Magrini, A.; Cirelli, E.; Bellucci, S.; Mattei, M.; Rosato, N.; Bottini, N.; Bottini, M. In vivo targeting of intratumor regulatory T cells using PEG-modified single-walled carbon nanotubes. Bioconjug Chem. 2013, 24, 852–858. [Google Scholar] [CrossRef]
- Zanganeh, S.; Hutter, G.; Spitler, R.; Lenkov, O.; Mahmoudi, M.; Shaw, A.; Pajarinen, J.S.; Nejadnik, H.; Goodman, S.; Moseley, M.; et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat. Nanotechnol. 2016, 11, 986–994. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, L.; Li, Z.; Zhang, W.; Luo, F.; Chu, Y.; Chen, G. Glycocalyx-Mimicking Nanoparticles Improve Anti-PD-L1 Cancer Immunotherapy through Reversion of Tumor-Associated Macrophages. Biomacromolecules 2018, 19, 2098–2108. [Google Scholar] [CrossRef]
- He, W.; Liang, P.; Guo, G.; Huang, Z.; Niu, Y.; Dong, L.; Wang, C.; Zhang, J. Re-polarizing Myeloid-derived Suppressor Cells (MDSCs) with Cationic Polymers for Cancer Immunotherapy. Sci. Rep. 2016, 6, 24506. [Google Scholar] [CrossRef]
- Plebanek, M.P.; Bhaumik, D.; Bryce, P.J.; Thaxton, C.S. Scavenger Receptor Type B1 and Lipoprotein Nanoparticle Inhibit Myeloid-Derived Suppressor Cells. Mol. Cancer Ther. 2018, 17, 686–697. [Google Scholar] [CrossRef]
- Park, J.; Wrzesinski, S.H.; Stern, E.; Look, M.; Criscione, J.; Ragheb, R.; Jay, S.M.; Demento, S.L.; Agawu, A.; Licona Limon, P.; et al. Combination delivery of TGF-beta inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat. Mater. 2012, 11, 895–905. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Y.; Zhang, L.; Huang, L. Nanoparticle-delivered transforming growth factor-beta siRNA enhances vaccination against advanced melanoma by modifying tumor microenvironment. ACS Nano 2014, 8, 3636–3645. [Google Scholar] [CrossRef]
- Ye, Y.; Wang, J.; Hu, Q.; Hochu, G.M.; Xin, H.; Wang, C.; Gu, Z. Synergistic Transcutaneous Immunotherapy Enhances Antitumor Immune Responses through Delivery of Checkpoint Inhibitors. ACS Nano 2016, 10, 8956–8963. [Google Scholar] [CrossRef]
- Lu, J.; Liu, X.; Liao, Y.P.; Salazar, F.; Sun, B.; Jiang, W.; Chang, C.H.; Jiang, J.; Wang, X.; Wu, A.M.; et al. Nano-enabled pancreas cancer immunotherapy using immunogenic cell death and reversing immunosuppression. Nat. Commun. 2017, 8, 1811. [Google Scholar] [CrossRef]
- Alhallak, K.; Sun, J.; Muz, B.; Jeske, A.; O’Neal, J.; Ritchey, J.K.; Achilefu, S.; DiPersio, J.F.; Azab, A.K. Liposomal phytohemagglutinin: In vivo T-cell activator as a novel pan-cancer immunotherapy. J. Cell Mol. Med. 2022, 26, 940–944. [Google Scholar] [CrossRef]
- Li, S.Y.; Liu, Y.; Xu, C.F.; Shen, S.; Sun, R.; Du, X.J.; Xia, J.X.; Zhu, Y.H.; Wang, J. Restoring anti-tumor functions of T cells via nanoparticle-mediated immune checkpoint modulation. J. Control. Release Off. J. Control. Release Soc. 2016, 231, 17–28. [Google Scholar] [CrossRef]
- Emens, L.A.; Cruz, C.; Eder, J.P.; Braiteh, F.; Chung, C.; Tolaney, S.M.; Kuter, I.; Nanda, R.; Cassier, P.A.; Delord, J.P.; et al. Long-term Clinical Outcomes and Biomarker Analyses of Atezolizumab Therapy for Patients With Metastatic Triple-Negative Breast Cancer: A Phase 1 Study. JAMA Oncol. 2019, 5, 74–82. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- West, H.; McCleod, M.; Hussein, M.; Morabito, A.; Rittmeyer, A.; Conter, H.J.; Kopp, H.G.; Daniel, D.; McCune, S.; Mekhail, T.; et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): A multicentre, randomised, open-label, phase 3 trial. Lancet. Oncol. 2019, 20, 924–937. [Google Scholar] [CrossRef]
- Giannatempo, P.; Raggi, D.; Marandino, L.; Bandini, M.; Fare, E.; Calareso, G.; Colecchia, M.; Gallina, A.; Ross, J.S.; Alessi, A.; et al. Pembrolizumab and nab-paclitaxel as salvage therapy for platinum-treated, locally advanced or metastatic urothelial carcinoma: Interim results of the open-label, single-arm, phase II PEANUT study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 1764–1772. [Google Scholar] [CrossRef]
- Morgensztern, D.; Dols, M.C.; Ponce Aix, S.; Postmus, P.E.; Bennouna, J.; Fischer, J.R.; Juan-Vidal, O.; Stewart, D.J.; Ardizzoni, A.; Bhore, R.; et al. nab-Paclitaxel Plus Durvalumab in Patients with Previously Treated Advanced Stage Non-small Cell Lung Cancer (ABOUND.2L+). Front. Oncol. 2020, 10, 569715. [Google Scholar] [CrossRef]
- Bonvalot, S.; Rutkowski, P.L.; Thariat, J.; Carrere, S.; Ducassou, A.; Sunyach, M.P.; Agoston, P.; Hong, A.; Mervoyer, A.; Rastrelli, M.; et al. NBTXR3, a first-in-class radioenhancer hafnium oxide nanoparticle, plus radiotherapy versus radiotherapy alone in patients with locally advanced soft-tissue sarcoma (Act.In.Sarc): A multicentre, phase 2–3, randomised, controlled trial. Lancet Oncol. 2019, 20, 1148–1159. [Google Scholar] [CrossRef]
- Trujillo-Alonso, V.; Pratt, E.C.; Zong, H.; Lara-Martinez, A.; Kaittanis, C.; Rabie, M.O.; Longo, V.; Becker, M.W.; Roboz, G.J.; Grimm, J.; et al. FDA-approved ferumoxytol displays anti-leukaemia efficacy against cells with low ferroportin levels. Nat. Nanotechnol. 2019, 14, 616–622. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| NCT Number | Clinical Trial Phase | Type of Cancer | Current Status | Nanoparticles | Additional Treatments and Procedures |
|---|---|---|---|---|---|
| NCT03589339 | 1 |
| Recruiting | NBTXR3 | Nivolumab, pembrolizumab, and stereotactic ablative body radiotherapy (SABRT) |
| NCT05039632 | 1 and 2 |
| Not yet recruiting | NBTXR3 | Ipilimumab, nivolumab, and abscopal RT |
| NCT04484909 | 1 |
| Recruiting | NBTXR3 | RT |
| NCT04615013 | 1 |
| Recruiting | NBTXR3 | Capecitabine, carboplatin, docetaxel, fluorouracil, leucovorin, oxaliplatin, paclitaxel, and intensity-modulated RT |
| NCT02379845 | 2 and 3 |
| Completed | NBTXR3 | RT |
| NCT04862455 | 2 |
| Recruiting | NBTXR3 | Pembrolizumab, hypofractionated RT, and SBRT |
| NCT03464734 | 2 |
| Completed | Nanoparticle albumin-bound (Nab) paclitaxel | Pembrolizumab |
| NCT04247165 | 1 and 2 |
| Recruiting | Nab-paclitaxel | Gemcitabine, nivolumab, ipilimumab, and SBRT |
| NCT04132817 | 1 |
| Active, not recruiting | Nab-paclitaxel | Nivolumab and ipilimumab |
| NCT04929041 | 2 and 3 |
| Recruiting | Nab-paclitaxel | Carboplatin, ipilimumab, nivolumab, pembrolizumab, pemetrexed, and SBRT |
| NCT04148911 | 3 |
| Active, not recruiting | Nab-paclitaxel | Atezolizumab |
| NCT05266937 | 2 |
| Recruiting | Nab-paclitaxel | Atezolizumab and carboplatin |
| NCT04865250 | 2 |
| Recruiting | Nab-paclitaxel | Atezolizumab and carboplatin |
| NCT03456063 | 3 |
| Active, not recruiting | Nab-paclitaxel | Atezolizumab, pemetrexed, carboplatin, cisplatin, and gemcitabine |
| NCT05272696 | 2 |
| Not yet recruiting | Nab-paclitaxel | Cisplatin, pembrolizumab, adjuvant chemoradiotherapy, and surgery |
| NCT04297605 | 1 |
| Recruiting | Nab-paclitaxel | Pembrolizumab and pemetrexed |
| NCT04754815 | 2 |
| Withdrawn | Nab-paclitaxel | Pembrolizumab and pemetrexed |
| NCT02425891 | 3 |
| Completed | Nab-paclitaxel | Atezolizumab |
| NCT02367781 | 3 |
| Completed | Nab-paclitaxel | Atezolizumab, carboplatin, and pemetrexed |
| NCT02250326 | 2 |
| Active, not recruiting | Nab-paclitaxel | CC-486 and duravalumab |
| NCT04895358 | 3 |
| Recruiting | Nab-paclitaxel and liposomal doxorubicin | Pembrolizumab, capecitabine, and dextrose |
| NCT03539328 | 2 |
| Unknown | Liposomal doxorubicin | Pembrolizumab, gemcitabine, and paclitaxel |
| NCT03591276 | 1 and 2 |
| Recruiting | Pegylated liposomal doxorubicin | Pembrolizumab |
| NCT05255666 | 2 |
| Recruiting | Liposomal Irinotecan | Pembrolizumab |
| NCT03596281 | 1 |
| Active, not recruiting | Pegylated liposomal doxorubicin | Pembrolizumab and bevacizumab |
| NCT03409198 | 2 |
| Completed | Pegylated liposomal doxorubicin | Ipilimumab, nivolumab, and cyclophosphamide |
| NCT04682847 | Unknown |
| Recruiting | Ferumoxytol | Adaptive Stereotactic RT |
| NCT04900792 | 1 |
| Not yet recruiting | Ferumoxytol | Ascorbate, temozolomide, and external beam RT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Domínguez, D.J.; López-Enríquez, S.; Alba, G.; Garnacho, C.; Jiménez-Cortegana, C.; Flores-Campos, R.; de la Cruz-Merino, L.; Hajji, N.; Sánchez-Margalet, V.; Hontecillas-Prieto, L. Cancer Nano-Immunotherapy: The Novel and Promising Weapon to Fight Cancer. Int. J. Mol. Sci. 2024, 25, 1195. https://doi.org/10.3390/ijms25021195
García-Domínguez DJ, López-Enríquez S, Alba G, Garnacho C, Jiménez-Cortegana C, Flores-Campos R, de la Cruz-Merino L, Hajji N, Sánchez-Margalet V, Hontecillas-Prieto L. Cancer Nano-Immunotherapy: The Novel and Promising Weapon to Fight Cancer. International Journal of Molecular Sciences. 2024; 25(2):1195. https://doi.org/10.3390/ijms25021195
Chicago/Turabian StyleGarcía-Domínguez, Daniel J., Soledad López-Enríquez, Gonzalo Alba, Carmen Garnacho, Carlos Jiménez-Cortegana, Rocío Flores-Campos, Luis de la Cruz-Merino, Nabil Hajji, Víctor Sánchez-Margalet, and Lourdes Hontecillas-Prieto. 2024. "Cancer Nano-Immunotherapy: The Novel and Promising Weapon to Fight Cancer" International Journal of Molecular Sciences 25, no. 2: 1195. https://doi.org/10.3390/ijms25021195