
Role of Immunotherapy in Sarcomas



Abstract
1. Introduction
2. Current Sarcoma Treatment Landscape
3. Cancer Immunotherapy
4. Immune Microenvironment and Biomarkers in Sarcoma
5. Immune Checkpoint Blockade
6. Combination of ICIs with Tyrosine Kinase Inhibitors
7. Combination of ICIs with Conventional Chemotherapy
8. Combination of Immunotherapy with Local Radiation Therapy
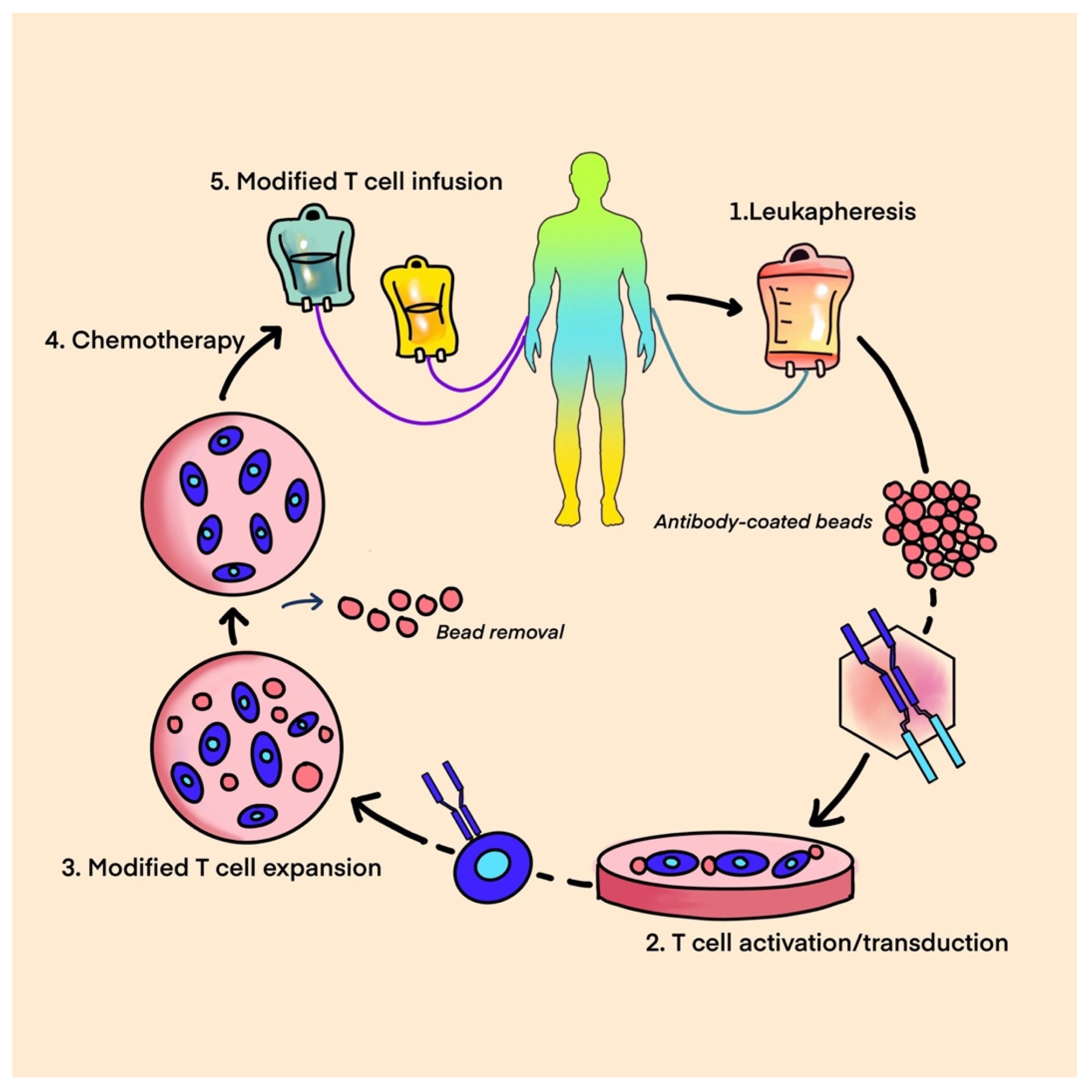
9. Adoptive Cell Therapy
- A.
- T-cell therapy.
- B.
- Chimeric antigen receptor T-cell therapy (CAR-T).
- C.
- T-cell receptor-based therapy (TCR).
- (1)
- T-Cell Therapy
- (2)
- CAR T-Cell Therapy
- An antigen-recognition domain—a single-chain variable fragment (scFv) as a part of a genetically engineered monoclonal antibody that targets the tumor antigen.
- A hinge that links a recognition site to the transmembrane domain bridging the membrane.
- An intracellular domain that facilitates T-cell receptor signaling [81].
- (3)
- TCR Therapy
10. Oncolytic Viruses
11. Cancer Vaccines
12. Cancer Targeted Antibodies
13. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Banks, L.B.; D’Angelo, S.P. The Role of Immunotherapy in the Management of Soft Tissue Sarcomas: Current Landscape and Future Outlook. J. Natl. Compr. Cancer Netw. 2022, 20, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Sbaraglia, M.; Bellan, E.; Dei Tos, A.P. The 2020 WHO Classification of Soft Tissue Tumours: News and perspectives. Pathol. J. Ital. Soc. Anat. Pathol. Diagn. Cytopathol. 2020, 113, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Savina, M.; Le Cesne, A.; Blay, J.Y.; Ray-Coquard, I.; Mir, O.; Toulmonde, M.; Cousin, S.; Terrier, P.; Ranchere-Vince, D.; Meeus, P.; et al. Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: The METASARC observational study. BMC Med. 2017, 15, 78. [Google Scholar] [CrossRef] [PubMed]
- Von Mehren, M.; Kane, J.M.; Agulnik, M.; Bui, M.M.; Carr-Ascher, J.; Choy, E.; Connelly, M.; Dry, S.; Ganjoo, K.N.; Gonzalez, R.J.; et al. Soft Tissue Sarcoma, Version 2.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 815–833. [Google Scholar] [CrossRef] [PubMed]
- Penel, N.; Bui, B.N.; Bay, J.-O.; Cupissol, D.; Ray-Coquard, I.; Piperno-Neumann, S.; Kerbrat, P.; Fournier, C.; Taieb, S.; Jimenez, M.; et al. Phase II Trial of Weekly Paclitaxel for Unresectable Angiosarcoma: The ANGIOTAX Study. J. Clin. Oncol. 2008, 26, 5269–5274. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.J.; Ravi, V.; Riedel, R.F.; Ganjoo, K.; Tine, B.A.V.; Chugh, R.; Cranmer, L.; Gordon, E.M.; Hornick, J.L.; Du, H.; et al. nab-Sirolimus for Patients with Malignant Perivascular Epithelioid Cell Tumors. J. Clin. Oncol. 2021, 39, 3660–3670. [Google Scholar] [CrossRef] [PubMed]
- Seddon, B.; Strauss, S.J.; Whelan, J.; Leahy, M.; Woll, P.J.; Cowie, F.; Rothermundt, C.; Wood, Z.; Benson, C.; Ali, N.; et al. Gemcitabine and docetaxel versus doxorubicin as first-line treatment in previously untreated advanced unresectable or metastatic soft-tissue sarcomas (GeDDiS): A randomised controlled phase 3 trial. Lancet Oncol. 2017, 18, 1397–1410. [Google Scholar] [CrossRef]
- Grünwald, V.; Karch, A.; Schuler, M.; Schöffski, P.; Kopp, H.-G.; Bauer, S.; Kasper, B.; Lindner, L.H.; Chemnitz, J.-M.; Crysandt, M.; et al. Randomized Comparison of Pazopanib and Doxorubicin as First-Line Treatment in Patients with Metastatic Soft Tissue Sarcoma Age 60 Years or Older: Results of a German Intergroup Study. J. Clin. Oncol. 2020, 38, 3555–3564. [Google Scholar] [CrossRef]
- Van der Graaf, W.T.; Blay, J.Y.; Chawla, S.P.; Kim, D.W.; Bui-Nguyen, B.; Casali, P.G.; Schoffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Mir, O.; Brodowicz, T.; Italiano, A.; Wallet, J.; Blay, J.Y.; Bertucci, F.; Chevreau, C.; Piperno-Neumann, S.; Bompas, E.; Salas, S.; et al. Safety and efficacy of regorafenib in patients with advanced soft tissue sarcoma (REGOSARC): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016, 17, 1732–1742. [Google Scholar] [CrossRef]
- Gounder, M.M.; Mahoney, M.R.; van Tine, B.A.; Ravi, V.; Attia, S.; Deshpande, H.A.; Gupta, A.A.; Milhem, M.M.; Conry, R.M.; Movva, S.; et al. Sorafenib for Advanced and Refractory Desmoid Tumors. N. Engl. J. Med. 2018, 379, 2417–2428. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, P.; Klimczak, A.; Ługowska, I.; Jagielska, B.; Wągrodzki, M.; Dębiec-Rychter, M.; Pieńkowska-Grela, B.; Świtaj, T. Long-term results of treatment of advanced dermatofibrosarcoma protuberans (DFSP) with imatinib mesylate—The impact of fibrosarcomatous transformation. Eur. J. Surg. Oncol. (EJSO) 2017, 43, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; von Mehren, M.; Blanke, C.D.; van den Abbeele, A.D.; Eisenberg, B.; Roberts, P.J.; Heinrich, M.C.; Tuveson, D.A.; Singer, S.; Janicek, M.; et al. Efficacy and Safety of Imatinib Mesylate in Advanced Gastrointestinal Stromal Tumors. N. Engl. J. Med. 2002, 347, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Misaghi, A.; Goldin, A.; Awad, M.; Kulidjian, A.A. Osteosarcoma: A comprehensive review. SICOT-J 2018, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Zöllner, S.K.; Amatruda, J.F.; Bauer, S.; Collaud, S.; de Álava, E.; DuBois, S.G.; Hardes, J.; Hartmann, W.; Kovar, H.; Metzler, M.; et al. Ewing Sarcoma-Diagnosis, Treatment, Clinical Challenges and Future Perspectives. J. Clin. Med. 2021, 10, 1685. [Google Scholar] [CrossRef] [PubMed]
- Gazendam, A.; Popovic, S.; Parasu, N.; Ghert, M. Chondrosarcoma: A Clinical Review. J. Clin. Med. 2023, 12, 2506. [Google Scholar] [CrossRef] [PubMed]
- Albarran, V.; Villamayor, M.L.; Chamorro, J.; Rosero, D.I.; Pozas, J.; San Roman, M.; Calvo, J.C.; Perez de Aguado, P.; Moreno, J.; Guerrero, P.; et al. Receptor Tyrosine Kinase Inhibitors for the Treatment of Recurrent and Unresectable Bone Sarcomas. Int. J. Mol. Sci. 2022, 23, 13784. [Google Scholar] [CrossRef]
- FDA.gov. FDA Grants Approval to Atezolizumab for Alveolar Soft Part Sarcoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-approval-atezolizumab-alveolar-soft-part-sarcoma#:~:text=On%20December%209%2C%202022%2C%20the%20Food%20and%20Drug,unresectable%20or%20metastatic%20alveolar%20soft%20part%20sarcoma%20%28ASPS%29 (accessed on 5 October 2023).
- Groisberg, R.; Hong, D.S.; Behrang, A.; Hess, K.; Janku, F.; Piha-Paul, S.; Naing, A.; Fu, S.; Benjamin, R.; Patel, S.; et al. Characteristics and outcomes of patients with advanced sarcoma enrolled in early phase immunotherapy trials. J. Immunother. Cancer 2017, 5, 100. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Petitprez, F.; de Reyniès, A.; Keung, E.Z.; Chen, T.W.; Sun, C.M.; Calderaro, J.; Jeng, Y.M.; Hsiao, L.P.; Lacroix, L.; Bougoüin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef]
- Sautès-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Bessede, A.; Pulido, M.; Bompas, E.; Piperno-Neumann, S.; Chevreau, C.; Penel, N.; Bertucci, F.; Toulmonde, M.; Bellera, C.; et al. Pembrolizumab in soft-tissue sarcomas with tertiary lymphoid structures: A phase 2 PEMBROSARC trial cohort. Nat. Med. 2022, 28, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Liu, S.; Luo, X.; Sun, Y.; Li, X.; Luo, K.; Liao, S.; Li, F.; Liang, J.; Zhan, X.; et al. A novel molecular signature for predicting prognosis and immunotherapy response in osteosarcoma based on tumor-infiltrating cell marker genes. Front. Immunol. 2023, 14, 1150588. [Google Scholar] [CrossRef] [PubMed]
- Kostine, M.; Briaire-de Bruijn, I.H.; Cleven, A.H.G.; Vervat, C.; Corver, W.E.; Schilham, M.W.; van Beelen, E.; van Boven, H.; Haas, R.L.; Italiano, A.; et al. Increased infiltration of M2-macrophages, T-cells and PD-L1 expression in high grade leiomyosarcomas supports immunotherapeutic strategies. Oncoimmunology 2018, 7, e1386828. [Google Scholar] [CrossRef] [PubMed]
- Keung, E.Z.; Burgess, M.; Salazar, R.; Parra, E.R.; Rodrigues-Canales, J.; Bolejack, V.; van Tine, B.A.; Schuetze, S.M.; Attia, S.; Riedel, R.F.; et al. Correlative Analyses of the SARC028 Trial Reveal an Association Between Sarcoma-Associated Immune Infiltrate and Response to Pembrolizumab. Clin. Cancer Res. 2020, 26, 1258–1266. [Google Scholar] [CrossRef]
- Atılgan, A.O.; Tepeoğlu, M.; Özen, Ö.; Reyhan, A.N.H.; Ayhan, A. The expression of programmed death-ligand 1 and programmed death-ligand 2 in endometrial carcinosarcoma: Correlation with mismatch repair protein expression status, tumor-infiltrating lymphocyte infiltration, and clinical outcomes. Ann. Diagn. Pathol. 2023, 65, 152137. [Google Scholar] [CrossRef]
- Ok Atılgan, A.; Yılmaz Akçay, E.; Özen, Ö.; Haberal Reyhan, A.N.; Ayhan, A. The Overexpression of Programmed Death-Ligand 2 in Uterine Adenosarcoma: Correlation with High-Grade Morphology, Mutant Type TP53 Expression and Clinical Outcomes. Int. J. Surg. Pathol. 2023, 31, 352–364. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Maki, R.G.; Jungbluth, A.A.; Gnjatic, S.; Schwartz, G.K.; D’Adamo, D.R.; Keohan, M.L.; Wagner, M.J.; Scheu, K.; Chiu, R.; Ritter, E.; et al. A Pilot Study of Anti-CTLA4 Antibody Ipilimumab in Patients with Synovial Sarcoma. Sarcoma 2013, 2013, 168145. [Google Scholar] [CrossRef]
- Ben-Ami, E.; Barysauskas, C.M.; Solomon, S.; Tahlil, K.; Malley, R.; Hohos, M.; Polson, K.; Loucks, M.; Severgnini, M.; Patel, T.; et al. Immunotherapy with single agent nivolumab for advanced leiomyosarcoma of the uterus: Results of a phase 2 study. Cancer 2017, 123, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Burgess, M.A.; Bolejack, V.; Schuetze, S.; Tine, B.A.V.; Attia, S.; Riedel, R.F.; Hu, J.S.; Davis, L.E.; Okuno, S.H.; Priebat, D.A.; et al. Clinical activity of pembrolizumab (P) in undifferentiated pleomorphic sarcoma (UPS) and dedifferentiated/pleomorphic liposarcoma (LPS): Final results of SARC028 expansion cohorts. J. Clin. Oncol. 2019, 37, 11015. [Google Scholar] [CrossRef]
- Blay, J.-Y.; Penel, N.; Ray-Coquard, I.L.; Cousin, S.; Bertucci, F.; Bompas, E.; Eymard, J.-C.; Saada-Bouzid, E.; Soulie, P.; Boudou-Rouquette, P.; et al. High clinical activity of pembrolizumab in chordoma, alveolar soft part sarcoma (ASPS) and other rare sarcoma histotypes: The French AcSé pembrolizumab study from Unicancer. J. Clin. Oncol. 2021, 39, 11520. [Google Scholar] [CrossRef]
- Italiano, A.; Bellera, C.; D’Angelo, S. PD1/PD-L1 targeting in advanced soft-tissue sarcomas: A pooled analysis of phase II trials. J. Hematol. Oncol. 2020, 13, 55. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, X.; Liu, J.; Liang, D.; Zhao, M.; Yu, W.; Chen, P. Nivolumab plus ipilimumab versus nivolumab in individuals with treatment-naive programmed death-ligand 1 positive metastatic soft tissue sarcomas: A multicentre retrospective study. BMC Cancer 2021, 21, 108. [Google Scholar] [CrossRef] [PubMed]
- Lussier, D.M.; Johnson, J.L.; Hingorani, P.; Blattman, J.N. Combination immunotherapy with α-CTLA-4 and α-PD-L1 antibody blockade prevents immune escape and leads to complete control of metastatic osteosarcoma. J. Immunother. Cancer 2015, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Mahoney, M.R.; van Tine, B.A.; Atkins, J.; Milhem, M.M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef]
- Uboha, N.V.; Milhem, M.M.; Kovacs, C.; Amin, A.; Magley, A.; Purkayastha, D.D.; Piha-Paul, S.A. Phase II study of spartalizumab (PDR001) and LAG525 in advanced solid tumors and hematologic malignancies. J. Clin. Oncol. 2019, 37, 2553. [Google Scholar] [CrossRef]
- Naqash, A.R.; Coyne, G.H.O.S.; Moore, N.; Sharon, E.; Takebe, N.; Fino, K.K.; Ferry-Galow, K.V.; Hu, J.S.; Tine, B.A.V.; Burgess, M.A.; et al. Phase II study of atezolizumab in advanced alveolar soft part sarcoma (ASPS). J. Clin. Oncol. 2021, 39, 11519. [Google Scholar] [CrossRef]
- Chen, A.P.; Sharon, E.; O’Sullivan-Coyne, G.; Moore, N.; Foster, J.C.; Hu, J.S.; van Tine, B.A.; Conley, A.P.; Read, W.L.; Riedel, R.F.; et al. Atezolizumab for Advanced Alveolar Soft Part Sarcoma. N. Engl. J. Med. 2023, 389, 911–921. [Google Scholar] [CrossRef]
- Zhou, M.; Bui, N.; Bolleddu, S.; Lohman, M.; Becker, H.-C.; Ganjoo, K. Nivolumab plus ipilimumab for soft tissue sarcoma: A single institution retrospective review. Immunotherapy 2020, 12, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Delyon, J.; Biard, L.; Renaud, M.; Resche-Rigon, M.; Le Goff, J.; Dalle, S.; Heidelberger, V.; da Meda, L.; Toullec, L.; Carcelain, G.; et al. PD-1 blockade with pembrolizumab in classic or endemic Kaposi’s sarcoma: A multicentre, single-arm, phase 2 study. Lancet Oncol. 2022, 23, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Zer, A.I.O.; Yosef, L.; Avram, D.; Jacobi, O.; Fenig, E.; Kurman, N.; Peretz, I.; Shamai, S.; Merimsky, O.; Ben-Ami, E.; et al. Phase II single-arm study of nivolumab and ipilimumab (Nivo/Ipi) in previously treated classical Kaposi sarcoma (cKS). Ann. Oncol. 2022, 33, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Somaiah, N.; Conley, A.P.; Parra, E.R.; Lin, H.; Amini, B.; Solis Soto, L.; Salazar, R.; Barreto, C.; Chen, H.; Gite, S.; et al. Durvalumab plus tremelimumab in advanced or metastatic soft tissue and bone sarcomas: A single-centre phase 2 trial. Lancet Oncol. 2022, 23, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Bahleda, R.; Wagner, A.J.; Burgess, M.A.; Junker, N.; Chisamore, M.; Peterson, P.; Szpurka, A.M.; Ceccarelli, M.; Tap, W.D. Results of an Open-label, Phase Ia/b Study of Pembrolizumab plus Olaratumab in Patients with Unresectable, Locally Advanced, or Metastatic Soft-Tissue Sarcoma. Clin. Cancer Res. 2023, 29, 3320–3328. [Google Scholar] [CrossRef]
- Paoluzzi, L.; Cacavio, A.; Ghesani, M.; Karambelkar, A.; Rapkiewicz, A.; Weber, J.; Rosen, G. Response to anti-PD1 therapy with nivolumab in metastatic sarcomas. Clin. Sarcoma Res. 2016, 6, 24. [Google Scholar] [CrossRef]
- Wilky, B.A.; Trucco, M.M.; Subhawong, T.K.; Florou, V.; Park, W.; Kwon, D.; Wieder, E.D.; Kolonias, D.; Rosenberg, A.E.; Kerr, D.A.; et al. Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: A single-centre, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 837–848. [Google Scholar] [CrossRef]
- Xie, L.; Xu, J.; Sun, X.; Guo, W.; Gu, J.; Liu, K.; Zheng, B.; Ren, T.; Huang, Y.; Tang, X.; et al. Apatinib plus camrelizumab (anti-PD1 therapy, SHR-1210) for advanced osteosarcoma (APFAO) progressing after chemotherapy: A single-arm, open-label, phase 2 trial. J. Immunother. Cancer 2020, 8, e000798. [Google Scholar] [CrossRef]
- Palmerini, E.; Lopez-Pousa, A.; Grignani, G.; Redondo, A.; Hindi, N.; Stacchiotti, S.; Sebio, A.; Lopez-Martin, J.A.; Morales, C.M.V.; Martinez-Trufero, J.; et al. IMMUNOSARC: A collaborative Spanish (GEIS) and Italian (ISG) sarcoma groups phase I/II trial of sunitinib and nivolumab in advanced soft tissue and bone sarcoma: Results from the phase II part, bone sarcoma cohort. J. Clin. Oncol. 2020, 38, 11522. [Google Scholar] [CrossRef]
- Martin-Broto, J.; Hindi, N.; Grignani, G.; Martinez-Trufero, J.; Redondo, A.; Valverde, C.; Stacchiotti, S.; Lopez-Pousa, A.; D’Ambrosio, L.; Gutierrez, A.; et al. Nivolumab and sunitinib combination in advanced soft tissue sarcomas: A multicenter, single-arm, phase Ib/II trial. J. Immunother. Cancer 2020, 8, e001561. [Google Scholar] [CrossRef]
- Kim, H.S.; Cho, H.J.; Yun, K.-H.; Lee, Y.H.; Kim, S.H.; Baek, W.; Jeon, M.K. Durvalumab and pazopanib in patients with advanced soft tissue sarcoma: A single-center, single-arm, phase 2 trial. J. Clin. Oncol. 2021, 39, 11551. [Google Scholar] [CrossRef]
- Cousin, S.; Bellera, C.; Guegan, J.P.; Valentin, T.; Bahleda, R.; Metges, J.P.; Cassier, P.A.; Cantarel, C.; Spalato Ceruso, M.; Kind, M.; et al. 1494P Regomune—A phase II study of regorafenib + avelumab in solid tumors: Results of the soft tissue sarcoma (STS) cohort. Ann. Oncol. 2022, 33, S1230. [Google Scholar] [CrossRef]
- Grilley-Olson, J.E.; Allred, J.B.; Schuetze, S.; Davis, E.J.; Wagner, M.J.; Poklepovic, A.S.; Waechter, B.; Riedel, R.F.; Welliver, M.X.; Berg, S.A.; et al. A multicenter phase II study of cabozantinib + nivolumab for patients (pts) with advanced angiosarcoma (AS) previously treated with a taxane (Alliance A091902). J. Clin. Oncol. 2023, 41, 11503. [Google Scholar] [CrossRef]
- Eulo, V.; Toeniskoetter, J.; Ruff, T.; Luo, J.; Kemp, L.; Tellez, C.M.; Weiss, M.C.; Hirbe, A.C.; Meyer, C.F.; Elias, A.D.; et al. Randomized phase II trial of cabozantinib combined with PD-1 and CTLA-4 inhibition versus cabozantinib in metastatic soft tissue sarcoma. J. Clin. Oncol. 2023, 41, LBA11504. [Google Scholar] [CrossRef]
- Toulmonde, M.; Penel, N.; Adam, J.; Chevreau, C.; Blay, J.Y.; Le Cesne, A.; Bompas, E.; Piperno-Neumann, S.; Cousin, S.; Grellety, T.; et al. Use of PD-1 Targeting, Macrophage Infiltration, and IDO Pathway Activation in Sarcomas: A Phase 2 Clinical Trial. JAMA Oncol. 2018, 4, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Nathenson, M.; Choy, E.; Carr, N.D.; Hibbard, H.D.; Mazzola, E.; Catalano, P.J.; Thornton, K.A.; Morgan, J.A.; Cote, G.M.; Merriam, P.; et al. Phase II study of eribulin and pembrolizumab in patients (pts) with metastatic soft tissue sarcomas (STS): Report of LMS cohort. J. Clin. Oncol. 2020, 38, 11559. [Google Scholar] [CrossRef]
- Smrke, A.; Ostler, A.; Napolitano, A.; Vergnano, M.; Asare, B.; Fotiadis, N.; Thway, K.; Zaidi, S.; Miah, A.B.; van der Graaf, W.; et al. 1526MO GEMMK: A phase I study of gemcitabine (gem) and pembrolizumab (pem) in patients (pts) with leiomyosarcoma (LMS) and undifferentiated pleomorphic sarcoma UPS). Ann. Oncol. 2021, 32, S1114. [Google Scholar] [CrossRef]
- Wagner, M.J.; Zhang, Y.; Cranmer, L.D.; Loggers, E.T.; Black, G.; McDonnell, S.; Maxwell, S.; Johnson, R.; Moore, R.; Hermida de Viveiros, P.; et al. A Phase 1/2 Trial Combining Avelumab and Trabectedin for Advanced Liposarcoma and Leiomyosarcoma. Clin. Cancer Res. 2022, 28, 2306–2312. [Google Scholar] [CrossRef]
- Toulmonde, M.; Brahmi, M.; Giraud, A.; Chakiba, C.; Bessede, A.; Kind, M.; Toulza, E.; Pulido, M.; Albert, S.; Guégan, J.-P.; et al. Trabectedin plus Durvalumab in Patients with Advanced Pretreated Soft Tissue Sarcoma and Ovarian Carcinoma (TRAMUNE): An Open-Label, Multicenter Phase Ib Study. Clin. Cancer Res. 2022, 28, 1765–1772. [Google Scholar] [CrossRef]
- Adnan, N.; Sekhon, S.; Chawla, S.P.; Kim, T.T.; Chua-Alcala, V.S.; Fernando, M.; Ahari, A.; Feske, W.; Quon, D.V.; Gordon, E.M. GALLANT: A phase 2 study using metronomic gemcitabine, doxorubicin, nivolumab, and docetaxel as second/third-line therapy for advanced sarcoma (NCT04535713). J. Clin. Oncol. 2022, 40, 11518. [Google Scholar] [CrossRef]
- Andreou, D.; Flörcken, A.; Groß, T.; Richter, S.; Kessler, T.; Kortüm, M.; Schmidt, C.A.; Kasper, B.; Wardelmann, E.; Benedict, A.; et al. Efficacy and safety of nivolumab and trabectedin in pretreated patients with advanced soft tissue sarcomas (STS): Results of a phase II trial of the German Interdisciplinary Sarcoma Group (GISG-15, NitraSarc). J. Clin. Oncol. 2023, 41, 11500. [Google Scholar] [CrossRef]
- Beveridge, R.D.; Moura, D.; Ramos, R.; Martinez-Trufero, J.; Carrasco-Garcia, I.; Lopez-Pousa, A.; Billalabeitia, E.G.; Gutierrez, A.; Jurado, J.C.; Sebio, A.; et al. ImmunoSarc2: A Spanish Sarcoma Group (GEIS) phase Ib trial of doxorubicin and dacarbazine plus nivolumab in first line treatment of advanced leiomyosarcoma. J. Clin. Oncol. 2023, 41, 11502. [Google Scholar] [CrossRef]
- Pollack, S.M.; Redman, M.W.; Baker, K.K.; Wagner, M.J.; Schroeder, B.A.; Loggers, E.T.; Trieselmann, K.; Copeland, V.C.; Zhang, S.; Black, G.; et al. Assessment of Doxorubicin and Pembrolizumab in Patients with Advanced Anthracycline-Naive Sarcoma: A Phase 1/2 Nonrandomized Clinical Trial. JAMA Oncol. 2020, 6, 1778–1782. [Google Scholar] [CrossRef] [PubMed]
- Livingston, M.B.; Jagosky, M.H.; Robinson, M.M.; Ahrens, W.A.; Benbow, J.H.; Farhangfar, C.J.; Foureau, D.M.; Maxwell, D.M.; Baldrige, E.A.; Begic, X.; et al. Phase II Study of Pembrolizumab in Combination with Doxorubicin in Metastatic and Unresectable Soft-Tissue Sarcoma. Clin. Cancer Res. 2021, 27, 6424–6431. [Google Scholar] [CrossRef] [PubMed]
- Maleddu, A.; Mailhot, A.; Cartwright, C.; Gao, D.; Tellez, C.M.; Powers, K.; Kemp, L.; Therrien, N.; Patel, J.M.; Grossman, J.E.; et al. A single-arm, open-label phase 2 trial of doxorubicin plus zalifrelimab, a CTLA-4 inhibitor, with balstilimab, a PD-1 inhibitor, in patients with advanced/metastatic soft tissue sarcomas. J. Clin. Oncol. 2023, 41, 11501. [Google Scholar] [CrossRef]
- Gordon, E.M.; Chawla, S.P.; Tellez, W.A.; Younesi, E.; Thomas, S.; Chua-Alcala, V.S.; Chomoyan, H.; Valencia, C.; Brigham, D.A.; Moradkhani, A.; et al. SAINT: A Phase I/Expanded Phase II Study Using Safe Amounts of Ipilimumab, Nivolumab and Trabectedin as First-Line Treatment of Advanced Soft Tissue Sarcoma. Cancers 2023, 15, 906. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Yang, H.; Chon, H.J.; Kim, C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp. Mol. Med. 2020, 52, 1475–1485. [Google Scholar] [CrossRef]
- Fleuren, E.D.G.; Terry, R.L.; Meyran, D.; Omer, N.; Trapani, J.A.; Haber, M.; Neeson, P.J.; Ekert, P.G. Enhancing the Potential of Immunotherapy in Paediatric Sarcomas: Breaking the Immunosuppressive Barrier with Receptor Tyrosine Kinase Inhibitors. Biomedicines 2021, 9, 1798. [Google Scholar] [CrossRef]
- Cho, H.J.; Yun, K.-H.; Shin, S.-J.; Lee, Y.H.; Kim, S.H.; Baek, W.; Han, Y.D.; Kim, S.K.; Lee, J.; Cho, I.; et al. Abstract CT038: Comprehensive molecular characterization of clinical response to durvalumab plus pazopanib combination in patients with advanced soft tissue sarcomas: A phase 2 clinical trial. Cancer Res. 2023, 83, CT038. [Google Scholar] [CrossRef]
- Roulleaux Dugage, M.; Nassif, E.F.; Italiano, A.; Bahleda, R. Improving Immunotherapy Efficacy in Soft-Tissue Sarcomas: A Biomarker Driven and Histotype Tailored Review. Front. Immunol. 2021, 12, 775761. [Google Scholar] [CrossRef]
- Wagner, M.; He, Q.; Zhang, Y.; Cranmer, L.; Loggers, E.; McDonnell, S.; Maxwell, S.; Pollack, S. 796 A phase I/II trial combining avelumab and trabectedin for advanced liposarcoma and leiomyosarcoma. J. Immunother. Cancer 2020, 8, A476. [Google Scholar] [CrossRef]
- Roland, C.L.; Keung, E.Z.-Y.; Lazar, A.J.; Torres, K.E.; Wang, W.-L.; Guadagnolo, A.; Bishop, A.J.; Lin, H.Y.; Hunt, K.; Feig, B.W.; et al. Preliminary results of a phase II study of neoadjuvant checkpoint blockade for surgically resectable undifferentiated pleomorphic sarcoma (UPS) and dedifferentiated liposarcoma (DDLPS). J. Clin. Oncol. 2020, 38, 11505. [Google Scholar] [CrossRef]
- Keung, E.Z.; Tsai, J.-W.; Ali, A.M.; Cormier, J.N.; Bishop, A.J.; Guadagnolo, B.A.; Torres, K.E.; Somaiah, N.; Hunt, K.K.; Wargo, J.A.; et al. Analysis of the immune infiltrate in undifferentiated pleomorphic sarcoma of the extremity and trunk in response to radiotherapy: Rationale for combination neoadjuvant immune checkpoint inhibition and radiotherapy. OncoImmunology 2018, 7, e1385689. [Google Scholar] [CrossRef]
- Chandran, S.S.; Somerville, R.P.T.; Yang, J.C.; Sherry, R.M.; Klebanoff, C.A.; Goff, S.L.; Wunderlich, J.R.; Danforth, D.N.; Zlott, D.; Paria, B.C.; et al. Treatment of metastatic uveal melanoma with adoptive transfer of tumour-infiltrating lymphocytes: A single-centre, two-stage, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 792–802. [Google Scholar] [CrossRef]
- Zhao, Y.; Deng, J.; Rao, S.; Guo, S.; Shen, J.; Du, F.; Wu, X.; Chen, Y.; Li, M.; Chen, M.; et al. Tumor Infiltrating Lymphocyte (TIL) Therapy for Solid Tumor Treatment: Progressions and Challenges. Cancers 2022, 14, 4160. [Google Scholar] [CrossRef]
- Balch, C.M.; Riley, L.B.; Bae, Y.J.; Salmeron, M.A.; Platsoucas, C.D.; von Eschenbach, A.; Itoh, K. Patterns of human tumor-infiltrating lymphocytes in 120 human cancers. Arch. Surg. 1990, 125, 200–205. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Mullinax, J.E.; Hall, M.; Beatty, M.; Weber, A.M.; Sannasardo, Z.; Svrdlin, T.; Hensel, J.; Bui, M.; Richards, A.; Gonzalez, R.J.; et al. Expanded Tumor-infiltrating Lymphocytes from Soft Tissue Sarcoma Have Tumor-specific Function. J. Immunother. 2021, 44, 63–70. [Google Scholar] [CrossRef]
- Zhou, X.; Wu, J.; Duan, C.; Liu, Y. Retrospective Analysis of Adoptive TIL Therapy plus Anti-PD1 Therapy in Patients with Chemotherapy-Resistant Metastatic Osteosarcoma. J. Immunol. Res. 2020, 2020, 7890985. [Google Scholar] [CrossRef]
- Thanindratarn, P.; Dean, D.C.; Nelson, S.D.; Hornicek, F.J.; Duan, Z. Chimeric antigen receptor T (CAR-T) cell immunotherapy for sarcomas: From mechanisms to potential clinical applications. Cancer Treat. Rev. 2020, 82, 101934. [Google Scholar] [CrossRef]
- Richards, R.M.; Sotillo, E.; Majzner, R.G. CAR T Cell Therapy for Neuroblastoma. Front. Immunol. 2018, 9, 2380. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) –Specific Chimeric Antigen Receptor–Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef]
- Hegde, M.; DeRenzo, C.C.; Zhang, H.; Mata, M.; Gerken, C.; Shree, A.; Yi, Z.; Brawley, V.; Dakhova, O.; Wu, M.-F.; et al. Expansion of HER2-CAR T cells after lymphodepletion and clinical responses in patients with advanced sarcoma. J. Clin. Oncol. 2017, 35, 10508. [Google Scholar] [CrossRef]
- Tsimberidou, A.M.; van Morris, K.; Vo, H.H.; Eck, S.; Lin, Y.F.; Rivas, J.M.; Andersson, B.S. T-cell receptor-based therapy: An innovative therapeutic approach for solid tumors. J. Hematol. Oncol. 2021, 14, 102. [Google Scholar] [CrossRef]
- Akers, S.N.; Odunsi, K.; Karpf, A.R. Regulation of cancer germline antigen gene expression: Implications for cancer immunotherapy. Future Oncol. 2010, 6, 717–732. [Google Scholar] [CrossRef]
- Kakimoto, T.; Matsumine, A.; Kageyama, S.; Asanuma, K.; Matsubara, T.; Nakamura, T.; Iino, T.; Ikeda, H.; Shiku, H.; Sudo, A. Immunohistochemical expression and clinicopathological assessment of the cancer testis antigens NY-ESO-1 and MAGE-A4 in high-grade soft-tissue sarcoma. Oncol. Lett. 2019, 17, 3937–3943. [Google Scholar] [CrossRef]
- Iura, K.; Kohashi, K.; Ishii, T.; Maekawa, A.; Bekki, H.; Otsuka, H.; Yamada, Y.; Yamamoto, H.; Matsumoto, Y.; Iwamoto, Y.; et al. MAGEA4 expression in bone and soft tissue tumors: Its utility as a target for immunotherapy and diagnostic marker combined with NY-ESO-1. Virchows Arch. 2017, 471, 383–392. [Google Scholar] [CrossRef]
- Ramachandran, I.; Lowther, D.E.; Dryer-Minnerly, R.; Wang, R.; Fayngerts, S.; Nunez, D.; Betts, G.; Bath, N.; Tipping, A.J.; Melchiori, L.; et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J. Immunother. Cancer 2019, 7, 276. [Google Scholar] [CrossRef]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef]
- Pan, Q.; Weng, D.; Liu, J.; Han, Z.; Ou, Y.; Xu, B.; Peng, R.; Que, Y.; Wen, X.; Yang, J.; et al. Phase 1 clinical trial to assess safety and efficacy of NY-ESO-1-specific TCR T cells in HLA-A∗02:01 patients with advanced soft tissue sarcoma. Cell Rep. Med. 2023, 4, 101133. [Google Scholar] [CrossRef]
- Kawai, A.; Ishihara, M.; Nakamura, T.; Kitano, S.; Iwata, S.; Takada, K.; Emori, M.; Kato, K.; Endo, M.; Matsumoto, Y.; et al. Safety and Efficacy of NY-ESO-1 Antigen-Specific T-Cell Receptor Gene-Transduced T Lymphocytes in Patients with Synovial Sarcoma: A Phase I/II Clinical Trial. Clin. Cancer Res. 2023, 29, 5069–5078. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Tine, B.A.V.; Attia, S.; Blay, J.-Y.; Strauss, S.J.; Morales, C.M.V.; Razak, A.R.A.; Winkle, E.V.; Trivedi, T.; Biswas, S.; et al. SPEARHEAD-1: A phase 2 trial of afamitresgene autoleucel (Formerly ADP-A2M4) in patients with advanced synovial sarcoma or myxoid/round cell liposarcoma. J. Clin. Oncol. 2021, 39, 11504. [Google Scholar] [CrossRef]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef]
- Bai, Y.; Hui, P.; Du, X.; Su, X. Updates to the antitumor mechanism of oncolytic virus. Thorac. Cancer 2019, 10, 1031–1035. [Google Scholar] [CrossRef]
- Turnbull, S.; West, E.J.; Scott, K.J.; Appleton, E.; Melcher, A.; Ralph, C. Evidence for Oncolytic Virotherapy: Where Have We Got to and Where Are We Going? Viruses 2015, 7, 6291–6312. [Google Scholar] [CrossRef]
- Le Boeuf, F.; Selman, M.; Son, H.H.; Bergeron, A.; Chen, A.; Tsang, J.; Butterwick, D.; Arulanandam, R.; Forbes, N.E.; Tzelepis, F.; et al. Oncolytic Maraba Virus MG1 as a Treatment for Sarcoma. Int. J. Cancer 2017, 141, 1257–1264. [Google Scholar] [CrossRef]
- Monga, V.; Miller, B.J.; Tanas, M.; Boukhar, S.; Allen, B.; Anderson, C.; Stephens, L.; Hartwig, S.; Varga, S.; Houtman, J.; et al. Intratumoral talimogene laherparepvec injection with concurrent preoperative radiation in patients with locally advanced soft-tissue sarcoma of the trunk and extremities: Phase IB/II trial. J. Immunother. Cancer 2021, 9, e003119. [Google Scholar] [CrossRef]
- Kelly, C.M.; Antonescu, C.R.; Bowler, T.; Munhoz, R.; Chi, P.; Dickson, M.A.; Gounder, M.M.; Keohan, M.L.; Movva, S.; Dholakia, R.; et al. Objective Response Rate Among Patients with Locally Advanced or Metastatic Sarcoma Treated with Talimogene Laherparepvec in Combination with Pembrolizumab: A Phase 2 Clinical Trial. JAMA Oncol. 2020, 6, 402–408. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Agulnik, M.; Ryan, C.W.; Milhem, M.M.; George, S.; Jones, R.L.; Chmielowski, B.; Tine, B.A.V.; Tawbi, H.A.-H.; Elias, A.D.; et al. Trivalent ganglioside vaccine and immunologic adjuvant versus adjuvant alone in metastatic sarcoma patients rendered disease-free by surgery: A randomized phase 2 trial. J. Clin. Oncol. 2014, 32, 10520. [Google Scholar] [CrossRef]
- Kawaguchi, S.; Tsukahara, T.; Ida, K.; Kimura, S.; Murase, M.; Kano, M.; Emori, M.; Nagoya, S.; Kaya, M.; Torigoe, T.; et al. SYT-SSX breakpoint peptide vaccines in patients with synovial sarcoma: A study from the Japanese Musculoskeletal Oncology Group. Cancer Sci. 2012, 103, 1625–1630. [Google Scholar] [CrossRef]
- Chawla, S.P.; van Tine, B.A.; Pollack, S.M.; Ganjoo, K.N.; Elias, A.D.; Riedel, R.F.; Attia, S.; Choy, E.; Okuno, S.H.; Agulnik, M.; et al. Phase II Randomized Study of CMB305 and Atezolizumab Compared with Atezolizumab Alone in Soft-Tissue Sarcomas Expressing NY-ESO-1. J. Clin. Oncol. 2022, 40, 1291–1300. [Google Scholar] [CrossRef]
- Yu, R.K.; Tsai, Y.T.; Ariga, T.; Yanagisawa, M. Structures, biosynthesis, and functions of gangliosides—An overview. J. Oleo Sci. 2011, 60, 537–544. [Google Scholar] [CrossRef]
- Berois, N.; Osinaga, E. Glycobiology of neuroblastoma: Impact on tumor behavior, prognosis, and therapeutic strategies. Front. Oncol. 2014, 4, 114. [Google Scholar] [CrossRef]
- Lopez, P.H.; Schnaar, R.L. Gangliosides in cell recognition and membrane protein regulation. Curr. Opin. Struct. Biol. 2009, 19, 549–557. [Google Scholar] [CrossRef]
- Battula, V.L.; Shi, Y.; Evans, K.W.; Wang, R.Y.; Spaeth, E.L.; Jacamo, R.O.; Guerra, R.; Sahin, A.A.; Marini, F.C.; Hortobagyi, G.; et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J. Clin. Investig. 2012, 122, 2066–2078. [Google Scholar] [CrossRef]
- Krengel, U.; Bousquet, P.A. Molecular recognition of gangliosides and their potential for cancer immunotherapies. Front. Immunol. 2014, 5, 325. [Google Scholar] [CrossRef]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef]
- Cavdarli, S.; Groux-Degroote, S.; Delannoy, P. Gangliosides: The Double-Edge Sword of Neuro-Ectodermal Derived Tumors. Biomolecules 2019, 9, 311. [Google Scholar] [CrossRef]
- Yoshida, S.; Fukumoto, S.; Kawaguchi, H.; Sato, S.; Ueda, R.; Furukawa, K. Ganglioside G(D2) in small cell lung cancer cell lines: Enhancement of cell proliferation and mediation of apoptosis. Cancer Res. 2001, 61, 4244–4252. [Google Scholar]
- Valentino, L.; Moss, T.; Olson, E.; Wang, H.J.; Elashoff, R.; Ladisch, S. Shed tumor gangliosides and progression of human neuroblastoma. Blood 1990, 75, 1564–1567. [Google Scholar] [CrossRef]
- Yeh, S.D.; Larson, S.M.; Burch, L.; Kushner, B.H.; Laquaglia, M.; Finn, R.; Cheung, N.K. Radioimmunodetection of neuroblastoma with iodine-131-3F8: Correlation with biopsy, iodine-131-metaiodobenzylguanidine and standard diagnostic modalities. J. Nucl. Med. 1991, 32, 769–776. [Google Scholar]
- Bailey, K.; Cost, C.; Davis, I.; Glade-Bender, J.; Grohar, P.; Houghton, P.; Isakoff, M.; Stewart, E.; Laack, N.; Yustein, J.; et al. Emerging novel agents for patients with advanced Ewing sarcoma: A report from the Children’s Oncology Group (COG) New Agents for Ewing Sarcoma Task Force. F1000Research 2019, 8, 493. [Google Scholar] [CrossRef]
- Kailayangiri, S.; Altvater, B.; Meltzer, J.; Pscherer, S.; Luecke, A.; Dierkes, C.; Titze, U.; Leuchte, K.; Landmeier, S.; Hotfilder, M.; et al. The ganglioside antigen G(D2) is surface-expressed in Ewing sarcoma and allows for MHC-independent immune targeting. Br. J. Cancer 2012, 106, 1123–1133. [Google Scholar] [CrossRef]
- Roth, M.; Linkowski, M.; Tarim, J.; Piperdi, S.; Sowers, R.; Geller, D.; Gill, J.; Gorlick, R. Ganglioside GD2 as a therapeutic target for antibody-mediated therapy in patients with osteosarcoma. Cancer 2014, 120, 548–554. [Google Scholar] [CrossRef]
- Zhu, W.; Mao, X.; Wang, W.; Chen, Y.; Li, D.; Li, H.; Dou, P. Anti-ganglioside GD2 monoclonal antibody synergizes with cisplatin to induce endoplasmic reticulum-associated apoptosis in osteosarcoma cells. Pharmazie 2018, 73, 80–86. [Google Scholar] [CrossRef]
- Chang, H.R.; Cordon-Cardo, C.; Houghton, A.N.; Cheung, N.K.; Brennan, M.F. Expression of disialogangliosides GD2 and GD3 on human soft tissue sarcomas. Cancer 1992, 70, 633–638. [Google Scholar] [CrossRef]
- Saraf, A.J.; Dickman, P.S.; Hingorani, P. Disialoganglioside GD2 Expression in Pediatric Rhabdomyosarcoma: A Case Series and Review of the Literature. J. Pediatr. Hematol./Oncol. 2019, 41, 118–120. [Google Scholar] [CrossRef]
- Huang, C.Y.; Ye, Z.H.; Huang, M.Y.; Lu, J.J. Regulation of CD47 expression in cancer cells. Transl. Oncol. 2020, 13, 100862. [Google Scholar] [CrossRef]
- Ozaniak, A.; Smetanova, J.; Bartolini, R.; Rataj, M.; Capkova, L.; Hacek, J.; Fialova, M.; Krupickova, L.; Striz, I.; Lischke, R.; et al. A novel anti-CD47-targeted blockade promotes immune activation in human soft tissue sarcoma but does not potentiate anti-PD-1 blockade. J. Cancer Res. Clin. Oncol. 2023, 149, 3789–3801. [Google Scholar] [CrossRef]
- Theruvath, J.; Menard, M.; Smith, B.A.H.; Linde, M.H.; Coles, G.L.; Dalton, G.N.; Wu, W.; Kiru, L.; Delaidelli, A.; Sotillo, E.; et al. Anti-GD2 synergizes with CD47 blockade to mediate tumor eradication. Nat. Med. 2022, 28, 333–344. [Google Scholar] [CrossRef]
- Hingorani, P.; Krailo, M.; Buxton, A.; Hutson, P.; Sondel, P.M.; Diccianni, M.; Yu, A.; Morris, C.D.; Womer, R.B.; Crompton, B.; et al. Phase 2 study of anti-disialoganglioside antibody, dinutuximab, in combination with GM-CSF in patients with recurrent osteosarcoma: A report from the Children’s Oncology Group. Eur. J. Cancer 2022, 172, 264–275. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Trial/Design | Phase | Agent/ Intervention | Indication/Prior Lines of Treatment | Evaluated Patients (n) and Tumor Subtypes | ORR (%) | PFS (Weeks (w) or Months (m)) | OS (w or m) | Outcomes in Subtypes/Notes |
|---|---|---|---|---|---|---|---|---|
| ICI Monotherapy or Combination | ||||||||
| Maki et al., 2013 [31] | Phase II | ipilimumab | Locally recurrent or metastatic SS, at least 1 prior line treatment | 6 SS | 0% | 1.85 m | 8.75 m | |
| Tawbi et al. (SARC028) 2017 [30] | Phase II | pembrolizumab | Advanced/metastatic STS or bone sarcoma, at least 1 prior line treatment | 40 BS cohort (22 OS, 13 ES, 5 CS) | - | 8 w | 52 w | 1 PR in CS and 1 PR in OST |
| 40 STS cohort (10 LMS, 10 LPS, 10 SS, 10 UPS) | - | 18 w | 49 w | 1 CR and 3 PRs in UPS, <2 PRs in LPS, 1 PR in SS | ||||
| Ben-Ami et al., 2017 [32] | Phase II | nivolumab | Advanced or metastatic uterine LMS, at least 1 prior line of treatment | 12 LMS | 0% | 1.8 m | - | |
| D’Angelo et al. Alliance A091401 2018 [38] | Phase II | nivolumab/ipilimumab vs. nivolumab | Advanced or metastatic BS and STS, at least 1 prior line of treatment | Nivolumab/ipilimumab: 42 (3 AS, 4 BS, 14 LMS, 2 LPS, 6 SCS, 2 SS, 6 UPS/MFH, 1 unspecified sarcoma, 4 others) | 16% | 4.1 m | 10.7 m | Response in uterine LMS, non-uterine LMS, MFS, UPS/MFH, AS |
| Nivolumab: 43 (5 BS, 15 LMS, 3 LPS, 2 unspecified sarcoma, 5 SCS, 2 SS, 5 UPS, 6 others | 5% | 1.7 m | 10.7 m | 1 PR in ASPS and 1 PR in non-uterine LMS | ||||
| Uboha et al., 2019 [39] | Phase II | spartalizumab + LAG525 (anti-LAG3) | Advanced solid tumors and hematologic malignancies | 10 | CBR 40% | - | - | Sarcoma cohort did not meet the expansion criterion |
| Zhou et al., 2020 [42] | Retrospective | nivolumab + ipilimumab | Advanced or metastatic STS, 87% received at least 1 prior line | 38 (9 LMS, 8 Sarcoma NOS, 6 LPS, 5 MFS, 3 MPNST, 2 SFT, 1 Breast AS, 1 FDFP, 1 RMS, 1 SS) | 15% | 2.7 m | 12 m | CR in 1 MFS, 1 PR in each MPNST, SFT, MFS, DDLS, and sarcoma NOS |
| Naqash et al., 2021 [40] | Phase II | pembrolizumab | Advanced or metastatic ASPS | 43 | 37.2% | - | - | |
| Blay et al. French AcSé 2021 [34] | Phase II | pembrolizumab | Advanced rare sarcoma | 98 (34 chordoma, 14 ASPS, 11 SMRT, 8 DSCRT, 31 other histotypes) | 15.3 | 2.75 m | 19.7 m | Highest ORR in chordoma, ASPS, SMRT, DSCRT |
| Delyon et al., 2022 [43] | Phase II | pembrolizumab | Classic/endemic Kaposi sarcoma with extensive cutaneous extension, 71% had at least 1 prior line | 17 (8 classic KS and 9 endemic KS) | 71% | - | - | |
| Zer et al., 2022 [44] | Phase II | ipilimumab and nivolumab | Classic Kaposi sarcoma, at least 1 prior line of treatment | 11 | 45% | not reached | - | |
| Somaiah et al., 2022 [45] | Phase II | durvalumab + tremelimumab. | Advanced or metastatic sarcoma (BS and STS), 91% had at least 1 prior line | 57 (3 DDLPS, 2WDLPS, 1 PLS, 5 AS, 5 LMS, 5 UPS, 5 SS, 1 CDOS, 4 COS, 10 ASPS, 5 chordomas, 11 other sarcomas) | 12% | 2.8 m | 21.6 m | ASPS ORR 40% |
| ICI Combination with TKI | ||||||||
| Schoffski et al., 2016 [46] | Ia/Ib | pembrolizumab + olaratumab (monoclonal antibody against platelet derived growth factor receptor alpha) | Advanced or metastatic STS, 92% had at least 1 prior line | 28 | 21.4% | 2.7 m | 14.8 m | |
| Paoluzzie et al., 2016 [47] | Retrospective study | durvalumab + pazopanib | Metastatic STS and BS, median 2 prior lines of treatment | 28 (24 STS, 4 BS with 24 evaluable patients) | 10% | - | - | 3 PRs (1 DDCS with nivolimab alone), 1 EpS, 1 MOS) |
| Wilky et al., 2019 [48] | Phase II | pembrolizumab + axitinib | Advanced or metastatic STS, 81% with at least 1 prior line of treatment | 33 (12 ASPS, 6 LMS (4 uterine), 5 High-grade PS, 2 DDLPS, 8 other histotypes) | 25% | 4.7 m | 18.7 m | ASPS ORR 50% |
| Xie et al. APFAO trial 2020 [49] | Phase II | camrelizumab + apatinib | Advanced or metastatic OS, at least 1 prior line of treatment | 43 (OS including osteoblastic, chondroblastic, fibroblastic and small cell) | 20.1% | 6.2 m | 11.3 m | |
| Palmerini et al. IMMUNOSARC 2020 [50] | Phase II | nivolumab + sunitinib | Advanced BS cohort, at least 1 prior line of treatment | 40 (17 OS, 14 CS, 8 ES, 1 bone UPS, 4 DDCS) | 5% | 3.7 m | 14.2 m | 1 CR in DDCS and 1 PR in OS |
| Martin-Broto et al. IMMUNOSARC 2020 [51] | Phase I/II | nivolumab + sunitinib | Metastatic STS, at least 1 prior line of treatment | 52 (9 SS, 8 UPS, 7 clear cell sarcoma, 7 SFT, 7 EpS, 5 AS, 4 ESMCS, 4 ASPS, 1 EHET) | 21% | 5.6 m | - | 1 CR in AS, 2 PRs in ASPS, 1 PR in ESMCS and 1 PR in SS |
| Kim et al., 2021 [52] | Phase II | durvalumab + pazopanib | Advanced or metastatic STS, at least 1 prior line of treatment | 46 | 28.3% | 7.7 m | - | Objective responses in ASPS, AS, UPS, DSRCT |
| Cousin et al. REGOMUNE 2022 [53] | Phase II | avelumab + regorafenib | Advanced or metastatic STS, at least 1 prior line of treatment | 43 (22 LMS, 9 SS, 4 LPS, 4 UPS, 10 other subtypes) | 9.3% | 1.8 m | 15.1 m | |
| Allred et al. Alliance A091902 trial 2023 [54] | Phase II | nivolumab with carbozantinib | Advanced AS, previously treated | 18 (AS including 12 cutaneous, 1 liver, 2 breast, 6 others) | 72% | 9.6 m | 20.5 m | |
| Eulo et al., 2023 [55] | Phase II | nivolumab/ipilimumab + cabozantinib N/I + C | Metastatic STS that lacks translocation, at least 1 prior line of treatment | 69 (N/I + C arm) | 11% | 5.4 m | - | |
| 36 (C only arm) | 6% | 3.8 m | - | |||||
| ICI Combination with Chemotherapy | ||||||||
| Toulmonde et al., 2018 [56] | Phase II | pembrolizumab + metronomic cyclophosphamide | Advanced or metastatic STS, 97% with at least 1 prior line of treatment | 50 (15 LMS, 16 UPS, 16 other sarcomas, 10 GIST) | 2% | 1.4 m | - | |
| Italiano et al. Amended PEMBROSARC 2022 [23] | Phase II | pembrolizumab with metronomic cyclophosphamide | TLS-positive advanced STS, 63% had at least 1 prior line of treatment | 35 (12 WDLPS/DDLPS, 4 LMS, 6 UPS, 3 EpS, 10 other histotypes) | 30% | 6 m PFS 40% | - | PRs: 5 in DDLPS, 3 EpS, 1 LMS SDs: 6 DDLPS, 1 FMS, 1 MFS, 1 uterine LMS, 1 UPS |
| Nathenson et al., 2020 [57] | Phase II | pembrolizumab + eribulin | Metastatic STS, at least 1 prior line of treatment | 19 LMS (11 uterine LMS) | 5.3% | 11.1 w | - | |
| Smrke et al., 2021 [58] | Phase I | pembrolizumab + gemcitabine | Advanced or metastatic LMS, UPS | 13 (2 UPS, 11 LMS) | - | 5.1 m | - | LMS—DCR 73% (8 SDs, 3 PDs) UPS—DCR 100% (2 PRs) |
| Wagner et al., 2022 [59] | Phase I/II | avelumab + trabectedin | Advanced or metastatic LPS and LMS, 86% had at least 1 prior line of treatment | 35, only 23 evaluable (24 with LMS, 11 with LPS | 13% | 8.3 m | 27 m | LMS 4 PRs, 9 SDs LPS 7 SDs |
| Toulmonde et al., 2022 [60] | Phase Ib | durvalumab + trabectedin | Advanced or metastatic STS cohort, at least 1 prior line of treatment | 16 (6 LMS, 2 DDLPS, 8 others) | 7% | 12 m PFS 14.3% | - | |
| Adnan et al. Gallant trial 2022 [61] | Phase II | nivolumab + metronomic gemcitabine, doxorubicin, and docetaxel | Advanced or metastatic STS, at least 1 prior line of treatment | 39 (15 LMS, 4 PS, 4 SS, 3 LPS, 3 OS, 10 others) | 20.5% | 4.6 m | 6.2 m | mPFS 2 m historically in previously treated patients |
| Andreou et al. NITRA-SARC 2023 [62] | Phase II | nivolumab + trabectedin | Advanced or metastatic STS, at least 1 prior line of treatment | Group A—43 (28 LMS and 15 LPS) | - | 5.5 m | 18.7 m | |
| Group B—49 (12 UPS, 11 SCS, 6 FMS, 5 SS, 4 EpS) | - | 2.3 m | 5.6 m | |||||
| Beveridge et al. ImmunoSarc2 Cohort 7b 2023 [63] | Phase Ib | doxorubicin and dacarbazine plus nivolumab and nivolumab maintenance 1 year | Advanced or metastatic LMS, anthracycline naïve patients | 16 LMS | 56% | 8.67 m | - | |
| ICI as Front-line | ||||||||
| Pollack et al., 2020 [64] | Phase I/II | pembrolizumab + doxorubicin | Anthracycline naïve sarcoma Excluding ES, ARMS, ERMS, 76% with no prior line of treatment | 37 (11 LMS, 4 DDLPS, 3 CCCS, 3 UPS, 2 SFT, 2 ESS, 2 EHET, 8 other histotypes | 19% | 8.1 m | 27.6 m | |
| Livingston et al., 2021 [65] | Phase II | pembrolizumab + doxorubicin | Anthracycline Naïve advanced STS, 86.7% had no prior treatment | 28 (7 LPS, 10 LMS, 1 SS, 4 UPS, 2 AS, 6 other histotypes) | 36.7% | 5.7 m | 17 m | The most clinical benefit in UPS, EpAS, LMS, LPS |
| Maleddu et al., 2023 [66] | Phase II | doxorubicin + anti-CTLA-4 zalifrelimab and anti-PD1 balstilimab | Advanced or metastatic STS, no prior doxorubicin or ICI | 28 | 36% | 25.6 m | - | Responses seen in IS, AS, MPNST, LPS, LMS, ESS, UPS, and SEpF. |
| Gordon et al. SAINT Trial 2023 [67] | Phase I/II | ipilimumab + nivolumab and trabectedin | Advanced or metastatic STS, treatment naive | 101 (14 LPS, 26 LMS, 9 UPS, 7 RMS, 5 SS, 4 clear CS, 4 PS, 4 MFS, 3 PNST, 3 MLS, 2 carcinosarcoma, 2 DSRCT, 2 sarcoma NOS) | 25.3% | 6.7 m | 24.6 m | |
| Chen et al., 2021 [36] | Retrospective | nivolumab + ipilimumab vs. nivolumab | Metastatic STS (100% PD-L1-positive tumors: PD-L1 expression > 1%), treatment naive | 74—Nivolumab and ipilimumab arm 43 non-uterine LMS, 20 LPS, 11 SS) | - | 4.1 m | 12.2 m | |
| 76—Nivolumab arm (40 non-uterine LMS, 22 LPS, 14 SS) | - | 2.2 m | 9.2 m | |||||
| Phase | NCT Number/Trial Name | Status | Conditions | Interventions |
|---|---|---|---|---|
| ICI | ||||
| Phase I/II | NCT03138161 SAINT | Recruiting | Unresectable or metastatic STS as first-line treatment | trabectedin + ipilimumab + nivolumab |
| Phase II | NCT04095208 | Recruiting | Advanced or Metastatic STS (TLS+) | nivolumab + relatimab vs. nivolumab |
| Phase II | NCT04802876 ACROPOLI (SOLTI-1904) | Recruiting | Across multiple cancer types with PD1-high mRNA Expressing Tumors—Include Sarcoma Cohort | spartalizumab + tislelizumab |
| ICI with TKIs | ||||
| Phase II | NCT04784247 | Recruiting | Advanced STS | lenvatinib + pembrolizumab |
| Phase II | NCT05182164 | Recruiting | Advance sarcomas: ES, OS, UPS | pembrolizumab + carbozantinib |
| Phase II | NCT04551430 | Active, not recruiting | Metastatic STS | cabozantinib + nivolumab + ipilimumab |
| ICI with Chemotherapy | ||||
| Phase II | NCT03899805 | Active, not recruiting | STS (LPS, LMS, UPS) | eribulin + pembrolizumab |
| Phase II | NCT04535713 GALLANT | Recruiting | Advanced sarcoma | metronomic gemcitabine + doxorubicin + docetaxel + nivolumab |
| Phase I/II | NCT05876715 LINNOVATE | Recruiting | Advanced STS | lurbinectedin + nivolumab + ipilimumab |
| Phase I/II | NCT04577014 | Recruiting | Advanced STS | retifanlimab + gemcitabine + docetaxel |
| Phase II | NCT04028063 | Recruiting | Advanced STS | doxorubicin + zalifrelimab − AGEN1884 + balstilimab − AGEN2034 |
| ICI with Radiation Therapy | ||||
| Phase I | NCT05488366 | Recruiting | Metastatic STS | pembrolizumab + Radiation Therapy |
| Phase II | NCT03307616 | Active, no recruiting | Recurrent or resectable DDLPS and UPS before surgery | Nivolumab ± ipilimumab + Radiation Therapy |
| Phase I/II | NCT03116529 | Active, not recruiting | High-risk STS | durvalumab + tremelimumab + Radiation + Surgery |
| Trial Number | Phase | Intervention | Disease |
|---|---|---|---|
| NCT01953900 | Phase I | Anti-GD2 T-cells in combination with a varicella zoster vaccine and lymphodepleting chemotherapy | GD2-positive sarcoma and neuroblastoma in relapsed or refractory setting |
| NCT04995003 | Phase I | Anti-HER2 CAR T-cells in combination with an immune checkpoint inhibitor drug (pembrolizumab or nivolumab) | HER 2-positive Sarcoma in patients disease progression or recurrence after at least one prior systemic therapy |
| NCT02107963 | Phase I | Administering escalating doses of autologous anti-GD2-CAR T-cells | Osteosarcoma, GD2+ solid tumors that recurred or progressed on treatment |
| NCT00902044 | Phase I | Anti-HER2 CAR T-cells with fludarabine and cyclophosphamide | Refractory HER2-positive sarcoma or metastatic HER2-positive sarcoma with disease progression after receiving at least one prior systemic therapy |
| NCT03721068 | Phase I | Anti-GD2 CAR T-cells, fludarabine and cyclophosphamide | Relapsed refractory osteosarcoma and neuroblastoma |
| Clinical Trial | Phase | Intervention | Disease |
|---|---|---|---|
| NCT03462316 | Phase I | Anti-NY-ESO-1 (TCR Affinity-Enhancing Specific T-cell Therapy) | Advanced bone and soft-tissue sarcoma that failed first-line treatment |
| NCT05296564 | Phase I | Anti-HBI 0201-ESO TCRT (anti-NY-ESO-1 TCR-Gene Engineered Lymphocytes) | NY-ESO-1—Expressing Metastatic cancers (synovial sarcoma, STS, etc.) that failed first-line or second-line treatment, recurrence of disease, progression of disease |
| NCT03132922 | Phase I | Genetically Engineered Anti-MAGE-A4 | MAGE-A4-Positive Tumors (synovial sarcoma, myxoid round-cell liposarcoma) failed first line of therapy |
| NCT | Phase | Intervention | Disease |
|---|---|---|---|
| NCT01241162 | Phase I | Mature DC pulsed with peptides derived from NY-ESO-1, MAGE-A1, and MAGE-A3 for vaccine production. | Relapsed refractory Ewing sarcoma, osteogenic sarcoma, rhabdomyosarcoma or synovial sarcoma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalal, S.; Shan, K.S.; Thaw Dar, N.N.; Hussein, A.; Ergle, A. Role of Immunotherapy in Sarcomas. Int. J. Mol. Sci. 2024, 25, 1266. https://doi.org/10.3390/ijms25021266
Dalal S, Shan KS, Thaw Dar NN, Hussein A, Ergle A. Role of Immunotherapy in Sarcomas. International Journal of Molecular Sciences. 2024; 25(2):1266. https://doi.org/10.3390/ijms25021266
Chicago/Turabian StyleDalal, Shivani, Khine Swe Shan, Nyein Nyein Thaw Dar, Atif Hussein, and Alejandra Ergle. 2024. "Role of Immunotherapy in Sarcomas" International Journal of Molecular Sciences 25, no. 2: 1266. https://doi.org/10.3390/ijms25021266
APA StyleDalal, S., Shan, K. S., Thaw Dar, N. N., Hussein, A., & Ergle, A. (2024). Role of Immunotherapy in Sarcomas. International Journal of Molecular Sciences, 25(2), 1266. https://doi.org/10.3390/ijms25021266