
Missing Heritability in Albinism: Deep Characterization of a Hungarian Albinism Cohort Raises the Possibility of the Digenic Genetic Background of the Disease

 , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
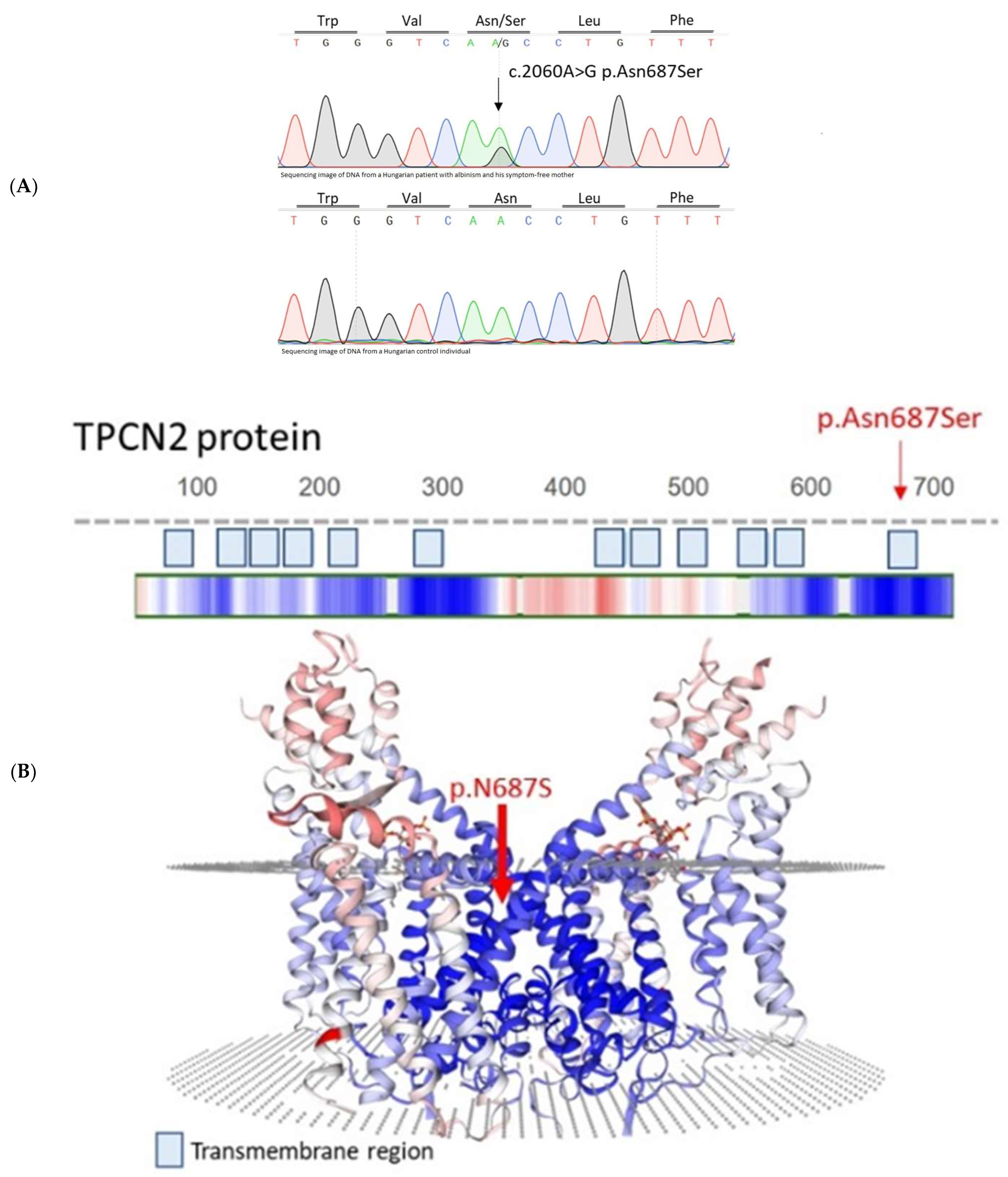
2. Results
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. The 22-Gene Panel for Albinism Genetics
4.3. Whole-Exome Sequencing (WES)
4.4. Chimaeric Constructs
4.5. Whole-Endolysosomal Patch-Clamp Experiments
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bakker, R.; Wagstaff, E.L.; Kruijt, C.C.; Emri, E.; van Karnebeek, C.D.M.; Hoffmann, M.B.; Brooks, B.P.; Boon, C.J.F.; Montoliu, L.; van Genderen, M.M.; et al. The retinal pigmentation pathway in human albinism: Not so black and white. Prog. Retin. Eye Res. 2022, 91, 101091. [Google Scholar] [CrossRef] [PubMed]
- AlAbdi, L.; Alshammari, M.; Helaby, R.; Khan, A.O.; Alkuraya, F.S. PMEL is mutated in oculocutaneous albinism. Hum. Genet. 2023, 142, 139–144. [Google Scholar] [CrossRef]
- Pavan, W.J.; Sturm, R.A. The Genetics of Human Skin and Hair Pigmentation. Annu. Rev. Genom. Hum. Genet. 2019, 20, 41–72. [Google Scholar] [CrossRef] [PubMed]
- Arveiler, B.; Lasseaux, E.; Morice-Picard, F. Clinique et génétique de l’albinisme [Clinical and genetic aspects of albinism]. Presse Med. 2017, 46, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Lasseaux, E.; Plaisant, C.; Michaud, V.; Pennamen, P.; Trimouille, A.; Gaston, L.; Monfermé, S.; Lacombe, D.; Rooryck, C.; Morice-Picard, F.; et al. Molecular characterization of a series of 990 index patients with albinism. Pigment. Cell Melanoma Res. 2018, 31, 466–474. [Google Scholar] [CrossRef]
- Arveiler, B.; Michaud, V.; Lasseaux, E. Albinism: An Underdiagnosed Condition. J. Investig. Dermatol. 2020, 140, 1449–1451. [Google Scholar] [CrossRef]
- Michaud, V.; Lasseaux, E.; Green, D.J.; Gerrard, D.T.; Plaisant, C.; Fitzgerald, T.; Birney, E.; Arveiler, B.; Black, G.C.; Sergouniotis, P.I.; et al. The contribution of common regulatory and protein-coding TYR variants to the genetic architecture of albinism. Nat. Commun. 2022, 13, 3939. [Google Scholar] [CrossRef]
- Fernández, A.; Hayashi, M.; Garrido, G.; Montero, A.; Guardia, A.; Suzuki, T.; Montoliu, L. Genetics of non-syndromic and syndromic oculocutaneous albinism in human and mouse. Pigment. Cell Melanoma Res. 2021, 34, 786–799. [Google Scholar] [CrossRef]
- Simeonov, D.R.; Wang, X.; Wang, C.; Sergeev, Y.; Dolinska, M.; Bower, M.; Fischer, R.; Winer, D.; Dubrovsky, G.; Balog, J.Z.; et al. orgDNA Variations in Oculocutaneous Albinism: An UpdatedMutation List and Current Outstanding Issues in MolecularDiagnostics. Hum. Mutat. 2013, 34, 827–835. [Google Scholar] [CrossRef]
- Maroilley, T.; Tarailo-Graovac, M. Uncovering Missing Heritability in Rare Diseases. Genes 2019, 10, 275. [Google Scholar] [CrossRef]
- Nagy, N.; Dubois, A.; Szell, M.; Rajan, N. Genetic Testing in CYLD Cutaneous Syndrome: An Update. Appl. Clin. Genet. 2021, 14, 427–444. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, Z.; Wang, Y.; Qi, Z.; Bai, D.; Wang, C.; Chen, Y.; Xu, W.; Zhu, X.; Jeon, J.; et al. A gain-of-function TPC2 variant R210C increases affinity to PI(3,5)P2 and causes lysosome acidification and hypopigmentation. Nat. Commun. 2023, 14, 226. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Yuan, Y.; Gunaratne, G.S.; Rahman, T.; Marchant, J.S. Activation of endo-lysosomal two-pore channels by NAADP and PI(3,5)P2. Five things to know. Cell Calcium 2022, 103, 102543. [Google Scholar] [CrossRef] [PubMed]
- She, J.; Zeng, W.; Guo, J.; Chen, Q.; Bai, X.C.; Jiang, Y. Structural mechanisms of phospholipid activation of the human TPC2 channel. eLife 2019, 8, e45222. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, X.; Dong, X.P.; Samie, M.; Li, X.; Cheng, X.; Goschka, A.; Shen, D.; Zhou, Y.; Harlow, J.; et al. TPC proteins are phosphoinositide- activated sodium-selective ion channels in endosomes and lysosomes. Cell 2012, 151, 372–383. [Google Scholar] [CrossRef]
- Böck, J.; Krogsaeter, E.; Passon, M.; Chao, Y.-K.; Sharma, S.; Grallert, H.; Peters, A.; Grimm, C. Human genome diversity data reveal that L564P is the predominant TPC2 variant and a prerequisite for the blond hair associated M484L gain-of-function effect. PLoS Genet. 2021, 17, e1009236. [Google Scholar] [CrossRef]
- Chao, Y.K.; Schludi, V.; Chen, C.C.; Butz, E.; Nguyen, O.N.P.; Müller, M.; Krüger, J.; Kammerbauer, C.; Ben-Johny, M.; Vollmar, A.M.; et al. TPC2 polymorphisms associated with a hair pigmentation phenotype in humans result in gain of channel function by independent mechanisms. Proc. Natl. Acad. Sci. USA 2017, 114, E8595–E8602. [Google Scholar] [CrossRef]
- Lin, S.; Sanches-Bretano, A.; Leslie, J.; Williams, K.B.; Lee, H.; Thomas, N.H.; Callaway, J.; Deline, J.; Ratnayaka, J.A.; Baralle, D.; et al. Evidence that the Ser192Tyr/Arg402Gln in cis Tyrosinase gene haplotype is a disease-causing allele in oculocutaneous albinism type 1B (OCA1B). npj Genom. Med. 2022, 7, 2. [Google Scholar] [CrossRef]
- Ambrosio, A.L.; Boyle, J.A.; Aradi, A.E.; Christian, K.A.; Di Pietro, S.M. TPC2 controls pigmentation by regulating melanosome pH and size. Proc. Natl. Acad. Sci. USA 2016, 113, 5622–5627. [Google Scholar] [CrossRef]
- Bellono, N.W.; Escobar, I.E.; Lefkovith, A.J.; Marks, M.S.; Oancea, E. An intracellular anion channel critical for pigmentation. eLife 2014, 3, e04543. [Google Scholar] [CrossRef]
- Deltas, C. Digenic inheritance and genetic modifiers. Clin. Genet. 2018, 93, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Artero, E.; Morice-Picard, F.; Bremond-Gignac, D.; Drumare-Bouvet, I.; Duncombe-Poulet, C.; Leclerc-Mercier, S.; Dufresne, H.; Kaplan, J.; Jouanne, B.; Arveiler, B.; et al. Management of albinism: French guidelines for diagnosis and care. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 1449–1459. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Ruiz, N.; Lao, O.; Aróstegu, J.; Laayouni, H.; Casals, F. Assessing the digenic model in rare disorders using population sequencing data. Eur. J. Hum. Genet. 2022, 30, 1439–1443. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhang, L.; Long, Q.; Jiang, H.; Li, M. An accurate prediction model of digenic interaction for estimatingpathogenic gene pairs of human disease. Comput. Struct. Biotechnol. J. 2022, 20, 3639–3652. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Pathogenic Variants | Common Polymorphisms | |||||||
|---|---|---|---|---|---|---|---|---|
| Patients | Mutation 1 | Mutation 2 | Gene | Type | L374F SLC45A2 | S192Y TYR | R402Q TYR | R305W OCA2 |
| 1 | p.Gln487Ter | p.Gly409Asp | SLC45A2 | OCA4 | homozygous | |||
| 2 | p.Val367Ile | p.Gly198Asp | SLC45A2 | OCA4 | homozygous | heterozygous | ||
| 3 | p.Asn489Asp | p.Val443Ile | OCA2 | OCA2 | heterozygous | heterozygous | heterozygous | heterozygous |
| 4 | p.Arg217Gln | p.Ala490fs | TYR | OCA1 | homozygous | heterozygous | ||
| 5 | p.Arg217Gln | p.Ala490fs | TYR | OCA1 | homozygous | heterozygous | ||
| 6 | p.Val183Leu | p.Arg402Ter | TYR | OCA1 | homozygous | heterozygous | ||
| 7 | p.Pro21Ser | p.Met96fs | TYR | OCA1 | heterozygous | heterozygous | ||
| 8 | p.Pro21Ser | p.Met96fs | TYR | OCA1 | heterozygous | heterozygous | ||
| 9 | p.Arg217Gln | p.Arg402Ter | TYR | OCA1 | homozygous | heterozygous | heterozygous | heterozygous |
| 10 | p.Pro21Ser | p.Val183Leu | TYR | OCA1 | homozygous | homozygous | heterozygous | |
| 11 | p.Tyr245Leufs | P.Gly321Alafs | HSP1 | HSP1 | homozygous | heterozygous | ||
| 12 | ND | ND | ND | ND | homozygous | homozygous | heterozygous | |
| 13 | ND | ND | ND | ND | homozygous | heterozygous | ||
| 14 | ND | ND | ND | ND | homozygous | heterozygous | ||
| 15 | ND | ND | ND | ND | homozygous | heterozygous | ||
| 16 * | ND | ND | ND | ND | homozygous | heterozygous | heterozygous | |
| 17 | ND | ND | ND | ND | homozygous | homozygous | heterozygous | |
| Variant | N687S TPCN2 | S192Y TYR | R402Q TYR | R305W OCA2 | L374F SLC45A2 |
|---|---|---|---|---|---|
| Patient No. 16 | heterozygous | WT | heterozygous | heterozygous | homozygous |
| Mother of the patient | heterozygous | WT | heterozygous | WT | homozygous |
| Father of the patient | WT | heterozygous | WT | heterozygous | homozygous |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagy, N.; Pal, M.; Kun, J.; Galik, B.; Urban, P.; Medvecz, M.; Fabos, B.; Neller, A.; Abdolreza, A.; Danis, J.; et al. Missing Heritability in Albinism: Deep Characterization of a Hungarian Albinism Cohort Raises the Possibility of the Digenic Genetic Background of the Disease. Int. J. Mol. Sci. 2024, 25, 1271. https://doi.org/10.3390/ijms25021271
Nagy N, Pal M, Kun J, Galik B, Urban P, Medvecz M, Fabos B, Neller A, Abdolreza A, Danis J, et al. Missing Heritability in Albinism: Deep Characterization of a Hungarian Albinism Cohort Raises the Possibility of the Digenic Genetic Background of the Disease. International Journal of Molecular Sciences. 2024; 25(2):1271. https://doi.org/10.3390/ijms25021271
Chicago/Turabian StyleNagy, Nikoletta, Margit Pal, Jozsef Kun, Bence Galik, Peter Urban, Marta Medvecz, Beata Fabos, Alexandra Neller, Aliasgari Abdolreza, Judit Danis, and et al. 2024. "Missing Heritability in Albinism: Deep Characterization of a Hungarian Albinism Cohort Raises the Possibility of the Digenic Genetic Background of the Disease" International Journal of Molecular Sciences 25, no. 2: 1271. https://doi.org/10.3390/ijms25021271
APA StyleNagy, N., Pal, M., Kun, J., Galik, B., Urban, P., Medvecz, M., Fabos, B., Neller, A., Abdolreza, A., Danis, J., Szabo, V., Yang, Z., Fenske, S., Biel, M., Gyenesei, A., Adam, E., & Szell, M. (2024). Missing Heritability in Albinism: Deep Characterization of a Hungarian Albinism Cohort Raises the Possibility of the Digenic Genetic Background of the Disease. International Journal of Molecular Sciences, 25(2), 1271. https://doi.org/10.3390/ijms25021271