Abstract
Porphyromonas gingivalis (P. gingivalis) is a key pathogen of periodontitis. Increasing evidence shows that P. gingivalis signals to mitochondria in periodontal cells, including gingival epithelial cells, gingival fibroblast cells, immune cells, etc. Mitochondrial dysfunction affects the cellular state and participates in periodontal inflammatory response through the aberrant release of mitochondrial contents. In the current review, it was summarized that P. gingivalis induced mitochondrial dysfunction by altering the mitochondrial metabolic state, unbalancing mitochondrial quality control, prompting mitochondrial reactive oxygen species (ROS) production, and regulating mitochondria-mediated apoptosis. This review outlines the impacts of P. gingivalis and its virulence factors on the mitochondrial function of periodontal cells and their role in periodontitis.
1. Introduction
Periodontitis is defined as a chronic inflammatory disease under the effect of dysbiotic plaque biofilms, leading to the destruction of periodontal tissues, the resorption of alveolar bone, and even the loosening and loss of teeth [1,2]. Since periodontitis is a multifactorial disease, its treatment is constantly being explored and refined. A critical appraisal of authoritative sources proposes that the periodontitis risk assessment should look at nonclinical indicators (e.g., microbiological, immunological, genetic, and epigenetic) in addition to relying on clinical factors [3]. Immunology in periodontitis revealed that neutrophils are early responders in the immunopathologic reaction to subgingival biofilm and attract macrophages, dendritic cells, and T cells to the foci [4]. In the periodontitis process, the struggle between microbial infection and host immune system defense determines the degree and persistence of inflammatory infiltration and the enriched type of immune cells [5]. Moreover, the imbalanced immune homeostasis in periodontitis is believed to be the pathologic basis for its association with systemic diseases such as diabetes [6], obesity [2], and atherosclerotic cardiovascular disease [7]. Of note, it has been affirmed that the disturbances of the host immune response triggered by the dysbiotic microbiome are the underlying pathological mechanisms of low-grade systemic inflammation elicited by periodontitis [8].
Periodontal flora colonizes the subgingival region and transforms from harmless to virulent under certain circumstances, with Porphyromonas gingivalis (P. gingivalis) considered to be the keystone bacterium. Previous studies have focused on the host cell functional alteration by P. gingivalis and its virulence factors at the cellular and molecular levels, and the molecular pathways involved in this inflammatory immune response [9,10,11]. Currently, there is a rising interest from an organelle perspective in exploring the pathology of periodontitis, especially mitochondria, as the important cellular energy production factories driving normal cellular activity.
Govindaraj et al. revealed that 60% of patients with chronic periodontitis have different forms of mitochondrial structural abnormalities in their gingival tissue [12]. Furthermore, according to a complete mitochondrial DNA (mtDNA) sequence analysis, they found the inflamed tissue was more prone to mtDNA mutations and mitochondrial dysfunction [12]. An animal experiment observed that periodontium cells from periodontitis mice displayed a significantly severer level of impaired mitochondrial oxidative phosphorylation function and oxidative stress than the control group [13]. In a vitro study, similar disruptions in mitochondrial structure and function were proven in human gingival fibroblasts (HGFs) from patients with chronic periodontitis [14]. More importantly, the extra stimulation of lipopolysaccharide (LPS) from P. gingivalis exacerbated mitochondrial abnormalities in inflamed HGFs and induced these abnormalities in the healthy HGFs, indicating the mitochondrial dysfunction triggered by P. gingivalis may play a vital part in the pathogenesis of periodontitis [14]. Also, changes in the functional mitochondrial molecules can influence the state of periodontal cells. For example, abnormal enhancement of mitochondrial permeability transition pores was found to aggravate the pro-inflammatory activation of macrophages [15].
Additionally, mitochondrial dysfunction may be a shared pathogenic background in periodontal comorbidities. A cross-sectional study demonstrated that a mitochondrion-derived biomarker of mitochondrial dysfunction, the blood sample concentration of methylmalonic acid, may act as a bridging indicator in the association between periodontitis and cognitive impairment in older adults aged ≥60 years [16]. Similarly, mitochondrial oxidative deregulation was proved to be a mutual systemic cytopathological feature of periodontitis and type 2 diabetes [17,18]. Moreover, there is evidence to suggest that the application of prominent polyphenolic compounds that optimize mitochondrial function, such as resveratrol, ameliorated the destruction of renal structures in mice with periodontitis [19] and alleviated the ferroptosis of alveolar osteocytes in mice with diabetic periodontitis [20]. In light of the mitochondrial pathway being so extensively involved in the pathological process of periodontitis, it has received mounting interest in exploring how the crucial periodontal pathogen, P. gingivalis, disrupts mitochondria.
The effect of P. gingivalis on specific functional mitochondrial molecules varies from its distinct virulence factors. And how these mitochondrial changes caused by P. gingivalis are involved in the periodontal inflammatory process is not yet clear. In this review, we presented how mitochondrial metabolic transformations and mitochondrial signaling molecules, such as mitochondrial DNA, mitochondrial ROS, and mitochondrial-mediated apoptotic effectors, were involved in periodontitis. In addition, mitochondrial dysfunction caused by P. gingivalis was described and summarized in an attempt to better understand how P. gingivalis destabilizes periodontal homeostasis.
2. Porphyromonas gingivalis and Periodontitis
P. gingivalis is a gram-negative anaerobe mainly existing in the subgingival biofilms. The diverse virulence factors of P. gingivalis, including LPS, fimbriae, gingipain, capsules, etc., act as adhesive or destructive agents independently or synergistically mediating the periodontal inflammation and periodontal tissue damage [9]. P. gingivalis causes extensive destruction to various cells in periodontal tissue, with pro-inflammatory response as the main manifestation. P. gingivalis adheres to and invades cells through the collaboration of different virulence factors, perturbing the structural and functional integrity of the gingival epithelium, the first defensive line of periodontium [21]. P. gingivalis also penetrates the epithelium thus attacking the deeper cells of the periodontal tissue. Under P. gingivalis infection, gingival fibroblasts, as the essential components of the periodontium, undergo a shift to a pro-inflammatory phenotype, recruiting leukocytes, secreting protein hydrolysing enzymes, and inducing osteoclast formation [22]. It can directly induce monocyte/macrophage differentiation into osteoclasts through the receptor activator of the nuclear factor-kappa B (NF-κB) ligand pathway, thus promoting the absorption of the bone mineral matrix and collagen matrix [23]. Meanwhile, P. gingivalis and its virulence factors prevent osteoblast differentiation and bone formation [24,25]. More importantly, P. gingivalis elicits a massive and widespread release of pro-inflammatory factors from immune cells [26,27] and promotes the expansion of immune-suppressive cells [28] as a way to expand its inflammatory damaging and immune evasion effects. When pro-inflammatory cytokines, such as interleukin (IL)-1, -6, -11,-17, and tumor necrosis factor-alpha (TNF-α), achieve critical concentrations, the inflammatory response leads to periodontal tissue damage [29]. It is worth mentioning that in addition to its local effects in the periodontal tissues, P. gingivalis can, in some cases, invade distant organs through haematogenous dissemination, causing damage to other tissue [30]. In a word, P. gingivalis interferes with multiple periodontal cells.
3. Mitochondrial Involvement in Periodontal Inflammation
3.1. Mitochondrial Structure and Function
As central regulators of aerobic energy production, mitochondria rely primarily on electron transfer and proton gradients which occur across the mitochondrial membrane to influence cellular homeostasis, generating sufficient ATP in the process. Mitochondria are double membrane organelles. In the mitochondrial matrix, the mtDNA is present in multiple copies and packed into compact particles, termed nucleoids. The outer mitochondrial membrane (OMM) separates the mitochondria from the cytosol and is generally permeable serving as a platform for communication between signaling molecules. The inner mitochondrial membrane (IMM) delimits the mitochondrial matrix and folds inward into deeply convoluted cristae for expanding the surface to ensure that mitochondrial electron transport chain (ETC) complexes attached to the membrane can conform to the needs of cellular energy production. And the ETC complexes contain NADH-ubiquinone oxidoreductase (complex I), succinate-ubiquinone oxidoreductase (complex II), ubiquinol-cytochrome c reductase (complex III), cytochrome c oxidase (complex IV), as well as the FoF1-ATP synthase (ATP synthase or complex V) and two mobile electron carriers, coenzyme Q (CoQ) and cytochrome c (cyt c) [31]. Compared to the OMM, the IMM is impermeable to support the transmembrane proton gradient and therefore requires a specific mitochondrial carrier family to transport solutes [32]. When subjected to stress or injury, the mitochondrial structure and the location and function of mitochondrial proteins shift. These changes result in aberrant alterations in mitochondrial molecules and shifts in mitochondrial metabolic types, thus affecting the phenotype of immune cells and taking part in the inflammatory response.
3.2. Mitochondria Are Involved in the Inflammatory Response
Mitochondrial ROS (mtROS), efflux of mitochondrial contents, and mitochondria-dependent apoptotic molecules are involved in the activation of inflammatory pathways. The activation of inflammasome and polarization of immune cells correlate with mtROS. mtROS mainly includes oxygen radicals that arise from the excessive electrons transferring to oxygen during mitochondrial respiration which can bind to lipids, proteins, and DNA, causing oxidative damage and corresponding molecular dysfunction [33]. Studies have confirmed that mtROS interacted directly with NOD-like receptor thermal protein domain associated protein 3 (NLRP3), triggering its activation [34,35]. Although mitochondria are not the only source responsible for ROS production, evidence has confirmed that mitochondria-targeted ROS inhibitors attenuate the NLRP3 inflammatory response more effectively than pan-ROS inhibitors [36]. In addition, mtROS are engaged in the macrophage polarization towards a pro-inflammatory MI phenotype and impair lysosomal function [37].
As an instrumental and fragile functional mitochondrial molecule, mtDNA is susceptible to mtROS and acts as an endogenous inflammatory stimulus. Numerous mitochondrial constituents and metabolites can function as damage-associated molecular patterns (DAMPs) to promote inflammation when released into the cytosol or extracellular milieu. Above all of the DAMPs, mtDNA is one of the most widely and valuably studied. Upon cellular stress or mitochondrial insult, mtDNA can be released into the cytoplasm via three main pathways responsible for the regulation of the permeability of mitochondrial membrane, including Bax/Bak, the voltage-dependent anion channel oligomers, and the mitochondrial permeability transition pore (mPTP) [38]. In the cytoplasm, mtDNA can be detected by multiple pattern recognition receptors, including NLRPs, toll-like receptors (TLRs), and the cyclic GMP/AMP synthase–stimulator of interferon genes systems, triggering aberrant pro-inflammatory and type I interferon (IFN) responses [38]. Since mtDNA is spatially close to the mitochondrial ETC, it is vulnerable to oxidative attack. The oxidized mtDNA turns into fragmentation and generates a specific NLRP3 ligand, ultimately promoting an increase in inflammatory mediators, including IL-1β, IL-18, and IFN [39]. Another study exploring the relationship between mtDNA and NLRP3 pointed out that newly synthesized mtDNAs were essential activators of macrophage NLRP3, which correlated with the fact that they had not yet been packaged by histones to form stable nucleoids and thus are highly susceptible to oxidative damage [40].
Furthermore, mitochondria-mediated apoptosis was revealed as a player in microbial pathogen-triggered inflammation [41]. The activated Bax and Bak pores on the OMM lead to the release of mitochondrial pro-apoptotic factors and the subsequent cell death, whereas the anti-apoptotic effector B cell lymphoma 2 (Bcl-2) serves as a ruling checkpoint during the process [42]. Mitochondria-dependent apoptosis of osteoblasts and human gingival epithelial cells can be initiated under inflammatory factors stimulation, such as cytokines IFN-γ, TNF-α, and transforming growth factor β1 [43,44]. However, the apoptotic process appears oppositely in the real periodontitis condition. Bulut et al. revealed that a high rate of positive expression of Bcl-2 has been reported in the inflammatory gingival tissues of patients with generalized aggressive periodontitis [45]. Similarly, up-regulation of anti-apoptosis member genes of oral neutrophils was identified in patients with chronic periodontitis compared with healthy counterparts by examining the transcriptome [46], indicating that delayed apoptosis may be behind the localized retention and accumulation of inflammatory cells, resulting in progressive periodontal destruction.
3.3. Mitochondrial Energy Metabolism Shifts with Periodontal Condition
Cellular respiration comprises glycolysis in the cytoplasm, mitochondrial tricarboxylic acid (TCA) cycle, and mitochondrial oxidative phosphorylation (OXPHOS). OXPHOS occurring in the ETC utilizes the reducing equivalents generated by the TCA cycle to transfer electrons to O2, forming H2O and actuating the transmembrane movement of protons to power the synthesis of ATP, thereby completing oxidative phosphorylation and producing sufficient energy to drive cellular functions. The shifts between glycolysis, which is anaerobic and produces only a fairly small amount of ATP, and OXPHOS, which is aerobic and responsible for most of the required ATP, affect cellular activity. More specifically, bioenergetic metabolic transformations are closely correlated with functional changes in osteoblasts and immune cells.
Several studies showed that macrophages and Th17 cells with OXPHOS as the main metabolic phenotype seemed to exhibit anti-periodontitis properties [47,48]. It was found that Th17 cells cultured under OXPHOS conditions are resistant to apoptotic cell death compared to those under glycolytic conditions [47]. Moreover, the conversion of pro-inflammatory M1 macrophages to anti-inflammatory M2 macrophages promoted bone formation in canine periodontal tissue defects, and this macrophage polarization change was proven to be accompanied by a shift in cellular metabolism from glycolysis and ROS generation to oxidative phosphorylation [48]. The mechanism behind the synchronization of glycolysis with increased ROS synthesis may be paralleled by the fact that, following the dominance of glycolytic ATP synthesis, the mitochondrial membrane potential (MMP) generated by the oxidative phosphorylation complexes across the IMM via proton pumping is no longer employed to drive ATP synthesis, resulting in a high MMP and increased ROS synthesis [49]. Further research confirmed that P. gingivalis induced a shift in macrophage metabolism from OXPHOS to glycolysis which was supported by enhanced lactate release, decreased mitochondrial oxygen consumption, and increased ROS production [50] (Figure 1). However, the mineralization of osteoblasts is dominated by glycolysis, while there is a simultaneous rise in OXPHOS and glycolysis during the initial stages of osteoblast differentiation [51,52]. Accordingly, the struggle between oxidative phosphorylation and glycolysis cannot simply be concluded as a good or bad thing, but a more important consideration is under which metabolic environment the cell can maximize its reparative function or prevent the damage caused by periodontitis.
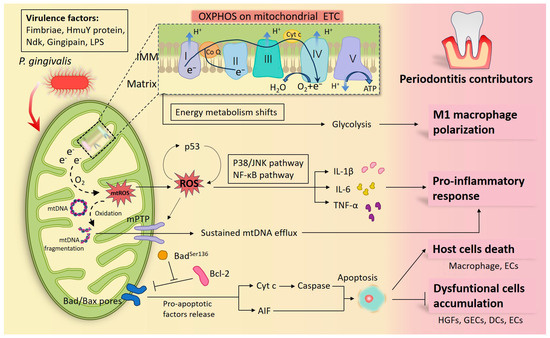
Figure 1.
Diagram of how Porphyromonas gingivalis (P. gingivalis) participates in periodontal inflammation by interfering with mitochondria. P. gingivalis has multiple virulence factors that can launch an attack at the host cells. When macrophages are infected, the center of the cellular energy metabolism transfers from oxidative phosphorylation (OXPHOS) into glycolysis, and macrophages differentiate toward the pro-inflammatory M1 phenotypes. Under defective OXPHOS, abnormal electrons released from the electron transport chain (ETC) drive reactive oxygen species (ROS) formation and oxidize mitochondrial DNA (mtDNA) into fragmentation. ROS propel the release of pro-inflammatory factors by activating the P38/JNK and NF-κB signaling pathways and forming a feedback loop with p53. Additionally sustained mtDNA efflux through the ROS/mitochondrial permeability transition pore (mPTP) pathway was found under P. gingivalis infection, which also contributes to inflammation. In addition, plenty of studies have confirmed that by modulating mitochondria-associated apoptotic molecules, P. gingivalis and its virulence factors inhibited periodontal cell apoptosis, thereby prolonging periodontal inflammation, such as in human gingival fibroblasts (HGFs), gingival epithelial cells (GECs), dendritic cell (DCs), peripheral blood mononuclear cells (PBMCs), epithelial cells (ECs), etc. Nonetheless, P. gingivalis promoting apoptosis in a mitochondrial manner has also been documented in some host cells, such as in macrophages and ECs, as a way to indicate its toxicity. Together, these cytopathic alterations illustrate the inflammatory damage in infected periodontal tissues.
As reputed by-products of mitochondrial energy metabolism, ROS are important condemnable mediators of P. gingivalis-induced periodontitis. For example, HGFs derived from patients with aggressive periodontitis exhibited higher levels of mtROS compared with those from the healthy group [53]. In vitro, LPS from P. gingivalis increased mtROS levels of HGFs isolated from a healthy human [54]. In addition, extensive research has verified that ROS served as key pathogenic mediators of systemic diseases, such as diabetes [55,56], atherosclerosis [57], hypertension [58], cardiovascular disease [59], benign prostatic hyperplasia [60], etc. So, it is no surprise that ROS elevation is a shared pathological link between systemic diseases and periodontitis, as P. gingivalis can cause oxidative stress in the arterial endothelium [61] and brain endothelial cells [62]. In parallel with affecting cellular activity, ROS also interfere with subcellular organelle functions. For instance, ROS generated from mitochondria were proven to directly modulate mitochondrial dynamics [63], mitochondrial biogenesis [64], and mitochondrial-dependent apoptosis [65]. Notably, the reduction of ROS was used as an effective indicator for periodontitis treatments, and significant alleviation and partial restorations in alveolar bone loss were observed in these trials [65,66]. Taken together, mitochondria serve as hubs for immune signaling and the generation of corresponding effectors in periodontitis, implicating the necessity of exploring the pathogenic pathway of P. gingivalis from the molecular level of mitochondria.
4. Porphyromonas gingivalis Causes Mitochondrial Dysfunction
4.1. Porphyromonas gingivalis Induces Mitochondrial Quality Control Imbalance
Given the main duty of mitochondria in energy production, they are exposed to high amounts of ROS making them particularly vulnerable to mtDNA mutations and protein misfolding, so they have evolved a quality control system to guarantee mitochondrial quantity and quality to match metabolic demand and maintain mitochondria homeostasis [67]. Mitochondrial quality control runs through the life cycle of mitochondria, starting with the growth and division of pre-existing organelles (mitochondrial biogenesis) and ending with the degradation of impaired or surplus organelles by mitophagy [68]. Mitochondria also undergo frequent fusion and fission events (mitochondrial dynamics) resulting in multiple scattered mitochondria or interconnected mitochondrial networks [68]. A growing wealth of evidence shows that P. gingivalis dysregulated mitochondrial quality control in periodontal cells, causing impaired cellular energy metabolism (Figure 2).
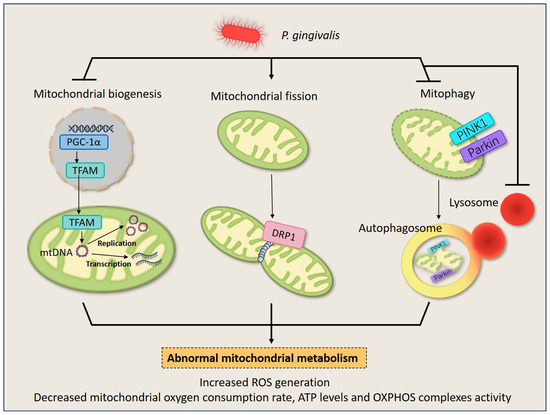
Figure 2.
P. gingivalis induces mitochondrial quality control imbalance in HGFs and macrophages. Upon P. gingivalis infection, the life cycle of mitochondria turns into chaos. P. gingivalis down-regulates mitochondrial biogenesis by inhibiting the expression of PGC1-α and TFAM. It also stimulates mitochondrial fission by inducing DRP1 expression, leading to mitochondrial fragments. Additionally, P. gingivalis blocks mitophagy by suppressing PINK1/Parkin expression and intracellular lysosome quantity, and results in the failure of dysfunctional mitochondria elimination. The dysregulation of mitochondrial homeostasis culminates in a decrease in overall mitochondrial mass and disruption of energy metabolism, therefore deranging normal cellular behavior.
4.1.1. Porphyromonas gingivalis Suppresses Mitochondrial Biogenesis and Disrupts mtDNA Homeostasis
The normal activity of cells and organelles depends on the production and interaction of specific proteins. Only a small proportion of mitochondrial proteins are encoded by the mitochondrial genome, whereas most proteins are encoded by the nuclear genome and require a coordinated import mechanism to send these proteins into the mitochondria and to assist with the eventual folding and assembly of proteins into functional complexes. So the occurrence of mitochondrial biogenesis relies on the up-regulation of transcription factors, as a way to promote sufficient mitochondrial protein production to ensure energy metabolism [69]. The major regulators include peroxisome proliferators-activated receptor γ coactivator alpha (PGC-1α), and the ultimate regulatory effector, mitochondrial transcription factor A (TFAM), which regulates the level of mtDNA replication and transcription [69]. It should be specified that though few proteins are encoded by mtDNA, they include the 13 polypeptides that constitute the ETC and a small amount of RNA required for the synthesis of mitochondrial proteins; therefore, abnormalities in mtDNA homeostasis, including aberrant mtDNA localization and copy number, as well as mtDNA damage and fragmentation, also account for mitochondrial mass deficiency [70]. There is a case to suggest that mitochondrial biogenesis disorders participate in the development of periodontitis. Miyazaki et al. confirmed that a decline in mitochondrial biogenesis mediated by TFAM deficiency led to ATP depletion and aggravated osteoclast apoptosis and bone resorption activity [71].
Studies have reported that P. gingivalis-LPS induced disorder of mitochondrial biogenesis and disruption of mtDNA homeostasis in HGFs [14,64,72]. It was found that P. gingivalis-LPS infection led to a decrease in the protein levels of phosphorylated PGC-1α and TFAM in HGFs and was accompanied by a decline in mitochondrial oxygen consumption rate and ATP levels [64]. Moreover, fewer mtDNA copy number was discovered in the P. gingivalis-LPS treated HGFs [14], implying that mtDNA-based protein transcriptional expression may be affected. Also, in response to P. gingivalis-LPS stimulation, significant efflux of mtDNA into the cytoplasm and out of the cell via a ROS/mPTP pathway was observed in primary HGFs, and the efflux lasted even in the passaged cells removed from the stimulus [72], suggesting that the spread and expansion of periodontal inflammation might be partially attributable to the persistent mtDNA efflux from infected cells and the activation of corresponding inflammatory pathways (Figure 1).
4.1.2. Porphyromonas gingivalis Promotes Mitochondrial Fission
Within the cytoplasm, mitochondria are not all isolated organelles but also joined to form larger networks or distributed unevenly to match the local energy demands of the cell [73]. The changes in the morphology and distribution of mitochondria are mediated by fusion and fission, which are collectively referred to as mitochondrial dynamics. In mammalian cells, the fusion of mitochondria is regulated by mitofusin 1 (MFN1), MFN2, and optic atrophy protein 1, while the fission is primarily carried out by the dynamin-related protein 1 (DRP1) oligomerizing in the membrane [74]. The fragmentations brought by the over-divided mitochondria are believed to be part of the possible causes of OXPHOS failure, since deletion of mtDNA and abnormal activity of the respiratory chain complex may occur during this process [75].
P. gingivalis disrupts mitochondrial energy synthesis by stimulating DRP1-dependent mitochondrial fission. Experiments have confirmed that P. gingivalis-LPS promoted total DRP1 and phosphorylated-DRP1 (Ser616) and decreased MFN2 in mouse gingival tissue and HGFs, leading to shorter mitochondrial length and even fragmentation [54]. Furthermore, it was found that DRP1-knockdown rescued the mitochondrial fragmentation and the significant reduction in complex I activity and ATP levels brought by P. gingivalis-LPS infection in HGFs [76]. Moreover, the role of P. gingivalis on mitochondrial fission has also been verified in vascular endothelial cells, but with dissimilarities in the specific regulatory genes [77]. Specifically, up-regulation of p-DRP1 was detected, whereas total DRP1 and three fusion proteins remained unchanged [77]. Despite differences in the detection and regulation of specific mitochondrial fission and fusion proteins across studies, DRP1-mediated mitochondrial fission appears to be the main effect of P. gingivalis. Still, more research is needed to distinguish the effects of P. gingivalis and its various virulence factors on mitochondrial dynamics-related proteins in periodontal cells.
4.1.3. Porphyromonas gingivalis Inhibits Mitophagy
Mitophagy involves distinct steps to remove defective or superfluous mitochondria, during which mitochondria are decorated with poly-ubiquitin chains, engulfed by autophagosomes, and degraded following lysosomal fusion [78,79]. So far, mitochondrial receptors including proteins containing an LIR motif (Nix, BNIP3, and FUNDC1) and the ubiquitin-relying PINK1–Parkin axis have been confirmed to be associated with mitophagy [80]. However, not all of them were proven to be differentially expressed in periodontitis. What has been documented is that P. gingivalis affects macrophages by restraining the classical PTEN-induced kinase 1 (PINK1)-dependent mitophagy.
Jiang et al. uncovered that the mRNA and protein levels of PINK1 and Parkin were significantly decreased in both inflammatory gingival tissues and bone marrow-derived macrophages (BMDMs) infected by P. gingivalis [81], suggesting that the mitophagy mechanism was impaired in periodontitis. It was subsequently revealed that mitophagy inducers dexmedetomidine, urolithin A, and resveratrol suppressed the production of IL-1β, IL-6, TNF-α, and ROS of P. gingivalis-triggered BMDMs; however, knockdown of PINK1 failed to have a pro-inflammatory effect [81]. These observations indicated that mitophagy inhibited by P. gingivalis was involved in the pro-inflammatory transformation of macrophages, but may not necessarily via a PINK1-mediated pathway.
Nevertheless, the successful activation of mitophagy also requires the coordination of lysosomes. A study demonstrated that while P. gingivalis promoted the expression of PINK1–Parkin in oral epithelial cells, it caused an intracellular lysosome absence and a significant reduction in the co-localization of PINK1-labeled mitochondria and lysosomes [82]. It hinted that P. gingivalis inhibited the ultimate degradation of dysfunctional mitochondria by posing an effect on lysosomes [82].
4.2. Crosstalk between Porphyromonas gingivalis and ROS
ROS are generated during mitochondrial oxidative metabolism as well as in cellular response to xenobiotics, cytokines, and bacterial invasion. Although the main ROS production comes from mitochondria, organelles like peroxisomal and endoplasmic reticular could also produce ROS [83]. ROS includes oxygen ions/O2•, free radicals (superoxide/O2− and hydroxyl radicals), and peroxides (hydrogen peroxide/H2O2) [84]. At the very beginning, ROS was simply thought to be a kind of toxic by-product in an aerobic environment, but gradually more studies proved their specific role as intracellular signaling molecules contributing to retrograde redox signaling from the organelle to the cytosol and nucleus, regulating a variety of homeostatic cellular functions [85,86]. ROS can activate signaling pathways to initiate biological processes in physiological and pathological conditions [87]. More remarkably, when ROS production overwhelms the cellular antioxidant defense system, oxidative stress occurs [88]. Oxidative stress has long been regarded as the pathological environment of periodontitis. P. gingivalis contributes to the formation of an oxidative stress environment in periodontitis by increasing mtROS production.
Mitochondria are the major source of intracellular ROS, playing a role as an important intermediate mediator of P. gingivalis-activated inflammation [89]. A study revealed that the increase in the concentration of mtROS preceded the expression of inflammatory cytokines in HGFs under P. gingivalis-LPS treatment, and further experiments demonstrated that elimination of mtROS successfully suppressed the LPS-induced p38/JNK signaling pathway and NF-κB signaling pathway, thus decreasing the expression of IL-1β, IL-6, and TNF-α [89]. Studies further characterized that the increase and mitochondrial translocation in p53 formed a feedback loop with ROS, together participating in the inflammatory response above [90,91] (Figure 1). Moreover, while P. gingivalis triggers inflammation by inducing mtROS, mtROS also act as central pathogenic factors evoking dysfunction and aberrant activity of cells. Bullón revealed that mtROS modulate cellular activity in P. gingivalis-LPS infected HGFs by down-regulating mitochondrial biogenesis and contributing to cell death [64]. Nonetheless, whether the elevated mtROS are caused by intact P. gingivalis or by certain virulence factors remains elusive. For example, Liu et al. proved the supernatant of P. gingivalis did not induce ROS in human oral epithelial cells, but the invasion of the intact P. gingivalis did induce ROS [82].
Although P. gingivalis appears to trigger oxidative stress by stimulating an increase in ROS, causing periodontal damage, ROS themselves own an antibacterial effect. Generally, ROS generation can be applied by host cells as a defensive mechanism to combat intracellular pathogens. West et al. identified that macrophages generated mtROS to resist bacterial invasion through the direct communication of activated TLRs with mitochondrial Complex I [92]. However, P. gingivalis seems to have some specificity in avoiding oxidative damage. P. gingivalis can sense the oxidative stress in the environment and potentiate its gene expression concerned with antioxidant regulators, including OxyR and RprY [93]. And studies have proved that the haem layer on the surface of P. gingivalis serves as a buffer layer against oxidative attacks [94,95]. Moreover, these antioxidant responses of P. gingivalis were further amplified in plaque biofilms [96,97].
The causes of the oxidative stress state in the periodontal microenvironment may be comprehensive and not limited to P. gingivalis-induced ROS generation. Thus, the role of P. gingivalis on host cells in the context of oxidative stress seemed to be quite complicated and somehow controversial. On the one hand, oxidative stress aggravates the aggressiveness of P. gingivalis against host cells. Knowles et al. revealed that gingipain exacerbated the suppression of transcriptional function in oral epithelial cells under oxidative stress [98]. And macrophage endotoxin tolerance to P. gingivalis-LPS was hindered under an oxidative stress environment, therefore contributing to periodontitis progression [99]. On the other hand, P. gingivalis also restrains cellular exogenous oxidative stress to some extent, prolonging its pathogenic effect. It was found that when gingival epithelial cells (GECs) underwent oxidative stress in response to extracellular ATP stimulation, P. gingivalis-Nucleoside-diphosphate-kinase (Ndk) infection up-regulated the antioxidant glutathione and inhibited cytosolic and mitochondrial ROS generation thus blocking the oxidative stress and maintaining its persistence intracellularly [100].
Intriguingly, based on the dual pro-inflammatory and antimicrobial effects of ROS, new treatments have emerged to suppress periodontal bacteria and improve cellular status by appropriately promoting oxidative stress in the periodontal microenvironment. Photobiomodulation, an emerging periodontal treatment, converts cellular metabolism into mitochondrial OXPHOS and enhances cellular antioxidant activity by inducing a modest boost in ROS [101]. Evidence has demonstrated that treating periodontitis tissue of beagles with photobiomodulation decreased the concentration of all dominant pathogens, including P. gingivalis, Fusobacterium nucleatum, and Corynebacterium [102].
4.3. Porphyromonas gingivalis Influences Mitochondria-Mediated Apoptosis
Cell apoptosis is initiated by extracellular and intracellular signals via two main pathways, the death receptors and the mitochondria-mediated pathways [103]. The collapse of mitochondrial inner transmembrane potential and mitochondrial outer membrane permeabilization (MOMP) play a primary and central role in mitochondria-mediated apoptosis, causing uncontrolled mitochondrial energy metabolism, activation, and spillover of mitochondrial pro-apoptotic effectors [104,105]. There are a host of mitochondrial pro-apoptotic factors presented in mitochondria and released into the cytoplasm upon induction of apoptosis, including cyt c, apoptosis inducing factor (AIF), Smac/DIABLO, and several procaspases (such as procaspase-2, -3, and -9) [106]. It is worth noting that cyt c release is one of the most common and persuasive indicators of mitochondria-mediated apoptosis, whereas relatively few studies have been conducted on the AIF-release-determined pathway. AIF is unique in that when released as a pro-apoptotic factor, it translocates directly to the nucleus and mediates DNA fragmentation through the binding of nucleic acid endonucleases, distinguishing itself from the caspase-dependent apoptosis pathways mediated by cyt c release and death receptor [107,108]. The effect of P. gingivalis on mitochondria-mediated apoptosis is inconsistent in terms of different cell types and virulence factors (Figure 1).
It was reported P. gingivalis suppressed mitochondria-mediated apoptosis. Bax/Bak and Bcl-2-like proteins, when activated, are translocated from the cytosol to the OMM, where they undergo conformational rearrangement and assembly to regulate the MOMP and the release of mitochondrial-apoptotic mediators [42,109]. By down-regulating the expression of pro-apoptotic protein Bax and up-regulating the anti-apoptotic protein Bcl-2, P. gingivalis tended to keep the cells active at 24 h [110]. Moreover, subsequent studies confirmed that P. gingivalis inhibited mitochondria-mediated apoptosis of primary GECs through the activation of the phosphatidylinositol 3-kinase/Akt pathway which mediated the up-regulation of the phosphorylated BadSer136 preventing the pro-apoptotic protein Bad from competitively binding to Bax against Bcl-2 [111,112,113]. Similarly, Desta et al. found pretreatment of HGFs with P. gingivalis attenuated TNF-α-stimulated apoptosis, and when AIF was down-regulated by siRNA, cell death was reduced dramatically [114].
Nevertheless, studies on the role played by P. gingivalis-specific virulence factors in the resistance of mitochondria-mediated apoptosis have also been inconsistent in both periodontal and non-periodontal cells. Though, it was demonstrated that P. gingivalis similarly suppressed dendritic cell apoptosis by promoting Bcl-2, the different fimbriae phenotypes (i.e., fimA and Mfa1) exemplified marked differences in their regulatory roles [115]. Intriguingly, Mao et al. pointed out that the anti-apoptotic activity by P. gingivalis in hGECs did not require the presence of fimbriae [116]. In addition, P. gingivalis HmuY protein, which helps P. gingivalis uptake heme for nutrition, delays apoptosis in peripheral blood mononuclear cells via a Bcl-2-regulated way, thereby prolonging the release of cellular inflammatory factors [117,118]. As in non-periodontal cells, Lee et al. demonstrated that P. gingivalis-Ndk inhibited mitochondria-mediated apoptosis in adipocytes by directly binding to and phosphorylating the Heat-shock-protein-27 and the ndk-deficient P. gingivalis did not ameliorate staurosporine-induced apoptosis, showing the irreplaceable role of P. gingivalis-ndk [119]. Surprisingly, Boisvert et al. inhibited staurosporine-induced apoptosis in laryngeal cancer epithelial cells simply with purified gingipain adhesin peptide A44, which may be related to cell specificity [120]. In conclusion, the anti-apoptotic manifestation of P. gingivalis benefits its intracellular survival and prolongs the inflammatory state brought by infected cells.
Even with copious studies above showing that P. gingivalis inhibited apoptosis in the mitochondrial pathway, the phenomenon of mitochondria-associated apoptosis caused by P. gingivalis has also been observed in some cells. THP-1 macrophages showed a shift in mitochondria-associated apoptotic proteins towards pro-apoptosis in response to P. gingivalis stimulation [121]. In addition, several studies demonstrated that butyric acid, metabolites of P. gingivalis, caused mitochondria-dependent apoptosis in HGFs, but the ultimate nuclear DNA damage severity and apoptosis sensitivity correlated with the duration and concentration of butyric acid, and inflammatory environment [122,123]. Likewise, the apoptotic effect of butyric acid has also been observed in nerve cells, which may account for neuropathic pain in the varying stages of periodontitis [124]. It has also been shown that P. gingivalis induced apoptosis in epithelial cells through the AIF release pathway [125]. Moreover, mitochondria-dependent apoptosis under P. gingivalis was also detected in tissues outside the periodontal correlation, including cardiomyocytes [126], vascular endothelial cells [127], adipocytes [128], and skin fibroblasts [129]. The initiation of mitochondria-mediated apoptosis by P. gingivalis reflects more of its dual effects in causing a total collapse of cellular structure and function while suppressing chronic bacterial infection accumulation.
5. Conclusions
The mitochondrial pathway is an inextricable link in the pathology of periodontitis due to P. gingivalis. It causes inflammatory destruction of periodontal tissues and bone resorption by invading intracellularly, penetrating the epithelial barrier, and attacking fibroblasts and immune cells in the deeper tissues. Under certain circumstances, P. gingivalis enters the blood circulation to influence the development of systemic diseases. Mitochondrial homeostasis has an imperative role in the periodontal environment.
In summary, P. gingivalis triggers an imbalance in mitochondrial quality control, leading to an accumulation of defective mitochondria, and a sustained mtROS and efflux of DAMPs which trigger inflammatory responses. Moreover, P. gingivalis owns anti-mitochondrial-mediated apoptotic properties, therefore maintaining the local accumulation of pro-inflammatory cells. However, the anti-apoptotic effects of P. gingivalis were not presented in all cells, and the molecular mechanisms underlying the mitochondrial dysfunction caused by P. gingivalis have not been fully investigated. Additionally, studies on how to ameliorate P. gingivalis-associated periodontitis by intervening with mitochondria will be instrumental in further optimizing mitochondria-targeted periodontitis therapy.
Author Contributions
Conceptualization, S.L. and D.Z.; methodology, T.X. and N.L.; writing—original draft preparation, T.X., Q.Z. and A.J.; writing—review and editing, D.Z., S.L. and Y.P.; visualization, S.L., J.Z. and Y.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China, grant number 81970943.
Acknowledgments
We thank the authors of references for providing the data for the final manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Papapanou, P.N.; Sanz, M.; Buduneli, N.; Dietrich, T.; Feres, M.; Fine, D.H.; Flemmig, T.F.; Garcia, R.; Giannobile, W.V.; Graziani, F.; et al. Periodontitis: Consensus report of workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J. Clin. Periodontol. 2018, 45, S162–S170. [Google Scholar] [CrossRef] [PubMed]
- Jepsen, S.; Suvan, J.; Deschner, J. The association of periodontal diseases with metabolic syndrome and obesity. Periodontology 2000 2020, 83, 125–153. [Google Scholar] [CrossRef] [PubMed]
- Farina, R.; Lopez, R.; Simonelli, A.; Trombelli, L. Accuracy and applicability of periodontitis risk assessment tools: A critical appraisal. Periodontology 2000 2023, 1–18. [Google Scholar] [CrossRef] [PubMed]
- El-Awady, A.R.; Elashiry, M.; Morandini, A.C.; Meghil, M.M.; Cutler, C.W. Dendritic cells a critical link to alveolar bone loss and systemic disease risk in periodontitis: Immunotherapeutic implications. Periodontology 2000 2022, 89, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, C.; Abdalla, H.; Sulliman, S.; Rojas, P.; Wu, Y.-C.; Almarhoumi, R.; Huang, R.-Y.; Galindo, M.; Vernal, R.; Kantarci, A. RvE1 Impacts the Gingival Inflammatory Infiltrate by Inhibiting the T Cell Response in Experimental Periodontitis. Front. Immunol. 2021, 12, 664756. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Xin, Z.; Gao, S.; Li, Y.; Guo, S.; Fu, Y.; Xu, R.; Wang, D.; Cheng, J.; Liu, L.; et al. SIRT6-regulated macrophage efferocytosis epigenetically controls inflammation resolution of diabetic periodontitis. Theranostics 2023, 13, 231–249. [Google Scholar] [CrossRef] [PubMed]
- Irwandi, R.A.; Chiesa, S.T.; Hajishengallis, G.; Papayannopoulos, V.; Deanfield, J.E.; D’aiuto, F. The Roles of Neutrophils Linking Periodontitis and Atherosclerotic Cardiovascular Diseases. Front. Immunol. 2022, 13, 915081. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Chavakis, T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat. Rev. Immunol. 2021, 21, 426–440. [Google Scholar] [CrossRef]
- Xu, W.; Zhou, W.; Wang, H.; Liang, S. Roles of Porphyromonas gingivalis and its Virulence factors in periodontitis. Adv. Protein Chem. Struct. Biol. 2020, 120, 45–84. [Google Scholar] [CrossRef]
- Jia, L.; Han, N.; Du, J.; Guo, L.; Luo, Z.; Liu, Y. Pathogenesis of Important Virulence Factors of Porphyromonas gingivalis via Toll-Like Receptors. Front. Cell. Infect. Microbiol. 2019, 9, 262. [Google Scholar] [CrossRef]
- Cheat, B.; Torrens, C.; Foda, A.; Baroukh, B.; Sadoine, J.; Slimani, L.; Witko-Sarsat, V.; Huck, O.; Gosset, M.; Bouchet, J. NLRP3 Is Involved in Neutrophil Mobilization in Experimental Periodontitis. Front. Immunol. 2022, 13, 839929. [Google Scholar] [CrossRef] [PubMed]
- Govindaraj, P.; Khan, N.A.; Gopalakrishna, P.; Chandra, R.V.; Vanniarajan, A.; Reddy, A.A.; Singh, S.; Kumaresan, R.; Srinivas, G.; Singh, L.; et al. Mitochondrial dysfunction and genetic heterogeneity in chronic periodontitis. Mitochondrion 2011, 11, 504–512. [Google Scholar] [CrossRef]
- Sun, X.; Mao, Y.; Dai, P.; Li, X.; Gu, W.; Wang, H.; Wu, G.; Ma, J.; Huang, S. Mitochondrial dysfunction is involved in the aggravation of periodontitis by diabetes. J. Clin. Periodontol. 2017, 44, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, X.; Xue, F.; Zheng, M.; Luan, Q. Abnormal mitochondrial structure and function are retained in gingival tissues and human gingival fibroblasts from patients with chronic periodontitis. J. Periodontal Res. 2022, 57, 94–103. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Liu, F.; Li, M.; Ren, M.; Wang, X.; Deng, Y.; Wu, X.; Li, Y.; Yang, S.; Song, J. Mitochondrial Calcium Ion Nanogluttons Alleviate Periodontitis via Controlling mPTPs. Adv. Healthc. Mater. 2023, 12, e2203106. [Google Scholar] [CrossRef]
- Li, A.; Du, M.; Chen, Y.; Marks, L.A.; Visser, A.; Xu, S.; Tjakkes, G.E. Periodontitis and cognitive impairment in older adults: The mediating role of mitochondrial dysfunction. J. Periodontol. 2022, 93, 1302–1313. [Google Scholar] [CrossRef]
- Ferreira, I.L.; Costa, S.; Moraes, B.J.; Costa, A.; Fokt, O.; Marinho, D.; Alves, V.; Baptista, I.P.; Rego, A.C. Mitochondrial and Redox Changes in Periodontitis and Type 2 Diabetes Human Blood Mononuclear Cells. Antioxidants 2023, 12, 226. [Google Scholar] [CrossRef]
- Portes, J.; Bullón, B.; Quiles, J.L.; Battino, M.; Bullón, P. Diabetes Mellitus and Periodontitis Share Intracellular Disorders as the Main Meeting Point. Cells 2021, 10, 2411. [Google Scholar] [CrossRef]
- Li, X.; Liu, X.C.; Ding, X.; Liu, X.M.; Cao, N.B.; Deng, Y.; Hou, Y.B.; Yu, W.X. Resveratrol protects renal damages induced by periodontitis via preventing mitochondrial dysfunction in rats. Oral Dis. 2023, 29, 1812–1825. [Google Scholar] [CrossRef]
- Li, Y.; Huang, Z.; Pan, S.; Feng, Y.; He, H.; Cheng, S.; Wang, L.; Wang, L.; Pathak, J.L. Resveratrol Alleviates Diabetic Periodontitis-Induced Alveolar Osteocyte Ferroptosis Possibly via Regulation of SLC7A11/GPX4. Nutrients 2023, 15, 2115. [Google Scholar] [CrossRef]
- Andrian, E.; Grenier, D.; Rouabhia, M. Porphyromonas gingivalis-epithelial cell interactions in periodontitis. J. Dent. Res. 2006, 85, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Wielento, A.; Lagosz-Cwik, K.; Potempa, J.; Grabiec, A. The Role of Gingival Fibroblasts in the Pathogenesis of Periodontitis. J. Dent. Res. 2023, 102, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; An, H.-J.; Kim, J.-Y.; Kim, W.-H.; Gwon, M.-G.; Kim, H.-J.; Han, S.M.; Park, I.; Park, S.C.; Leem, J.; et al. Bee venom attenuates Porphyromonas gingivalis and RANKL-induced bone resorption with osteoclastogenic differentiation. Food Chem. Toxicol. 2019, 129, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Maekawa, T.; Domon, H.; Sirisereephap, K.; Isono, T.; Hirayama, S.; Hiyoshi, T.; Sasagawa, K.; Takizawa, F.; Maeda, T.; et al. Erythromycin Restores Osteoblast Differentiation and Osteogenesis Suppressed by Porphyromonas gingivalis Lipopolysaccharide. Pharmaceuticals 2023, 16, 303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Swearingen, E.B.; Ju, J.; Rigney, T.; Tribble, G.D. Porphyromonas gingivalis invades osteoblasts and inhibits bone formation. Microbes Infect. 2010, 12, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Yoshizawa-Smith, S.; Glowacki, A.; Maltos, K.; Pacheco, C.; Shehabeldin, M.; Mulkeen, M.; Myers, N.; Chong, R.; Verdelis, K.; et al. Induction of M2 Macrophages Prevents Bone Loss in Murine Periodontitis Models. J. Dent. Res. 2019, 98, 200–208. [Google Scholar] [CrossRef]
- Zhang, L.; Gao, L.; Xu, C.; Li, X.; Wang, P.; Zhang, C.; Zhao, C. Porphyromonas gingivalis lipopolysaccharide promotes T- helper 17 cell differentiation from human CD4+ naïve T cells via toll-like receptor-2 in vitro. Arch. Oral Biol. 2019, 107, 104483. [Google Scholar] [CrossRef]
- Su, L.; Xu, Q.; Zhang, P.; Michalek, S.M.; Katz, J. Phenotype and Function of Myeloid-Derived Suppressor Cells Induced by Porphyromonas gingivalis Infection. Infect. Immun. 2017, 85, e00213-17. [Google Scholar] [CrossRef]
- Cochran, D.L. Inflammation and Bone Loss in Periodontal Disease. J. Periodontol. 2008, 79, 1569–1576. [Google Scholar] [CrossRef]
- Aleksijević, L.H.; Aleksijević, M.; Škrlec, I.; Šram, M.; Šram, M.; Talapko, J. Porphyromonas gingivalis Virulence Factors and Clinical Significance in Periodontal Disease and Coronary Artery Diseases. Pathogens 2022, 11, 1173. [Google Scholar] [CrossRef]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, J.J.; Kunji, E.R. The SLC25 Mitochondrial Carrier Family: Structure and Mechanism. Trends Biochem. Sci. 2020, 45, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Bayr, H. Reactive oxygen species. Crit. Care Med. 2005, 33, S498–S501. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, M.; Chen, Z.; Yu, Y.; Shi, H.; Yu, Y.; Wang, Y.; Chen, R.; Ge, J. Mitochondrial calpain-1 activates NLRP3 inflammasome by cleaving ATP5A1 and inducing mitochondrial ROS in CVB3-induced myocarditis. Basic Res. Cardiol. 2022, 117, 40. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-L.; Zhu, X.-H.; Ran, L.; Lang, H.-D.; Yi, L.; Mi, M.-T. Trimethylamine-N-Oxide Induces Vascular Inflammation by Activating the NLRP3 Inflammasome Through the SIRT3-SOD2-mtROS Signaling Pathway. J. Am. Heart Assoc. 2017, 6, e006347. [Google Scholar] [CrossRef] [PubMed]
- Hseu, Y.-C.; Tseng, Y.-F.; Pandey, S.; Shrestha, S.; Lin, K.-Y.; Lin, C.-W.; Lee, C.-C.; Huang, S.-T.; Yang, H.-L. Coenzyme Q0 Inhibits NLRP3 Inflammasome Activation through Mitophagy Induction in LPS/ATP-Stimulated Macrophages. Oxidative Med. Cell. Longev. 2022, 2022, 4266214. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Chen, Y.; Peng, T.; Li, L.; Zhu, W.; Liu, F.; Liu, S.; An, X.; Luo, R.; Cheng, J.; et al. Mitochondrial ROS-induced lysosomal dysfunction impairs autophagic flux and contributes to M1 macrophage polarization in a diabetic condition. Clin. Sci. 2019, 133, 1759–1777. [Google Scholar] [CrossRef]
- Luna-Sánchez, M.; Bianchi, P.; Quintana, A. Mitochondria-Induced Immune Response as a Trigger for Neurodegeneration: A Pathogen from Within. Int. J. Mol. Sci. 2021, 22, 8523. [Google Scholar] [CrossRef]
- Xian, H.; Watari, K.; Sanchez-Lopez, E.; Offenberger, J.; Onyuru, J.; Sampath, H.; Ying, W.; Hoffman, H.M.; Shadel, G.S.; Karin, M. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity 2022, 55, 1370–1385.e8. [Google Scholar] [CrossRef]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.-J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. Available online: https://pubmed.ncbi.nlm.nih.gov/30046112/ (accessed on 2 October 2023). [CrossRef]
- Deo, P.; Chow, S.H.; Han, M.-L.; Speir, M.; Huang, C.; Schittenhelm, R.B.; Dhital, S.; Emery, J.; Li, J.; Kile, B.T.; et al. Mitochondrial dysfunction caused by outer membrane vesicles from Gram-negative bacteria activates intrinsic apoptosis and inflammation. Nat. Microbiol. 2020, 5, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.D.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, M.; Hiroi, M.; Kanegae, H.; Ohmori, Y. Costimulation of Murine Osteoblasts with Interferon-γ and Tumor Necrosis Factor-α Induces Apoptosis through Downregulation of Bcl-2 and Release of Cytochrome c from Mitochondria. Mediat. Inflamm. 2018, 2018, 3979606. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, T.; Fujita, T.; Kajiya, M.; Matsuda, S.; Ouhara, K.; Shiba, H.; Kurihara, H. Involvement of smad2 and Erk/Akt cascade in TGF-β1-induced apoptosis in human gingival epithelial cells. Cytokine 2015, 75, 165–173. [Google Scholar] [CrossRef]
- Bulut, Ş.; Uslu, H.; Özdemir, B.H.; Bulut, Ö.E. Expression of caspase-3, p53 and Bcl-2 in generalized aggressive periodontitis. Head Face Med. 2006, 2, 17. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lakschevitz, F.S.; Aboodi, G.M.; Glogauer, M. Oral neutrophil transcriptome changes result in a pro-survival phenotype in periodontal diseases. PLoS ONE 2013, 8, e68983. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.S.; Mbah, N.E.; Shan, M.; Loesel, K.; Lin, L.; Sajjakulnukit, P.; Correa, L.O.; Andren, A.; Lin, J.; Hayashi, A.; et al. OXPHOS promotes apoptotic resistance and cellular persistence in TH17 cells in the periphery and tumor microenvironment. Sci. Immunol. 2022, 7, eabm8182. [Google Scholar] [CrossRef]
- He, X.-T.; Li, X.; Zhang, M.; Tian, B.-M.; Sun, L.-J.; Bi, C.-S.; Deng, D.-K.; Zhou, H.; Qu, H.-L.; Wu, C.; et al. Role of molybdenum in material immunomodulation and periodontal wound healing: Targeting immunometabolism and mitochondrial function for macrophage modulation. Biomaterials 2022, 283, 121439. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef]
- Fleetwood, A.J.; Lee, M.K.; Singleton, W.; Achuthan, A.; Lee, M.-C.; O’Brien-Simpson, N.M.; Cook, A.D.; Murphy, A.J.; Dashper, S.G.; Reynolds, E.C.; et al. Metabolic Remodeling, Inflammasome Activation, and Pyroptosis in Macrophages Stimulated by Porphyromonas gingivalis and Its Outer Membrane Vesicles. Front. Cell. Infect. Microbiol. 2017, 7, 351. [Google Scholar] [CrossRef]
- Guntur, A.R.; Le, P.T.; Farber, C.R.; Rosen, C.J. Bioenergetics during calvarial osteoblast differentiation reflect strain differences in bone mass. Endocrinology 2014, 155, 1589–1595. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Peng, Y.; Huang, X.; Xiao, J.; Ma, L.; Liu, H.; Huang, H.; Yang, Z.; Wang, C.; Wang, X.; et al. Glycometabolic reprogramming in cementoblasts: A vital target for enhancing cell mineralization. FASEB J. 2023, 37, e23241. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, X.; Luan, Q.-X. Hyperresponsiveness of human gingival fibroblasts from patients with aggressive periodontitis against bacterial lipopolysaccharide. Exp. Ther. Med. 2021, 21, 417. [Google Scholar] [CrossRef] [PubMed]
- Bullon, P.; Cordero, M.D.; Quiles, J.L.; del Carmen Ramirez-Tortosa, M.; Gonzalez-Alonso, A.; Alfonsi, S.; García-Marín, R.; de Miguel, M.; Battino, M. Autophagy in periodontitis patients and gingival fibroblasts: Unraveling the link between chronic diseases and inflammation. BMC Med. 2012, 10, 122. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.M.; Park, S.-H. Regulation of reactive oxygen species by phytochemicals for the management of cancer and diabetes. Crit. Rev. Food Sci. Nutr. 2022, 63, 5911–5936. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Narayanan, S.; Xu, C.; Angelstig, S.E.; Grünler, J.; Zhao, A.; Di Toro, A.; Bernardi, L.; Mazzone, M.; Carmeliet, P.; et al. Repression of hypoxia-inducible factor-1 contributes to increased mitochondrial reactive oxygen species production in diabetes. eLife 2022, 11, e70714. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liu, S.; Zeng, X.; Guo, Z.; Chen, D.; Li, S.; Tian, Z.; Qu, Y. Reduction of Reactive Oxygen Species Accumulation Using Gadolinium-Doped Ceria for the Alleviation of Atherosclerosis. ACS Appl. Mater. Interfaces 2023, 15, 10414–10425. [Google Scholar] [CrossRef] [PubMed]
- Dianat, M.; Radan, M.; Mard, S.A.; Sohrabi, F.; Saryazdi, S.S.N. Contribution of reactive oxygen species via the OXR1 signaling pathway in the pathogenesis of monocrotaline-induced pulmonary arterial hypertension: The protective role of Crocin. Life Sci. 2020, 256, 117848. [Google Scholar] [CrossRef]
- Luptak, I.; Qin, F.; Sverdlov, A.L.; Pimentel, D.R.; Panagia, M.; Croteau, D.; Siwik, D.A.; Bachschmid, M.M.; He, H.; Balschi, J.A.; et al. Energetic Dysfunction Is Mediated by Mitochondrial Reactive Oxygen Species and Precedes Structural Remodeling in Metabolic Heart Disease. Antioxid. Redox Signal. 2019, 31, 539–549. [Google Scholar] [CrossRef]
- Fang, C.; Wu, L.; Zhao, M.-J.; Deng, T.; Gu, J.-M.; Guo, X.-P.; Li, C.; Li, W.; Zeng, X.-T. Periodontitis Exacerbates Benign Prostatic Hyperplasia through Regulation of Oxidative Stress and Inflammation. Oxidative Med. Cell. Longev. 2021, 2021, 2094665. [Google Scholar] [CrossRef]
- Xie, M.; Tang, Q.; Nie, J.; Zhang, C.; Zhou, X.; Yu, S.; Sun, J.; Cheng, X.; Dong, N.; Hu, Y.; et al. BMAL1-Downregulation Aggravates Porphyromonas Gingivalis -Induced Atherosclerosis by Encouraging Oxidative Stress. Circ. Res. 2020, 126, e15–e29. [Google Scholar] [CrossRef]
- Charoensaensuk, V.; Chen, Y.-C.; Lin, Y.-H.; Ou, K.-L.; Yang, L.-Y.; Lu, D.-Y. Porphyromonas gingivalis Induces Proinflammatory Cytokine Expression Leading to Apoptotic Death through the Oxidative Stress/NF-κB Pathway in Brain Endothelial Cells. Cells 2021, 10, 3033. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Ji, Y.; Zhao, S.; Li, H.; Jiang, Y.; Mao, J.; Chen, Y.; Zhang, X.; Mao, Y.; Sun, X.; et al. Crosstalk between reactive oxygen species and Dynamin-related protein 1 in periodontitis. Free Radic. Biol. Med. 2021, 172, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Bullón, P.; Román-Malo, L.; Marín-Aguilar, F.; Alvarez-Suarez, J.M.; Giampieri, F.; Battino, M.; Cordero, M.D. Lipophilic antioxidants prevent lipopolysaccharide-induced mitochondrial dysfunction through mitochondrial biogenesis improvement. Pharmacol. Res. 2015, 91, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Yang, W.; Wang, C.; Qin, W.; Ming, J.; Zhang, M.; Qian, H.; Jiao, T. Methylene Blue-Mediated Photodynamic Therapy Induces Macrophage Apoptosis via ROS and Reduces Bone Resorption in Periodontitis. Oxidative Med. Cell. Longev. 2019, 2019, 1529520. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jiang, Y.; Mao, J.; Ren, X.; Ji, Y.; Mao, Y.; Chen, Y.; Sun, X.; Pan, Y.; Ma, J.; et al. Hydroxytyrosol prevents periodontitis-induced bone loss by regulating mitochondrial function and mitogen-activated protein kinase signaling of bone cells. Free Radic. Biol. Med. 2021, 176, 298–311. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Popov, L. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef]
- Lee, H.-C.; Wei, Y.-H. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int. J. Biochem. Cell Biol. 2005, 37, 822–834. [Google Scholar] [CrossRef]
- Miyazaki, T.; Iwasawa, M.; Nakashima, T.; Mori, S.; Shigemoto, K.; Nakamura, H.; Katagiri, H.; Takayanagi, H.; Tanaka, S. Intracellular and extracellular ATP coordinately regulate the inverse correlation between osteoclast survival and bone resorption. J. Biol. Chem. 2012, 287, 37808–37823. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Y.; Shi, Q.; Wang, X.; Zou, P.; Zheng, M.; Luan, Q. Mitochondrial DNA Efflux Maintained in Gingival Fibroblasts of Patients with Periodontitis through ROS/mPTP Pathway. Oxidative Med. Cell. Longev. 2022, 2022, 1000213. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.J.; Berridge, M.J.; Lipp, P.; Bootman, M.D. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 2002, 21, 1616–1627. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef] [PubMed]
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Liu, B.; Sun, J.; Zhang, X.; Zhu, Z.; Wang, H.; Xiao, A.; Gan, X. Perturbation of mitochondrial dynamics links to the aggravation of periodontitis by diabetes. J. Histotechnol. 2023, 46, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Dong, Q.; Luo, Y.; Liu, Y.; Gao, L.; Pan, Y.; Zhang, D. Porphyromonas gingivalis infection promotes mitochondrial dysfunction through Drp1-dependent mitochondrial fission in endothelial cells. Int. J. Oral Sci. 2021, 13, 28. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Harper, J.W.; Ordureau, A.; Heo, J.-M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Zhang, Y.; Yao, Y.; Qiu, X.; Wang, G.; Hu, Z.; Chen, S.; Wu, Z.; Yuan, N.; Gao, H.; Wang, J.; et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat. Immunol. 2019, 20, 433–446. [Google Scholar] [CrossRef]
- Jiang, K.; Li, J.; Jiang, L.; Li, H.; Lei, L. PINK1-mediated mitophagy reduced inflammatory responses to Porphyromonas gingivalis in macrophages. Oral Dis. 2022, 29, 3665–3676. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1111/odi.14286 (accessed on 2 December 2022). [CrossRef]
- Liu, M.; Shao, J.; Zhao, Y.; Ma, B.; Ge, S. Porphyromonas gingivalis Evades Immune Clearance by Regulating Lysosome Efflux. J. Dent. Res. 2023, 102, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Morrell, C.N. Reactive Oxygen Species: Finding the right balance. Circ. Res. 2008, 103, 571–572. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Oxygen radicals and signaling. Curr. Opin. Cell Biol. 1998, 10, 248–253. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Zheng, M.; Luan, Q.X. Mitochondrial reactive oxygen species mediate the lipopolysaccharide-induced pro-inflammatory response in human gingival fibroblasts. Exp. Cell Res. 2016, 347, 212–221. [Google Scholar] [CrossRef]
- Liu, J.; Wang, X.; Zheng, M.; Luan, Q. Oxidative stress in human gingival fibroblasts from periodontitis versus healthy counterparts. Oral Dis. 2023, 29, 1214–1225. [Google Scholar] [CrossRef]
- Liu, J.; Zeng, J.; Wang, X.; Zheng, M.; Luan, Q. P53 mediates lipopolysaccharide-induced inflammation in human gingival fibroblasts. J. Periodontol. 2018, 89, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef]
- Henry, L.G.; McKenzie, R.M.; Robles, A.; Fletcher, H.M.; Xie, H.; Fitzpatrick, R.E.; Wijeyewickrema, L.C.; Pike, R.N.; Aquaro, S.; Scopelliti, F.; et al. Oxidative stress resistance in Porphyromonas gingivalis. Future Microbiol. 2012, 7, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.A.; McKenzie, R.; McLean, L.; Sowers, L.C.; Fletcher, H.M. 8-oxo-7,8-dihydroguanine is removed by a nucleotide excision repair-like mechanism in Porphyromonas gingivalis W83. J. Bacteriol. 2004, 186, 7697–7703. [Google Scholar] [CrossRef] [PubMed]
- Smalley, J.W.; Birss, A.J.; Silver, J. The periodontal pathogen Porphyromonas gingivalis harnesses the chemistry of the mu-oxo bishaem of iron protoporphyrin IX to protect against hydrogen peroxide. FEMS Microbiol. Lett. 2000, 183, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Romero-Lastra, P.; Sánchez, M.C.; Llama-Palacios, A.; Figuero, E.; Herrera, D.; Sanz, M. Gene expression of Porphyromonas gingivalis ATCC 33277 when growing in an in vitro multispecies biofilm. PLoS ONE 2019, 14, e0221234. [Google Scholar] [CrossRef]
- Zhu, B.; Macleod, L.C.; Newsome, E.; Liu, J.; Xu, P. Aggregatibacter actinomycetemcomitans mediates protection of Porphyromonas gingivalis from Streptococcus sanguinis hydrogen peroxide production in multi-species biofilms. Sci. Rep. 2019, 9, 4944. [Google Scholar] [CrossRef]
- Knowles, A.A.; Campbell, S.G.; Cross, N.A.; Stafford, P. Dysregulation of Stress-Induced Translational Control by Porphyromonas gingivalis in Host Cells. Microorganisms 2023, 11, 606. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Yamamoto, Y.; Egashira, K.; Sato, A.; Kondo, Y.; Saiki, S.; Kimura, M.; Chikazawa, T.; Ishigami, A.; Murakami, S. Oxidative Stress Inhibits Endotoxin Tolerance and May Affect Periodontitis. J. Dent. Res. 2023, 102, 331–339. [Google Scholar] [CrossRef]
- Choi, C.H.; Spooner, R.; DeGuzman, J.; Koutouzis, T.; Ojcius, D.M.; Yilmaz, Ö. Porphyromonas gingivalis-nucleoside-diphosphate-kinase inhibits ATP-induced reactive-oxygen-species via P2X7 receptor/NADPH-oxidase signalling and contributes to persistence. Cell. Microbiol. 2013, 15, 961–976. [Google Scholar] [CrossRef]
- Amaroli, A.; Ravera, S.; Baldini, F.; Benedicenti, S.; Panfoli, I.; Vergani, L. Photobiomodulation with 808-nm diode laser light promotes wound healing of human endothelial cells through increased reactive oxygen species production stimulating mitochondrial oxidative phosphorylation. Lasers Med. Sci. 2019, 34, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sun, T.; Liu, C.; Cao, Y.; Liu, X. Photobiomodulation (450 nm) alters the infection of periodontitis bacteria via the ROS/MAPK/mTOR signaling pathway. Free Radic. Biol. Med. 2020, 152, 838–853. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Dallaporta, B.; Resche-Rigon, M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 1998, 60, 619–642. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Wischhof, L.; Scifo, E.; Ehninger, D.; Bano, D. AIFM1 beyond cell death: An overview of this OXPHOS-inducing factor in mitochondrial diseases. EBioMedicine 2022, 83, 104231. [Google Scholar] [CrossRef]
- Novo, N.; Ferreira, P.; Medina, M. The apoptosis-inducing factor family: Moonlighting proteins in the crosstalk between mitochondria and nuclei. IUBMB Life 2021, 73, 568–581. [Google Scholar] [CrossRef]
- Flores-Romero, H.; Dadsena, S.; García-Sáez, A.J. Mitochondrial pores at the crossroad between cell death and inflammatory signaling. Mol. Cell 2023, 83, 843–856. [Google Scholar] [CrossRef]
- Nakhjiri, S.F.; Park, Y.; Yilmaz, O.; Chung, W.O.; Watanabe, K.; El-Sabaeny, A.; Park, K.; Lamont, R.J. Inhibition of epithelial cell apoptosis by Porphyromonas gingivalis. FEMS Microbiol. Lett. 2001, 200, 145–149. [Google Scholar] [CrossRef]
- Wang, Y.; Dong, Y.; Zhang, W.; Wang, Y.; Jao, Y.; Liu, J.; Zhang, M.; He, H. AMPK/mTOR/p70S6K axis prevents apoptosis of Porphyromonas gingivalis-infected gingival epithelial cells via BadSer136 phosphorylation. Apoptosis 2023, 28, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, O.; Jungas, T.; Verbeke, P.; Ojcius, D.M. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect. Immun. 2004, 72, 3743–3751. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Jermanus, C.; Barbetta, B.; Choi, C.; Verbeke, P.; Ojcius, D.; Yilmaz, Ö. Porphyromonas gingivalis infection sequesters pro-apoptotic Bad through Akt in primary gingival epithelial cells. Mol. Oral Microbiol. 2010, 25, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Desta, T.; Graves, D.T. Fibroblast apoptosis induced by Porphyromonas gingivalis is stimulated by a gingipain and caspase-independent pathway that involves apoptosis-inducing factor. Cell. Microbiol. 2007, 9, 2667–2675. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Meghil, M.M.; Tawfik, O.K.; Elashiry, M.; Rajendran, M.; Arce, R.M.; Fulton, D.J.; Schoenlein, P.V.; Cutler, C.W. Disruption of Immune Homeostasis in Human Dendritic Cells via Regulation of Autophagy and Apoptosis by Porphyromonas gingivalis. Front. Immunol. 2019, 10, 2286. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Park, Y.; Hasegawa, Y.; Tribble, G.D.; James, C.E.; Handfield, M.; Stavropoulos, M.F.; Yilmaz, Ö.; Lamont, R.J. Intrinsic apoptotic pathways of gingival epithelial cells modulated by Porphyromonas gingivalis. Cell. Microbiol. 2007, 9, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Carvalho-Filho, P.C.; Trindade, S.C.; Olczak, T.; Sampaio, G.P.; Oliveira-Neto, M.G.; Santos, H.A.; Pereira, B.F.P.; Moura-Costa, L.; Xavier, M.T.; Meyer, R. Porphyromonas gingivalis HmuY stimulates expression of Bcl-2 and Fas by human CD3+ T cells. BMC Microbiol. 2013, 13, 206. [Google Scholar] [CrossRef]
- Carvalho-Filho, P.C.; Moura-Costa, L.F.; Pimentel, A.C.M.; Lopes, M.P.P.; Freitas, S.A.; Miranda, P.M.; Costa, R.S.; Figueirêdo, C.A.V.; Meyer, R.; Gomes-Filho, I.S.; et al. Apoptosis Transcriptional Profile Induced by Porphyromonas gingivalis HmuY. Mediat. Inflamm. 2019, 2019, 6758159. [Google Scholar] [CrossRef]
- Lee, J.; Roberts, J.S.; Atanasova, K.R.; Chowdhury, N.; Yilmaz, Ö. A novel kinase function of a nucleoside-diphosphate-kinase homologue in Porphyromonas gingivalis is critical in subversion of host cell apoptosis by targeting heat-shock protein 27. Cell. Microbiol. 2018, 20, e12825. [Google Scholar] [CrossRef]
- Boisvert, H.; Duncan, M.J. Translocation of Porphyromonas gingivalis gingipain adhesin peptide A44 to host mitochondria prevents apoptosis. Infect. Immun. 2010, 78, 3616–3624. [Google Scholar] [CrossRef]
- Che, C.; Liu, J.; Yang, J.; Ma, L.; Bai, N.; Zhang, Q. Osteopontin is essential for IL-1β production and apoptosis in peri-implantitis. Clin. Implant. Dent. Relat. Res. 2018, 20, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Shirasugi, M.; Nishioka, K.; Yamamoto, T.; Nakaya, T.; Kanamura, N. Normal human gingival fibroblasts undergo cytostasis and apoptosis after long-term exposure to butyric acid. Biochem. Biophys. Res. Commun. 2017, 482, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Kurita-Ochiai, T.; Seto, S.; Suzuki, N.; Yamamoto, M.; Otsuka, K.; Abe, K.; Ochiai, K. Butyric acid induces apoptosis in inflamed fibroblasts. J. Dent. Res. 2008, 87, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Seki, K.; Cueno, M.E.; Kamio, N.; Saito, Y.; Kamimoto, A.; Kurita-Ochiai, T.; Ochiai, K. Varying butyric acid amounts induce different stress- and cell death-related signals in nerve growth factor-treated PC12 cells: Implications in neuropathic pain absence during periodontal disease progression. Apoptosis 2016, 21, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, J.; Lin, L.; Zhao, H.; Miao, L.; Pan, Y. Porphyromonas gingivalis degrades integrin β1 and induces AIF-mediated apoptosis of epithelial cells. Infect. Dis. 2019, 51, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Shiheido, Y.; Maejima, Y.; Suzuki, J.-I.; Aoyama, N.; Kaneko, M.; Watanabe, R.; Sakamaki, Y.; Wakayama, K.; Ikeda, Y.; Akazawa, H.; et al. Porphyromonas gingivalis, a periodontal pathogen, enhances myocardial vulnerability, thereby promoting post-infarct cardiac rupture. J. Mol. Cell. Cardiol. 2016, 99, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Sheets, S.M.; Potempa, J.; Travis, J.; Casiano, C.A.; Fletcher, H.M. Gingipains from Porphyromonas gingivalis W83 induce cell adhesion molecule cleavage and apoptosis in endothelial cells. Infect. Immun. 2005, 73, 1543–1552. [Google Scholar] [CrossRef]
- Lv, Y.-T.; Zeng, J.-J.; Lu, J.-Y.; Zhang, X.-Y.; Xu, P.-P.; Su, Y. Porphyromonas gingivalis lipopolysaccharide (Pg-LPS) influences adipocytes injuries through triggering XBP1 and activating mitochondria-mediated apoptosis. Adipocyte 2021, 10, 28–37. [Google Scholar] [CrossRef]
- Bullon, P.; Cordero, M.D.; Quiles, J.L.; Morillo, J.M.; del Carmen Ramirez-Tortosa, M.; Battino, M. Mitochondrial dysfunction promoted by Porphyromonas gingivalis lipopolysaccharide as a possible link between cardiovascular disease and periodontitis. Free Radic. Biol. Med. 2011, 50, 1336–1343. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).