Mouse Model of Nitrogen Mustard Ocular Surface Injury Characterization and Sphingolipid Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
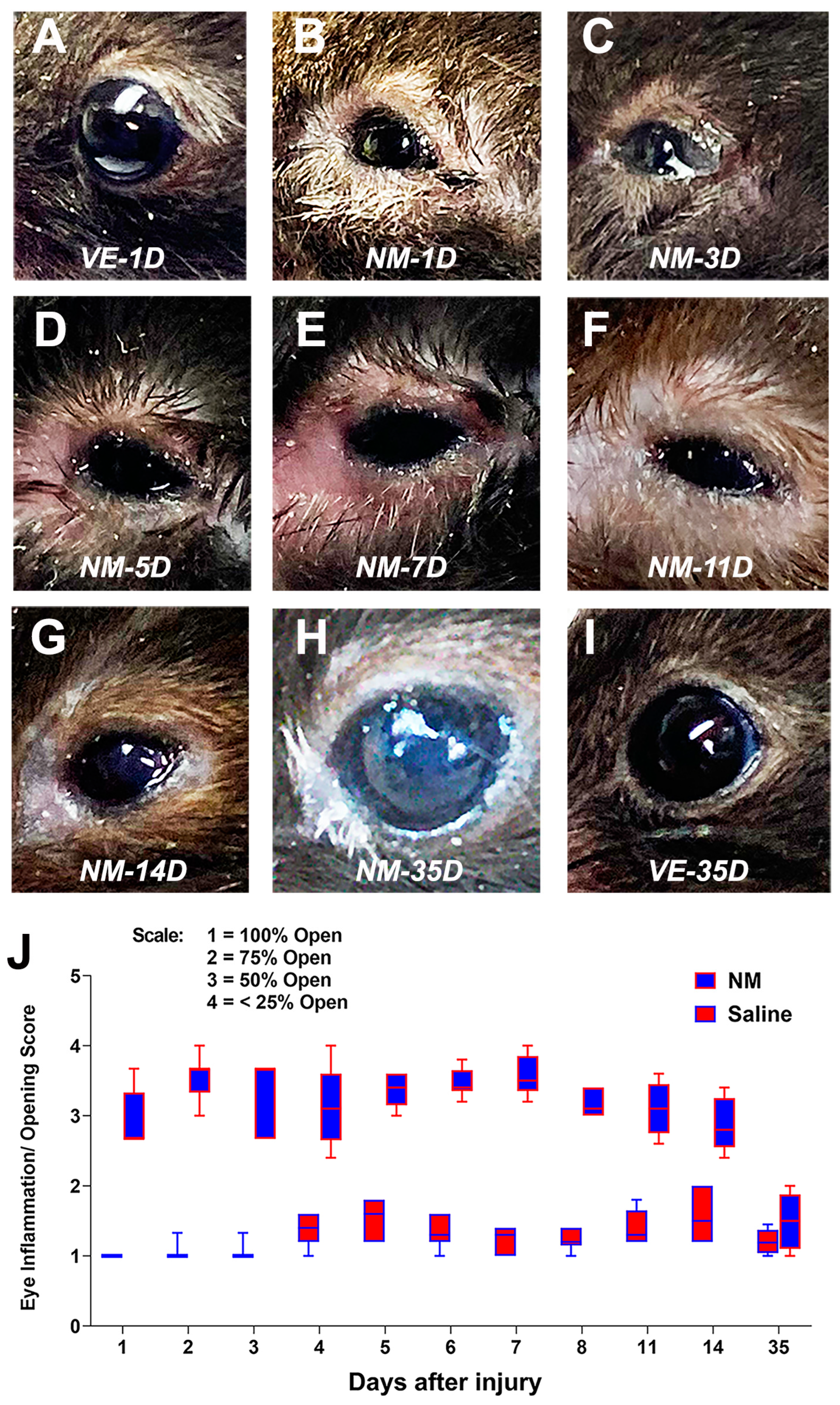
2.1. Developing a Mouse Model of NM-Induced Ocular Surface Injury
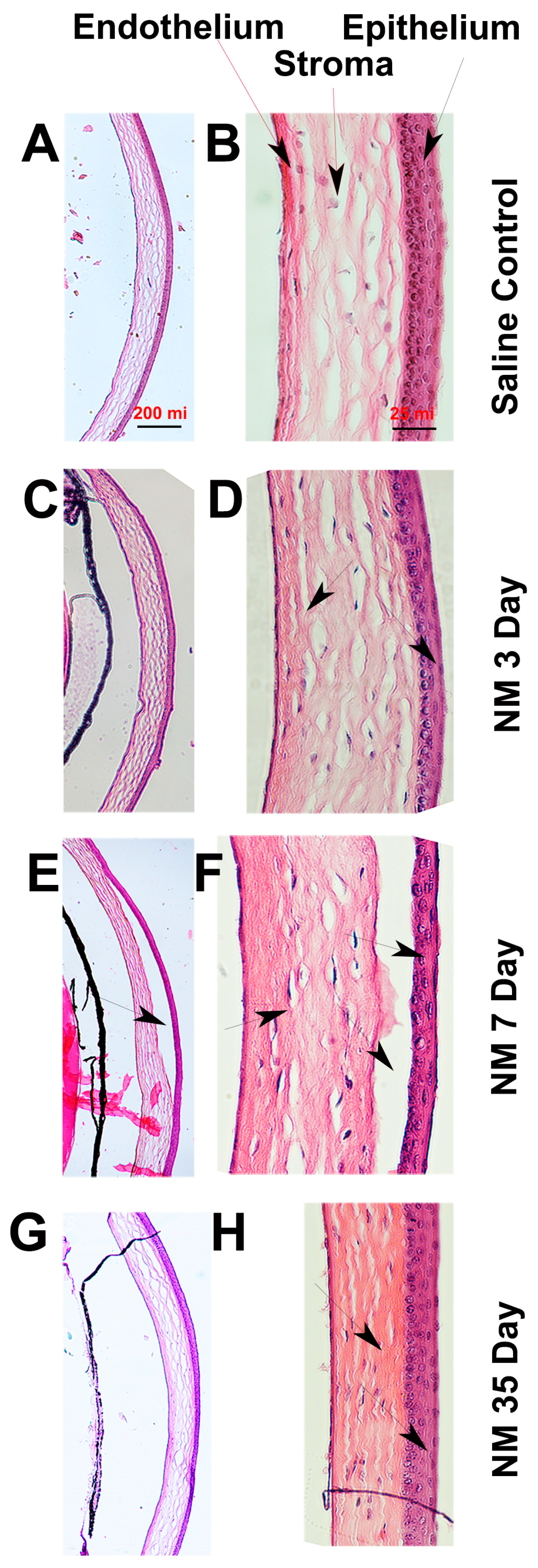
2.2. Histopathological Characterization of NM-Injured Mouse Corneas
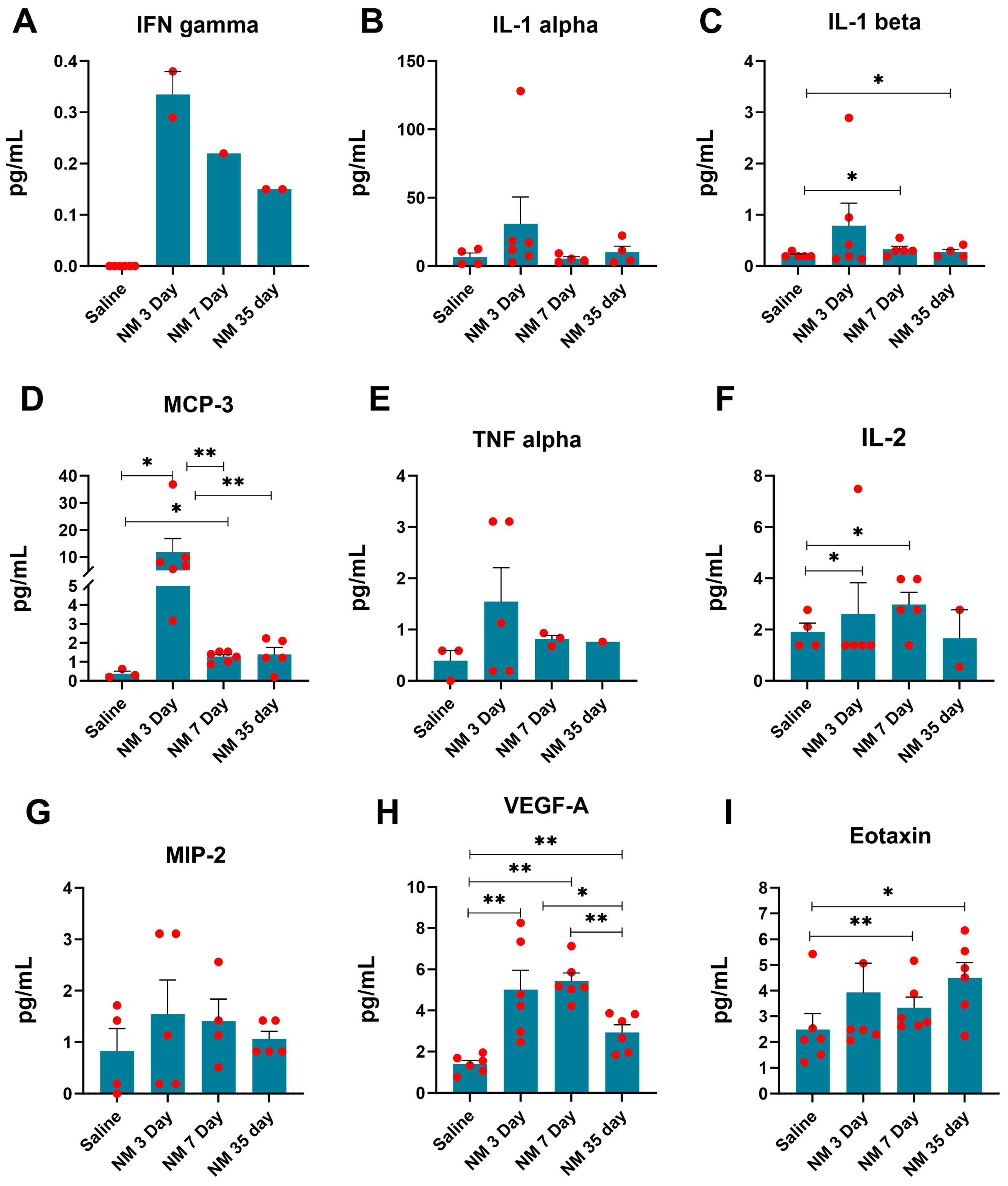
2.3. Characterization of Inflammatory Markers following NM Injury
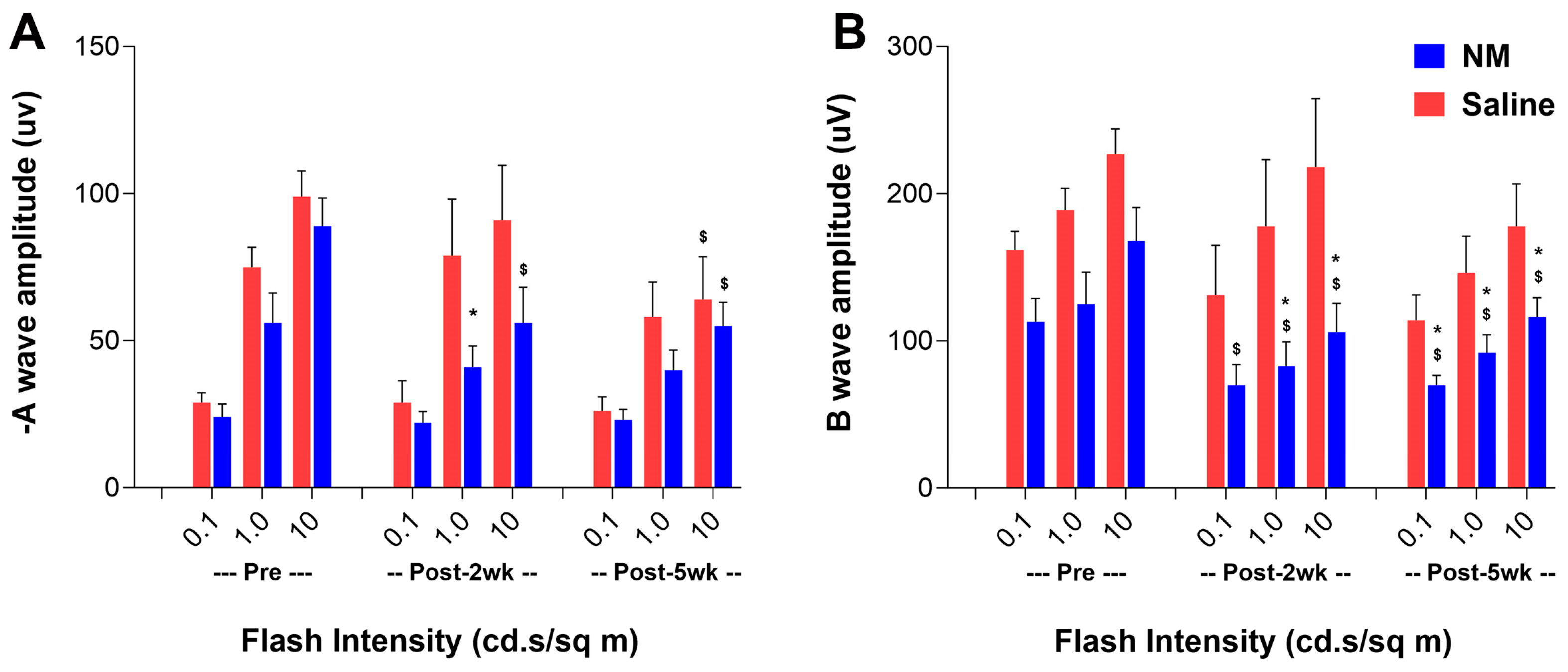
2.4. Effect of NM-Induced Ocular Surface Injury on the Visual Functions
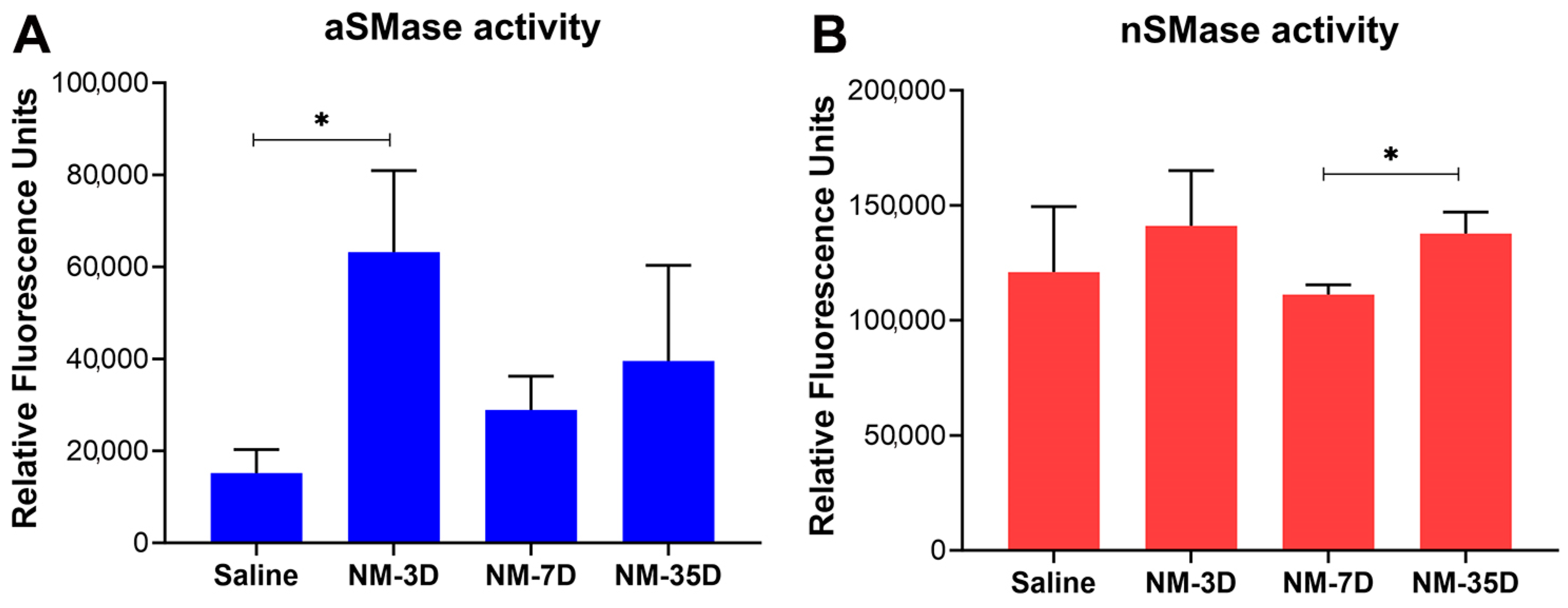
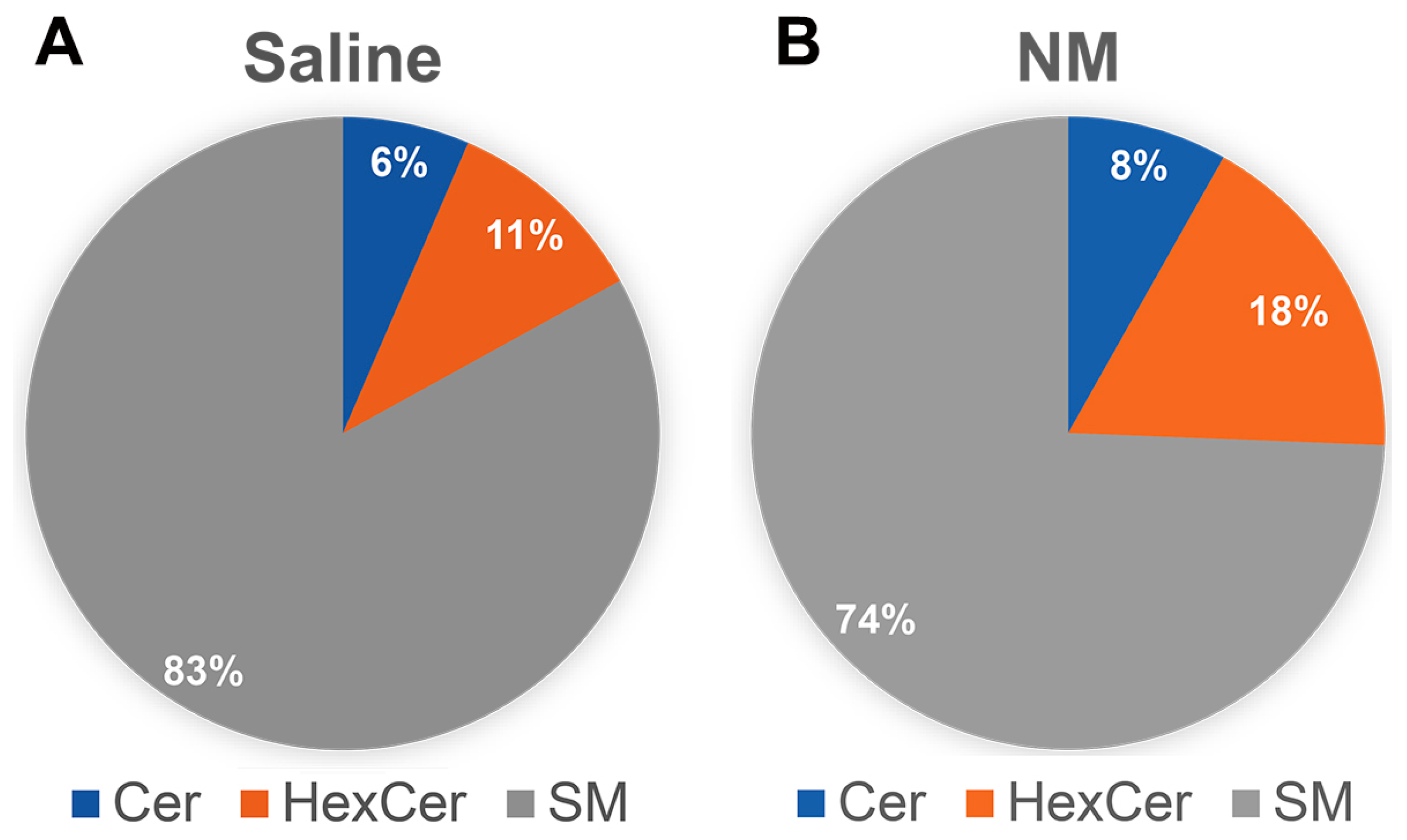
2.5. Characterization of Sphingolipid Signaling in NM-Induced Ocular Surface Injury
3. Discussion
4. Materials and Methods
4.1. Animal Care
4.2. Ocular Surface Injury by NM and Clinical Characterization of NM-Induced Injury
4.3. Histopathological Characterization of NM-Induced Injury
4.4. Multiplex Analysis of the Inflammatory Markers of the Ocular Surface following NM Injury
4.5. Analysis of Visual Function by Optokinetic Nystagmus (OKN) following NM Injury
4.6. Analysis of Retinal Function by Electroretinogram (ERG) following NM Injury
4.7. Analysis of the Activity of the Sphingomyelinase Enzyme (SMase) following NM Injury
4.8. Sphingolipid Analysis of the Ocular Surface following NM Injury
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hutchinson, R. Weapons of Mass Destruction: The No-Nonsense Guide to Nuclear, Chemical and Biological Weapons Today; Hachette UK: London, UK, 2011. [Google Scholar]
- Panahi, Y.; Roshandel, D.; Sadoughi, M.M.; Ghanei, M.; Sahebkar, A. Sulfur Mustard-Induced Ocular Injuries: Update on Mechanisms and Management. Curr. Pharm. Des. 2017, 23, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- WHO. Public Health Response to Biological and Chemical Weapons: WHO Guidance; World Health Organization: Geneva, Switzerland, 2004. [Google Scholar]
- Ghabili, K.; Agutter, P.S.; Ghanei, M.; Ansarin, K.; Panahi, Y.; Shoja, M.M. Sulfur mustard toxicity: History, chemistry, pharmacokinetics, and pharmacodynamics. Crit. Rev. Toxicol. 2011, 41, 384–403. [Google Scholar] [CrossRef] [PubMed]
- Ghabili, K.; Agutter, P.S.; Ghanei, M.; Ansarin, K.; Shoja, M.M. Mustard gas toxicity: The acute and chronic pathological effects. J. Appl. Toxicol. 2010, 30, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Papirmeister, B.; Feister, A.J.; Robinson, S.I.; Ford, R.D. Medical Defense against Mustard Gas: Toxic Mechanisms and Pharmacological Implications; CRC Press: Boca Raton, FL, USA, 1991. [Google Scholar]
- Araj, H.; Tseng, H.; Yeung, D.T. Supporting discovery and development of medical countermeasures for chemical injury to eye and skin. Exp. Eye Res. 2022, 221, 109156. [Google Scholar] [CrossRef] [PubMed]
- Yeung, D.T.; Araj, H.; Harper, J.R.; Platoff, G.E., Jr. Considerations in developing medical countermeasures against chemical ocular toxicity. Toxicol. Lett. 2020, 334, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Kehe, K.; Balszuweit, F.; Emmler, J.; Kreppel, H.; Jochum, M.; Thiermann, H. Sulfur mustard research—Strategies for the development of improved medical therapy. Eplasty 2008, 8, e32. [Google Scholar] [PubMed]
- Araj, H.; Tumminia, S.J.; Yeung, D.T. Ocular Surface—Merging Challenges and Opportunities. Transl. Vis. Sci. Technol. 2020, 9, 3. [Google Scholar] [CrossRef]
- Goswami, D.G.; Kant, R.; Ammar, D.A.; Kumar, D.; Enzenauer, R.W.; Petrash, J.M.; Tewari-Singh, N.; Agarwal, R. Acute corneal injury in rabbits following nitrogen mustard ocular exposure. Exp. Mol. Pathol. 2019, 110, 104275. [Google Scholar] [CrossRef]
- Kadar, T.; Dachir, S.; Cohen, M.; Gutman, H.; Cohen, L.; Brandeis, R.; Horwitz, V.; Amir, A. Prolonged impairment of corneal innervation after exposure to sulfur mustard and its relation to the development of delayed limbal stem cell deficiency. Cornea 2013, 32, e44–e50. [Google Scholar] [CrossRef]
- Kadar, T.; Cohen, M.; Cohen, L.; Fishbine, E.; Sahar, R.; Brandeis, R.; Dachir, S.; Amir, A. Endothelial cell damage following sulfur mustard exposure in rabbits and its association with the delayed-onset ocular lesions. Cutan. Ocul. Toxicol. 2013, 32, 115–123. [Google Scholar] [CrossRef]
- Kadar, T.; Dachir, S.; Cohen, L.; Sahar, R.; Fishbine, E.; Cohen, M.; Turetz, J.; Gutman, H.; Buch, H.; Brandeis, R.; et al. Ocular injuries following sulfur mustard exposure--pathological mechanism and potential therapy. Toxicology 2009, 263, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Kadar, T.; Turetz, J.; Fishbine, E.; Sahar, R.; Chapman, S.; Amir, A. Characterization of acute and delayed ocular lesions induced by sulfur mustard in rabbits. Curr. Eye Res. 2001, 22, 42–53. [Google Scholar] [CrossRef] [PubMed]
- McNutt, P.M.; Kelly, K.E.M.; Altvater, A.C.; Nelson, M.R.; Lyman, M.E.; O’Brien, S.; Conroy, M.T.; Ondeck, C.A.; Bodt, S.M.L.; Wolfe, S.E.; et al. Dose-dependent emergence of acute and recurrent corneal lesions in sulfur mustard-exposed rabbit eyes. Toxicol. Lett. 2021, 341, 33–42. [Google Scholar] [CrossRef] [PubMed]
- McNutt, P.M.; Nguyen, D.L.; Nelson, M.R.; Lyman, M.E.; Eisen, M.M.; Ondeck, C.A.; Wolfe, S.E.; Pagarigan, K.T.; Mangkhalakhili, M.C.; Kniffin, D.M.; et al. Corneal Endothelial Cell Toxicity Determines Long-Term Outcome After Ocular Exposure to Sulfur Mustard Vapor. Cornea 2020, 39, 640–648. [Google Scholar] [CrossRef] [PubMed]
- McNutt, P.; Tuznik, K.; Nelson, M.; Adkins, A.; Lyman, M.; Glotfelty, E.; Hughes, J.; Hamilton, T. Structural, morphological, and functional correlates of corneal endothelial toxicity following corneal exposure to sulfur mustard vapor. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6735–6744. [Google Scholar] [CrossRef]
- McNutt, P.; Hamilton, T.; Nelson, M.; Adkins, A.; Swartz, A.; Lawrence, R.; Milhorn, D. Pathogenesis of acute and delayed corneal lesions after ocular exposure to sulfur mustard vapor. Cornea 2012, 31, 280–290. [Google Scholar] [CrossRef]
- Milhorn, D.; Hamilton, T.; Nelson, M.; McNutt, P. Progression of ocular sulfur mustard injury: Development of a model system. Ann. N. Y. Acad. Sci. 2010, 1194, 72–80. [Google Scholar] [CrossRef]
- Ruff, A.L.; Jarecke, A.J.; Hilber, D.J.; Rothwell, C.C.; Beach, S.L.; Dillman, J.F., 3rd. Development of a mouse model for sulfur mustard-induced ocular injury and long-term clinical analysis of injury progression. Cutan. Ocul. Toxicol. 2013, 32, 140–149. [Google Scholar] [CrossRef]
- Tewari-Singh, N.; Goswami, D.G.; Kant, R.; Ammar, D.A.; Kumar, D.; Enzenauer, R.W.; Casillas, R.P.; Croutch, C.R.; Petrash, J.M.; Agarwal, R. Histopathological and Molecular Changes in the Rabbit Cornea From Arsenical Vesicant Lewisite Exposure. Toxicol. Sci. 2017, 160, 420–428. [Google Scholar] [CrossRef]
- Alemi, H.; Dehghani, S.; Forouzanfar, K.; Surico, P.L.; Narimatsu, A.; Musayeva, A.; Sharifi, S.; Wang, S.; Dohlman, T.H.; Yin, J.; et al. Insights into mustard gas keratopathy-characterizing corneal layer-specific changes in mice exposed to nitrogen mustard. Exp. Eye Res. 2023, 236, 109657. [Google Scholar] [CrossRef]
- An, S.; Shen, X.; Anwar, K.; Ashraf, M.; Lee, H.; Koganti, R.; Ghassemi, M.; Djalilian, A.R. Therapeutic Potential of Mesenchymal Stem Cell-Secreted Factors on Delay in Corneal Wound Healing by Nitrogen Mustard. Int. J. Mol. Sci. 2022, 23, 11510. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, M.; Baharnoori, S.M.; Cheraqpour, K.; Momenaei, B.; Mirshahi, R.; Chow, C.; Shahjahan, S.; Nguyen, T.; Ashraf, M.J.; Huang, X.; et al. Cellular senescence implication in mustard keratopathy. Exp. Eye Res. 2023, 233, 109565. [Google Scholar] [CrossRef]
- Soleimani, M.; Mirzaei, A.; Cheraqpour, K.; Baharnoori, S.M.; Arabpour, Z.; Ashraf, M.J.; Ghassemi, M.; Djalilian, A.R. The Potential of Mesenchymal Stem/Stromal Cell Therapy in Mustard Keratopathy: Discovering New Roads to Combat Cellular Senescence. Cells 2023, 12, 2744. [Google Scholar] [CrossRef] [PubMed]
- Quinville, B.M.; Deschenes, N.M.; Ryckman, A.E.; Walia, J.S. A Comprehensive Review: Sphingolipid Metabolism and Implications of Disruption in Sphingolipid Homeostasis. Int. J. Mol. Sci. 2021, 22, 5793. [Google Scholar] [CrossRef]
- Green, C.D.; Maceyka, M.; Cowart, L.A.; Spiegel, S. Sphingolipids in metabolic disease: The good, the bad, and the unknown. Cell Metab. 2021, 33, 1293–1306. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.V.; Basu, S.K.; Qaladize, B.; Grambergs, R.; Rotstein, N.P.; Mandal, N. Sphingolipids as critical players in retinal physiology and pathology. J. Lipid Res. 2021, 62, 100037. [Google Scholar] [CrossRef] [PubMed]
- Alshaikh, R.A.; Ryan, K.B.; Waeber, C. Sphingosine 1-phosphate, a potential target in neovascular retinal disease. Br. J. Ophthalmol. 2022, 106, 1187–1195. [Google Scholar] [CrossRef]
- Chen, H.; Chan, A.Y.; Stone, D.U.; Mandal, N.A. Beyond the cherry-red spot: Ocular manifestations of sphingolipid-mediated neurodegenerative and inflammatory disorders. Surv. Ophthalmol. 2014, 59, 64–76. [Google Scholar] [CrossRef]
- Gronert, K. Lipid autacoids in inflammation and injury responses: A matter of privilege. Mol. Interv. 2008, 8, 28–35. [Google Scholar] [CrossRef]
- Kalish, B.T.; Kieran, M.W.; Puder, M.; Panigrahy, D. The growing role of eicosanoids in tissue regeneration, repair, and wound healing. Prostaglandins Other Lipid Mediat. 2013, 104–105, 130–138. [Google Scholar] [CrossRef]
- Ljubimov, A.V.; Saghizadeh, M. Progress in corneal wound healing. Prog. Retin. Eye Res. 2015, 49, 17–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Hu, X.; Qi, X.; Di, G.; Zhang, Y.; Wang, Q.; Zhou, Q. Resolvin D1 promotes corneal epithelial wound healing and restoration of mechanical sensation in diabetic mice. Mol. Vis. 2018, 24, 274–285. [Google Scholar] [PubMed]
- Chiurchiu, V.; Leuti, A.; Maccarrone, M. Bioactive Lipids and Chronic Inflammation: Managing the Fire Within. Front. Immunol. 2018, 9, 38. [Google Scholar] [CrossRef]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Charkoftaki, G.; Jester, J.V.; Thompson, D.C.; Vasiliou, V. Nitrogen mustard-induced corneal injury involves the sphingomyelin-ceramide pathway. Ocul. Surf. 2018, 16, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Goswami, D.G.; Mishra, N.; Kant, R.; Agarwal, C.; Ammar, D.A.; Petrash, J.M.; Tewari-Singh, N.; Agarwal, R. Effect of dexamethasone treatment at variable therapeutic windows in reversing nitrogen mustard-induced corneal injuries in rabbit ocular in vivo model. Toxicol. Appl. Pharmacol. 2022, 437, 115904. [Google Scholar] [CrossRef] [PubMed]
- Amini, H.; Solaymani-Dodaran, M.; Mousavi, B.; Alam Beladi, S.N.; Soroush, M.R.; Abolghasemi, J.; Vahedian-Azimi, A.; Salesi, M.; Guest, P.C.; Sahebkar, A.; et al. Long-term Health Outcomes Among Survivors Exposed to Sulfur Mustard in Iran. JAMA Netw. Open 2020, 3, e2028894. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, H.; Javadi, M.A.; Ardestani, S.K.; Mahmoudi, M.; Pourfarzam, S.; Mahdavi, M.R.V.; Yarmohammadi, M.E.; Baradaran-Rafii, A.; Jadidi, K.; Shariatpanahi, S.; et al. Alteration in inflammatory mediators in seriously eye-injured war veterans, long-term after sulfur mustard exposure. Int. Immunopharmacol. 2020, 80, 105897. [Google Scholar] [CrossRef]
- Gore, A.; Kadar, T.; Dachir, S.; Horwitz, V. Therapeutic measures for sulfur mustard-induced ocular injury. Toxicol. Lett. 2021, 340, 58–66. [Google Scholar] [CrossRef]
- Javadi, M.A.; Jafarinasab, M.R.; Feizi, S.; Karimian, F.; Negahban, K. Management of Mustard Gas-Induced Limbal Stem Cell Deficiency and Keratitis. Ophthalmology 2011, 118, 1272–1281. [Google Scholar] [CrossRef]
- Biswas, S.; Bhattacherjee, P.; Paterson, C.A. Prostaglandin E2 receptor subtypes, EP1, EP2, EP3 and EP4 in human and mouse ocular tissues—A comparative immunohistochemical study. Prostaglandins Leukot. Essent. Fat. Acids 2004, 71, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.D.; Gilroy, D.W.; Serhan, C.N. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 2014, 40, 315–327. [Google Scholar] [CrossRef]
- Kumar, R.K.; Thomas, P.S.; Seetoo, D.-Q.; Herbert, C.; McKenzie, A.N.J.; Foster, P.S.; Lloyd, A.R. Eotaxin Expression by Epithelial Cells and Plasma Cells in Chronic Asthma. Lab. Investig. 2002, 82, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Minshall, E.M.; Cameron, L.; Lavigne, F.; Leung, D.Y.M.; Hamilos, D.; Garcia-Zepada, E.A.; Rothenberg, M.; Luster, A.D.; Hamid, Q. Eotaxin mRNA and Protein Expression in Chronic Sinusitis and Allergen-induced Nasal Responses in Seasonal Allergic Rhinitis. Am. J. Respir. Cell Mol. Biol. 1997, 17, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T. Lipid mediators in health and disease: Enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 123–150. [Google Scholar] [CrossRef] [PubMed]
- Flitter, B.A.; Fang, X.; Matthay, M.A.; Gronert, K. The potential of lipid mediator networks as ocular surface therapeutics and biomarkers. Ocul. Surf. 2021, 19, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Paranjpe, V.; Galor, A.; Grambergs, R.; Mandal, N. The role of sphingolipids in meibomian gland dysfunction and ocular surface inflammation. Ocul. Surf. 2022, 26, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Makide, K.; Kitamura, H.; Sato, Y.; Okutani, M.; Aoki, J. Emerging lysophospholipid mediators, lysophosphatidylserine, lysophosphatidylthreonine, lysophosphatidylethanolamine and lysophosphatidylglycerol. Prostaglandins Other Lipid Mediat. 2009, 89, 135–139. [Google Scholar] [CrossRef]
- El Alwani, M.; Wu, B.X.; Obeid, L.M.; Hannun, Y.A. Bioactive sphingolipids in the modulation of the inflammatory response. Pharmacol. Ther. 2006, 112, 171–183. [Google Scholar] [CrossRef]
- Gomez-Munoz, A.; Presa, N.; Gomez-Larrauri, A.; Rivera, I.G.; Trueba, M.; Ordonez, M. Control of inflammatory responses by ceramide, sphingosine 1-phosphate and ceramide 1-phosphate. Prog. Lipid Res. 2016, 61, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Albeituni, S.; Stiban, J. Roles of Ceramides and Other Sphingolipids in Immune Cell Function and Inflammation. Adv. Exp. Med. Biol. 2019, 1161, 169–191. [Google Scholar] [CrossRef] [PubMed]
- Nixon, G.F. Sphingolipids in inflammation: Pathological implications and potential therapeutic targets. Br. J. Pharmacol. 2009, 158, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Schwandner, R.; Wiegmann, K.; Bernardo, K.; Kreder, D.; Kronke, M. TNF receptor death domain-associated proteins TRADD and FADD signal activation of acid sphingomyelinase. J. Biol. Chem. 1998, 273, 5916–5922. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, M.; Singh, S.; Jaffrezou, J.P.; Aggarwal, B.B. Acidic sphingomyelinase-generated ceramide is needed but not sufficient for TNF-induced apoptosis and nuclear factor-kappa B activation. J. Immunol. 1996, 157, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Reiss, L.K.; Raffetseder, U.; Gibbert, L.; Drescher, H.K.; Streetz, K.L.; Schwarz, A.; Martin, C.; Uhlig, S.; Adam, D. Reevaluation of Lung Injury in TNF-Induced Shock: The Role of the Acid Sphingomyelinase. Mediators Inflamm. 2020, 2020, 3650508. [Google Scholar] [CrossRef] [PubMed]
- Stiles, M.; Qi, H.; Sun, E.; Tan, J.; Porter, H.; Allegood, J.; Chalfant, C.E.; Yasumura, D.; Matthes, M.T.; LaVail, M.M.; et al. Sphingolipid profile alters in retinal dystrophic P23H-1 rats and systemic FTY720 can delay retinal degeneration. J. Lipid Res. 2016, 57, 818–831. [Google Scholar] [CrossRef]
- Wilkerson, J.L.; Basu, S.K.; Stiles, M.A.; Prislovsky, A.; Grambergs, R.C.; Nicholas, S.; Karamichos, D.; Allegood, J.C.; Proia, R.L.; Mandal, N. Ablation of Sphingosine kinase 1 Protects Cornea from Neovascularization in a Mouse Corneal Injury Model. Cells 2022, 11, 2914. [Google Scholar] [CrossRef]
- Galor, A.; Sanchez, V.; Jensen, A.; Burton, M.; Maus, K.; Stephenson, D.; Chalfant, C.; Mandal, N. Meibum sphingolipid composition is altered in individuals with meibomian gland dysfunction-a side by side comparison of Meibum and Tear Sphingolipids. Ocul. Surf. 2022, 23, 87–95. [Google Scholar] [CrossRef]
- Stephenson, D.J.; MacKnight, H.P.; Hoeferlin, L.A.; Washington, S.L.; Sawyers, C.; Archer, K.J.; Strauss, J.F., 3rd; Walsh, S.W.; Chalfant, C.E. Bioactive lipid mediators in plasma are predictors of preeclampsia irrespective of aspirin therapy. J. Lipid Res. 2023, 64, 100377. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basu, S.K.; Prislovsky, A.; Lenchik, N.; Stephenson, D.J.; Agarwal, R.; Chalfant, C.E.; Mandal, N. Mouse Model of Nitrogen Mustard Ocular Surface Injury Characterization and Sphingolipid Signaling. Int. J. Mol. Sci. 2024, 25, 742. https://doi.org/10.3390/ijms25020742
Basu SK, Prislovsky A, Lenchik N, Stephenson DJ, Agarwal R, Chalfant CE, Mandal N. Mouse Model of Nitrogen Mustard Ocular Surface Injury Characterization and Sphingolipid Signaling. International Journal of Molecular Sciences. 2024; 25(2):742. https://doi.org/10.3390/ijms25020742
Chicago/Turabian StyleBasu, Sandip K., Amanda Prislovsky, Nataliya Lenchik, Daniel J. Stephenson, Rajesh Agarwal, Charles E. Chalfant, and Nawajes Mandal. 2024. "Mouse Model of Nitrogen Mustard Ocular Surface Injury Characterization and Sphingolipid Signaling" International Journal of Molecular Sciences 25, no. 2: 742. https://doi.org/10.3390/ijms25020742