


The Current and Promising Oral Delivery Methods for Protein- and Peptide-Based Drugs

 , , , and
, , , and
Abstract
:1. Introduction
2. Oral Administration of PPs
2.1. Advantages of Oral Administration
2.2. Types of Peptides
2.3. Penetration of Mucus Membranes—A Lesson from Viruses and Prions
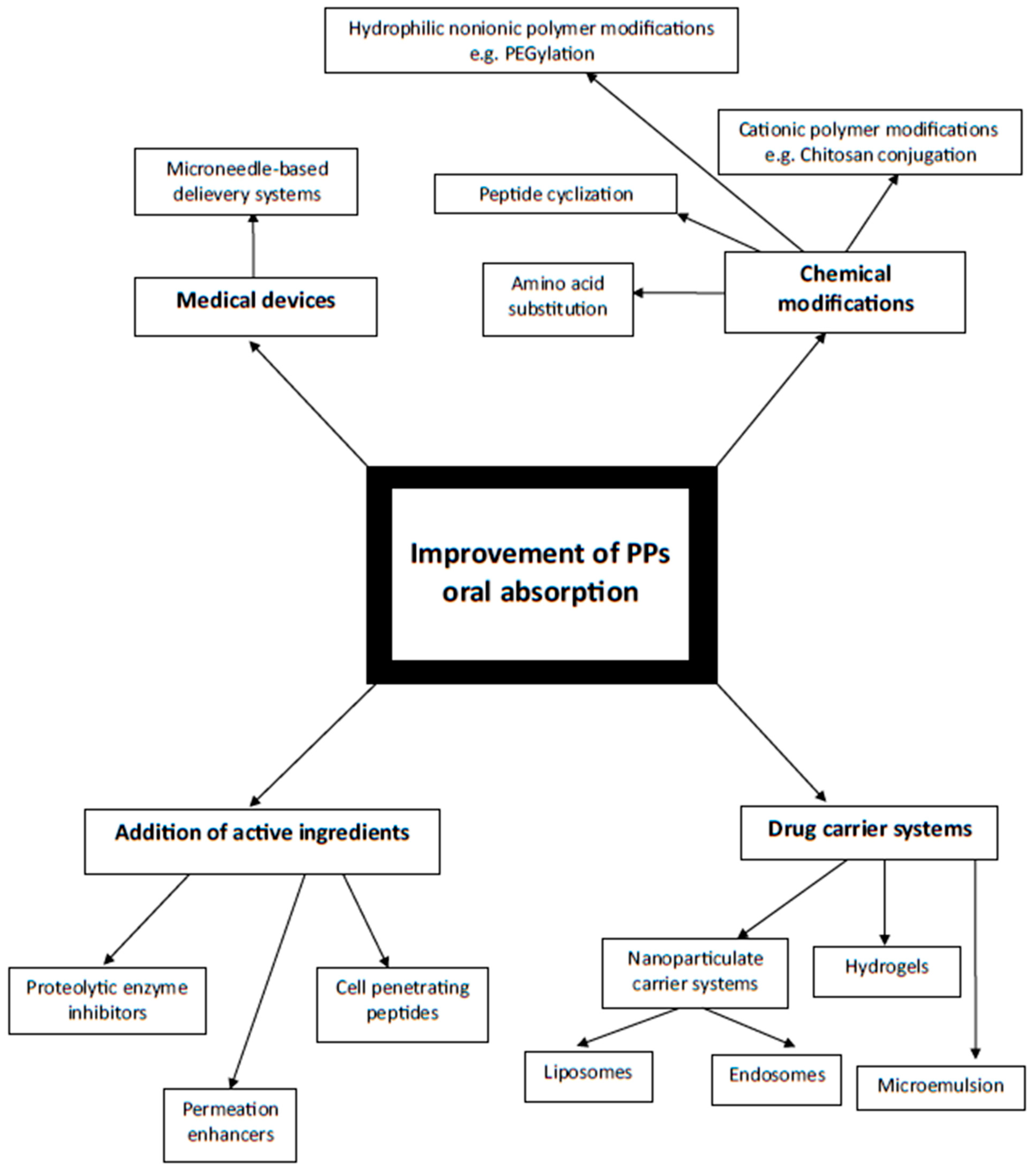
3. Difficulties Associated with Oral Administration and Methods of Their Resolution
3.1. Main Factors Affecting Absorption from Digestive System
3.1.1. pH in GI Tract
3.1.2. Digestive Enzymes
3.1.3. Mucus
3.1.4. Epithelium
3.2. Potential Solutions to the Oral PPDs Delivery Issues
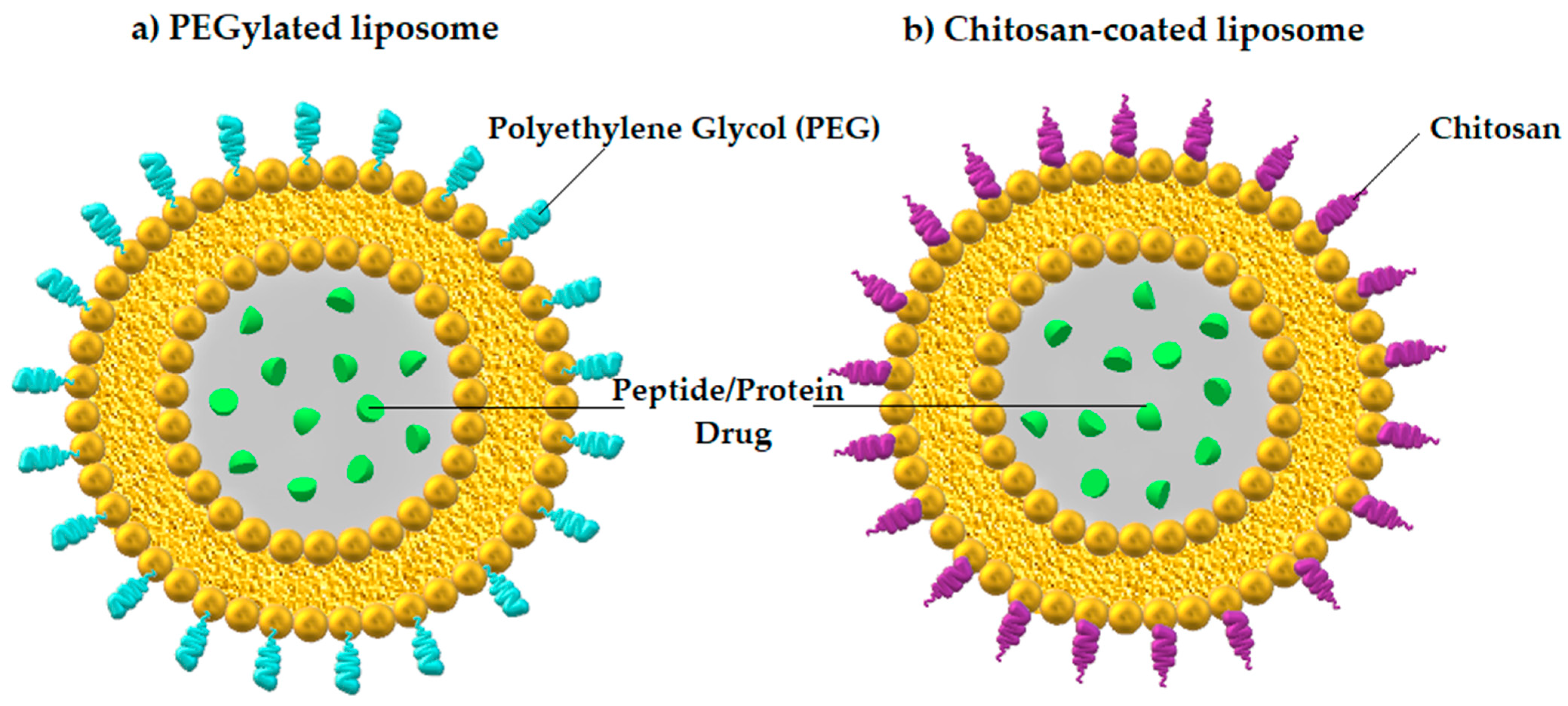
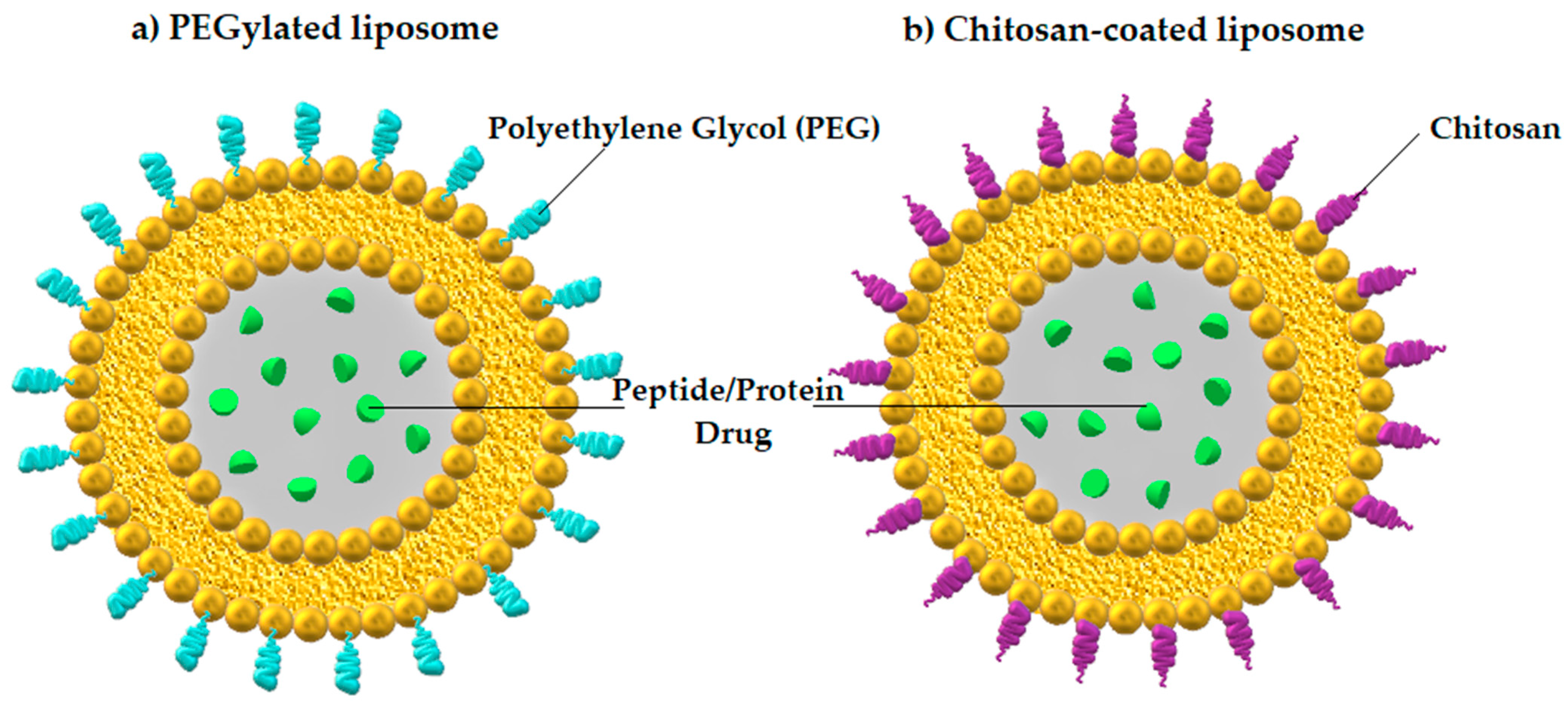
3.2.1. Nanoparticles
3.2.2. Transport Channels
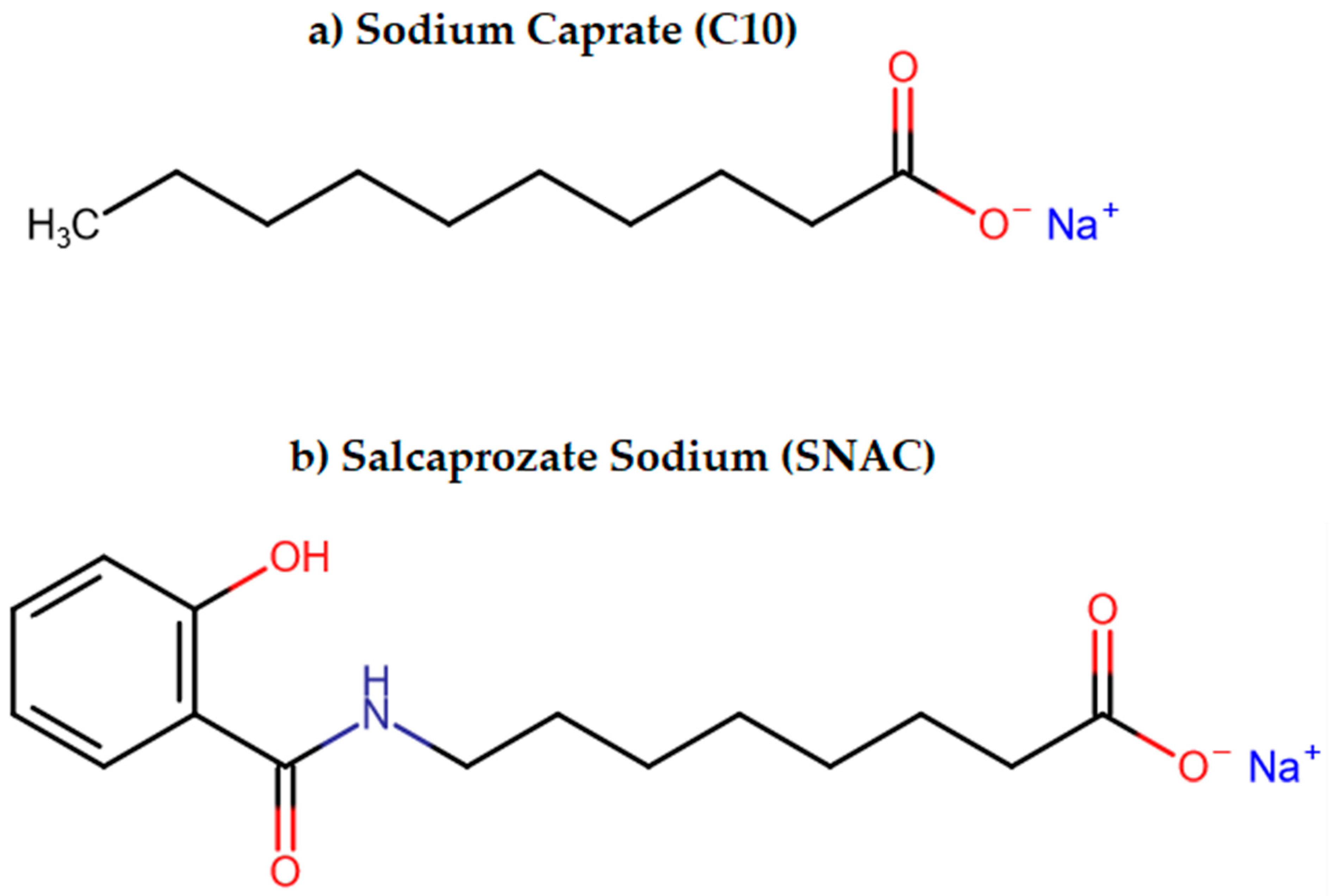
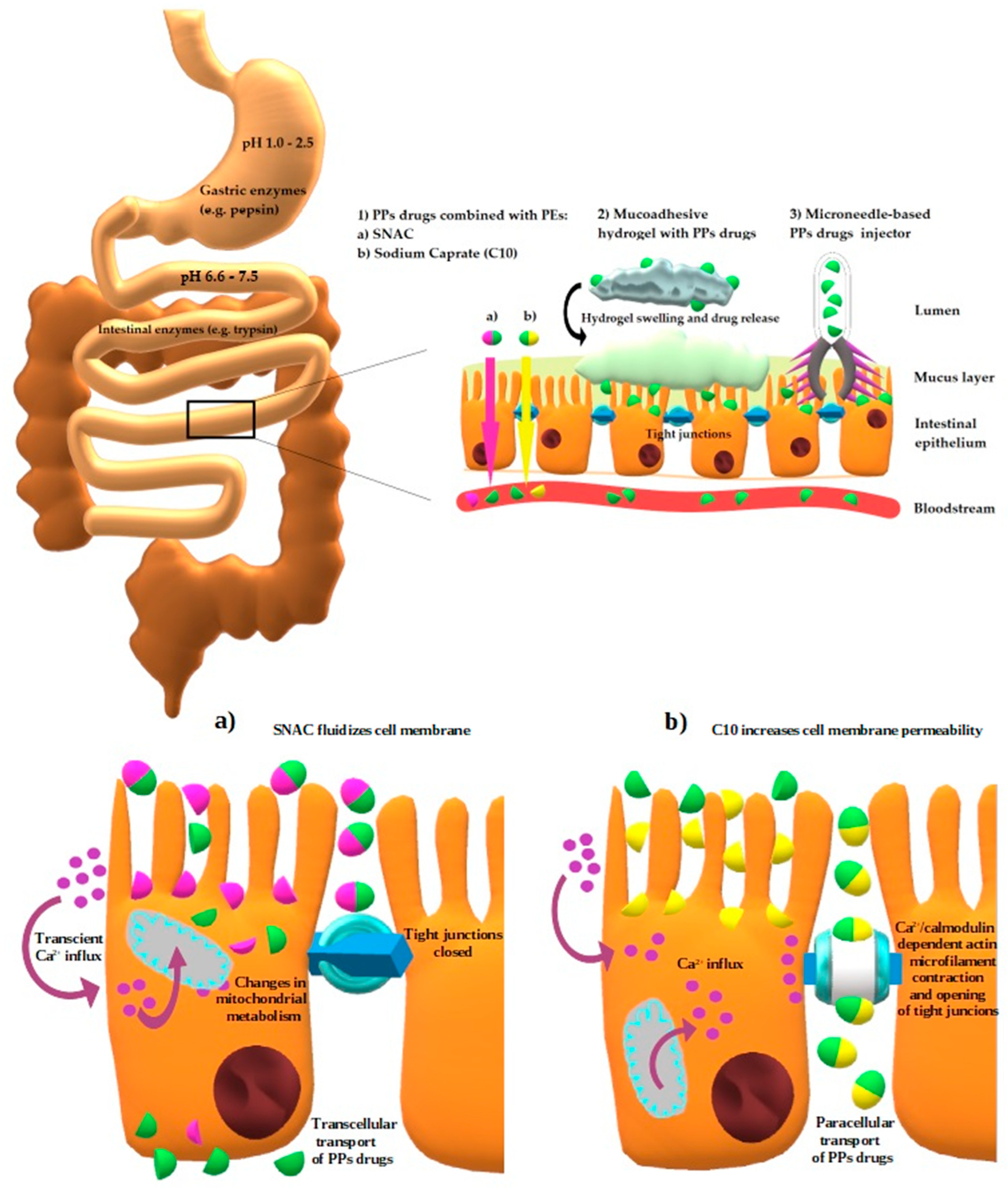
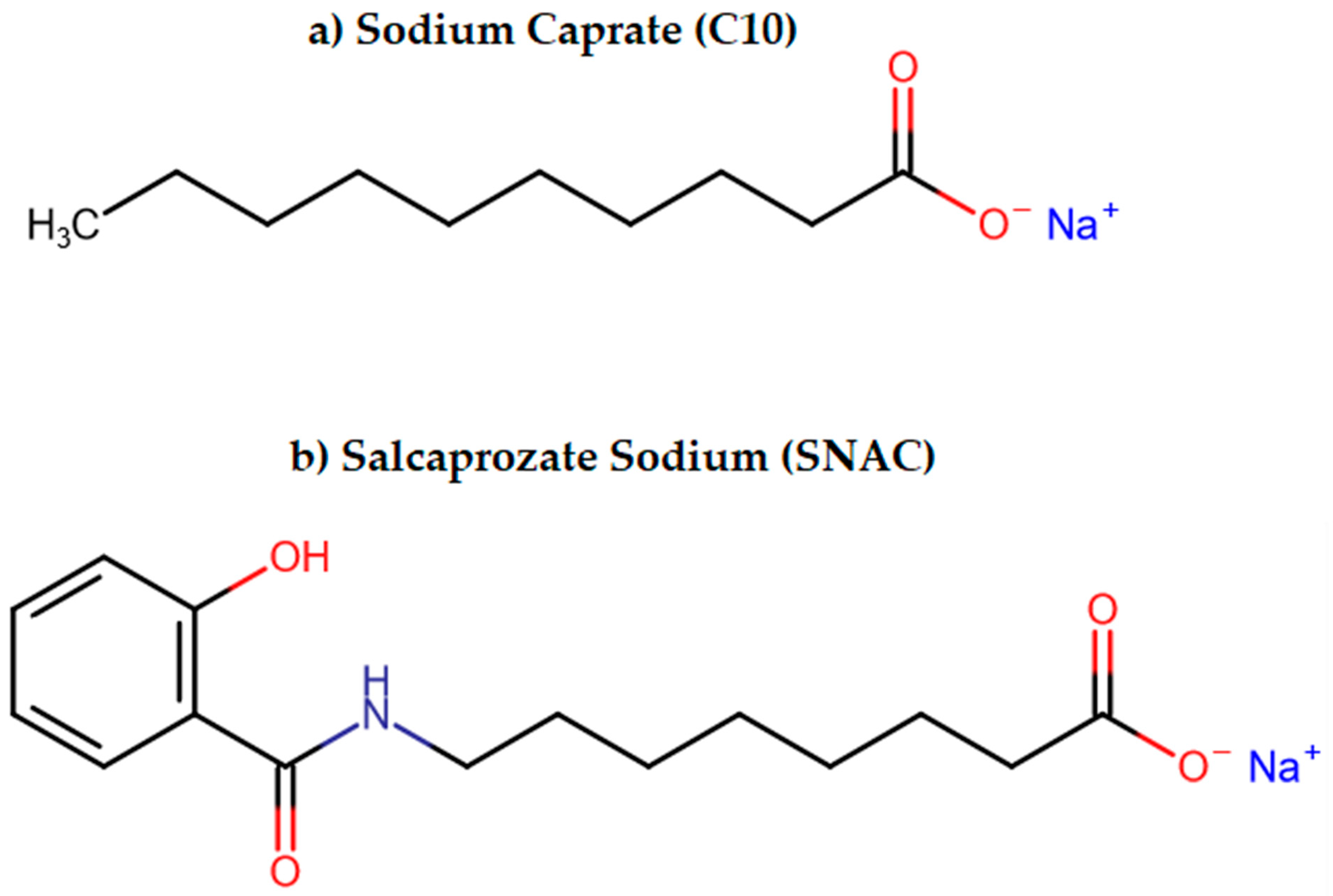
3.2.3. Permeation Enhancers (PEs)
3.2.4. Peptide Cyclization and Substitutions of AAs
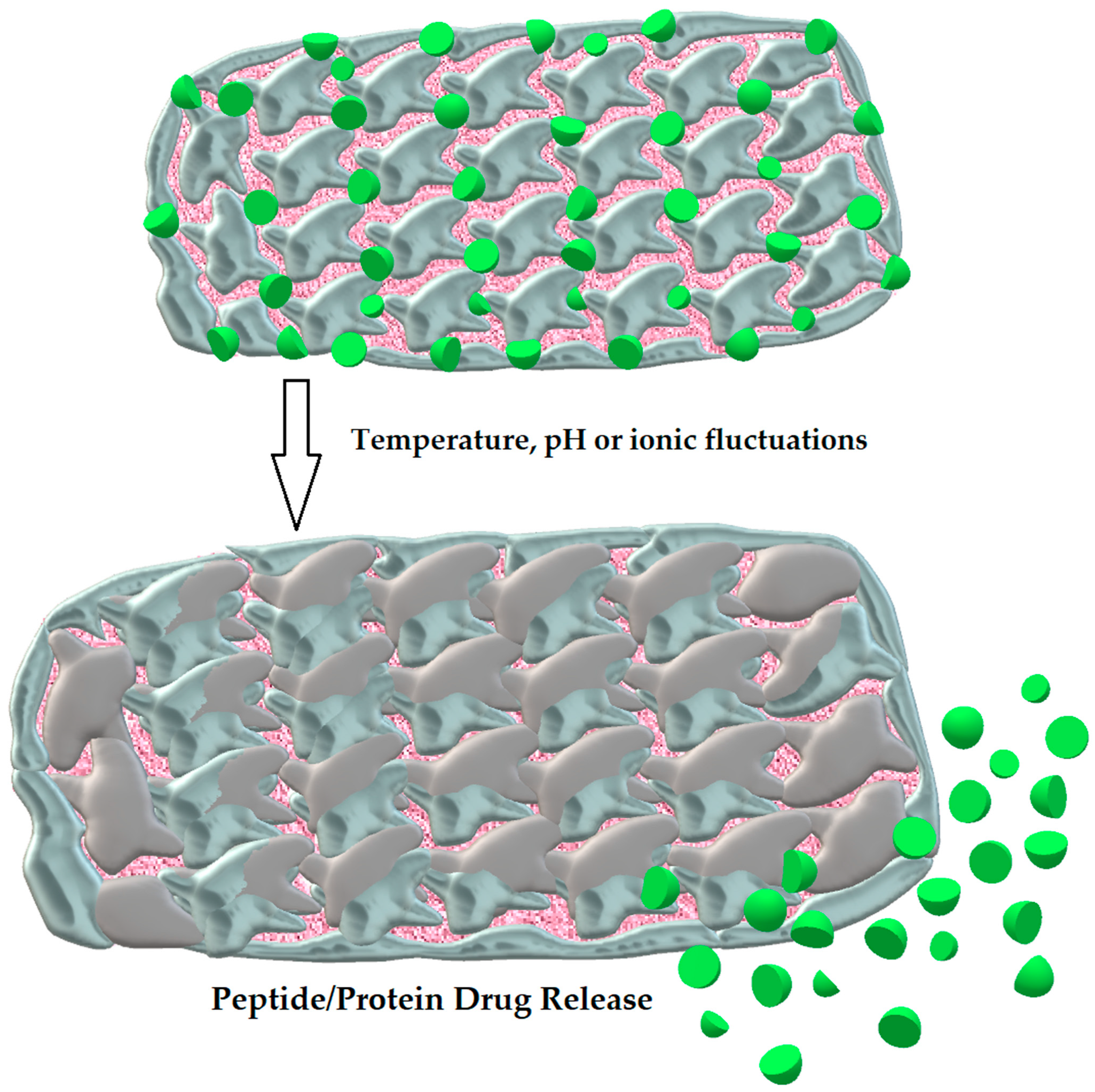
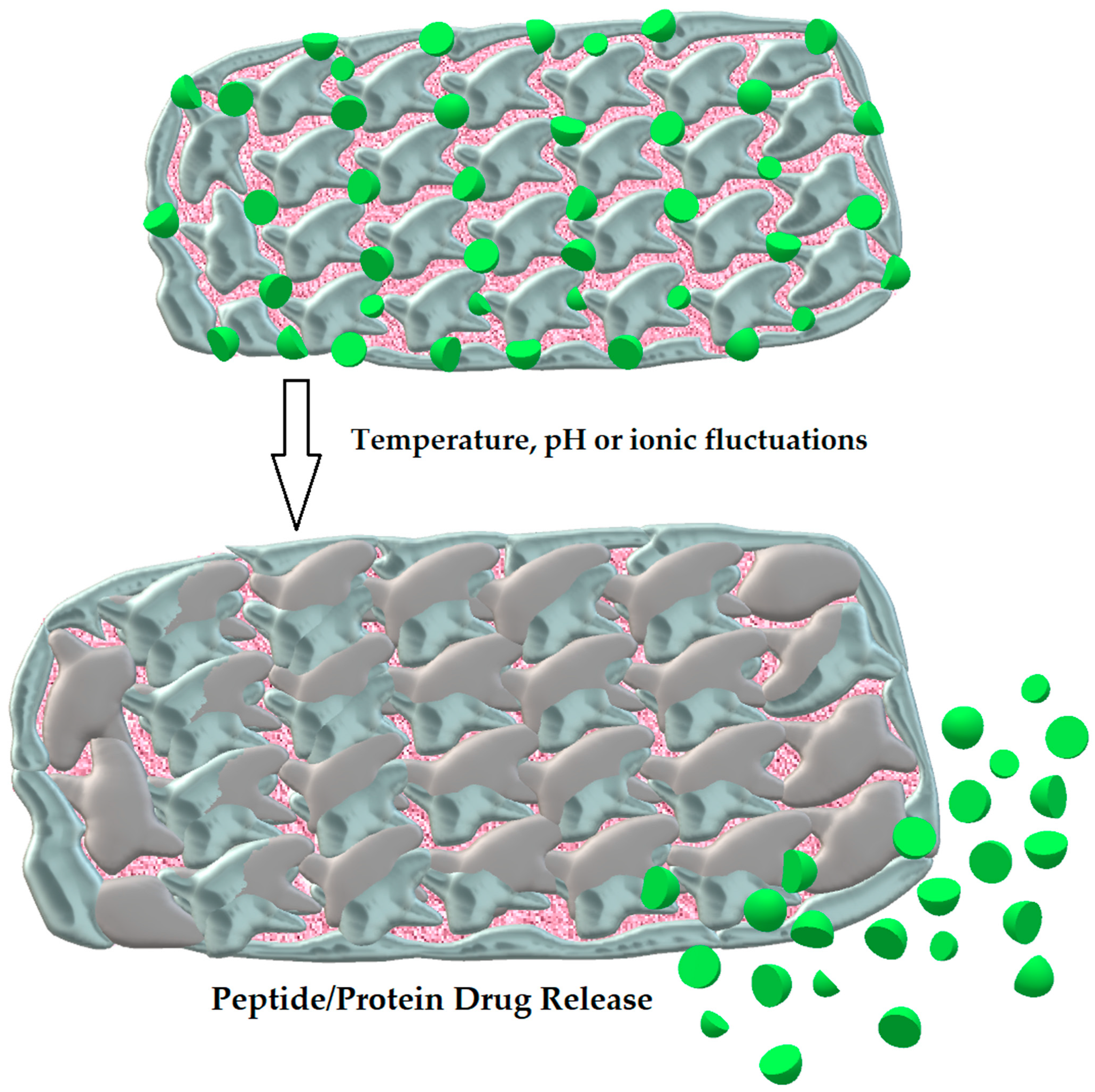
3.2.5. Hydrogels
3.2.6. Microneedles
3.2.7. Microemulsion
3.2.8. Proteolytic Enzyme Inhibitors
3.2.9. Cell-Penetrating Peptides
3.2.10. Bacteria-Mediated Therapy
4. Oral PPs and Oligonucleotide Therapeutics Available on Medical Market or Previously Used in Therapy
4.1. Examples of Currently Used Oral PPs
4.1.1. Cyclosporine
4.1.2. Insulin
4.1.3. Semaglutide (GLP-1 Analog)
4.1.4. Desmopressin
4.1.5. Octreotide
4.1.6. Orally Delivered Agents Targeting Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9)
5. Ongoing Clinical Trials Which May Be Meaningful in Respect of Oral Therapy Implementation
6. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAs | Amino acids |
| ACTH | Adrenocorticotropic hormone |
| ASBT | Apical sodium-dependent bile acid transporter |
| ASOs | Antisense oligonucleotides |
| AUC | Area under the curve |
| BMI | Body mass index |
| C-CPE | Clostridium perfringens enterotoxin |
| Cmax | Peak blood concentration |
| CNS | Central nervous system |
| CPPs | Cell-penetrating peptides |
| CVD | Cardiovascular disease |
| DC-LIPs | Deoxycholic acid and chitosan conjugate-modified liposomes |
| DDAVP | Desmopressin acetate |
| EC | European Commission |
| EDTA | Ethylenediaminetetraacetic acid |
| EMA | European Medicines Agency |
| FDA | Food and Drug Administration |
| GI | Gastrointestinal |
| GLP-1 | Glucagon-like peptide-1 |
| GLP-1RAs | Glucagon-like peptide-1 receptor agonists |
| GnRH | Gonadotropin-releasing hormone |
| IGF-1R | Insulin-like growth factor type 1 receptor |
| LDL-Rs | Low density lipoprotein receptors |
| LUMI | Luminal unfolding microneedle injector |
| MCT-1 | Monocarboxylate transporter 1 |
| NF-κB | Nuclear factor kappa B |
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 |
| PEG | Polyethylene glycol |
| PEs | Permeation enhancers |
| pHPMA | poly[N-(2-hydroxypropyl)methacrylamide] |
| pI | Isoelectric point |
| PKC | Protein kinase C |
| PPDs | Peptide and protein drugs |
| PPs | Peptides and proteins |
| PrPSc | Scrapie isoform of the prion protein |
| PTH | Parathyroid hormone |
| PVA | Poly(vinyl alcohol) |
| SGF | Simulated gastric fluid |
| siRNA | small interfering RNA |
| SMEDDSs | Self-microemulsifying drug-delivery systems |
| SNAC | Salcaprozate sodium |
| SNEDDSs | Self-nanoemulsifying drug-delivery systems |
| TJs | Tight junctions |
| Tmax | Time to peak blood concentration |
| TNF-alpha | Tumor necrosis factor alpha |
| WHO | World Health Organization |
References
- Rengasamy, K.R.R.; Khan, H.; Ahmad, I.; Lobine, D.; Mahomoodally, F.; Suroowan, S.; Hassan, S.T.S.; Xu, S.; Patel, S.; Daglia, M.; et al. Bioactive peptides and proteins as alternative antiplatelet drugs. Med. Res. Rev. 2019, 39, 2153–2171. [Google Scholar] [CrossRef] [PubMed]
- Jain, D.; Mahammad, S.S.; Singh, P.P.; Kodipyaka, R. A review on parenteral delivery of peptides and proteins. Drug Dev. Ind. Pharm. 2019, 45, 1403–1420. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.W.; Hil-lal, T.A.; Byun, Y. Strategies for non-invasive delivery of biologics. J. Drug Target. 2012, 20, 481–501. [Google Scholar] [CrossRef] [PubMed]
- International Union of Pure and Applied Chemistry and International Union of Biochemistry Joint Commission on Biochemical Nomenclature. Nomenclature and Symbolism for Amino Acids and Peptides; IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN): Research Triangle Park, NC, USA, 1984. [Google Scholar]
- Di, L. Strategic approaches to optimizing peptide ADME properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Chen, Z.; Paul, P.K.; Lu, Y.; Wu, W.; Qi, J. Oral delivery of proteins and peptides: Challenges, status quo and future perspectives. Acta Pharm. Sin. B 2021, 11, 2416–2448. [Google Scholar] [CrossRef] [PubMed]
- Brayden, D.J.; Alonso, M.-J. Oral delivery of peptides: Opportunities and issues for translation. Adv. Drug Deliv. Rev. 2016, 106, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.-F.; Zhu, L.-L.; Chen, M.; Xu, H.-M.; Wang, H.-F.; Feng, X.-Q.; Zhu, X.-P.; Zhou, Q. The optimal choice of medication administration route regarding intravenous, intramuscular, and subcutaneous injection. Patient Prefer. Adherence 2015, 9, 923–942. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Gokarn, Y.; Mitragotri, S. Non-invasive delivery strategies for biologics. Nat. Rev. Drug Discov. 2019, 18, 19–40. [Google Scholar] [CrossRef]
- Wong, C.Y.; Martinez, J.; Dass, C.R. Oral delivery of insulin for treatment of diabetes: Status quo, challenges and opportunities. J. Pharm. Pharmacol. 2016, 68, 1093–1108. [Google Scholar] [CrossRef]
- Koziolek, M.; Grimm, M.; Becker, D.; Iordanov, V.; Zou, H.; Shimizu, J.; Wanke, C.; Garbacz, G.; Weitschies, W. Investigation of ph and temperature profiles in the GI tract of fasted human subjects using the intellicap(®) system. J. Pharm. Sci. 2015, 104, 2855–2863. [Google Scholar] [CrossRef]
- Gracia, R.; Yus, C.; Abian, O.; Mendoza, G.; Irusta, S.; Sebastian, V.; Andreu, V.; Arruebo, M. Enzyme structure and function protection from gastrointestinal degradation using enteric coatings. Int. J. Biol. Macromol. 2018, 119, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Boronikolos, G.C.; Menge, B.A.; Schenker, N.; Breuer, T.G.K.; Otte, J.-M.; Heckermann, S.; Schliess, F.; Meier, J.J. Upper gastrointestinal motility and symptoms in individuals with diabetes, prediabetes and normal glucose tolerance. Diabetologia 2015, 58, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Sinha, V.; Singh, A.; Kumar, R.V.; Singh, S.; Kumria, R.; Bhinge, J. Oral colon-specific drug delivery of protein and peptide drugs. Crit. Rev. Ther. Drug Carrier Syst. 2007, 24, 63–92. [Google Scholar] [CrossRef] [PubMed]
- Bohley, M.; Haunberger, A.; Goepferich, A.M. Intracellular availability of poorly soluble drugs from lipid nanocapsules. Eur. J. Pharm. Biopharm. 2019, 139, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Renukuntla, J.; Vadlapudi, A.D.; Patel, A.; Boddu, S.H.S.; Mitra, A.K. Approaches for enhancing oral bioavailability of peptides and proteins. Int. J. Pharm. 2013, 447, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.; Gomez-Orellana, I. Challenges for the oral delivery of macromolecules. Nat. Rev. Drug Discov. 2003, 2, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Mathieu, C.; Gillard, P.; Benhalima, K. Insulin analogues in type 1 diabetes mellitus: Getting better all the time. Nat. Rev. Endocrinol. 2017, 13, 385–399. [Google Scholar] [CrossRef]
- Sawyer, W.H.; Manning, M. Synthetic analogs of oxytocin and the vasopressins. Annu. Rev. Pharmacol. 1973, 13, 5–17. [Google Scholar] [CrossRef]
- Ensign, L.M.; Cone, R.; Hanes, J. Oral drug delivery with polymeric nanoparticles: The gastrointestinal mucus barriers. Adv. Drug Deliv. Rev. 2012, 64, 557–570. [Google Scholar] [CrossRef]
- Maisel, K.; Ensign, L.; Reddy, M.; Cone, R.; Hanes, J. Effect of surface chemistry on nanoparticle interaction with gastrointestinal mucus and distribution in the gastrointestinal tract following oral and rectal administration in the mouse. J. Control. Release 2015, 197, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Huckaby, J.T.; Lai, S.K. PEGylation for enhancing nanoparticle diffusion in mucus. Adv. Drug Deliv. Rev. 2018, 124, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Zhu, X.; Liu, M.; Li, L.; Zhong, J.; Sun, W.; Zhang, Z.; Huang, Y. Overcoming the diffusion barrier of mucus and absorption barrier of epithelium by self-assembled nanoparticles for oral delivery of insulin. ACS Nano 2015, 9, 2345–2356. [Google Scholar] [CrossRef]
- Wang, A.; Yang, T.; Fan, W.; Yang, Y.; Zhu, Q.; Guo, S.; Zhu, C.; Yuan, Y.; Zhang, T.; Gan, Y. Protein corona liposomes achieve efficient oral insulin delivery by overcoming mucus and epithelial barriers. Adv. Healthc. Mater. 2019, 8, e1801123. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.K.; Farrell, S.O. (Eds.) Biochemistry; Brooks/Cole Publishing Company: Pacific Grove, CA, USA, 2011; ISBN 9780840068583. [Google Scholar]
- Bradford, B.M.; Reizis, B.; Mabbott, N.A. Oral Prion Disease Pathogenesis Is Impeded in the Specific Absence of CXCR5-Expressing Dendritic Cells. J. Virol. 2017, 91, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Urayama, A.; Concha-Marambio, L.; Khan, U.; Bravo-Alegria, J.; Kharat, V.; Soto, C. Prions efficiently cross the intestinal barrier after oral administration: Study of the bioavailability, and cellular and tissue distribution in vivo. Sci. Rep. 2016, 6, 32338. [Google Scholar] [CrossRef]
- Pauletti, G.M.; Gangwar, S.; Knipp, G.T.; Nerurkar, M.M.; Okumu, F.W.; Tamura, K.; Siahaan, T.J.; Borchardt, R.T. Structural requirements for intestinal absorption of peptide drugs. J. Control. Release 1996, 41, 3–17. [Google Scholar] [CrossRef]
- Xu, Y.; Zheng, Y.; Wu, L.; Zhu, X.; Zhang, Z.; Huang, Y. Novel Solid Lipid Nanoparticle with Endosomal Escape Function for Oral Delivery of Insulin. ACS Appl. Mater. Interfaces 2018, 10, 9315–9324. [Google Scholar] [CrossRef]
- Sun, L.; Liu, Z.; Tian, H.; Le, Z.; Liu, L.; Leong, K.W.; Mao, H.-Q.; Chen, Y. Scalable Manufacturing of Enteric Encapsulation Systems for Site-Specific Oral Insulin Delivery. Biomacromolecules 2019, 20, 528–538. [Google Scholar] [CrossRef]
- Geary, R.S.; Khatsenko, O.; Bunker, K.; Crooke, R.; Moore, M.; Burckin, T.; Truong, L.; Sasmor, H.; Levin, A.A. Absolute bioavailability of 2′-O-(2-methoxyethyl)-modified antisense oligonucleotides following intraduodenal instillation in rats. J. Pharmacol. Exp. Ther. 2001, 296, 898–904. [Google Scholar]
- Uddin, M.N.; Patel, N.J.; Bhowmik, T.; D’Souza, B.; Akalkotkar, A.; Etzlar, F.; Oettinger, C.W.; D’Souza, M. Enhanced bioavailability of orally administered antisense oligonucleotide to nuclear factor kappa B mRNA after microencapsulation with albumin. J. Drug Target. 2013, 21, 450–457. [Google Scholar] [CrossRef]
- Fallingborg, J. Intraluminal pH of the human gastrointestinal tract. Dan. Med. Bull. 1999, 46, 183–196. [Google Scholar] [PubMed]
- Baliga, S.; Muglikar, S.; Kale, R. Salivary pH: A diagnostic biomarker. J. Indian Soc. Periodontol. 2013, 17, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.F.; Pye, G.; Bramley, R.; Clark, A.G.; Dyson, T.J.; Hardcastle, J.D. Measurement of gastrointestinal pH profiles in normal ambulant human subjects. Gut 1988, 29, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Nguyen, T.H.; Stüben, M.; Scheid, P. Demonstration of a pH gradient in the gastric gland of the acid-secreting guinea pig mucosa. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G597–G604. [Google Scholar] [CrossRef]
- Williams, C. Occurrence and significance of gastric colonization during acid-inhibitory therapy. Best Pract. Res. Clin. Gastroenterol. 2001, 15, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, R.; Inoue, K.Y.; Nishino, K.; Yamasaki, S. Intestinal and fecal pH in human health. Front. Microbiom. 2023, 2, 1192316. [Google Scholar] [CrossRef]
- Goto, Y.; Fink, A.L. Conformational states of beta-lactamase: Molten-globule states at acidic and alkaline pH with high salt. Biochemistry 1989, 28, 945–952. [Google Scholar] [CrossRef]
- Tokmakov, A.A.; Kurotani, A.; Sato, K.-I. Protein pi and intracellular localization. Front. Mol. Biosci. 2021, 8, 775736. [Google Scholar] [CrossRef]
- Cheng, Y.K.; Pettitt, B.M. Stabilities of double- and triple-strand helical nucleic acids. Prog. Biophys. Mol. Biol. 1992, 58, 225–257. [Google Scholar] [CrossRef]
- López-Otín, C.; Bond, J.S. Proteases: Multifunctional enzymes in life and disease. J. Biol. Chem. 2008, 283, 30433–30437. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Y.; Dong, P.; An, R.; Xue, C.; Ge, Y.; Wei, L.; Liang, X. Digestion of nucleic acids starts in the stomach. Sci. Rep. 2015, 5, 11936. [Google Scholar] [CrossRef] [PubMed]
- Gavhane, Y.N.; Yadav, A.V. Loss of orally administered drugs in GI tract. Saudi Pharm. J. 2012, 20, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Ozorio, L.; Mellinger-Silva, C.; Cabral, L.M.C.; Jardin, J.; Boudry, G.; Dupont, D. The Influence of Peptidases in Intestinal Brush Border Membranes on the Absorption of Oligopeptides from Whey Protein Hydrolysate: An Ex Vivo Study Using an Ussing Chamber. Foods 2020, 9, 1415. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Watanabe, E.; Kawashima, Y.; Plichta, D.R.; Wang, Z.; Ujike, M.; Ang, Q.Y.; Wu, R.; Furuichi, M.; Takeshita, K.; et al. Identification of trypsin-degrading commensals in the large intestine. Nature 2022, 609, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Bansil, R.; Turner, B.S. Mucin structure, aggregation, physiological functions and biomedical applications. Curr. Opin. Colloid Interface Sci 2006, 11, 164–170. [Google Scholar] [CrossRef]
- Bansil, R.; Turner, B.S. The biology of mucus: Composition, synthesis and organization. Adv. Drug Deliv. Rev. 2018, 124, 3–15. [Google Scholar] [CrossRef]
- Gendler, S.J.; Spicer, A.P. Epithelial mucin genes. Annu. Rev. Physiol. 1995, 57, 607–634. [Google Scholar] [CrossRef]
- Lai, S.K.; Wang, Y.-Y.; Wirtz, D.; Hanes, J. Micro- and macrorheology of mucus. Adv. Drug Deliv. Rev. 2009, 61, 86–100. [Google Scholar] [CrossRef]
- Bernkop-Schnürch, A.; Fragner, R. Investigations into the Diffusion Behaviour of Polypeptides in Native Intestinal Mucus with Regard to their Peroral Administration. Pharm. Pharmacol. Commun. 1996, 2, 361–363. [Google Scholar]
- Desai, M.A.; Mutlu, M.; Vadgama, P. A study of macromolecular diffusion through native porcine mucus. Experientia 1992, 48, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Boegh, M.; Nielsen, H.M. Mucus as a barrier to drug delivery—Understanding and mimicking the barrier properties. Basic Clin. Pharmacol. Toxicol. 2015, 116, 179–186. [Google Scholar] [CrossRef]
- Allaire, J.M.; Crowley, S.M.; Law, H.T.; Chang, S.-Y.; Ko, H.-J.; Vallance, B.A. The intestinal epithelium: Central coordinator of mucosal immunity. Trends Immunol. 2018, 39, 677–696. [Google Scholar] [CrossRef] [PubMed]
- Capaldo, C.T.; Powell, D.N.; Kalman, D. Layered defense: How mucus and tight junctions seal the intestinal barrier. J. Mol. Med. 2017, 95, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Okumura, R.; Takeda, K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp. Mol. Med. 2017, 49, e338. [Google Scholar] [CrossRef] [PubMed]
- Vancamelbeke, M.; Vermeire, S. The intestinal barrier: A fundamental role in health and disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Van Itallie, C.M. Tight junctions and the molecular basis for regulation of paracellular permeability. Am. J. Physiol. 1995, 269, G467–G475. [Google Scholar] [CrossRef]
- Liu, C.; Kou, Y.; Zhang, X.; Cheng, H.; Chen, X.; Mao, S. Strategies and industrial perspectives to improve oral absorption of biological macromolecules. Expert Opin. Drug Deliv. 2018, 15, 223–233. [Google Scholar] [CrossRef]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Zhong, J.; Xia, B.; Shan, S.; Zheng, A.; Zhang, S.; Chen, J.; Liang, X.-J. High-quality milk exosomes as oral drug delivery system. Biomaterials 2021, 277, 121126. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Shi, J.; Xie, J.; Wang, Y.; Sun, J.; Liu, T.; Zhao, Y.; Zhao, X.; Wang, X.; Ma, Y.; et al. Large-scale generation of functional mRNA-encapsulating exosomes via cellular nanoporation. Nat. Biomed. Eng. 2020, 4, 69–83. [Google Scholar] [CrossRef]
- Rufino-Ramos, D.; Albuquerque, P.R.; Carmona, V.; Perfeito, R.; Nobre, R.J.; Pereira de Almeida, L. Extracellular vesicles: Novel promising delivery systems for therapy of brain diseases. J. Control. Release 2017, 262, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Wiklander, O.P.B.; Brennan, M.Á.; Lötvall, J.; Breakefield, X.O.; El Andaloussi, S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019, 11, eaav8521. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.K.; Aqil, F.; Jeyabalan, J.; Spencer, W.A.; Beck, J.; Gachuki, B.W.; Alhakeem, S.S.; Oben, K.; Munagala, R.; Bondada, S.; et al. Milk-derived exosomes for oral delivery of paclitaxel. Nanomedicine 2017, 13, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Aqil, F.; Tyagi, N.; Gupta, R. Milk exosomes: A biogenic nanocarrier for small molecules and macromolecules to combat cancer. Am. J. Reprod. Immunol. 2021, 85, e13349. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wang, L.; Liu, X.; Bai, Y.; Wu, R.; Li, X.; Mao, Y.; Zhang, L.; Zheng, Y.; Gong, T.; et al. Milk-derived exosomes exhibit versatile effects for improved oral drug delivery. Acta Pharm. Sin. B 2022, 12, 2029–2042. [Google Scholar] [CrossRef] [PubMed]
- Barile, L.; Vassalli, G. Exosomes: Therapy delivery tools and biomarkers of diseases. Pharmacol. Ther. 2017, 174, 63–78. [Google Scholar] [CrossRef]
- Kooijmans, S.A.A.; Aleza, C.G.; Roffler, S.R.; van Solinge, W.W.; Vader, P.; Schiffelers, R.M. Display of GPI-anchored anti-EGFR nanobodies on extracellular vesicles promotes tumour cell targeting. J. Extracell. Vesicles 2016, 5, 31053. [Google Scholar] [CrossRef]
- Choi, D.-S.; Kim, D.-K.; Kim, Y.-K.; Gho, Y.S. Proteomics, transcriptomics and lipidomics of exosomes and ectosomes. Proteomics 2013, 13, 1554–1571. [Google Scholar] [CrossRef]
- Blanchard, N.; Lankar, D.; Faure, F.; Regnault, A.; Dumont, C.; Raposo, G.; Hivroz, C. TCR activation of human T cells induces the production of exosomes bearing the TCR/CD3/zeta complex. J. Immunol. 2002, 168, 3235–3241. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Barreiro, O.; Gordon-Alonso, M.; Sala-Valdés, M.; Sánchez-Madrid, F. Tetraspanin-enriched microdomains: A functional unit in cell plasma membranes. Trends Cell Biol. 2009, 19, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Olusanya, T.O.B.; Haj Ahmad, R.R.; Ibegbu, D.M.; Smith, J.R.; Elkordy, A.A. Liposomal drug delivery systems and anticancer drugs. Molecules 2018, 23, 907. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Bisht, B.; Dutta, S.; Paul, M.K. Current advances in the use of exosomes, liposomes, and bioengineered hybrid nanovesicles in cancer detection and therapy. Acta Pharmacol. Sin. 2022, 43, 2759–2776. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Xia, D.; Li, X.; Zhu, Q.; Yu, H.; Zhu, C.; Gan, Y. Comparative study of Pluronic® F127-modified liposomes and chitosan-modified liposomes for mucus penetration and oral absorption of cyclosporine A in rats. Int. J. Pharm. 2013, 449, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and challenges of liposome assisted drug delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef]
- Singh, A.; Neupane, Y.R.; Shafi, S.; Mangla, B.; Kohli, K. PEGylated liposomes as an emerging therapeutic platform for oral nanomedicine in cancer therapy: In vitro and in vivo assessment. J. Mol. Liq. 2020, 303, 112649. [Google Scholar] [CrossRef]
- Kumar, A.; Vimal, A.; Kumar, A. Why Chitosan? From properties to perspective of mucosal drug delivery. Int. J. Biol. Macromol. 2016, 91, 615–622. [Google Scholar] [CrossRef]
- de Barros, M.M.V.D.; Santos, S.A.A.R.; Araújo, J.R.C.; Barroso, L.K.V.; Benevides, S.C.; Magalhães, F.E.A.; Tavares, K.C.S.; de Azevedo, M.R.; de Oliveira, A.C.M.-M.; Silva, A.R.A.E.; et al. Development of a nanoformulation for oral protein administration: Characterization and preclinical orofacial antinociceptive effect. AAPS PharmSciTech 2022, 23, 239. [Google Scholar] [CrossRef]
- Mikušová, V.; Mikuš, P. Advances in Chitosan-Based Nanoparticles for Drug Delivery. Int. J. Mol. Sci. 2021, 22, 9652. [Google Scholar] [CrossRef]
- Balakrishnan, A.; Polli, J.E. Apical sodium dependent bile acid transporter (ASBT, SLC10A2): A potential prodrug target. Mol. Pharm. 2006, 3, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Bagre, A.P.; Jain, K.; Jain, N.K. Alginate coated chitosan core shell nanoparticles for oral delivery of enoxaparin: In vitro and in vivo assessment. Int. J. Pharm. 2013, 456, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Bin, W.; Tu, B.; Li, X.; Wang, W.; Liao, S.; Sun, C. A delivery system for oral administration of proteins/peptides through bile acid transport channels. J. Pharm. Sci. 2019, 108, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Liu, M.; Shan, W.; Zhu, X.; Li, L.; Zhang, Z.; Huang, Y. Bioinspired butyrate-functionalized nanovehicles for targeted oral delivery of biomacromolecular drugs. J. Control. Release 2017, 262, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Maher, S.; Brayden, D.J. Formulation strategies to improve the efficacy of intestinal permeation enhancers. Adv. Drug Deliv. Rev. 2021, 177, 113925. [Google Scholar] [CrossRef] [PubMed]
- Maher, S.; Brayden, D.J.; Casettari, L.; Illum, L. Application of permeation enhancers in oral delivery of macromolecules: An update. Pharmaceutics 2019, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, M.; Yoshida, T.; Kakutani, H.; Yagi, K. Targeting tight junction proteins-significance for drug development. Drug Discov. Today 2008, 13, 180–186. [Google Scholar] [CrossRef]
- Tomita, M.; Hayashi, M.; Awazu, S. Absorption-enhancing mechanism of EDTA, caprate, and decanoylcarnitine in Caco-2 cells. J. Pharm. Sci. 1996, 85, 608–611. [Google Scholar] [CrossRef]
- Kondoh, M.; Masuyama, A.; Takahashi, A.; Asano, N.; Mizuguchi, H.; Koizumi, N.; Fujii, M.; Hayakawa, T.; Horiguchi, Y.; Watanbe, Y. A novel strategy for the enhancement of drug absorption using a claudin modulator. Mol. Pharmacol. 2005, 67, 749–756. [Google Scholar] [CrossRef]
- Maher, S.; Mrsny, R.J.; Brayden, D.J. Intestinal permeation enhancers for oral peptide delivery. Adv. Drug Deliv. Rev. 2016, 106, 277–319. [Google Scholar] [CrossRef]
- Maher, S.; Heade, J.; McCartney, F.; Waters, S.; Bleiel, S.B.; Brayden, D.J. Effects of surfactant-based permeation enhancers on mannitol permeability, histology, and electrogenic ion transport responses in excised rat colonic mucosae. Int. J. Pharm. 2018, 539, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Zupančič, O.; Bernkop-Schnürch, A. Lipophilic peptide character—What oral barriers fear the most. J. Control. Release 2017, 255, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Twarog, C.; Fattah, S.; Heade, J.; Maher, S.; Fattal, E.; Brayden, D.J. Intestinal Permeation Enhancers for Oral Delivery of Macromolecules: A Comparison between Salcaprozate Sodium (SNAC) and Sodium Caprate (C10). Pharmaceutics 2019, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- McCartney, F.; Gleeson, J.P.; Brayden, D.J. Safety concerns over the use of intestinal permeation enhancers: A mini-review. Tissue Barriers 2016, 4, e1176822. [Google Scholar] [CrossRef] [PubMed]
- Aroda, V.R.; Blonde, L.; Pratley, R.E. A new era for oral peptides: SNAC and the development of oral semaglutide for the treatment of type 2 diabetes. Rev. Endocr. Metab. Disord. 2022, 23, 979–994. [Google Scholar] [CrossRef]
- Brayden, D.J.; Maher, S. Transient Permeation Enhancer® (TPE®) technology for oral delivery of octreotide: A technological evaluation. Expert Opin. Drug Deliv. 2021, 18, 1501–1512. [Google Scholar] [CrossRef]
- Zhang, R.-Y.; Thapa, P.; Espiritu, M.J.; Menon, V.; Bingham, J.-P. From nature to creation: Going around in circles, the art of peptide cyclization. Bioorg. Med. Chem. 2018, 26, 1135–1150. [Google Scholar] [CrossRef]
- Conibear, A.C.; Chaousis, S.; Durek, T.; Rosengren, K.J.; Craik, D.J.; Schroeder, C.I. Approaches to the stabilization of bioactive epitopes by grafting and peptide cyclization. Biopolymers 2016, 106, 89–100. [Google Scholar] [CrossRef]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef]
- Kremsmayr, T.; Aljnabi, A.; Blanco-Canosa, J.B.; Tran, H.N.T.; Emidio, N.B.; Muttenthaler, M. On the utility of chemical strategies to improve peptide gut stability. J. Med. Chem. 2022, 65, 6191–6206. [Google Scholar] [CrossRef]
- Nielsen, D.S.; Shepherd, N.E.; Xu, W.; Lucke, A.J.; Stoermer, M.J.; Fairlie, D.P. Orally absorbed cyclic peptides. Chem. Rev. 2017, 117, 8094–8128. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Guo, C.; Liu, L.; Zhao, L.; Zhang, Y.; Yin, T.; He, H.; Gou, J.; Pan, B.; Tang, X. Progress and prospects of polysaccharide-based nanocarriers for oral delivery of proteins/peptides. Carbohydr. Polym. 2023, 312, 120838. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, R.; Wang, Y.; Zhang, Y.; Yu, J.; Gu, Z. Recent advances in oral and transdermal protein delivery systems. Angew. Chem. Int. Ed 2023, 62, e202214795. [Google Scholar] [CrossRef] [PubMed]
- Hubálek, F.; Refsgaard, H.H.F.; Gram-Nielsen, S.; Madsen, P.; Nishimura, E.; Münzel, M.; Brand, C.L.; Stidsen, C.E.; Claussen, C.H.; Wulff, E.M.; et al. Molecular engineering of safe and efficacious oral basal insulin. Nat. Commun. 2020, 11, 3746. [Google Scholar] [CrossRef] [PubMed]
- Correa, S.; Grosskopf, A.K.; Lopez Hernandez, H.; Chan, D.; Yu, A.C.; Stapleton, L.M.; Appel, E.A. Translational applications of hydrogels. Chem. Rev. 2021, 121, 11385–11457. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zhang, S.; Wu, F.; Li, D.; Zhang, X.; Chen, W.; Xing, B. Rational design of nanogels for overcoming the biological barriers in various administration routes. Angew. Chem. Int. Ed. 2021, 60, 14760–14778. [Google Scholar] [CrossRef]
- Koetting, M.C.; Guido, J.F.; Gupta, M.; Zhang, A.; Peppas, N.A. pH-responsive and enzymatically-responsive hydrogel microparticles for the oral delivery of therapeutic proteins: Effects of protein size, crosslinking density, and hydrogel degradation on protein delivery. J. Control. Release 2016, 221, 18–25. [Google Scholar] [CrossRef]
- Hu, Y.; Gao, S.; Lu, H.; Ying, J.Y. Acid-Resistant and Physiological pH-Responsive DNA Hydrogel Composed of A-Motif and i-Motif toward Oral Insulin Delivery. J. Am. Chem. Soc. 2022, 144, 5461–5470. [Google Scholar] [CrossRef]
- Traverso, G.; Schoellhammer, C.M.; Schroeder, A.; Maa, R.; Lauwers, G.Y.; Polat, B.E.; Anderson, D.G.; Blankschtein, D.; Langer, R. Microneedles for drug delivery via the gastrointestinal tract. J. Pharm. Sci. 2015, 104, 362–367. [Google Scholar] [CrossRef]
- Abramson, A.; Caffarel-Salvador, E.; Khang, M.; Dellal, D.; Silverstein, D.; Gao, Y.; Frederiksen, M.R.; Vegge, A.; Hubálek, F.; Water, J.J.; et al. An ingestible self-orienting system for oral delivery of macromolecules. Science 2019, 363, 611–615. [Google Scholar] [CrossRef]
- Abramson, A.; Frederiksen, M.R.; Vegge, A.; Jensen, B.; Poulsen, M.; Mouridsen, B.; Jespersen, M.O.; Kirk, R.K.; Windum, J.; Hubálek, F.; et al. Oral delivery of systemic monoclonal antibodies, peptides and small molecules using gastric auto-injectors. Nat. Biotechnol. 2022, 40, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Industrial Applications of Microemulsions—Google Books; CRC Press: Boca Raton, FL, USA, 1996; ISBN 9780824797959.
- Cao, X.; Zhu, Q.; Wang, Q.-L.; Adu-Frimpong, M.; Wei, C.-M.; Weng, W.; Bao, R.; Wang, Y.-P.; Yu, J.-N.; Xu, X.M. Improvement of Oral Bioavailability and Anti-Tumor Effect of Zingerone Self-Microemulsion Drug Delivery System. J. Pharm. Sci. 2021, 110, 2718–2727. [Google Scholar] [CrossRef] [PubMed]
- Erel, G.; Kotmakçı, M.; Akbaba, H.; Sözer Karadağlı, S.; Kantarcı, A.G. Nanoencapsulated chitosan nanoparticles in emulsion-based oral delivery system: In vitro and in vivo evaluation of insulin loaded formulation. J. Drug Deliv. Sci. Technol. 2016, 36, 161–167. [Google Scholar] [CrossRef]
- Momoh, M.A.; Franklin, K.C.; Agbo, C.P.; Ugwu, C.E.; Adedokun, M.O.; Anthony, O.C.; Chidozie, O.E.; Okorie, A.N. Microemulsion-based approach for oral delivery of insulin: Formulation design and characterization. Heliyon 2020, 6, e03650. [Google Scholar] [CrossRef] [PubMed]
- Choonara, B.F.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; du Toit, L.C.; Pillay, V. A review of advanced oral drug delivery technologies facilitating the protection and absorption of protein and peptide molecules. Biotechnol. Adv. 2014, 32, 1269–1282. [Google Scholar] [CrossRef]
- Laskowski, M.; Haessler, H.A.; Miech, R.P.; Peanasky, R.J.; Laskowski, M. Effect of trypsin inhibitor on passage of insulin across the intestinal barrier. Science 1958, 127, 1115–1116. [Google Scholar] [CrossRef]
- Kassell, B.; Radicevic, M.; Berlow, S.; Peanasky, R.J.; Laskowski, M. The basic trypsin inhibitor of bovine pancreas. i. an improved method of preparation and amino acid composition. J. Biol. Chem. 1963, 238, 3274–3279. [Google Scholar] [CrossRef]
- Ziv, E.; Lior, O.; Kidron, M. Absorption of protein via the intestinal wall. A quantitative model. Biochem. Pharmacol. 1987, 36, 1035–1039. [Google Scholar] [CrossRef]
- Yoshiteru, U.; Yoshinori, N.; Akiko, K.; Kanji, T. Effect of organic acids, trypsin inhibitors and dietary protein on the pharmacological activity of recombinant human granulocyte colony-stimulating factor (rhG-CSF) in rats. Int. J. Pharm. 1992, 81, 133–141. [Google Scholar] [CrossRef]
- Fujii, S.; Yokoyama, T.; Ikegaya, K.; Sato, F.; Yokoo, N. Promoting effect of the new chymotrypsin inhibitor FK-448 on the intestinal absorption of insulin in rats and dogs. J. Pharm. Pharmacol. 1985, 37, 545–549. [Google Scholar] [CrossRef]
- Zhou, M.; Zou, X.; Cheng, K.; Zhong, S.; Su, Y.; Wu, T.; Tao, Y.; Cong, L.; Yan, B.; Jiang, Y. The role of cell-penetrating peptides in potential anti-cancer therapy. Clin. Transl. Med. 2022, 12, e822. [Google Scholar] [CrossRef] [PubMed]
- Walrant, A.; Cardon, S.; Burlina, F.; Sagan, S. Membrane Crossing and Membranotropic Activity of Cell-Penetrating Peptides: Dangerous Liaisons? Acc. Chem. Res. 2017, 50, 2968–2975. [Google Scholar] [CrossRef] [PubMed]
- Jafari, S.; Maleki Dizaj, S.; Adibkia, K. Cell-penetrating peptides and their analogues as novel nanocarriers for drug delivery. Bioimpacts 2015, 5, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Tréhin, R.; Merkle, H.P. Chances and pitfalls of cell penetrating peptides for cellular drug delivery. Eur. J. Pharm. Biopharm. 2004, 58, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Rehmani, S.; Dixon, J.E. Oral delivery of anti-diabetes therapeutics using cell penetrating and transcytosing peptide strategies. Peptides 2018, 100, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Akin, D.; Sturgis, J.; Ragheb, K.; Sherman, D.; Burkholder, K.; Robinson, J.P.; Bhunia, A.K.; Mohammed, S.; Bashir, R. Bacteria-mediated delivery of nanoparticles and cargo into cells. Nat. Nanotechnol. 2007, 2, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhang, X.; Han, S.; Dou, Y.; Liu, M.; Zhang, L.; Guo, J.; Shi, Q.; Gong, G.; Wang, R.; et al. Yeast Microcapsule-Mediated Targeted Delivery of Diverse Nanoparticles for Imaging and Therapy via the Oral Route. Nano Lett. 2017, 17, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Qin, W.; Xu, W.; Huang, F.; Xie, X.; Wang, F.; Ma, L.; Zhang, C. Bacteria-Mediated Tumor Therapy via Photothermally-Programmed Cytolysin A Expression. Small 2021, 17, e2102932. [Google Scholar] [CrossRef]
- Hu, Q.; Wu, M.; Fang, C.; Cheng, C.; Zhao, M.; Fang, W.; Chu, P.K.; Ping, Y.; Tang, G. Engineering nanoparticle-coated bacteria as oral DNA vaccines for cancer immunotherapy. Nano Lett. 2015, 15, 2732–2739. [Google Scholar] [CrossRef]
- Fan, J.-X.; Li, Z.-H.; Liu, X.-H.; Zheng, D.-W.; Chen, Y.; Zhang, X.-Z. Bacteria-Mediated Tumor Therapy Utilizing Photothermally-Controlled TNF-α Expression via Oral Administration. Nano Lett. 2018, 18, 2373–2380. [Google Scholar] [CrossRef]
- Luo, C.-H.; Huang, C.-T.; Su, C.-H.; Yeh, C.-S. Bacteria-Mediated Hypoxia-Specific Delivery of Nanoparticles for Tumors Imaging and Therapy. Nano Lett. 2016, 16, 3493–3499. [Google Scholar] [CrossRef]
- Claesen, J.; Fischbach, M.A. Synthetic microbes as drug delivery systems. ACS Synth. Biol. 2015, 4, 358–364. [Google Scholar] [CrossRef]
- Mahmood, A.; Bernkop-Schnürch, A. SEDDS: A game changing approach for the oral administration of hydrophilic macromolecular drugs. Adv. Drug Deliv. Rev. 2019, 142, 91–101. [Google Scholar] [CrossRef]
- Dokania, S.; Joshi, A.K. Self-microemulsifying drug delivery system (SMEDDS)--challenges and road ahead. Drug Deliv. 2015, 22, 675–690. [Google Scholar] [CrossRef]
- Multiple Dose Trial Examining Dose Range, Escalation and Efficacy of Oral Semaglutide in Subjects with Type 2 Diabetes. Available online: https://www.clinicaltrials.gov/study/NCT01923181?tab=results (accessed on 22 November 2023).
- Gasthuys, E.; Vermeulen, A.; Croubels, S.; Millecam, J.; Schauvliege, S.; van Bergen, T.; De Bruyne, P.; Vande Walle, J.; Devreese, M. Population pharmacokinetic modeling of a desmopressin oral lyophilisate in growing piglets as a model for the pediatric population. Front. Pharmacol. 2018, 9, 41. [Google Scholar] [CrossRef]
- Tuvia, S.; Atsmon, J.; Teichman, S.L.; Katz, S.; Salama, P.; Pelled, D.; Landau, I.; Karmeli, I.; Bidlingmaier, M.; Strasburger, C.J.; et al. Oral octreotide absorption in human subjects: Comparable pharmacokinetics to parenteral octreotide and effective growth hormone suppression. J. Clin. Endocrinol. Metab. 2012, 97, 2362–2369. [Google Scholar] [CrossRef]
- Aguirre, T.A.S.; Teijeiro-Osorio, D.; Rosa, M.; Coulter, I.S.; Alonso, M.J.; Brayden, D.J. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv. Drug Deliv. Rev. 2016, 106, 223–241. [Google Scholar] [CrossRef]
- Garg, S.K.; Rewers, A.H.; Akturk, H.K. Ever-Increasing Insulin-Requiring Patients Globally. Diabetes Technol. Ther. 2018, 20, S21–S24. [Google Scholar] [CrossRef]
- Colombo, D.; Ammirati, E. Cyclosporine in transplantation—A history of converging timelines. J. Biol. Regul. Homeost. Agents 2011, 25, 493–504. [Google Scholar]
- Wells, G.; Haguenauer, D.; Shea, B.; Suarez-Almazor, M.E.; Welch, V.A.; Tugwell, P. Cyclosporine for rheumatoid arthritis. Cochrane Database Syst. Rev. 2000, 1998, CD001083. [Google Scholar] [CrossRef] [PubMed]
- Meyrier, A. Treatment of idiopathic nephrotic syndrome with cyclosporine A. J. Nephrol. 1997, 10, 14–24. [Google Scholar]
- Kutlubay, Z.; Erdogan, B.Ç.; Engin, B.; Serdaroglu, S. Cyclosporine in Dermatology. Skinmed 2016, 14, 105–109. [Google Scholar]
- Russell, G.; Graveley, R.; Seid, J.; al-Humidan, A.K.; Skjodt, H. Mechanisms of action of cyclosporine and effects on connective tissues. Semin. Arthritis Rheum. 1992, 21, 16–22. [Google Scholar] [CrossRef]
- Patel, D.; Wairkar, S. Recent advances in cyclosporine drug delivery: Challenges and opportunities. Drug Deliv. Transl. Res. 2019, 9, 1067–1081. [Google Scholar] [CrossRef]
- Czogalla, A. Oral cyclosporine A--the current picture of its liposomal and other delivery systems. Cell. Mol. Biol. Lett. 2009, 14, 139–152. [Google Scholar] [CrossRef]
- Guada, M.; Lasa-Saracíbar, B.; Lana, H.; Dios-Viéitez, M.d.C.; Blanco-Prieto, M.J. Lipid nanoparticles enhance the absorption of cyclosporine A through the gastrointestinal barrier: In vitro and in vivo studies. Int. J. Pharm. 2016, 500, 154–161. [Google Scholar] [CrossRef]
- Choc, M.G. Bioavailability and pharmacokinetics of cyclosporine formulations: Neoral vs Sandimmune. Int. J. Dermatol. 1997, 36 (Suppl. S1), 1–6. [Google Scholar] [CrossRef]
- Venkataram, S.; Awni, W.M.; Jordan, K.; Rahman, Y.E. Pharmacokinetics of two alternative dosage forms for cyclosporine: Liposomes and intralipid. J. Pharm. Sci. 1990, 79, 216–219. [Google Scholar] [CrossRef]
- Zhang, X.; Yi, Y.; Qi, J.; Lu, Y.; Tian, Z.; Xie, Y.; Yuan, H.; Wu, W. Controlled release of cyclosporine A self-nanoemulsifying systems from osmotic pump tablets: Near zero-order release and pharmacokinetics in dogs. Int. J. Pharm. 2013, 452, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhou, Y.Q.; Potharaju, S.; Lou, H.; Johnson, J. Development of a self micro-emulsifying tablet of cyclosporine-A by the liquisolid compact technique. Int. J. Pharm. Sci. Res. 2011, 2, 2299–2308. [Google Scholar]
- Lee, J.H.; Park, T.G.; Choi, H.K. Development of oral drug delivery system using floating microspheres. J. Microencapsul. 1999, 16, 715–729. [Google Scholar] [CrossRef]
- Malaekeh-Nikouei, B.; Sajadi Tabassi, S.A.; Jaafari, M.R. Preparation, characterization, and mucoadhesive properties of chitosan-coated microspheres encapsulated with cyclosporine A. Drug Dev. Ind. Pharm. 2008, 34, 492–498. [Google Scholar] [CrossRef] [PubMed]
- IDF Diabetes Atlas|Tenth Edition. Available online: https://diabetesatlas.org/ (accessed on 22 November 2023).
- Macedo, A.; Filipe, P.; Thomé, N.G.; Vieira, J.; Oliveira, C.; Teodósio, C.; Ferreira, R.; Roque, L.; Fonte, P. A brief overview of the oral delivery of insulin as an alternative to the parenteral delivery. Curr. Mol. Med. 2020, 20, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Brayden, D.J. The centenary of the discovery of insulin: An update on the quest for oral delivery. Front. Drug. Deliv. 2021, 1, 726675. [Google Scholar] [CrossRef]
- Sims, E.K.; Carr, A.L.J.; Oram, R.A.; DiMeglio, L.A.; Evans-Molina, C. 100 years of insulin: Celebrating the past, present and future of diabetes therapy. Nat. Med. 2021, 27, 1154–1164. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. Advances in oral peptide therapeutics. Nat. Rev. Drug Discov. 2020, 19, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Halberg, I.B.; Lyby, K.; Wassermann, K.; Heise, T.; Zijlstra, E.; Plum-Mörschel, L. Efficacy and safety of oral basal insulin versus subcutaneous insulin glargine in type 2 diabetes: A randomised, double-blind, phase 2 trial. Lancet Diabetes Endocrinol. 2019, 7, 179–188. [Google Scholar] [CrossRef]
- Khedkar, A.; Lebovitz, H.; Fleming, A.; Cherrington, A.; Jose, V.; Athalye, S.N.; Vishweswaramurthy, A. Impact of Insulin Tregopil and Its Permeation Enhancer on Pharmacokinetics of Metformin in Healthy Volunteers: Randomized, Open-Label, Placebo-Controlled, Crossover Study. Clin. Transl. Sci. 2019, 12, 276–282. [Google Scholar] [CrossRef]
- Khedkar, A.; Iyer, H.; Anand, A.; Verma, M.; Krishnamurthy, S.; Savale, S.; Atignal, A. A dose range finding study of novel oral insulin (IN-105) under fed conditions in type 2 diabetes mellitus subjects. Diabetes Obes. Metab. 2010, 12, 659–664. [Google Scholar] [CrossRef]
- Lebovitz, H.E.; Fleming, A.; Cherrington, A.D.; Joshi, S.; Athalye, S.N.; Loganathan, S.; Vishweswaramurthy, A.; Panda, J.; Marwah, A. Efficacy and safety of Tregopil, a novel, ultra-rapid acting oral prandial insulin analog, as part of a basal-bolus regimen in type 2 diabetes: A randomized, active-controlled phase 2/3 study. Expert Opin. Pharmacother. 2022, 23, 1855–1863. [Google Scholar] [CrossRef]
- Geho, W.B.; Geho, H.C.; Lau, J.R.; Gana, T.J. Hepatic-directed vesicle insulin: A review of formulation development and preclinical evaluation. J. Diabetes Sci. Technol. 2009, 3, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Barnett, A.H.; Bellary, S. Inhaled human insulin (Exubera): Clinical profile and patient considerations. Vasc. Health Risk Manag. 2007, 3, 83–91. [Google Scholar] [PubMed]
- Al-Tabakha, M.M. Future prospect of insulin inhalation for diabetic patients: The case of Afrezza versus Exubera. J. Control. Release 2015, 215, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, L. The failure of exubera: Are we beating a dead horse? J. Diabetes Sci. Technol. 2008, 2, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Klonoff, D.C. Afrezza inhaled insulin: The fastest-acting FDA-approved insulin on the market has favorable properties. J. Diabetes Sci. Technol. 2014, 8, 1071–1073. [Google Scholar] [CrossRef] [PubMed]
- Rave, K.; Heise, T.; Pfützner, A.; Boss, A.H. Coverage of postprandial blood glucose excursions with inhaled technosphere insulin in comparison to subcutaneously injected regular human insulin in subjects with type 2 diabetes. Diabetes Care 2007, 30, 2307–2308. [Google Scholar] [CrossRef] [PubMed]
- Crudden, C.; Girnita, A.; Girnita, L. Targeting the IGF-1R: The Tale of the Tortoise and the Hare. Front. Endocrinol. 2015, 6, 64. [Google Scholar] [CrossRef]
- Wilson, L.M.; Castle, J.R. Recent advances in insulin therapy. Diabetes Technol. Ther. 2020, 22, 929–936. [Google Scholar] [CrossRef]
- Gardner, H.; Hamdy, O. Oral GLP1 analog: Where does the tide go? Clin. Med. Insights Endocrinol. Diabetes 2020, 13, 1179551420984130. [Google Scholar] [CrossRef]
- Antza, C.; Nirantharakumar, K.; Doundoulakis, I.; Tahrani, A.A.; Toulis, K.A. The development of an oral GLP-1 receptor agonist for the management of type 2 diabetes: Evidence to date. Drug Des. Devel. Ther. 2019, 13, 2985–2996. [Google Scholar] [CrossRef]
- American Diabetes Association Professional Practice Committee. Cardiovascular Disease and Risk Management: Standards of Medical Care in Diabetes-2022. Diabetes Care 2022, 45, S144–S174. [Google Scholar] [CrossRef]
- Lincoff, A.M.; Brown-Frandsen, K.; Colhoun, H.M.; Deanfield, J.; Emerson, S.S.; Esbjerg, S.; Hardt-Lindberg, S.; Hovingh, G.K.; Kahn, S.E.; Kushner, R.F.; et al. SELECT Trial Investigators Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes. N. Engl. J. Med. 2023, 389, 2221–2232. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. LEADER Steering Committee; LEADER Trial Investigators Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef]
- Aroda, V.R.; Rosenstock, J.; Terauchi, Y.; Jeppesen, O.L.E.; Christiansen, E.; Hertz, C.L.; Haluzik, M. Effect and safety of oral semaglutide monotherapy in type 2 diabetes—Pioneer 1 trial. Diabetes 2018, 67, 2-LB. [Google Scholar] [CrossRef]
- Pieber, T.R.; Bode, B.; Mertens, A.; Cho, Y.M.; Christiansen, E.; Hertz, C.L.; Wallenstein, S.O.R.; Buse, J.B. PIONEER 7 investigators Efficacy and safety of oral semaglutide with flexible dose adjustment versus sitagliptin in type 2 diabetes (PIONEER 7): A multicentre, open-label, randomised, phase 3a trial. Lancet Diabetes Endocrinol. 2019, 7, 528–539. [Google Scholar] [CrossRef]
- Mosenzon, O.; Blicher, T.M.; Rosenlund, S.; Eriksson, J.W.; Heller, S.; Hels, O.H.; Pratley, R.; Sathyapalan, T.; Desouza, C. PIONEER 5 Investigators Efficacy and safety of oral semaglutide in patients with type 2 diabetes and moderate renal impairment (PIONEER 5): A placebo-controlled, randomised, phase 3a trial. Lancet Diabetes Endocrinol. 2019, 7, 515–527. [Google Scholar] [CrossRef]
- Zinman, B.; Aroda, V.R.; Buse, J.B.; Cariou, B.; Harris, S.B.; Hoff, S.T.; Pedersen, K.B.; Tarp-Johansen, M.J.; Araki, E.; For the Pioneer 8 Investigators. 985-P: Oral Semaglutide as Add-On to Insulin in T2D: PIONEER 8. Diabetes 2019, 68, 985-P. [Google Scholar] [CrossRef]
- Husain, M.; Birkenfeld, A.L.; Donsmark, M.; Dungan, K.; Eliaschewitz, F.G.; Franco, D.R.; Jeppesen, O.K.; Lingvay, I.; Mosenzon, O.; Pedersen, S.D.; et al. PIONEER 6 Investigators Oral Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2019, 381, 841–851. [Google Scholar] [CrossRef]
- Buckley, S.T.; Bækdal, T.A.; Vegge, A.; Maarbjerg, S.J.; Pyke, C.; Ahnfelt-Rønne, J.; Madsen, K.G.; Schéele, S.G.; Alanentalo, T.; Kirk, R.K.; et al. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Sci. Transl. Med. 2018, 10, eaar7047. [Google Scholar] [CrossRef]
- Manning, M.; Balaspiri, L.; Moehring, J.; Haldar, J.; Sawyer, W.H. Synthesis and some pharmacological properties of deamino(4-threonine,8-D-arginine)vasopressin and deamino(8-D-arginine)vasopressin, highly potent and specific antidiuretic peptides, and (8-D-arginine)vasopressin and deamino-arginine-vasopressin. J. Med. Chem. 1976, 19, 842–845. [Google Scholar] [CrossRef] [PubMed]
- Michelet, R.; Dossche, L.; De Bruyne, P.; Colin, P.; Boussery, K.; Vande Walle, J.; Van Bocxlaer, J.; Vermeulen, A. Effects of food and pharmaceutical formulation on desmopressin pharmacokinetics in children. Clin. Pharmacokinet. 2016, 55, 1159–1170. [Google Scholar] [CrossRef]
- Available online: www.medsafe.govt.nz/profs/datasheet/m/Minirintab.pdf (accessed on 29 November 2023).
- Brayden, D.J.; Hill, T.A.; Fairlie, D.P.; Maher, S.; Mrsny, R.J. Systemic delivery of peptides by the oral route: Formulation and medicinal chemistry approaches. Adv. Drug Deliv. Rev. 2020, 157, 2–36. [Google Scholar] [CrossRef]
- Dec, A.; Niemiec, A.; Wojciechowska, E.; Maligłówka, M.; Bułdak, Ł.; Bołdys, A.; Okopień, B. Inclisiran-A Revolutionary Addition to a Cholesterol-Lowering Therapy. Int. J. Mol. Sci. 2023, 24, 6858. [Google Scholar] [CrossRef]
- Gennemark, P.; Walter, K.; Clemmensen, N.; Rekić, D.; Nilsson, C.A.M.; Knöchel, J.; Hölttä, M.; Wernevik, L.; Rosengren, B.; Kakol-Palm, D.; et al. An oral antisense oligonucleotide for PCSK9 inhibition. Sci. Transl. Med. 2021, 13, eabe9117. [Google Scholar] [CrossRef]
- Johns, D.G.; Campeau, L.-C.; Banka, P.; Bautmans, A.; Bueters, T.; Bianchi, E.; Branca, D.; Bulger, P.G.; Crevecoeur, I.; Ding, F.-X.; et al. Orally bioavailable macrocyclic peptide that inhibits binding of PCSK9 to the low density lipoprotein receptor. Circulation 2023, 148, 144–158. [Google Scholar] [CrossRef]
- Burnett, J.R.; Hooper, A.J. MK-0616: An oral PCSK9 inhibitor for hypercholesterolemia treatment. Expert Opin. Investig. Drugs 2023, 32, 873–878. [Google Scholar] [CrossRef]
- Sandimmun Neoral—Summary of Product Characteristics, Labelling and Package Leaflet. Available online: https://www.ema.europa.eu/en/documents/referral/sandimmun-neoral-article-30-referral-annex-iii_en.pdf (accessed on 3 January 2024).
- Rybelsus, INN-Semaglutide—Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/rybelsus-epar-product-information_en.pdf (accessed on 3 January 2024).
- Mycapssa, INN-Octreotide Acetate: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/mycapssa-epar-product-information_en.pdf (accessed on 3 January 2024).
- AFREZZA® (Insulin Human) Inhalation Powder: Highlights of Prescribing Information. Available online: https://www.afrezza.com/pdf/Full-Prescribing-Information.pdf (accessed on 3 January 2024).
- A Phase I Study to Evaluate the Pharmacokinetics of Parathyroid Hormone (1-34) Administered Orally via RaniPillTM Capsule. Available online: https://clinicaltrials.gov/study/NCT05164614?term=nct05164614&rank=1#study-overview (accessed on 22 November 2023).
- An Evaluation of the Pharmacokinetics and Pharmacodynamics of Oral Parathyroid Hormone [PTH (1-34)] and NATPARA® in Patients with Hypoparathyroidism. Available online: https://clinicaltrials.gov/study/NCT03516773?term=nct03516773&rank=1 (accessed on 22 November 2023).
- An Open-Label Dose-Finding Study to Evaluate the Pharmacodynamic (PD) Profiles and Efficacy of Different Dosing Regimens of Leuprolide Oral Tablets (Ovarest®) in Women with Endometriosis. Available online: https://clinicaltrials.gov/study/NCT05096065?term=nct05096065&rank=1 (accessed on 22 November 2023).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
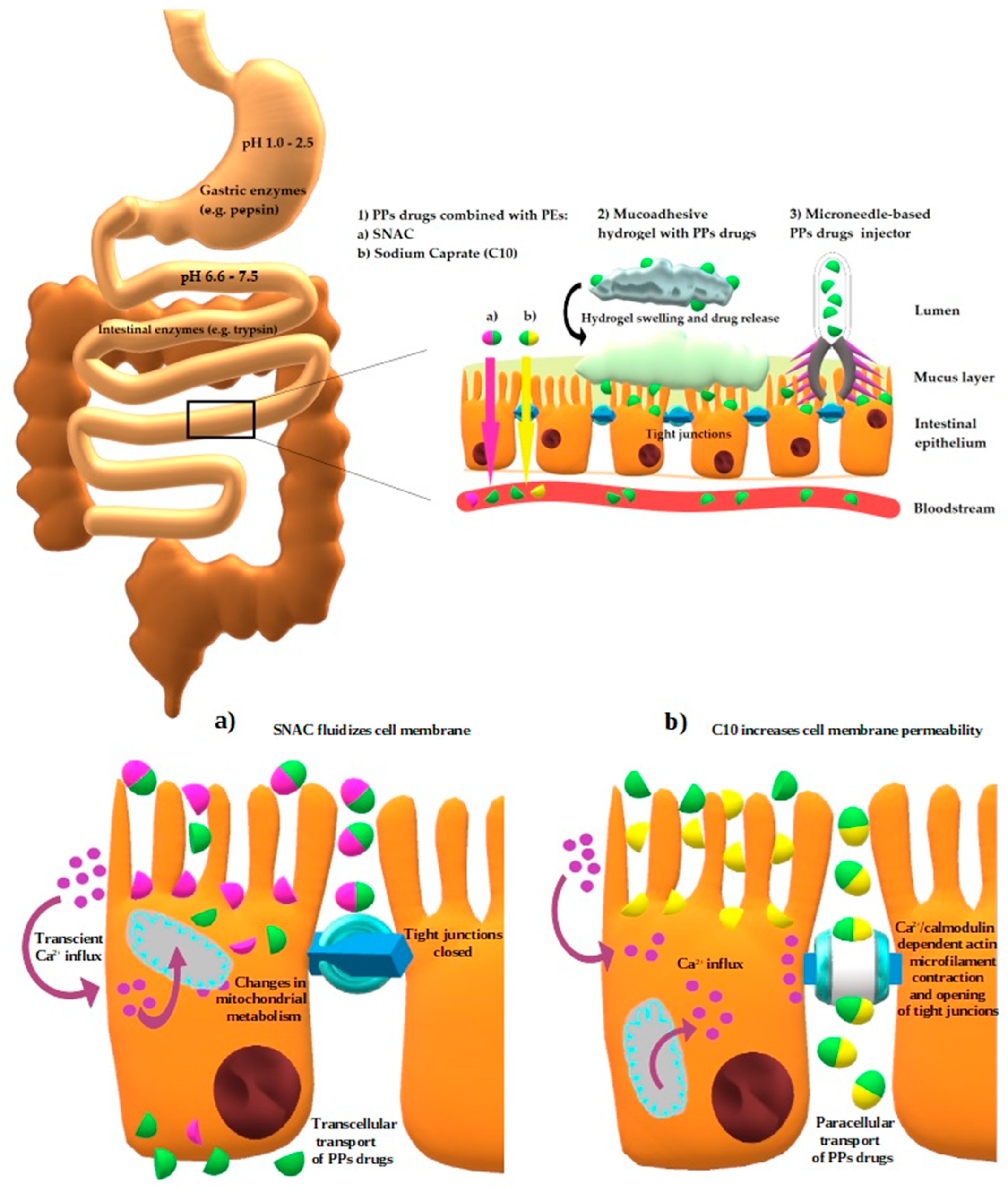{kind=link}
| Delivery Approach | Description | Outcome | PPDs Example |
|---|---|---|---|
| Nanoparticles (Liposomes) | Vesicular systems with ability to adhere to the mucus of gut (mucoadhesive type) or penetrate across the mucus barrier (mucus-penetrating type) [77] | Enhanced mucus-penetrating capability [77] | Insulin |
| Transport Channels | Particles mediating traffic across membranes [85] | Overcoming intestinal epithelial barrier [85] | Insulin |
| Permeation Enhancers | Chemical compounds facilitating penetration through gastric or intestinal epithelium [87] | Increased paracellular/transcellular absorption [87] | Octreotide |
| Peptide Cyclization and Substitutions of AAs | Structural modifications | Improved enzymatic stability [99] | Desmopressin, Insulin [99,106] |
| Hydrogels | Three-dimensional molecules with hydrophilic and mucoadhesive properties [108] | Long-lasting release of drug, prevention from proteolytic degradation [108] | Insulin [108] |
| Microneedles | Polymeric, microscopic needles [111] Gastric auto-injector [113] | Physical barriers penetration (both mucous and epithelium in the GI tract) [111,113] | Insulin [112] Adalimumab, Semaglutide-like GLP-1 analog, Insulin [113] |
| Microemulsion | Dispersed components including oily and water phases, surfactant and cosurfactant [114,115] | Reduction of interfacial tension and induction of intermolecular forces (surfactant), stabilization of hydrophobic drugs (cosurfactant), solubilization ensuring [114,115] | Cyclosporine |
| Proteolytic Enzyme Inhibitors | Substances decreasing enzymatic activity of proteases in GI tract [118] | Prevention of drug degradation [118] | Insulin |
| Cell-Penetrating Peptides | Short peptides able to deliver attached molecules through biological membranes penetration [124,125] | Permeability enhancement [124,125] | Insulin |
| Bacteria-mediated Therapy | Modified microorganisms (e.g., using biotechnological methods such as plasmid modifications) [131] | Bacteria capable of producing specific PPDs, selective drug delivery [131] | TNF-alpha [133] |
| Substance | Trade Name (Company) | Approval Date | Indications | Technology | Pharmacokinetics |
|---|---|---|---|---|---|
| Cyclosporine | Sandimmun® Neoral® (Novartis, Basel, Switzerland) [148] | 1995 (Neoral®) | Immunosuppression after transplantation, rheumatoid arthritis, nephrotic syndrome, psoriasis, toxic epider- mal necrolysis, atopic dermatitis [144] | Microemulsion [148] | Bioavailability of Neoral®: 20–50%, approximately 29% higher than Sandimmun® with 59% higher Cmax. Comparable concentration of cyclosporine in whole blood. Peak blood concentration within 1–2 h. Average volume of distribution—3.5 L/kg. Mainly liver metabolism via cytochrome P450. Biliary excretion, only 6% in the urine. Terminal half-life increase from 6.3 h to 20.4 h in case of severe liver dysfunction [193]. |
| Semaglutide | Rybelsus® (Novo Nordisk) [179] | 2019 | Type 2 diabetes mellitus [179] | Permeation Enhancer [179] | Oral dose 14 mg daily comparable to subcutaneous 0.5 mg once weekly. Only 1% bioavailability after oral administration, decreased by food or large amounts of water intake. Maximum plasma concentration after 1 h. Estimated absolute volume of distribution around 8.0 L. Excretion via the urine and stool. Approximately 1 week elimination half-life. Detectable in circulation for about 5 weeks [194]. |
| Desmopressin acetate (DDAVP) | Minirin® (Ferring Pharmaceuticals) [187] | 2008 | Central diabetes insipidus, Nocturnal enuresis [186] | Chemical Modifications [186] | Bioavailability 0.25% of sublingual form. Cmax 14, 30 and 65 pg/mL (for 200, 400 and 800 µg dose). Tmax 0.5–2.0 h after use. Half-life—2 h [187]. |
| Octreotide | Mycapssa® (Chiasma, Needham, MA, USA) [195] | 2020 | Acromegaly, Neuroendocrine Tumors [161] | Permeation Enhancer [179] | AUC of 20 mg oral octreotide acetate (single dose) comparable to a single subcutaneous dose (0.1 mg). Cmax 22–33% lower than subcutaneous form. Longer absorption time: peak concentrations 1.67–2.5 h after oral dose compared to 0.5 h after subcutaneous. Food decreases absorption by 90%. Elimination mainly via the stool and 32% by the urine. Similar to the subcutaneous form half-life (2.66 h and 2.27 h) [195]. |
| Inhalable Insulin | Afrezza® (MannKind Corporation) [171] | 2014 | Diabetes mellitus | Technosphere® microparticles [171] | Dose-dependent proportional increase in AUC up to 48 units. Intrapatient variations 16% of AUC and 21% of Cmax. Tmax 10–20 min after inhalation (4–48 units of Afrezza®). Apparent terminal half-life between 120 and 206 min [196]. |
| Subject of the Study | Study Start Date | Study Type | Details | Results |
|---|---|---|---|---|
| RT-102 oral optimized formulation of PTH(1-34) | 21 February 2022 | Prospective, single-center, open-label, phase I study [197] | PTH administered orally via the RaniPill® capsule, active comparator group receiving PTH subcutaneously | No results posted yet |
| EnteroBio’s oral PTH(1-34) (EB612(EBP05)) | 17 June 2018 | Randomized, active comparator, two-part, partial crossover design study [198] | Administered in patients with primary hypoparathyroidism, compared to NATPARA® | No results posted yet |
| Ovarest® Leuprolide oral tablets | 18 March 2022 | Open-label, non-randomized, phase II dose-finding study [199] | Determination of efficacy and pharmacodynamics of Ovarest®, minimally effective dose compared to Lupron Depot, safety and tolerability of the long-term administration [199] | No results posted yet |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicze, M.; Borówka, M.; Dec, A.; Niemiec, A.; Bułdak, Ł.; Okopień, B. The Current and Promising Oral Delivery Methods for Protein- and Peptide-Based Drugs. Int. J. Mol. Sci. 2024, 25, 815. https://doi.org/10.3390/ijms25020815
Nicze M, Borówka M, Dec A, Niemiec A, Bułdak Ł, Okopień B. The Current and Promising Oral Delivery Methods for Protein- and Peptide-Based Drugs. International Journal of Molecular Sciences. 2024; 25(2):815. https://doi.org/10.3390/ijms25020815
Chicago/Turabian StyleNicze, Michał, Maciej Borówka, Adrianna Dec, Aleksandra Niemiec, Łukasz Bułdak, and Bogusław Okopień. 2024. "The Current and Promising Oral Delivery Methods for Protein- and Peptide-Based Drugs" International Journal of Molecular Sciences 25, no. 2: 815. https://doi.org/10.3390/ijms25020815
APA StyleNicze, M., Borówka, M., Dec, A., Niemiec, A., Bułdak, Ł., & Okopień, B. (2024). The Current and Promising Oral Delivery Methods for Protein- and Peptide-Based Drugs. International Journal of Molecular Sciences, 25(2), 815. https://doi.org/10.3390/ijms25020815