
An Analysis of Genetic Polymorphisms in 76 Genes Related to the Development of Ovarian Tumors of Different Aggressiveness

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Distribution of Genetic Polymorphisms in Different Tumor Groups
2.2. Regression Analyses
2.3. Assessment of Relationship between Selected Gene Polymorphisms and Expression of Corresponding Proteins
3. Discussion
4. Materials and Methods
4.1. Patients and Clinicopathological Parameters
4.2. DNA Isolation and Quality Assessment
4.3. Construction of Total gDNA Libraries; 44-Gene Panel Enrichment and Verification; NGS Sequencing
4.4. Hot-Spot Panel Enrichment and Verification; NGS Sequencing
4.5. Bioinformatic Analyses
4.6. Verification of Selected Polymorphisms
4.7. Protein Concentration Measurement
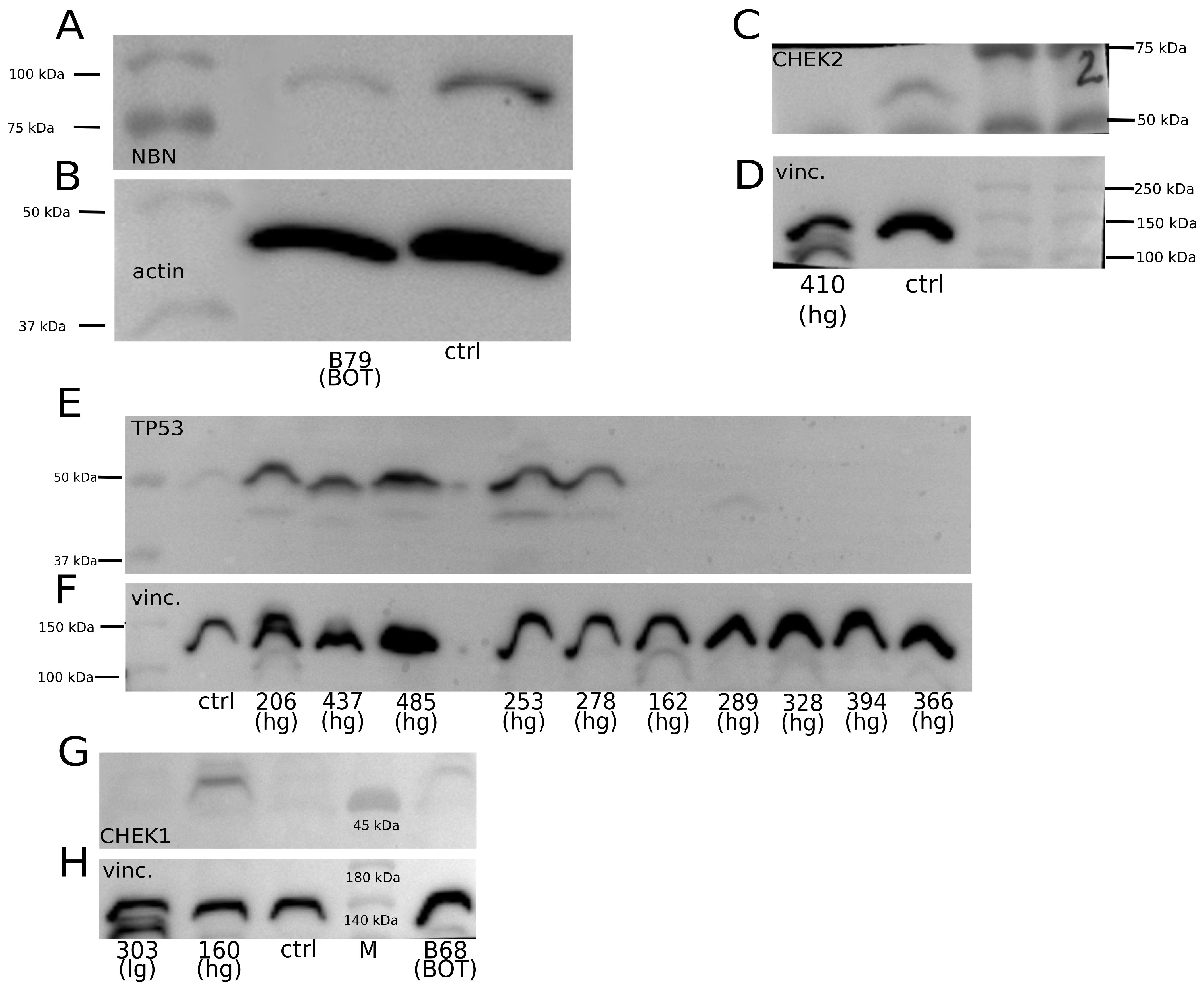
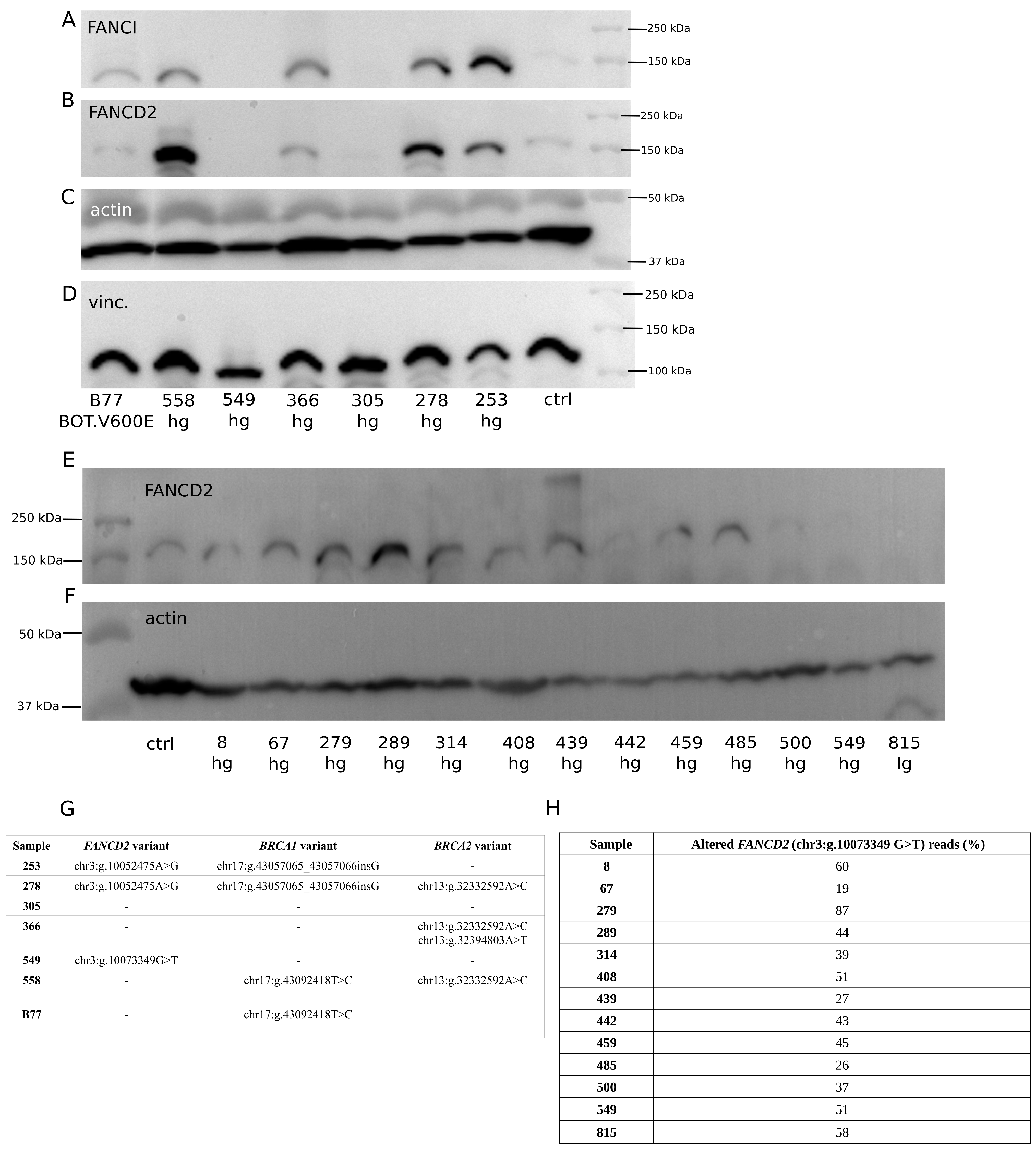
4.8. Western Blot (WB) Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arora, T.; Mullangi, S.; Lekkala, M.R. Ovarian Cancer; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Guo, T.; Dong, X.; Xie, S.; Zhang, L.; Zeng, P.; Zhang, L. Cellular Mechanism of Gene Mutations and Potential Therapeutic Targets in Ovarian Cancer. CMAR 2021, 13, 3081–3100. [Google Scholar] [CrossRef]
- Babaier, A.; Mal, H.; Alselwi, W.; Ghatage, P. Low-Grade Serous Carcinoma of the Ovary: The Current Status. Diagnostics 2022, 12, 458. [Google Scholar] [CrossRef]
- Wong, K.-K.; Bateman, N.W.; Ng, C.W.; Tsang, Y.T.M.; Sun, C.S.; Celestino, J.; Nguyen, T.V.; Malpica, A.; Hillman, R.T.; Zhang, J.; et al. Integrated Multi-Omic Analysis of Low-Grade Ovarian Serous Carcinoma Collected from Short and Long-Term Survivors. J. Transl. Med. 2022, 20, 606. [Google Scholar] [CrossRef]
- Hauptmann, S.; Friedrich, K.; Redline, R.; Avril, S. Ovarian Borderline Tumors in the 2014 WHO Classification: Evolving Concepts and Diagnostic Criteria. Virchows Arch. 2017, 470, 125–142. [Google Scholar] [CrossRef] [PubMed]
- Seong, S.J.; Kim, D.H.; Kim, M.K.; Song, T. Controversies in Borderline Ovarian Tumors. J. Gynecol. Oncol. 2015, 26, 343–349. [Google Scholar] [CrossRef]
- Niu, L.; Tian, H.; Xu, Y.; Cao, J.; Zhang, X.; Zhang, J.; Hou, J.; Lv, W.; Wang, J.; Xin, L.; et al. Recurrence Characteristics and Clinicopathological Results of Borderline Ovarian Tumors. BMC Women’s Health 2021, 21, 134. [Google Scholar] [CrossRef]
- Shih, K.; Zhou, Q.; Huh, J.; Morgan, J.; Iasonos, A.; Aghajanian, C.; Chi, D.; Barakat, R.; Abu-Rustum, N. Risk Factors for Recurrence of Ovarian Borderline Tumors. Gynecol. Oncol. 2011, 120, 480–484. [Google Scholar] [CrossRef]
- Bjørge, T.; Lie, A.K.; Hovig, E.; Gislefoss, R.E.; Hansen, S.; Jellum, E.; Langseth, H.; Nustad, K.; Tropé, C.G.; Dørum, A. BRCA1 Mutations in Ovarian Cancer and Borderline Tumours in Norway: A Nested Case–Control Study. Br. J. Cancer 2004, 91, 1829–1834. [Google Scholar] [CrossRef]
- Lakhani, S.R.; Manek, S.; Penault-Llorca, F.; Flanagan, A.; Arnout, L.; Merrett, S.; McGuffog, L.; Steele, D.; Devilee, P.; Klijn, J.G.M.; et al. Pathology of Ovarian Cancers in BRCA1 and BRCA2 Carriers. Clin. Cancer Res. 2004, 10, 2473–2481. [Google Scholar] [CrossRef]
- Singer, G.; Stöhr, R.; Cope, L.; Dehari, R.; Hartmann, A.; Cao, D.-F.; Wang, T.-L.; Kurman, R.J.; Shih, I.-M. Patterns of P53 Mutations Separate Ovarian Serous Borderline Tumors and Low- and High-Grade Carcinomas and Provide Support for a New Model of Ovarian Carcinogenesis: A Mutational Analysis with Immunohistochemical Correlation. Am. J. Surg. Pathol. 2005, 29, 218–224. [Google Scholar] [CrossRef]
- Mayr, D.; Hirschmann, A.; Löhrs, U.; Diebold, J. KRAS and BRAF Mutations in Ovarian Tumors: A Comprehensive Study of Invasive Carcinomas, Borderline Tumors and Extraovarian Implants. Gynecol. Oncol. 2006, 103, 883–887. [Google Scholar] [CrossRef]
- Grisham, R.N.; Iyer, G.; Garg, K.; Delair, D.; Hyman, D.M.; Zhou, Q.; Iasonos, A.; Berger, M.F.; Dao, F.; Spriggs, D.R.; et al. BRAF Mutation Is Associated with Early Stage Disease and Improved Outcome in Patients with Low-Grade Serous Ovarian Cancer. Cancer 2013, 119, 548–554. [Google Scholar] [CrossRef]
- Stemke-Hale, K.; Shipman, K.; Kitsou-Mylona, I.; de Castro, D.G.; Hird, V.; Brown, R.; Flanagan, J.; Hani Gabra, H.; Mills, G.B.; Agarwal, R.; et al. Frequency of Mutations and Polymorphisms in Borderline Ovarian Tumors of Known Cancer Genes. Mod. Pathol. 2013, 26, 544–552. [Google Scholar] [CrossRef]
- Ogrodniczak, A.; Menkiszak, J.; Gronwald, J.; Tomiczek-Szwiec, J.; Szwiec, M.; Cybulski, C.; Dębniak, T.; Huzarski, T.; Tołoczko-Grabarek, A.; Byrski, T.; et al. Association of Recurrent Mutations in BRCA1, BRCA2, RAD51C, PALB2, and CHEK2 with the Risk of Borderline Ovarian Tumor. Hered. Cancer Clin. Pract. 2022, 20, 11. [Google Scholar] [CrossRef]
- Ko, E.-A.; Kim, Y.-W.; Lee, D.; Choi, J.; Kim, S.; Seo, Y.; Bang, H.; Kim, J.-H.; Ko, J.-H. Expression of Potassium Channel Genes Predicts Clinical Outcome in Lung Cancer. Korean J. Physiol. Pharmacol. Off. J. Korean Physiol. Soc. Korean Soc. Pharmacol. 2019, 23, 529. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Szafron, L.A.; Iwanicka-Nowicka, R.; Podgorska, A.; Bonna, A.M.; Sobiczewski, P.; Kupryjanczyk, J.; Szafron, L.M. The Clinical Significance of CRNDE Gene Methylation, Polymorphisms, and CRNDEP Micropeptide Expression in Ovarian Tumors. Int. J. Mol. Sci. 2024, 25, 7531. [Google Scholar] [CrossRef]
- Griffith, O.L.; Spies, N.C.; Anurag, M.; Griffith, M.; Luo, J.; Tu, D.; Yeo, B.; Kunisaki, J.; Miller, C.A.; Krysiak, K.; et al. The Prognostic Effects of Somatic Mutations in ER-Positive Breast Cancer. Nat. Commun. 2018, 9, 3476. [Google Scholar] [CrossRef]
- Vias, M.; Morrill Gavarró, L.; Sauer, C.M.; Sanders, D.A.; Piskorz, A.M.; Couturier, D.-L.; Ballereau, S.; Hernando, B.; Schneider, M.P.; Hall, J.; et al. High-Grade Serous Ovarian Carcinoma Organoids as Models of Chromosomal Instability. eLife 2023, 12, e83867. [Google Scholar] [CrossRef]
- Smith, P.; Bradley, T.; Gavarró, L.M.; Goranova, T.; Ennis, D.P.; Mirza, H.B.; De Silva, D.; Piskorz, A.M.; Sauer, C.M.; Al-Khalidi, S.; et al. The Copy Number and Mutational Landscape of Recurrent Ovarian High-Grade Serous Carcinoma. Nat. Commun. 2023, 14, 4387. [Google Scholar] [CrossRef]
- Kupryjanczyk, J.; Bell, D.A.; Dimeo, D.; Beauchamp, R.; Thor, A.D.; Yandell, D.W. P53 Gene Analysis of Ovarian Borderline Tumors and Stage I Carcinomas. Hum. Pathol. 1995, 26, 387–392. [Google Scholar] [CrossRef]
- Katabuchi, H.; Tashiro, H.; Cho, K.R.; Kurman, R.J.; Hedrick Ellenson, L. Micropapillary Serous Carcinoma of the Ovary: An Immunohistochemical and Mutational Analysis of P53. Int. J. Gynecol. Pathol. 1998, 17, 54–60. [Google Scholar] [CrossRef]
- Testa, U.; Petrucci, E.; Pasquini, L.; Castelli, G.; Pelosi, E. Ovarian Cancers: Genetic Abnormalities, Tumor Heterogeneity and Progression, Clonal Evolution and Cancer Stem Cells. Medicines 2018, 5, 16. [Google Scholar] [CrossRef]
- KAPA HyperPETE. Available online: https://sequencing.roche.com/us/en/products/product-category/primer-extension-based-target-enrichment.html (accessed on 7 June 2024).
- Kang, E.Y.; Cheasley, D.; LePage, C.; Wakefield, M.J.; da Cunha Torres, M.; Rowley, S.; Salazar, C.; Xing, Z.; Allan, P.; Bowtell, D.D.L.; et al. Refined Cut-off for TP53 Immunohistochemistry Improves Prediction of TP53 Mutation Status in Ovarian Mucinous Tumors: Implications for Outcome Analyses. Mod. Pathol. 2021, 34, 194–206. [Google Scholar] [CrossRef]
- Kupryjańczyk, J.; Szymańska, T.; Madry, R.; Timorek, A.; Stelmachów, J.; Karpińska, G.; Rembiszewska, A.; Ziółkowska, I.; Kraszewska, E.; Debniak, J.; et al. Evaluation of Clinical Significance of TP53, BCL-2, BAX and MEK1 Expression in 229 Ovarian Carcinomas Treated with Platinum-Based Regimen. Br. J. Cancer 2003, 88, 848–854. [Google Scholar] [CrossRef]
- Ghezelayagh, T.S.; Pennington, K.P.; Norquist, B.M.; Khasnavis, N.; Radke, M.R.; Kilgore, M.R.; Garcia, R.L.; Lee, M.; Katz, R.; Leslie, K.K.; et al. Characterizing TP53 Mutations in Ovarian Carcinomas with and without Concurrent BRCA1 or BRCA2 Mutations. Gynecol. Oncol. 2021, 160, 786–792. [Google Scholar] [CrossRef]
- Lhotova, K.; Stolarova, L.; Zemankova, P.; Vocka, M.; Janatova, M.; Borecka, M.; Cerna, M.; Jelinkova, S.; Kral, J.; Volkova, Z.; et al. Multigene Panel Germline Testing of 1333 Czech Patients with Ovarian Cancer. Cancers 2020, 12, 956. [Google Scholar] [CrossRef]
- Flis, M.; Krawczyk, P.; Drogoń, I.; Kurek, K.; Kieszko, R.; Milanowski, J. The Effectiveness of Chemotherapy in Small Cell Lung Cancer Patients with BRCA2 Gene Mutation and Schwartz-Bartter Syndrome. Oncol. Clin. Pr. 2019, 15, 120–123. [Google Scholar] [CrossRef]
- Mylavarapu, S.; Das, A.; Roy, M. Role of BRCA Mutations in the Modulation of Response to Platinum Therapy. Front. Oncol. 2018, 8, 16. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different Roles in a Common Pathway of Genome Protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef]
- Moujaber, T.; Etemadmoghadam, D.; Kennedy, C.J.; Chiew, Y.-E.; Balleine, R.L.; Saunders, C.; Wain, G.V.; Gao, B.; Hogg, R.; Srirangan, S.; et al. BRAF Mutations in Low-Grade Serous Ovarian Cancer and Response to BRAF Inhibition. JCO Precis. Oncol. 2018, 2, 1–14. [Google Scholar] [CrossRef]
- Auner, V.; Kriegshäuser, G.; Tong, D.; Horvat, R.; Reinthaller, A.; Mustea, A.; Zeillinger, R. KRAS Mutation Analysis in Ovarian Samples Using a High Sensitivity Biochip Assay. BMC Cancer 2009, 9, 111. [Google Scholar] [CrossRef]
- Garziera, M.; Roncato, R.; Montico, M.; De Mattia, E.; Gagno, S.; Poletto, E.; Scalone, S.; Canzonieri, V.; Giorda, G.; Sorio, R.; et al. New Challenges in Tumor Mutation Heterogeneity in Advanced Ovarian Cancer by a Targeted Next-Generation Sequencing (NGS) Approach. Cells 2019, 8, 584. [Google Scholar] [CrossRef]
- Mutations in the KRAS Gene in Ovarian Tumors—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/19995707/ (accessed on 14 January 2024).
- Merz, V.; Gaule, M.; Zecchetto, C.; Cavaliere, A.; Casalino, S.; Pesoni, C.; Contarelli, S.; Sabbadini, F.; Bertolini, M.; Mangiameli, D.; et al. Targeting KRAS: The Elephant in the Room of Epithelial Cancers. Front. Oncol. 2021, 11, 638360. [Google Scholar] [CrossRef]
- Zhu, C.; Guan, X.; Zhang, X.; Luan, X.; Song, Z.; Cheng, X.; Zhang, W.; Qin, J.-J. Targeting KRAS Mutant Cancers: From Druggable Therapy to Drug Resistance. Mol. Cancer 2022, 21, 159. [Google Scholar] [CrossRef]
- Śmiech, M.; Leszczyński, P.; Kono, H.; Wardell, C.; Taniguchi, H. Emerging BRAF Mutations in Cancer Progression and Their Possible Effects on Transcriptional Networks. Genes 2020, 11, 1342. [Google Scholar] [CrossRef]
- Hunter, S.M.; Anglesio, M.S.; Ryland, G.L.; Sharma, R.; Chiew, Y.-E.; Rowley, S.M.; Doyle, M.A.; Li, J.; Gilks, C.B.; Moss, P.; et al. Molecular Profiling of Low Grade Serous Ovarian Tumours Identifies Novel Candidate Driver Genes. Oncotarget 2015, 6, 37663–37677. [Google Scholar] [CrossRef]
- Xing, D.; Suryo Rahmanto, Y.; Zeppernick, F.; Hannibal, C.G.; Kjaer, S.K.; Vang, R.; Shih, I.-M.; Wang, T.-L. Mutation of NRAS Is a Rare Genetic Event in Ovarian Low-Grade Serous Carcinoma. Hum. Pathol. 2017, 68, 87–91. [Google Scholar] [CrossRef]
- Champer, M.; Miller, D.; Kuo, D.Y.-S. Response to Trametinib in Recurrent Low-Grade Serous Ovarian Cancer with NRAS Mutation: A Case Report. Gynecol. Oncol. Rep. 2019, 28, 26–28. [Google Scholar] [CrossRef]
- Grill, C.; Larue, L. NRAS, NRAS, Which Mutation Is Fairest of Them All? J. Investig. Dermatol. 2016, 136, 1936–1938. [Google Scholar] [CrossRef]
- McCluggage, W.G. Ovarian Borderline Tumours: A Review with Comparison of Serous and Mucinous Types. Diagn. Histopathol. 2014, 20, 333–350. [Google Scholar] [CrossRef]
- Zhong, F.; Zhu, T.; Pan, X.; Zhang, Y.; Yang, H.; Wang, X.; Hu, J.; Han, H.; Mei, L.; Chen, D.; et al. Comprehensive Genomic Profiling of High-grade Serous Ovarian Carcinoma from Chinese Patients Identifies Co-occurring Mutations in the Ras/Raf Pathway with TP53. Cancer Med. 2019, 8, 3928–3935. [Google Scholar] [CrossRef]
- Bożyk, A.; Krawczyk, P.; Reszka, K.; Krukowska, K.; Kolak, A.; Mańdziuk, S.; Wojas-Krawczyk, K.; Ramlau, R.; Milanowski, J. Correlation between KRAS, NRAS and BRAF Mutations and Tumor Localizations in Patients with Primary and Metastatic Colorectal Cancer. Arch. Med. Sci. AMS 2022, 18, 1221–1230. [Google Scholar] [CrossRef]
- Alessandro, L.; Low, K.-J.E.; Abushelaibi, A.; Lim, S.-H.E.; Cheng, W.-H.; Chang, S.-K.; Lai, K.-S.; Sum, Y.W.; Maran, S. Identification of NRAS Diagnostic Biomarkers and Drug Targets for Endometrial Cancer-An Integrated in Silico Approach. Int. J. Mol. Sci. 2022, 23, 14285. [Google Scholar] [CrossRef]
- Sideris, M.; Emin, E.I.; Abdullah, Z.; Hanrahan, J.; Stefatou, K.M.; Sevas, V.; Emin, E.; Hollingworth, T.; Odejinmi, F.; Papagrigoriadis, S.; et al. The Role of KRAS in Endometrial Cancer: A Mini-Review. Anticancer Res. 2019, 39, 533–539. [Google Scholar] [CrossRef]
- He, M.; Breese, V.; Hang, S.; Zhang, C.; Xiong, J.; Jackson, C. BRAF V600E Mutations in Endometrial Adenocarcinoma. Diagn. Mol. Pathol. Am. J. Surg. Pathol. Part B 2013, 22, 35–40. [Google Scholar] [CrossRef]
- Gianni, P.; Matenoglou, E.; Geropoulos, G.; Agrawal, N.; Adnani, H.; Zafeiropoulos, S.; Miyara, S.J.; Guevara, S.; Mumford, J.M.; Molmenti, E.P.; et al. The Fanconi Anemia Pathway and Breast Cancer: A Comprehensive Review of Clinical Data. Clin. Breast Cancer 2022, 22, 10–25. [Google Scholar] [CrossRef]
- Del Valle, J.; Rofes, P.; Moreno-Cabrera, J.M.; López-Dóriga, A.; Belhadj, S.; Vargas-Parra, G.; Teulé, À.; Cuesta, R.; Muñoz, X.; Campos, O.; et al. Exploring the Role of Mutations in Fanconi Anemia Genes in Hereditary Cancer Patients. Cancers 2020, 12, 829. [Google Scholar] [CrossRef]
- Gajewski, S.; Hartwig, A. PARP1 Is Required for ATM-Mediated P53 Activation and P53-Mediated Gene Expression after Ionizing Radiation. Chem. Res. Toxicol. 2020, 33, 1933–1940. [Google Scholar] [CrossRef]
- Zuo, W.-W.; Zhao, C.-F.; Li, Y.; Sun, H.-Y.; Ma, G.-M.; Liu, Y.-P.; Kang, S. High Expression of PARP1 in Tumor and Stroma Cells Predicts Different Prognosis and Platinum Resistance in Patients With Advanced Epithelial Ovarian Cancer. Front. Oncol. 2022, 12, 931445. [Google Scholar] [CrossRef]
- Hockings, H.; Miller, R.E. The Role of PARP Inhibitor Combination Therapy in Ovarian Cancer. Ther. Adv. Med. Oncol. 2023, 15, 17588359231173183. [Google Scholar] [CrossRef]
- Miller, R.E.; El-Shakankery, K.H.; Lee, J.-Y. PARP Inhibitors in Ovarian Cancer: Overcoming Resistance with Combination Strategies. J. Gynecol. Oncol. 2022, 33, e44. [Google Scholar] [CrossRef]
- Wang, S.S.Y.; Jie, Y.E.; Cheng, S.W.; Ling, G.L.; Ming, H.V.Y. PARP Inhibitors in Breast and Ovarian Cancer. Cancers 2023, 15, 2357. [Google Scholar] [CrossRef]
- Anjali, K.; Singh, D.; Kumar, P.; Kumar, T.; Narayan, G.; Singh, S. PARP1 Rs1136410 (A/G) Polymorphism Is Associated with Early Age of Onset of Gallbladder Cancer. Eur. J. Cancer Prev. 2022, 31, 311–317. [Google Scholar] [CrossRef]
- Li, H.; Zha, Y.; Du, F.; Liu, J.; Li, X.; Zhao, X. Contributions of PARP-1 Rs1136410 C>T Polymorphism to the Development of Cancer. J. Cell. Mol. Med. 2020, 24, 14639–14644. [Google Scholar] [CrossRef]
- Ma, X.-B.; Wang, X.-J.; Wang, M.; Dai, Z.-M.; Jin, T.-B.; Liu, X.-H.; Kang, H.-F.; Lin, S.; Xu, P.; Dai, Z.-J. Impact of the PARP1 Rs1136410 and Rs3219145 Polymorphisms on Susceptibility and Clinicopathologic Features of Breast Cancer in a Chinese Population. Transl. Cancer Res. 2016, 5. [Google Scholar] [CrossRef]
- Postawski, K.; Monist, M.; Keith, G. PARP-1 Activity in Normal and Cancerous Human Endometrium and Its Relationship with Quantity of Abasic Sites (AP). Ginekol. Pol. 2011, 82, 16–21. [Google Scholar]
- Zhang, X.; Lu, X.; Akhter, S.; Georgescu, M.-M.; Legerski, R.J. FANCI Is a Negative Regulator of Akt Activation. Cell Cycle 2016, 15, 1134–1143. [Google Scholar] [CrossRef]
- Cai, Z.; Duan, Y.; Li, W.; Liu, Z.; Gong, Z.; Hong, S.; He, X.; Xuanyuan, X.; Chen, Y.; Bi, X.; et al. FANCI Serve as a Prognostic Biomarker Correlated with Immune Infiltrates in Skin Cutaneous Melanoma. Front. Immunol. 2023, 14, 1295831. [Google Scholar] [CrossRef]
- Fierheller, C.T.; Alenezi, W.M.; Serruya, C.; Revil, T.; Amuzu, S.; Bedard, K.; Subramanian, D.N.; Fewings, E.; Bruce, J.P.; Prokopec, S.; et al. Molecular Genetic Characteristics of FANCI, a Proposed New Ovarian Cancer Predisposing Gene. Genes 2023, 14, 277. [Google Scholar] [CrossRef]
- Fierheller, C.T.; Guitton-Sert, L.; Alenezi, W.M.; Revil, T.; Oros, K.K.; Gao, Y.; Bedard, K.; Arcand, S.L.; Serruya, C.; Behl, S.; et al. A Functionally Impaired Missense Variant Identified in French Canadian Families Implicates FANCI as a Candidate Ovarian Cancer-Predisposing Gene. Genome Med. 2021, 13, 186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hunter, T. Roles of Chk1 in Cell Biology and Cancer Therapy. Int. J. Cancer 2014, 134, 1013–1023. [Google Scholar] [CrossRef]
- McNeely, S.; Beckmann, R.; Bence Lin, A.K. CHEK Again: Revisiting the Development of CHK1 Inhibitors for Cancer Therapy. Pharmacol. Ther. 2014, 142, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fadaka, A.O.; Bakare, O.O.; Sibuyi, N.R.S.; Klein, A. Gene Expression Alterations and Molecular Analysis of CHEK1 in Solid Tumors. Cancers 2020, 12, 662. [Google Scholar] [CrossRef]
- Neizer-Ashun, F.; Bhattacharya, R. Reality CHEK: Understanding the Biology and Clinical Potential of CHK1. Cancer Lett. 2021, 497, 202–211. [Google Scholar] [CrossRef]
- Morden, C.R.; Farrell, A.C.; Sliwowski, M.; Lichtensztejn, Z.; Altman, A.D.; Nachtigal, M.W.; McManus, K.J. Chromosome Instability Is Prevalent and Dynamic in High-Grade Serous Ovarian Cancer Patient Samples. Gynecol. Oncol. 2021, 161, 769–778. [Google Scholar] [CrossRef]
- Creasman, W.J. Announcement, FIGO Stages 1988, Revisions. Gynecol. Oncol. 1989, 35, 125–127. [Google Scholar]
- Mehra, P.; Aditi, S.; Prasad, K.M.; Bariar, N.K. Histomorphological Analysis of Ovarian Neoplasms According to the 2020 WHO Classification of Ovarian Tumors: A Distribution Pattern in a Tertiary Care Center. Cureus 2023, 15, e38273. [Google Scholar] [CrossRef]
- Miller, A.B.; Hoogstraten, B.; Staquet, M.; Winkler, A. Reporting Results of Cancer Treatment. Cancer 1981, 47, 207–214. [Google Scholar] [CrossRef]
- Woroniecka, R.; Rymkiewicz, G.; Szafron, L.M.; Blachnio, K.; Szafron, L.A.; Bystydzienski, Z.; Pienkowska-Grela, B.; Borkowska, K.; Rygier, J.; Kotyl, A.; et al. Cryptic MYC Insertions in Burkitt Lymphoma: New Data and a Review of the Literature. PLoS ONE 2022, 17, e0263980. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Calculated Consequences. Available online: https://www.ensembl.org/info/genome/variation/prediction/predicted_data.html (accessed on 3 July 2021).
- Gerds, T.A.; Cai, T.; Schumacher, M. The Performance of Risk Prediction Models. Biom. J. 2008, 50, 457–479. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 44-GENE PANEL | ||||||
| Impact HIGH or MODERATE | ||||||
| Group Comparison and p-Value | ||||||
| Gene | BOT vs. BOT.V600E | BOT vs. lgOvCa | BOT vs. hgOvCa | BOT.V600E vs. lgOvCa | BOT.V600E vs. hgOvCa | lgOvCa vs. hgOvCa |
| TP53 | 5.67 × 10−31 (↑hgOvCa) | 1.23 × 10−18 (↑hgOvCa) | 1.8 × 10−9 (↑hgOvCa) | |||
| FANCB | 9.71 × 10−3 (↑BOT) | |||||
| SEM1 | 2.51 × 10−2 (↑BOT) | 1.01 × 10−2 (↑BOT) | ||||
| FANCA | 2.61 × 10−2 (↑BOT) | |||||
| FANCD2 | 4.97 × 10−2 (↑hgOvCa) | 1.52 × 10−2 (↑hgOvCa) | ||||
| BRCA2 | 1.47 × 10−2 (↑BOT) | |||||
| CHEK2 | 1.04 × 10−2 (↑BOT) | |||||
| MUTYH | 1.44 × 10−2 (↑BOT) | |||||
| RAD50 | 2.83 × 10−2 (↑BOT) | |||||
| Impact MODERATE | ||||||
| Group Comparison and p-Value | ||||||
| Gene | BOT vs. BOT.V600E | BOT vs. lgOvCa | BOT vs. hgOvCa | BOT.V600E vs. lgOvCa | BOT.V600E vs. hgOvCa | lgOvCa vs. hgOvCa |
| TP53 | 3.48 × 10−14 (↑hgOvCa) | 6.97 × 10−9 (↑hgOvCa) | 1.64 × 10−4 (↑hgOvCa) | |||
| BRCA1 | 2.76 × 10−2 (↑BOT) | |||||
| FANCB | 9.71 × 10−3 (↑BOT) | |||||
| SEM1 | 2.51 × 10−2 (↑BOT) | 1.01 × 10−2 (↑BOT) | ||||
| MUTYH | 3.8 × 10−2 (↑BOT) | |||||
| BRCA2 | 3.83 × 10−3 (↑BOT) | |||||
| CHEK2 | 5.94 × 10−3 (↑BOT) | |||||
| FANCA | 2.61 × 10−2 (↑BOT) | |||||
| FANCD2 | 4.97 × 10−2 (↑hgOvCa) | 1.52 × 10−2 (↑hgOvCa) | ||||
| RAD50 | 2.83 × 10−2 (↑BOT) | |||||
| PALB2 | 4.31 × 10−2 (↑BOT) | |||||
| ATM | 3.62 × 10−2 (↑lgOvCa) | |||||
| Impact HIGH | ||||||
| Group Comparison and p-Value | ||||||
| Gene | BOT vs. BOT.V600E | BOT vs. lgOvCa | BOT vs. hgOvCa | BOT.V600E vs. lgOvCa | BOT.V600E vs. hgOvCa | lgOvCa vs. hgOvCa |
| TP53 | 1.25 × 10−8 (↑hgOvCa) | 1.47 × 10−4 (↑hgOvCa) | 3.08 × 10−2 (↑hgOvCa) | |||
| BRCA1 | 1.25 × 10−7 (↑hgOvCa) | 6.01 × 10−4 (↑hgOvCa) | 3.4 × 10−2 (↑hgOvCa) | |||
| HOT-SPOT PANEL | ||||||
| Impact HIGH or MODERATE | ||||||
| Group Comparison and p-Value | ||||||
| Gene | BOT vs. BOT.V600E | BOT vs. lgOvCa | BOT vs. hgOvCa | BOT.V600E vs. lgOvCa | BOT.V600E vs. hgOvCa | lgOvCa vs. hgOvCa |
| TP53 | 1.01 × 10−29 (↑hgOvCa) | 7.62 × 10−18 (↑hgOvCa) | 2.35 × 10−7 (↑hgOvCa) | |||
| BRAF | 1.52 × 10−16 (↑BOT.V600E) | 1.08 × 10−8 (↑BOT.V600E) | 1.08 × 10−23 (↑BOT.V600E) | |||
| NRAS | 1.1 × 10−2 (↑lgOvCa) | 2.2 × 10−2 (↑lgOvCa) | 2.22 × 10−4 (↑lgOvCa) | |||
| BRCA1 | 1.08 × 10−4 (↑hgOvCa) | |||||
| FBXW7 | 3.67 × 10−2 (↑hgOvCa) | |||||
| KRAS | 6.44 × 10−5 (↑BOT) | 2.58 × 10−10 (↑BOT) | 5.13 × 10−3 (↑lgOvCa) | 2.77 × 10−3 (↑lgOvCa) | ||
| Impact MODERATE | ||||||
| Group Comparison and p-Value | ||||||
| Gene | BOT vs. BOT.V600E | BOT vs. lgOvCa | BOT vs. hgOvCa | BOT.V600E vs. lgOvCa | BOT.V600E vs. hgOvCa | lgOvCa vs. hgOvCa |
| TP53 | 2.27 × 10−15 (↑hgOvCa) | 1.66 × 10−9 (↑hgOvCa) | 8.26 × 10−5 (↑hgOvCa) | |||
| BRAF | 1.52 × 10−16 (↑BOT.V600E) | 1.08 × 10−8 (↑BOT.V600E) | 1.08 × 10−24 (↑BOT.V600E) | |||
| NRAS | 1.1 × 10−2 (↑lgOvCa) | 2.2 × 10−2 (↑lgOvCa) | 2.22 × 10−4 (↑lgOvCa) | |||
| KRAS | 6.44 × 10−5 (↑BOT) | 1.41 × 10−11 (↑BOT) | 5.13 × 10−3 (↑lgOvCa) | 1.11 × 10−3 (↑lgOvCa) | ||
| Impact HIGH | ||||||
| Group Comparison and p-Value | ||||||
| Gene | BOT vs. BOT.V600E | BOT vs. lgOvCa | BOT vs. hgOvCa | BOT.V600E vs. lgOvCa | BOT.V600E vs. hgOvCa | lgOvCa vs. hgOvCa |
| TP53 | 5.84 × 10−9 (↑hgOvCa) | 1.35 × 10−4 (↑hgOvCa) | ||||
| BRCA1 | 3.98 × 10−3 (↑hgOvCa) | |||||
| 44-Gene Panel | ||
| BOTS | ||
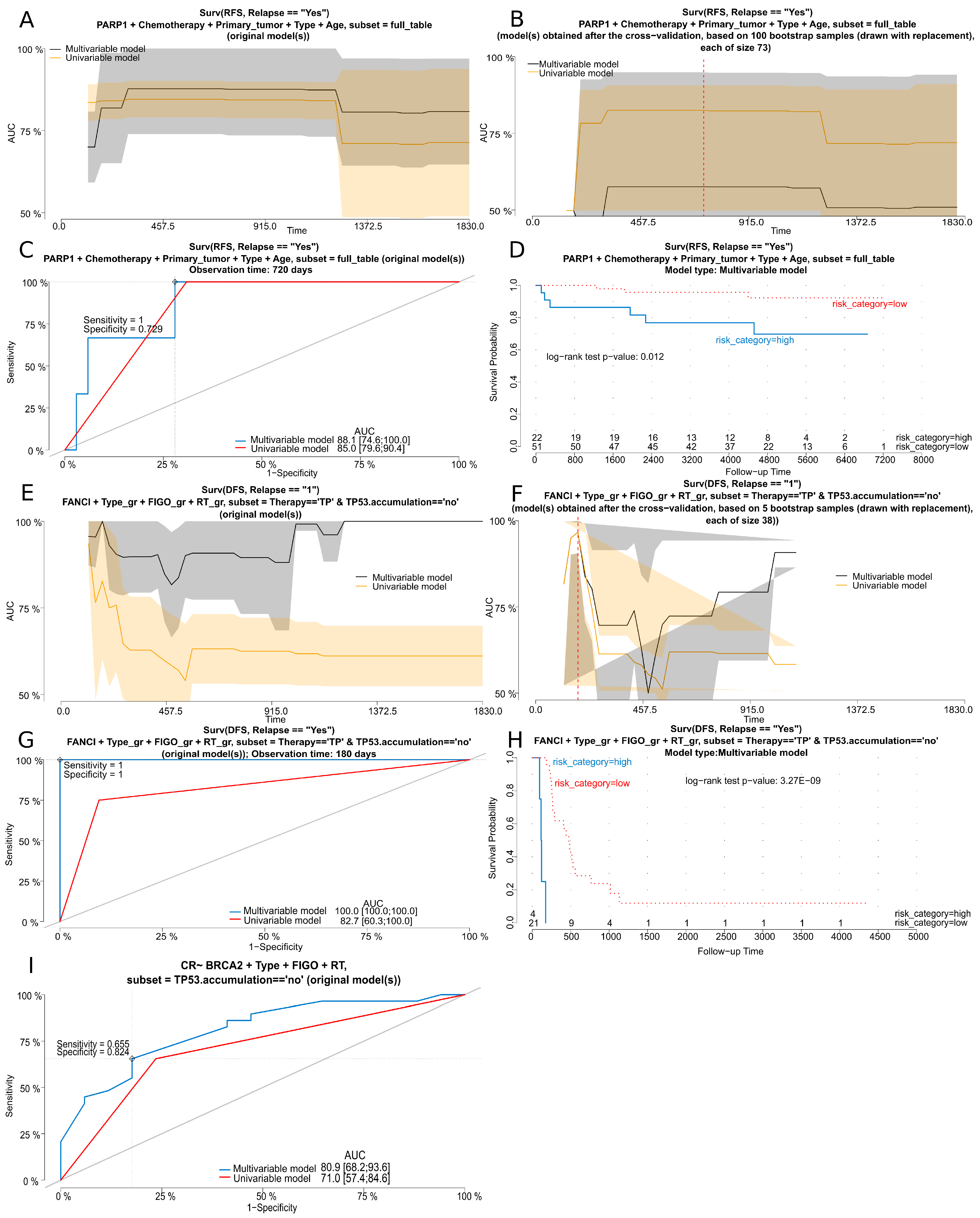
| RFS/relapse in the whole group of patients (full table) | HR [95% Cl] | p-value |
| PARP1 | 6.82 [1.584–29.39] | 0.01 |
| hgOvCa | ||
| DFS/relapse in the subgroup of patients treated with TP and without TP53 accumulation in tumors | HR [95% Cl] | p-value |
| FANCI | 40.02 [3.784–423.133] | 0.0022 |
| Residual tumor <2 cm vs. no residual tumor (0 cm) | 22.77 [2.061–251.608] | 0.01 |
| Residual tumor ≥2 cm vs. no residual tumor (0 cm) | 34.1 [2.547–456.619] | 0.0077 |
| CR in the subgroup of tumors without TP53 accumulation | OR [95% Cl] | p-value |
| BRCA2 | 7.06 [1.328–37.581] | 0.022 |
| OS/death in the whole group of patients (full table) | HR [95% Cl] | p-value |
| BRCA2 | 0.58 [0.399–0.85] | 0.005 |
| Residual tumor <2 cm vs. no residual tumor (0 cm) | 2.85 [1.654–4.903] | 1.6 × 10−4 |
| Residual tumor ≥2 cm vs. no residual tumor (0 cm) | 3.75 [2.058–6.821] | 1.55 × 10−5 |
| OS/death in patients with tumors without TP53 accumulation | HR [95% Cl] | p-value |
| BRCA2 | 0.42 [0.204–0.865] | 0.019 |
| Residual tumor ≥2 cm vs. no residual tumor (0 cm) | 4.63 [1.348–15.883] | 0.015 |
| OS/death in the subgroup of patients treated with TP | HR [95% Cl] | p-value |
| BRCA2 | 0.53 [0.337–0.84] | 0.007 |
| Residual tumor <2 cm vs. no residual tumor (0 cm) | 2.97 [1.616–5.471] | 4.6 × 10−4 |
| Residual tumor ≥2 cm vs. no residual tumor (0 cm) | 3.94 [1.944–7.986] | 1.4 × 10−4 |
| CR in the subgroup of patients treated with TP and without TP53 accumulation in tumors | OR [95% Cl] | p-value |
| BRCA2 | 6.73 [1.047–43.239] | 0.045 |
| PS in the subgroup of tumors without TP53 accumulation | OR [95% Cl] | p-value |
| BRCA2 | 8.23 [1.509–44.836] | 0.015 |
| PS in the subgroup of patients treated with TP and without TP53 accumulation in tumors | OR [95% Cl] | p-value |
| BRCA2 | 8.33 [1.251–55.476] | 0.028 |
| OS/death in the subgroup of patients treated with TP and without TP53 accumulation in tumors | HR [95% Cl] | p-value |
| FANCF | 0.15 [0.024–0.976] | 0.047 |
| Residual tumor <2 cm vs. no residual tumor (0 cm) | 3.69 [1.159–11.74] | 0.027 |
| Residual tumor ≥2 cm vs. no residual tumor (0 cm) | 7.75 [1.84–32.595] | 0.005 |
| Hot-Spot Panel | ||
| hgOvCa | ||
| OS/death in the subgroup of patients treated with TP and with TP53 accumulation in tumors | HR [95% Cl] | p-value |
| TSC2 | 2.52 [1.191–5.329] | 0.016 |
| Residual tumor <2 cm vs. no residual tumor (0 cm) | 2.86 [1.312–6.249] | 0.008 |
| Residual tumor ≥2 cm vs. no residual tumor (0 cm) | 2.61 [1.104–6.146] | 0.029 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szafron, L.A.; Sobiczewski, P.; Dansonka-Mieszkowska, A.; Kupryjanczyk, J.; Szafron, L.M. An Analysis of Genetic Polymorphisms in 76 Genes Related to the Development of Ovarian Tumors of Different Aggressiveness. Int. J. Mol. Sci. 2024, 25, 10876. https://doi.org/10.3390/ijms252010876
Szafron LA, Sobiczewski P, Dansonka-Mieszkowska A, Kupryjanczyk J, Szafron LM. An Analysis of Genetic Polymorphisms in 76 Genes Related to the Development of Ovarian Tumors of Different Aggressiveness. International Journal of Molecular Sciences. 2024; 25(20):10876. https://doi.org/10.3390/ijms252010876
Chicago/Turabian StyleSzafron, Laura A., Piotr Sobiczewski, Agnieszka Dansonka-Mieszkowska, Jolanta Kupryjanczyk, and Lukasz M. Szafron. 2024. "An Analysis of Genetic Polymorphisms in 76 Genes Related to the Development of Ovarian Tumors of Different Aggressiveness" International Journal of Molecular Sciences 25, no. 20: 10876. https://doi.org/10.3390/ijms252010876