
Myeloid GSK3α Deficiency Reduces Lesional Inflammation and Neovascularization during Atherosclerotic Progression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
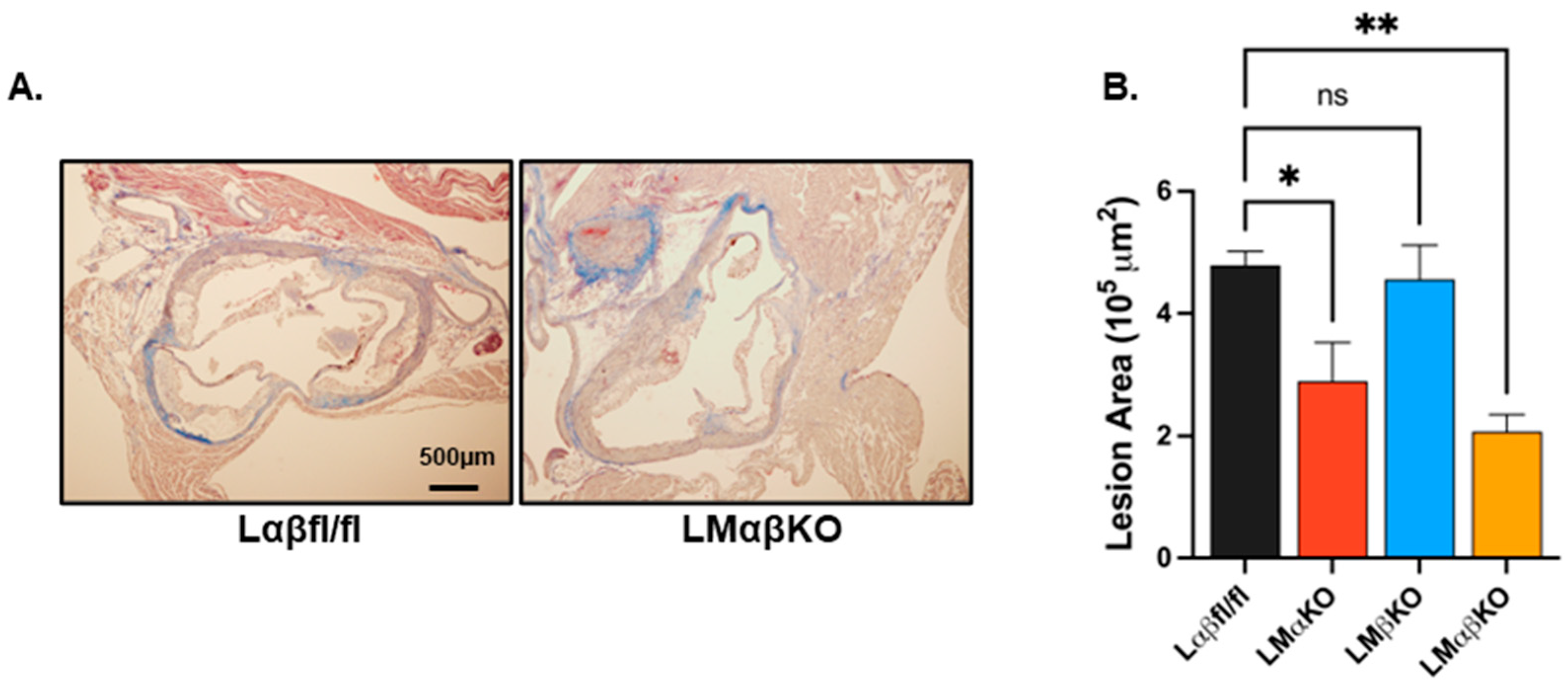
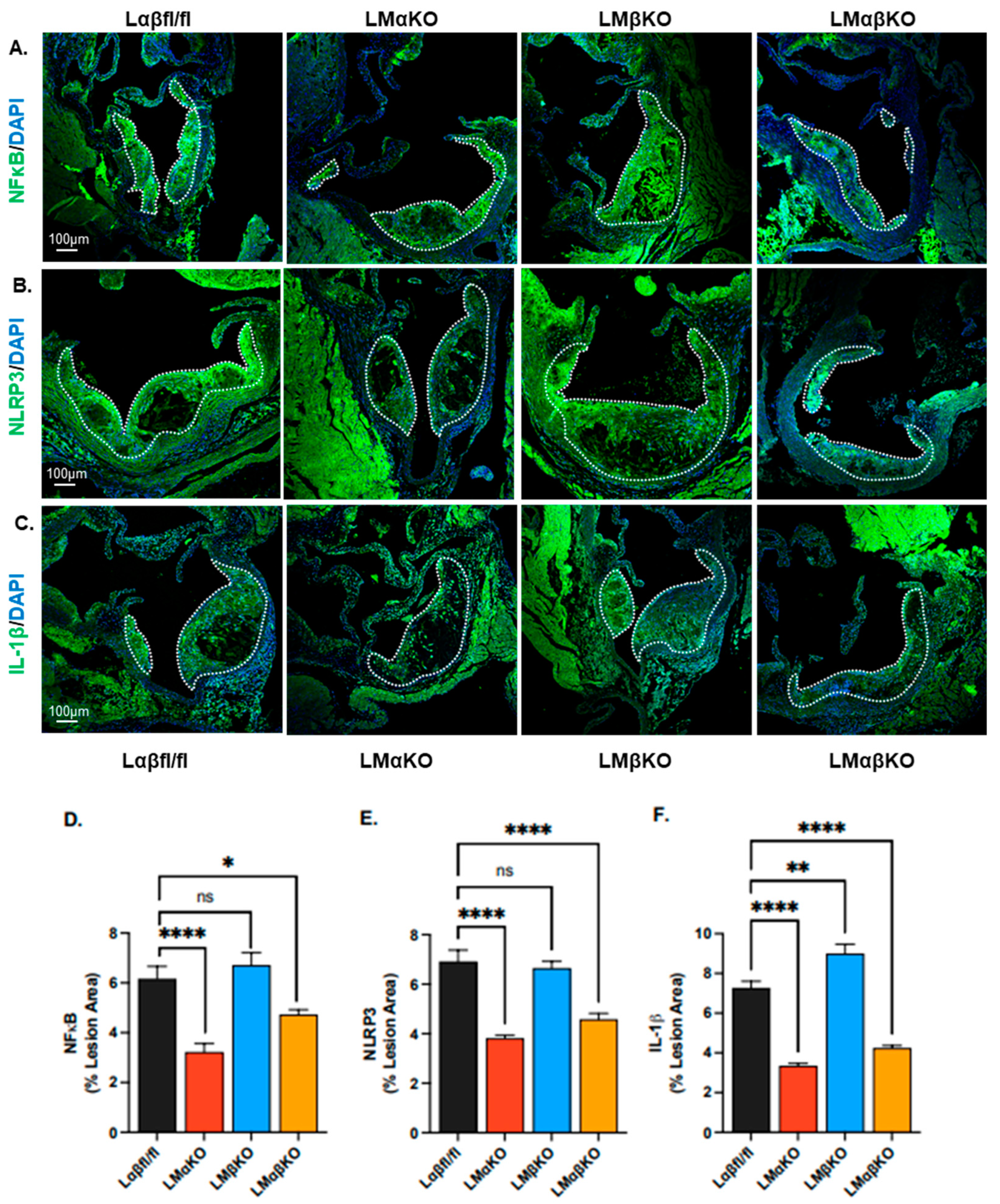
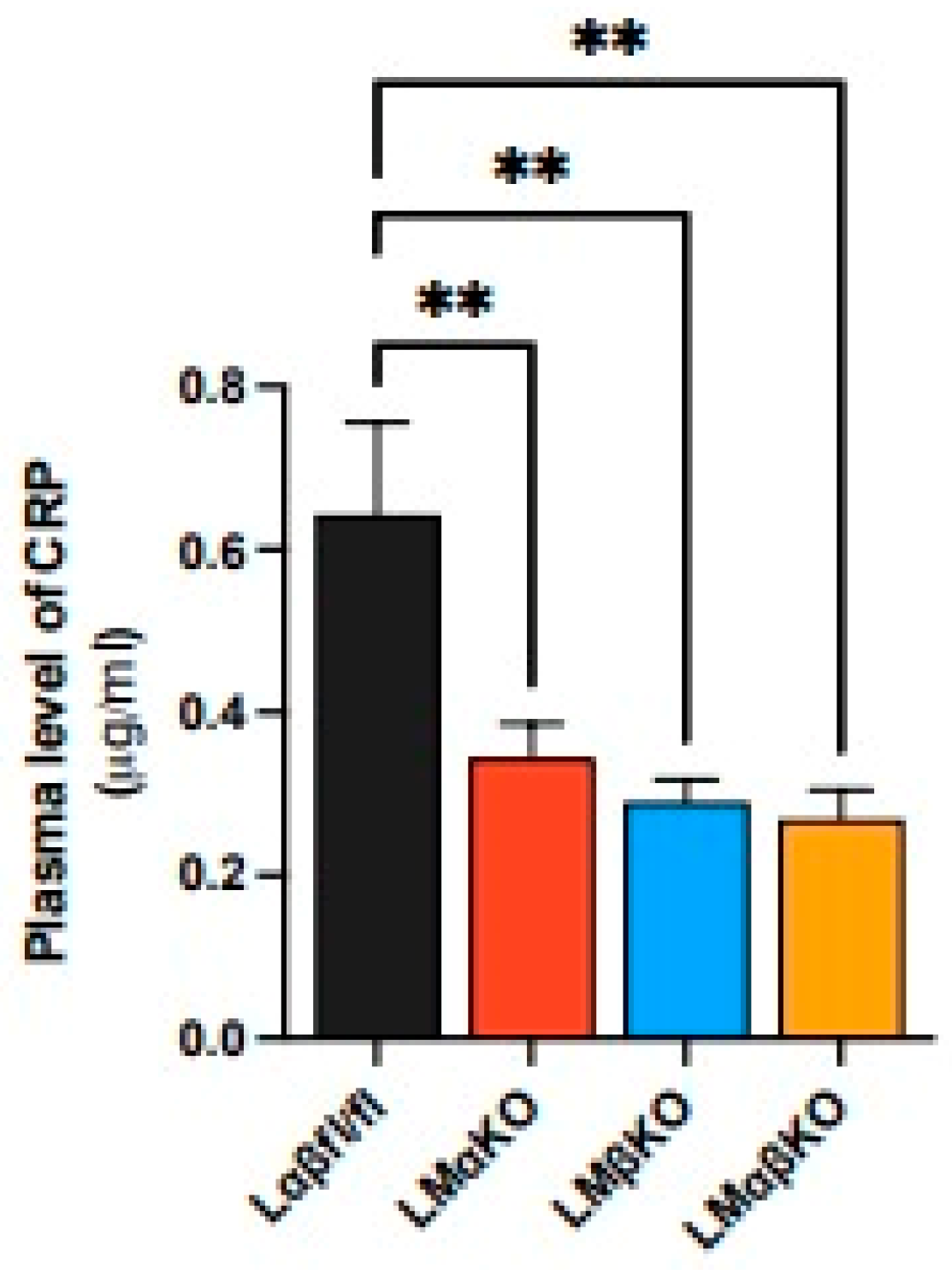
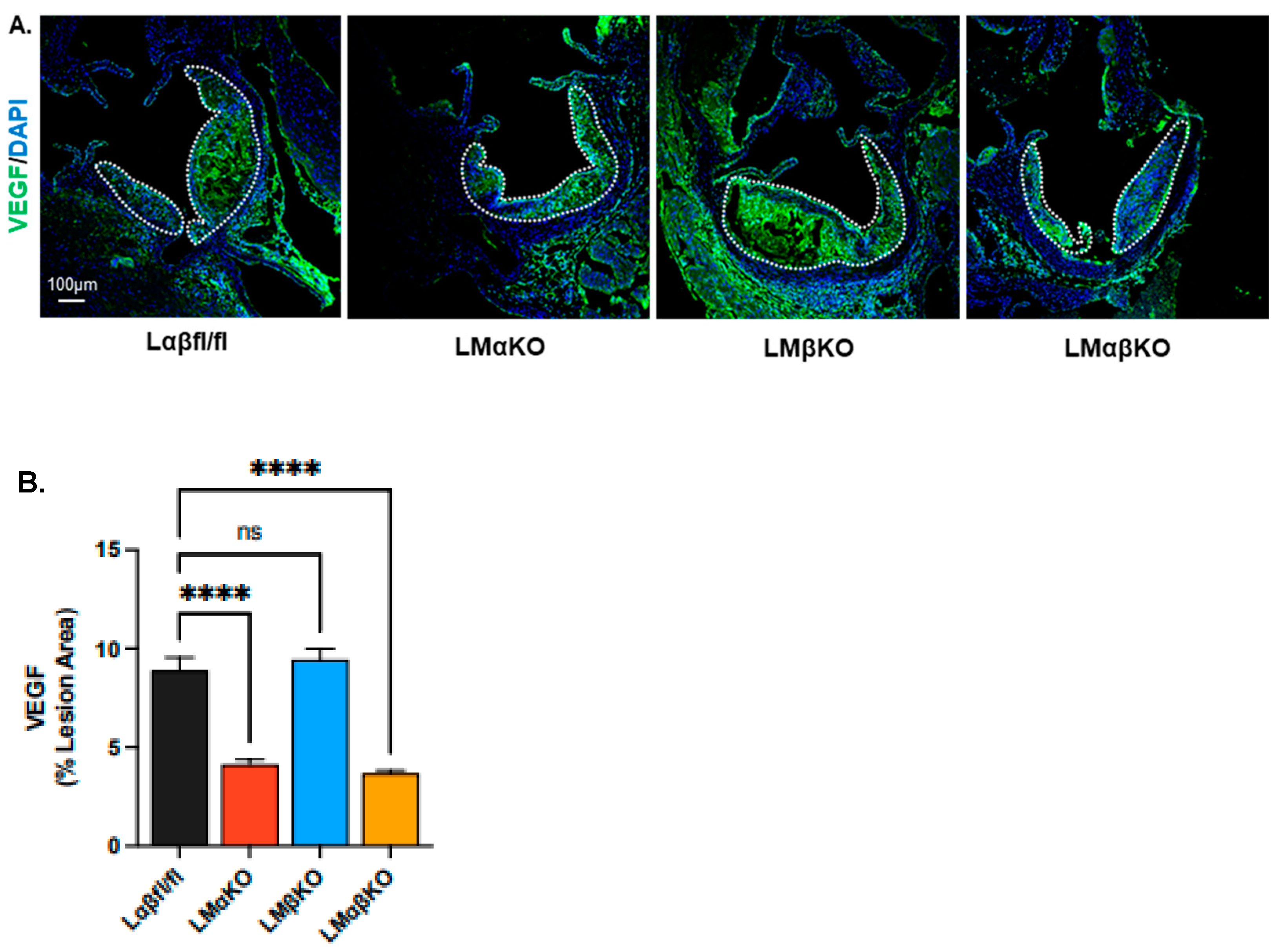
2.1. Myeloid-Specific GSK3α Deficiency Reduces Inflammation and Attenuates Atherosclerosis
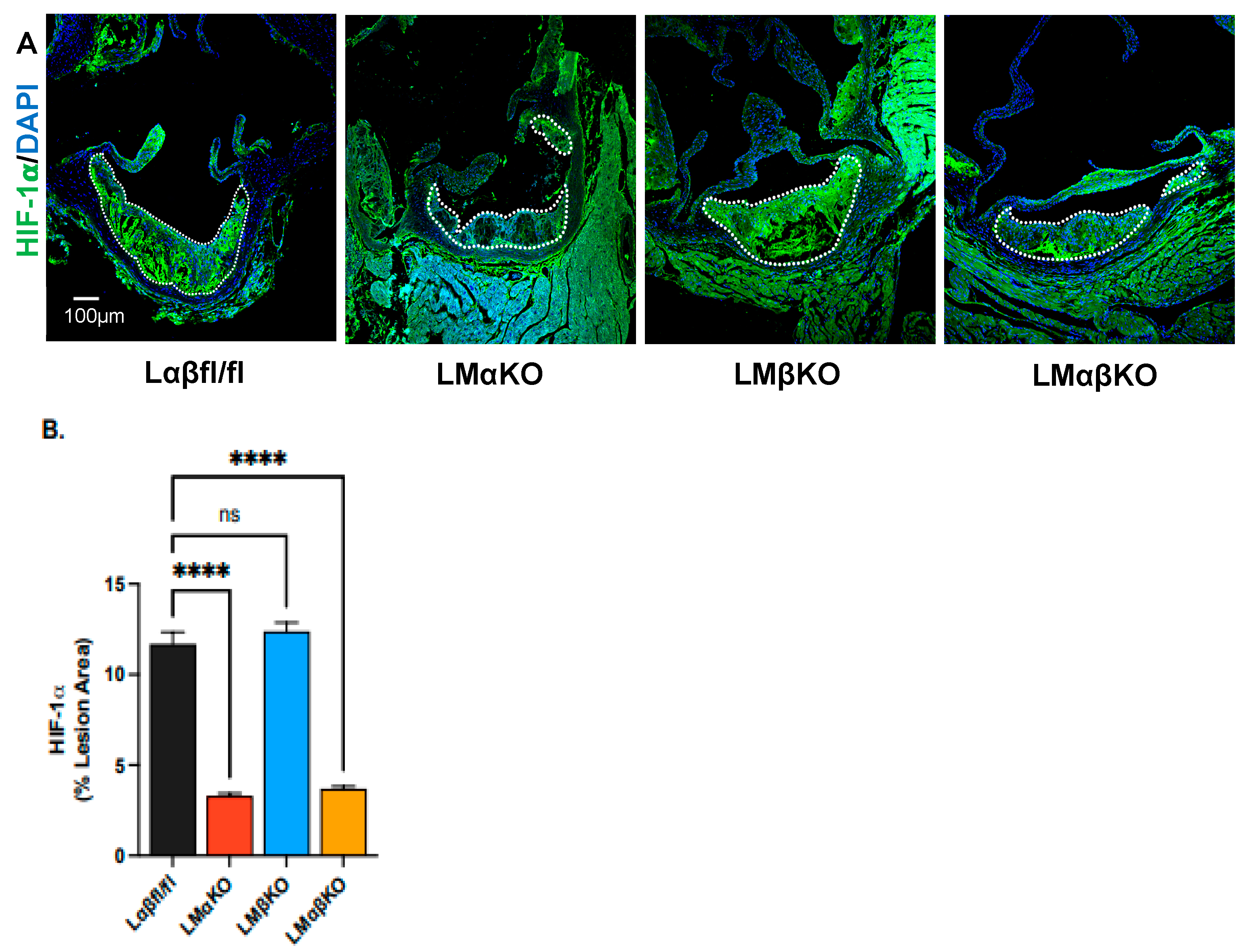
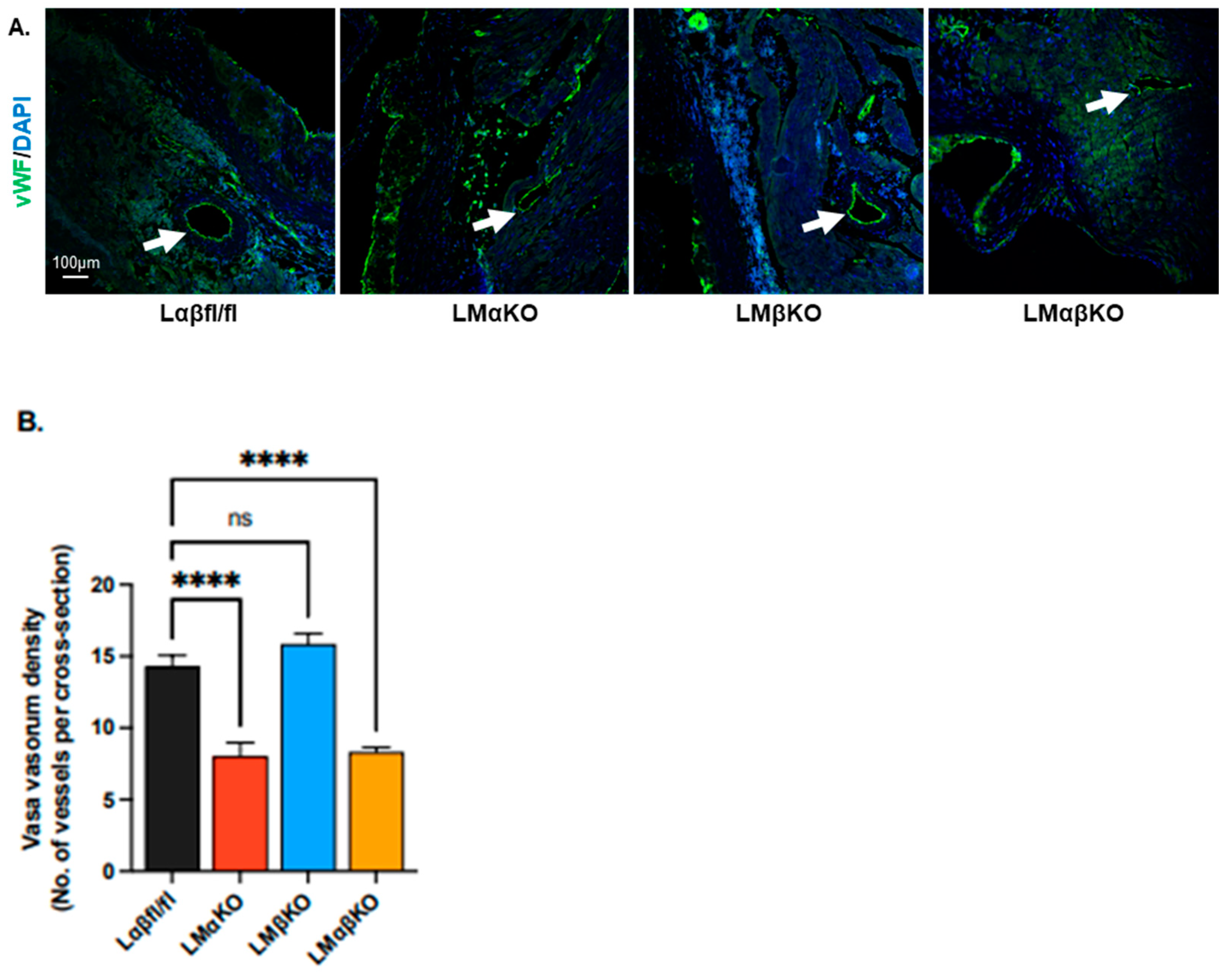
2.2. Myeloid-Specific GSK3α Deficiency Reduces Vasa Vasorum Density
3. Discussion
4. Materials and Methods
4.1. Mouse Models
4.2. Atherosclerotic Progression Model
4.3. Characterization of Aortic Lesions
4.4. Determination of Plasma CRP
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, A.; Patel, S.; McAlpine, C.; Werstuck, G. The role of endoplasmic reticulum stress-glycogen synthase kinase-3 signaling in atherogenesis. Int. J. Mol. Sci. 2018, 19, 1607. [Google Scholar] [CrossRef] [PubMed]
- Insull, W. The pathology of atherosclerosis: Plaque development and plaque responses to medical treatment. Am. J. Med. 2009, 122, S3–S14. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 1–18. [Google Scholar] [CrossRef]
- Gui, T.; Shimokado, A.; Sun, Y.; Akasaka, T.; Muragaki, Y. Diverse roles of macrophages in atherosclerosis: From inflammatory biology to Biomarker Discovery. Mediat. Inflamm. 2012, 2012, 693083. [Google Scholar] [CrossRef]
- Zmysłowski, A.; Szterk, A. Current knowledge on the mechanism of atherosclerosis and pro-atherosclerotic properties of oxysterols. Lipids Health Dis. 2017, 16, 188. [Google Scholar] [CrossRef]
- Bobryshev, Y.V.; Ivanova, E.A.; Chistiakov, D.A.; Nikiforov, N.G.; Orekhov, A.N. Macrophages and their role in atherosclerosis: Pathophysiology and transcriptome analysis. BioMed Res. Int. 2016, 2016, 9582430. [Google Scholar] [CrossRef]
- Patel, S.; Werstuck, G.H. Macrophage function and the role of GSK3. Int. J. Mol. Sci. 2022, 22, 2206. [Google Scholar] [CrossRef]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Bergheanu, S.C.; Bodde, M.C.; Jukema, J.W. Pathophysiology and treatment of atherosclerosis. Neth. Heart J. 2017, 25, 231–242. [Google Scholar] [CrossRef]
- McAlpine, C.S.; Bowes, A.J.; Khan, M.I.; Shi, Y.; Werstuck, G.H. Endoplasmic reticulum stress and glycogen synthase kinase-3β activation in apolipoprotein E-deficient mouse models of accelerated atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen Synthase Kinase-3 (GSK3): Inflammation, Diseases, and Therapeutics. Neurochem. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, J.; Di, L. Glycogen synthesis and beyond, a comprehensive review of GSK3 as a key regulator of metabolic pathways and a therapeutic target for treating metabolic diseases. Med. Res. Rev. 2022, 42, 946–982. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB signaling in inflammation. Signal Transduct. Target. Ther. 2021, 2, 17023. [Google Scholar] [CrossRef]
- Vitiello, L.; Spoletini, I.; Gorini, S.; Pontecorvo, L.; Ferrari, D.; Ferraro, E.; Stabile, E.; Caprio, M.; la Sala, A. Microvascular inflammation in atherosclerosis. IJC Metab. Endocr. 2014, 3, 1–7. [Google Scholar] [CrossRef]
- Hoffmeister, L.; Diekmann, M.; Brand, K.; Huber, R. GSK3, A Kinase Balancing Promotion and Resolution of Inflammation. Cells 2020, 9, 820. [Google Scholar] [CrossRef]
- Li, J.; Li, S.X.; Gao, X.H.; Zhao, L.F.; Du, J.; Wang, T.Y.; Wang, L.; Zhang, J.; Wang, H.Y.; Dong, R.; et al. HIF1A and VEGF regulate each other by competing endogenous RNA mechanism and involve in the pathogenesis of peritoneal fibrosis. Pathol.-Res. Pract. 2019, 215, 644–652. [Google Scholar] [CrossRef]
- Camaré, C.; Pucelle, M.; Nègre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef]
- Hippenstiel, S.; Krüll, M.; Ikemann, A.; Risau, W.; Clauss, M.; Suttorp, N. VEGF induces hyperpermeability by a direct action on endothelial cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1998, 274, L678–L684. [Google Scholar] [CrossRef]
- Barleon, B.; Sozzani, S.; Zhou, D.; Weich, H.A.; Mantovani, A.; Marme, D. Migration of Human Monocytes in Response to Vascular Endothelial Growth Factor (VEGF) Is Mediated via the VEGF Receptor flt-1. Blood 1996, 87, 3336–3343. [Google Scholar] [CrossRef]
- Wang, X.; Feuerstein, G.Z.; Gu, J.-L.; Lysko, P.G.; Yue, T.-L. Interleukin-1β induces expression of adhesion molecules in human vascular smooth muscle cells and enhances adhesion of leukocytes to smooth muscle cells. Atherosclerosis 1995, 115, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Skurk, C.; Thomas, S.R.; Bialik, A.; Suhara, T.; Kureishi, Y.; Birnbaum, M.; Keaney, J.F.; Walsh, K. Regulation of angiogenesis by glycogen synthase kinase-3β. J. Biol. Chem. 2002, 277, 41888–41896. [Google Scholar] [CrossRef] [PubMed]
- Skurk, C.; Maatz, H.; Rocnik, E.; Bialik, A.; Force, T.; Walsh, K. Glycogen-synthase Kinase3β/β-catenin axis promotes angiogenesis through activation of vascular endothelial growth factor signaling in endothelial cells. Circ. Res. 2005, 96, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Phukan, S.; Babu, V.; Kannoji, A.; Hariharan, R.; Balaji, V. GSK3β: Role in therapeutic landscape and development of modulators. Br. J. Pharmacol. 2010, 160, 1–19. [Google Scholar] [CrossRef]
- Murdaca, G.; Colombo, B.M.; Puppo, F. The role of Th17 lymphocytes in the autoimmune and chronic inflammatory diseases. Intern. Emerg. Med. 2011, 6, 487–495. [Google Scholar] [CrossRef]
- Patel, S.; Werstuck, G. Characterizing the Role of Glycogen Synthase Kinase-3α/β in Macrophage Polarization and the Regulation of Pro-Atherogenic Pathways in Cultured Ldlr−/− Macrophages. Front. Immunol. 2021, 12, 676752. [Google Scholar] [CrossRef]
- Venegas-Pino, D.E.; Banko, N.; Khan, M.I.; Shi, Y.; Werstuck, G.H. Quantitative analysis and characterization of atherosclerotic lesions in the murine aortic sinus. J. Vis. Exp. 2013, 82, 50933. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, S.; Shah, N.; D’Mello, B.; Lee, A.; Werstuck, G.H. Myeloid GSK3α Deficiency Reduces Lesional Inflammation and Neovascularization during Atherosclerotic Progression. Int. J. Mol. Sci. 2024, 25, 10897. https://doi.org/10.3390/ijms252010897
Patel S, Shah N, D’Mello B, Lee A, Werstuck GH. Myeloid GSK3α Deficiency Reduces Lesional Inflammation and Neovascularization during Atherosclerotic Progression. International Journal of Molecular Sciences. 2024; 25(20):10897. https://doi.org/10.3390/ijms252010897
Chicago/Turabian StylePatel, Sarvatit, Nisarg Shah, Brooke D’Mello, Anson Lee, and Geoff H. Werstuck. 2024. "Myeloid GSK3α Deficiency Reduces Lesional Inflammation and Neovascularization during Atherosclerotic Progression" International Journal of Molecular Sciences 25, no. 20: 10897. https://doi.org/10.3390/ijms252010897