
A Review of Biomarkers of Amyotrophic Lateral Sclerosis: A Pathophysiologic Approach

 , ,
, ,
Abstract
:1. Introduction
2. Pathophysiology of Amyotrophic Lateral Sclerosis
3. Biomarkers in Amyotrophic Lateral Sclerosis
3.1. Biomarkers Related to Proteinopathy
3.2. Biomarkers Related to Neurodegeneration
3.3. Biomarkers Related to Neuroinflammation
3.4. Biomarkers Related to Blood–Brain Barrier Disruption
3.5. Biomarkers Related to Syanptopathy
3.6. Biomarkers Related to Oxidative Stress
3.7. Biomarkers Related to Aberrant RNA Processing
3.8. Biomarkers Related to Muscle Changes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Talbott, E.O.; Malek, A.M.; Lacomis, D. Chapter 13—The Epidemiology of Amyotrophic Lateral Sclerosis. In Handbook of Clinical Neurology; Aminoff, M.J., Boller, F., Swaab, D.F., Eds.; Neuroepidemiology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 138, pp. 225–238. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Phukan, J.; Elamin, M.; Bede, P.; Jordan, N.; Gallagher, L.; Byrne, S.; Lynch, C.; Pender, N.; Hardiman, O. The Syndrome of Cognitive Impairment in Amyotrophic Lateral Sclerosis: A Population-Based Study. J. Neurol. Neurosurg. Psychiatry 2012, 83, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Barnwell, J.; Al-Chalabi, A.; Eisen, A. Young-Onset Amyotrophic Lateral Sclerosis: Historical and Other Observations. Brain 2012, 135 Pt 9, 2883–2891. [Google Scholar] [CrossRef] [PubMed]
- Goutman, S.A.; Chen, K.S.; Paez-Colasante, X.; Feldman, E.L. Emerging Understanding of the Genotype-Phenotype Relationship in Amyotrophic Lateral Sclerosis. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 148, pp. 603–623. [Google Scholar] [CrossRef]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel Genes Associated with Amyotrophic Lateral Sclerosis: Diagnostic and Clinical Implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic Lateral Sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef]
- Lanznaster, D.; Hergesheimer, R.C.; Bakkouche, S.E.; Beltran, S.; Vourc’h, P.; Andres, C.R.; Dufour-Rainfray, D.; Corcia, P.; Blasco, H. Aβ1-42 and Tau as Potential Biomarkers for Diagnosis and Prognosis of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 2911. [Google Scholar] [CrossRef]
- Klein, L.M.; Forshew, D.A. The Economic Impact of ALS. Neurology 1996, 47 (Suppl. 2), S126–S129. [Google Scholar] [CrossRef]
- Humpert, P.M.; Kopf, S.; Djuric, Z.; Laine, K.; Korosoglou, G.; Rudofsky, G.; Hamann, A.; Morcos, M.; von Eynatten, M.; Nawroth, P.P.; et al. Levels of Three Distinct P75 Neurotrophin Receptor Forms Found in Human Plasma Are Altered in Type 2 Diabetic Patients. Diabetologia 2007, 50, 1517–1522. [Google Scholar] [CrossRef]
- Pugdahl, K.; Camdessanché, J.-P.; Cengiz, B.; de Carvalho, M.; Liguori, R.; Rossatto, C.; Santos, M.O.; Vacchiano, V.; Johnsen, B. Gold Coast Diagnostic Criteria Increase Sensitivity in Amyotrophic Lateral Sclerosis. Clin. Neurophysiol. 2021, 132, 3183–3189. [Google Scholar] [CrossRef]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected Increase in Amyotrophic Lateral Sclerosis from 2015 to 2040. Nat Commun 2016, 7, 12408. [Google Scholar] [CrossRef]
- Miller, R.G.; Mitchell, J.D.; Moore, D.H. Riluzole for Amyotrophic Lateral Sclerosis (ALS)/Motor Neuron Disease (MND). Cochrane Database Syst. Rev. 2012, 65, CD001447. [Google Scholar] [CrossRef] [PubMed]
- Jack, J.; Chen, P. Overview of Current and Emerging Therapies for Amyotrophic Lateral Sclerosis. Am. J. Manag. Care 2020, 26, S191–S197. [Google Scholar] [CrossRef]
- Ketabforoush, A.H.M.E.; Chegini, R.; Barati, S.; Tahmasebi, F.; Moghisseh, B.; Joghataei, M.T.; Faghihi, F.; Azedi, F. The Promising Actor in the next Season of the Amyotrophic Lateral Sclerosis Treatment Series. Biomed. Pharmacother. 2023, 160, 114378. [Google Scholar] [CrossRef] [PubMed]
- Sironi, F.; De Marchi, F.; Mazzini, L.; Bendotti, C. Cell Therapy in ALS: An Update on Preclinical and Clinical Studies. Brain Res. Bull. 2023, 194, 64–81. [Google Scholar] [CrossRef]
- Cudkowicz, M.; Genge, A.; Maragakis, N.; Petri, S.; Berg, L.v.D.; Aho, V.V.; Sarapohja, T.; Kuoppamäki, M.; Garratt, C.; Al-Chalabi, A.; et al. Safety and Efficacy of Oral Levosimendan in People with Amyotrophic Lateral Sclerosis (the REFALS Study): A Randomised, Double-Blind, Placebo-Controlled Phase 3 Trial. Lancet Neurol. 2021, 20, 821–831. [Google Scholar] [CrossRef]
- Gendron, T.F.; Chew, J.; Stankowski, J.N.; Hayes, L.R.; Zhang, Y.-J.; Prudencio, M.; Carlomagno, Y.; Daughrity, L.M.; Jansen-West, K.; Perkerson, E.A.; et al. Poly(GP) Proteins Are a Useful Pharmacodynamic Marker for C9ORF72-Associated Amyotrophic Lateral Sclerosis. Sci. Transl. Med. 2017, 9, eaai7866. [Google Scholar] [CrossRef]
- Katzeff, J.S.; Bright, F.; Phan, K.; Kril, J.J.; Ittner, L.M.; Kassiou, M.; Hodges, J.R.; Piguet, O.; Kiernan, M.C.; Halliday, G.M.; et al. Biomarker Discovery and Development for Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Brain 2022, 145, 1598–1609. [Google Scholar] [CrossRef]
- Sproviero, D.; La Salvia, S.; Giannini, M.; Crippa, V.; Gagliardi, S.; Bernuzzi, S.; Diamanti, L.; Ceroni, M.; Pansarasa, O.; Poletti, A.; et al. Pathological Proteins Are Transported by Extracellular Vesicles of Sporadic Amyotrophic Lateral Sclerosis Patients. Front. Neurosci. 2018, 12, 487. [Google Scholar] [CrossRef]
- Beyer, L.; Günther, R.; Koch, J.C.; Klebe, S.; Hagenacker, T.; Lingor, P.; Biesalski, A.; Hermann, A.; Nabers, A.; Gold, R.; et al. TDP-43 as Structure-Based Biomarker in Amyotrophic Lateral Sclerosis. Ann. Clin. Transl. Neurol. 2021, 8, 271–277. [Google Scholar] [CrossRef]
- Pagliardini, V.; Pagliardini, S.; Corrado, L.; Lucenti, A.; Panigati, L.; Bersano, E.; Servo, S.; Cantello, R.; D’Alfonso, S.; Mazzini, L. Chitotriosidase and Lysosomal Enzymes as Potential Biomarkers of Disease Progression in Amyotrophic Lateral Sclerosis: A Survey Clinic-Based Study. J. Neurol. Sci. 2015, 348, 245–250. [Google Scholar] [CrossRef]
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chió, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Emerging Insights into the Complex Genetics and Pathophysiology of Amyotrophic Lateral Sclerosis. Lancet Neurol. 2022, 21, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Cordts, I.; Wachinger, A.; Scialo, C.; Lingor, P.; Polymenidou, M.; Buratti, E.; Feneberg, E. TDP-43 Proteinopathy Specific Biomarker Development. Cells 2023, 12, 597. [Google Scholar] [CrossRef] [PubMed]
- Jeon, G.S.; Shim, Y.-M.; Lee, D.-Y.; Kim, J.-S.; Kang, M.; Ahn, S.H.; Shin, J.-Y.; Geum, D.; Hong, Y.H.; Sung, J.-J. Pathological Modification of TDP-43 in Amyotrophic Lateral Sclerosis with SOD1 Mutations. Mol. Neurobiol. 2019, 56, 2007–2021. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, W.; Philips, T. The Changing Scene of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef]
- Batra, G.; Jain, M.; Singh, R.S.; Sharma, A.R.; Singh, A.; Prakash, A.; Medhi, B. Novel Therapeutic Targets for Amyotrophic Lateral Sclerosis. Indian J. Pharmacol. 2019, 51, 418. [Google Scholar] [CrossRef]
- Gong, Y.H.; Parsadanian, A.S.; Andreeva, A.; Snider, W.D.; Elliott, J.L. Restricted Expression of G86R Cu/Zn Superoxide Dismutase in Astrocytes Results in Astrocytosis But Does Not Cause Motoneuron Degeneration. J. Neurosci. 2000, 20, 660–665. [Google Scholar] [CrossRef]
- Bosch, L.V.D.; Vandenberghe, W.; Klaassen, H.; Houtte, E.V.; Robberecht, W. Calcium-Permeable AMPA Receptors and Selective Vulnerability of Motor Neurons. J. Neurol. Sci. 2000, 180, 29–34. [Google Scholar] [CrossRef]
- Kato, S.; Takikawa, M.; Nakashima, K.; Hirano, A.; Cleveland, D.W.; Kusaka, H.; Shibata, N.; Kato, M.; Nakano, I.; Ohama, E. New Consensus Research on Neuropathological Aspects of Familial Amyotrophic Lateral Sclerosis with Superoxide Dismutase 1 (SOD1) Gene Mutations: Inclusions Containing SOD1 in Neurons and Astrocytes. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 163–184. [Google Scholar] [CrossRef]
- Campanari, M.L.; García-Ayllón, M.-S.; Ciura, S.; Sáez-Valero, J.; Kabashi, E. Neuromuscular Junction Impairment in Amyotrophic Lateral Sclerosis: Reassessing the Role of Acetylcholinesterase. Front. Mol. Neurosci. 2016, 9, 160. [Google Scholar] [CrossRef]
- Cappello, V.; Francolini, M. Neuromuscular Junction Dismantling in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2017, 18, 2092. [Google Scholar] [CrossRef]
- Benigni, M.; Ricci, C.; Jones, A.R.; Giannini, F.; Al-Chalabi, A.; Battistini, S. Identification of miRNAs as Potential Biomarkers in Cerebrospinal Fluid from Amyotrophic Lateral Sclerosis Patients. Neuromol Med. 2016, 18, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Bittner, S.; Moccia, M.; Warnecke, T.; Ruck, T. Editorial: Pathophysiologic Insights From Biomarker Studies in Neurological Disorders. Front. Neurol. 2020, 11, 151. [Google Scholar] [CrossRef] [PubMed]
- Hanin, A.; Lambrecq, V.; Denis, J.A.; Imbert-Bismut, F.; Rucheton, B.; Lamari, F.; Bonnefont-Rousselot, D.; Demeret, S.; Navarro, V. Cerebrospinal Fluid and Blood Biomarkers of Status Epilepticus. Epilepsia 2020, 61, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Smith, D.H.; Blennow, K. Biomarkers of Mild Traumatic Brain Injury in Cerebrospinal Fluid and Blood. Nat. Rev. Neurol. 2013, 9, 201–210. [Google Scholar] [CrossRef]
- Blennow, K.; Hampel, H.; Weiner, M.; Zetterberg, H. Cerebrospinal Fluid and Plasma Biomarkers in Alzheimer Disease. Nat. Rev. Neurol. 2010, 6, 131–144. [Google Scholar] [CrossRef]
- Brunkhorst, R.; Pfeilschifter, W.; Foerch, C. Astroglial Proteins as Diagnostic Markers of Acute Intracerebral Hemorrhage-Pathophysiological Background and Clinical Findings. Transl. Stroke Res. 2010, 1, 246–251. [Google Scholar] [CrossRef]
- Panio, A.; Cava, C.; D’Antona, S.; Bertoli, G.; Porro, D. Diagnostic Circulating miRNAs in Sporadic Amyotrophic Lateral Sclerosis. Front. Med. 2022, 9, 861960. [Google Scholar] [CrossRef]
- Xu, Z.; Henderson, R.D.; David, M.; McCombe, P.A. Neurofilaments as Biomarkers for Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0164625. [Google Scholar] [CrossRef]
- Irwin, K.E.; Sheth, U.; Wong, P.C.; Gendron, T.F. Fluid Biomarkers for Amyotrophic Lateral Sclerosis: A Review. Mol. Neurodegener. 2024, 19, 9. [Google Scholar] [CrossRef]
- Sechidis, K.; Papangelou, K.; Metcalfe, P.D.; Svensson, D.; Weatherall, J.; Brown, G. Distinguishing Prognostic and Predictive Biomarkers: An Information Theoretic Approach. Bioinformatics 2018, 34, 3365–3376. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Polymenidou, M.; Cleveland, D.W. The Seeds of Neurodegeneration: Prion-like Spreading in ALS. Cell 2011, 147, 498–508. [Google Scholar] [CrossRef] [PubMed]
- De Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M.; et al. TDP-43 Proteinopathies: A New Wave of Neurodegenerative Diseases. J. Neurol. Neurosurg. Psychiatry 2021, 92, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Suk, T.R.; Rousseaux, M.W.C. The Role of TDP-43 Mislocalization in Amyotrophic Lateral Sclerosis. Mol. Neurodegener. 2020, 15, 45. [Google Scholar] [CrossRef]
- Roczniak-Ferguson, A.; Ferguson, S.M. Pleiotropic Requirements for Human TDP-43 in the Regulation of Cell and Organelle Homeostasis. Life Sci. Alliance 2019, 2, e201900358. [Google Scholar] [CrossRef]
- Hergesheimer, R.C.; Chami, A.A.; de Assis, D.R.; Vourc’h, P.; Andres, C.R.; Corcia, P.; Lanznaster, D.; Blasco, H. The Debated Toxic Role of Aggregated TDP-43 in Amyo-trophic Lateral Sclerosis: A Resolution in Sight? Brain 2019, 142, 1176–1194. [Google Scholar] [CrossRef]
- Tamaki, Y.; Ross, J.P.; Alipour, P.; Castonguay, C.-É.; Li, B.; Catoire, H.; Rochefort, D.; Urushitani, M.; Takahashi, R.; Sonnen, J.A.; et al. Spinal Cord Extracts of Amyotrophic Lateral Sclerosis Spread TDP-43 Pathology in Cerebral Organoids. PLoS Genet. 2023, 19, e1010606. [Google Scholar] [CrossRef]
- Smethurst, P.; Newcombe, J.; Troakes, C.; Simone, R.; Chen, Y.-R.; Patani, R.; Sidle, K. In Vitro Prion-like Behaviour of TDP-43 in ALS. Neurobiol. Dis. 2016, 96, 236–247. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Bell, S.; Wang, J.; Wen, S.; Baloh, R.H.; Appel, S.H. TDP-43 Activates Microglia through NF-κB and NLRP3 Inflammasome. Exp. Neurol. 2015, 273, 24–35. [Google Scholar] [CrossRef]
- Babazadeh, A.; Rayner, S.L.; Lee, A.; Chung, R.S. TDP-43 as a Therapeutic Target in Neurodegenerative Diseases: Focusing on Motor Neuron Disease and Frontotemporal Dementia. Ageing Res. Rev. 2023, 92, 102085. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A.; Neumann, M.; Baborie, A.; Sampathu, D.M.; Du Plessis, D.; Jaros, E.; Perry, R.H.; Trojanowski, J.Q.; Mann, D.M.A.; Lee, V.M.Y. A Harmonized Classification System for FTLD-TDP Pathology. Acta Neuropathol. 2011, 122, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Geser, F.; Winton, M.J.; Kwong, L.K.; Xu, Y.; Xie, S.X.; Igaz, L.M.; Garruto, R.M.; Perl, D.P.; Galasko, D.; Lee, V.M.-Y.; et al. Pathological TDP-43 in Parkinsonism–Dementia Complex and Amyotrophic Lateral Sclerosis of Guam. Acta Neuropathol. 2008, 115, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Kwong, L.K.; Sampathu, D.M.; Trojanowski, J.Q.; Lee, V.M.-Y. TDP-43 Proteinopathy in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis: Protein Misfolding Diseases Without Amyloidosis. Arch. Neurol. 2007, 64, 1388–1394. [Google Scholar] [CrossRef] [PubMed]
- Riemenschneider, H.; Simonetti, F.; Sheth, U.; Katona, E.; Roth, S.; Hutten, S.; Farny, D.; Michaelsen, M.; Nuscher, B.; Schmidt, M.K.; et al. Targeting the Glycine-Rich Domain of TDP-43 with Antibodies Prevents Its Aggregation in Vitro and Reduces Neurofilament Levels in Vivo. Acta Neuropathol. Commun. 2023, 11, 112. [Google Scholar] [CrossRef]
- Noto, Y.-I.; Shibuya, K.; Sato, Y.; Kanai, K.; Misawa, S.; Sawai, S.; Mori, M.; Uchiyama, T.; Isose, S.; Nasu, S.; et al. Elevated CSF TDP-43 Levels in Amyotrophic Lateral Sclerosis: Specificity, Sensitivity, and a Possible Prognostic Value. Amyotroph. Lateral Scler. 2011, 12, 140–143. [Google Scholar] [CrossRef]
- Hosokawa, M.; Arai, T.; Yamashita, M.; Tsuji, H.; Nonaka, T.; Masuda-Suzukake, M.; Tamaoka, A.; Hasegawa, M.; Akiyama, H. Differential Diagnosis of Amyotrophic Lateral Sclerosis from Guillain–Barré Syndrome by Quantitative Determination of TDP-43 in Cerebrospinal Fluid. Int. J. Neurosci. 2014, 124, 344–349. [Google Scholar] [CrossRef]
- Ren, Y.; Li, S.; Chen, S.; Sun, X.; Yang, F.; Wang, H.; Li, M.; Cui, F.; Huang, X. TDP-43 and Phosphorylated TDP-43 Levels in Paired Plasma and CSF Samples in Amyotrophic Lateral Sclerosis. Front. Neurol. 2021, 12, 663637. [Google Scholar] [CrossRef]
- Kasai, T.; Kojima, Y.; Ohmichi, T.; Tatebe, H.; Tsuji, Y.; Noto, Y.; Kitani-Morii, F.; Shinomoto, M.; Allsop, D.; Mizuno, T.; et al. Combined Use of CSF NfL and CSF TDP-43 Improves Diagnostic Performance in ALS. Ann. Clin. Transl. Neurol. 2019, 6, 2489–2502. [Google Scholar] [CrossRef]
- Kojima, Y.; Kasai, T.; Noto, Y.-I.; Ohmichi, T.; Tatebe, H.; Kitaoji, T.; Tsuji, Y.; Kitani-Morii, F.; Shinomoto, M.; Allsop, D.; et al. Amyotrophic lateral sclerosis: Correlations between fluid biomarkers of NfL, TDP-43, and tau, and clinical characteristics. PLoS ONE 2021, 16, e0260323. [Google Scholar] [CrossRef]
- Feneberg, E.; Oeckl, P.; Steinacker, P.; Verde, F.; Barro, C.; Van Damme, P.; Gray, E.; Grosskreutz, J.; Jardel, C.; Kuhle, J.; et al. Multicenter Evaluation of Neurofilaments in Early Symptom Onset Amyotrophic Lateral Sclerosis. Neurology 2018, 90, e22–e30. [Google Scholar] [CrossRef]
- Verstraete, E.; Kuiperij, H.B.; van Blitterswijk, M.M.; Veldink, J.H.; Schelhaas, H.J.; Berg, L.H.v.D.; Verbeek, M.M. TDP-43 Plasma Levels Are Higher in Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. 2012, 13, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Hishizawa, M.; Yamashita, H.; Akizuki, M.; Urushitani, M.; Takahashi, R. TDP-43 Levels Are Higher in Platelets from Patients with Sporadic Amyotrophic Lateral Sclerosis than in Healthy Controls. Neurochem. Int. 2019, 124, 41–45. [Google Scholar] [CrossRef]
- Gambino, C.M.; Ciaccio, A.M.; Sasso, B.L.; Giglio, R.V.; Vidali, M.; Agnello, L.; Ciaccio, M. The Role of TAR DNA Binding Protein 43 (TDP-43) as a CandiDate Biomarker of Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Diagnostics 2023, 13, 416. [Google Scholar] [CrossRef] [PubMed]
- Vu, L.T.; Bowser, R. Fluid-Based Biomarkers for Amyotrophic Lateral Sclerosis. Neurotherapeutics 2017, 14, 119–134. [Google Scholar] [CrossRef]
- Nolan, M.; Talbot, K.; Ansorge, O. Pathogenesis of FUS-Associated ALS and FTD: Insights from Rodent Models. Acta Neuropathol. Commun. 2016, 4, 99. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.M.; Al-Chalabi, A. Clinical Genetics of Amyotrophic Lateral Sclerosis: What Do We Really Know? Nat. Rev. Neurol. 2011, 7, 603–615. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.-C.; Liang, T.Y.; Mazur, C.; et al. Divergent Roles of ALS-Linked Proteins FUS/TLS and TDP-43 Intersect in Processing Long Pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Alirzayeva, H.; Loureiro, R.; Koyuncu, S.; Hommen, F.; Nabawi, Y.; Zhang, W.H.; Dao, T.T.P.; Wehrmann, M.; Lee, H.J.; Vilchez, D. ALS-FUS Mutations Cause Abnormal PARylation and Histone H1.2 Interaction, Leading to Pathological Changes. Cell Rep. 2024, 43, 114626. [Google Scholar] [CrossRef]
- Deng, H.; Gao, K.; Jankovic, J. The Role of FUS Gene Variants in Neurodegenerative Diseases. Nat. Rev. Neurol. 2014, 10, 337–348. [Google Scholar] [CrossRef]
- Scotter, E.L.; Chen, H.-J.; Shaw, C.E. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics 2015, 12, 352–363. [Google Scholar] [CrossRef]
- Pokrishevsky, E.; Grad, L.I.; Cashman, N.R. TDP-43 or FUS-Induced Misfolded Human Wild-Type SOD1 Can Propagate Intercellularly in a Prion-like Fashion. Sci. Rep. 2016, 6, 22155. [Google Scholar] [CrossRef] [PubMed]
- Grad, L.I.; Guest, W.C.; Yanai, A.; Pokrishevsky, E.; O’Neill, M.A.; Gibbs, E.; Semenchenko, V.; Yousefi, M.; Wishart, D.S.; Plotkin, S.S.; et al. Intermolecular Transmission of Superoxide Dismutase 1 Misfolding in Living Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16398–16403. [Google Scholar] [CrossRef]
- Wang, Q.; Johnson, J.L.; Agar, N.Y.R.; Agar, J.N. Protein Aggregation and Protein Instability Govern Familial Amyotrophic Lateral Sclerosis Patient Survival. PLoS Biol. 2008, 6, e170. [Google Scholar] [CrossRef]
- Blair, H.A. Tofersen: First Approval. Drugs 2023, 83, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- McCampbell, A.; Cole, T.; Wegener, A.J.; Tomassy, G.S.; Setnicka, A.; Farley, B.J.; Schoch, K.M.; Hoye, M.L.; Shabsovich, M.; Sun, L.; et al. Antisense Oligonucleotides Extend Survival and Reverse Decrement in Muscle Response in ALS Models. J. Clin. Investig. 2018, 128, 3558–3567. [Google Scholar] [CrossRef] [PubMed]
- Winer, L.; Srinivasan, D.; Chun, S.; Lacomis, D.; Jaffa, M.; Fagan, A.; Holtzman, D.M.; Wancewicz, E.; Bennett, C.F.; Bowser, R.; et al. SOD1 in Cerebral Spinal Fluid as a Pharmacodynamic Marker for Antisense Oligonucleotide Therapy. JAMA Neurol. 2013, 70, 201–207. [Google Scholar] [CrossRef]
- Miller, T.M.; Smith, R.A.; Kordasiewicz, H.; Kaspar, B.K. Gene-Targeted Therapies for the Central Nervous System. Arch. Neurol. 2008, 65, 447–451. [Google Scholar] [CrossRef]
- Urushitani, M.; Sik, A.; Sakurai, T.; Nukina, N.; Takahashi, R.; Julien, J.-P. Chromogranin-Mediated Secretion of Mutant Superoxide Dismutase Proteins Linked to Amyotrophic Lateral Sclerosis. Nat. Neurosci. 2006, 9, 108–118. [Google Scholar] [CrossRef]
- Zetterström, P.; Andersen, P.M.; Brännström, T.; Marklund, S.L. Misfolded Superoxide Dismutase-1 in CSF from Amyotrophic Lateral Sclerosis Patients. J. Neurochem. 2011, 117, 91–99. [Google Scholar] [CrossRef]
- Gertsman, I.; Wuu, J.; McAlonis-Downes, M.; Ghassemian, M.; Ling, K.; Rigo, F.; Bennett, F.; Benatar, M.; Miller, T.M.; Da Cruz, S. An Endogenous Peptide Marker Differentiates SOD1 Stability and Facilitates Pharmacodynamic Monitoring in SOD1 Amyotrophic Lateral Sclerosis. JCI Insight 2019, 4, e122768. [Google Scholar] [CrossRef]
- Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Shaw, P.J.; Sobue, G.; Bucelli, R.C.; Chiò, A.; Van Damme, P.; Ludolph, A.C.; Glass, J.D.; et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2022, 387, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Khouri, R.; Santos, G.S.; Soares, G.; Costa, J.M.; Barral, A.; Barral-Netto, M.; Van Weyenbergh, J. SOD1 Plasma Level as a Biomarker for Therapeutic Failure in Cutaneous Leishmaniasis. J. Infect. Dis. 2014, 210, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.-R.; Lu, T.-T.; Chang, H.-T.; Ge, X.; Huang, B.; Li, W.-M. Elevated Levels of Plasma Superoxide Dismutases 1 and 2 in Patients with Coronary Artery Disease. BioMed Res. Int. 2016, 2016, 3708905. [Google Scholar] [CrossRef] [PubMed]
- Kärkkäinen, J.; Selander, T.; Purdy, M.; Juvonen, P.; Eskelinen, M. Patients with Increased Levels of the Oxidative Stress Biomarker SOD1 Appear to Have Diminished Postoperative Pain After Midline Laparotomy: A Randomised Trial with Special Reference to Postoperative Pain Score (NRS). Anticancer. Res. 2018, 38, 1003–1008. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-Mediated ALS and FTD: Multiple Pathways to Disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Lee, K.-H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 2016, 167, 774–788. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Jansen-West, K.; Xu, Y.-F.; Gendron, T.F.; Bieniek, K.F.; Lin, W.-L.; Sasaguri, H.; Caulfield, T.; Hubbard, J.; Daughrity, L.; et al. Aggregation-Prone c9FTD/ALS Poly(GA) RAN-Translated Proteins Cause Neurotoxicity by Inducing ER Stress. Acta Neuropathol. 2014, 128, 505–524. [Google Scholar] [CrossRef]
- Sakae, N.; Bieniek, K.F.; Zhang, Y.-J.; Ross, K.; Gendron, T.F.; Murray, M.E.; Rademakers, R.; Petrucelli, L.; Dickson, D.W. Poly-GR Dipeptide Repeat Polymers Correlate with Neurodegeneration and Clinicopathological Subtypes in C9ORF72-Related Brain Disease. Acta Neuropathol. Commun. 2018, 6, 63. [Google Scholar] [CrossRef]
- Krishnan, G.; Raitcheva, D.; Bartlett, D.; Prudencio, M.; McKenna-Yasek, D.M.; Douthwright, C.; Oskarsson, B.E.; Ladha, S.; King, O.D.; Barmada, S.J.; et al. Poly(GR) and Poly(GA) in Cerebrospinal Fluid as Potential Biomarkers for C9ORF72-ALS/FTD. Nat. Commun. 2022, 13, 2799. [Google Scholar] [CrossRef]
- Lehmer, C.; Oeckl, P.; Weishaupt, J.H.; Volk, A.E.; Diehl-Schmid, J.; Schroeter, M.L.; Lauer, M.; Kornhuber, J.; Levin, J.; Fassbender, K.; et al. Poly-GP in Cerebrospinal Fluid Links C9orf72-associated Dipeptide Repeat Expression to the Asymptomatic Phase of ALS/FTD. EMBO Mol. Med. 2017, 9, 859–868. [Google Scholar] [CrossRef]
- Wilson, K.M.; Katona, E.; Glaria, I.; Carcolé, M.; Swift, I.J.; Sogorb-Esteve, A.; Heller, C.; Bouzigues, A.; Heslegrave, A.J.; Keshavan, A.; et al. Development of a Sensitive Trial-Ready Poly(GP) CSF Biomarker Assay for C9orf72-Associated Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2022, 93, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Ravits, J.M.; La Spada, A.R. ALS Motor Phenotype Heterogeneity, Focality, and Spread: Deconstructing Motor Neuron Degeneration. Neurology 2009, 73, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, P.; Blomquist, S.; Lührs, C.; Malmkvist, G.; Alling, C.; Solem, J.-O.; Ståhl, E. Neuron-Specific Enolase Increases in Plasma during and Immediately after Extracorporeal Circulation. Ann. Thorac. Surg. 2000, 69, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Isgrò, M.A.; Bottoni, P.; Scatena, R. Neuron-Specific Enolase as a Biomarker: Biochemical and Clinical Aspects. Adv. Exp. Med. Biol. 2015, 867, 125–143. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, A.; Hosokawa, T.; Nishioka, D.; Kotani, T.; Ishida, S.; Takeuchi, T.; Kimura, F.; Arawaka, S. Neuron-Specific Enolase Level Is a Useful Biomarker for Distinguishing Amyotrophic Lateral Sclerosis from Cervical Spondylotic Myelopathy. Sci. Rep. 2021, 11, 22827. [Google Scholar] [CrossRef]
- Hanin, A.; Denis, J.A.; Frazzini, V.; Cousyn, L.; Imbert-Bismut, F.; Rucheton, B.; Bonnefont-Rousselot, D.; Marois, C.; Lambrecq, V.; Demeret, S.; et al. Neuron Specific Enolase, S100-Beta Protein and Progranulin as Diagnostic Biomarkers of Status Epilepticus. J. Neurol. 2022, 269, 3752–3760. [Google Scholar] [CrossRef]
- Rech, T.H.; Vieira, S.; Nagel, F.; Brauner, J.; Scalco, R. Serum Neuron-Specific Enolase: A New Tool for Seizure Risk Monitoring after Status Epilepticus. Eur. J. Neurol. 2022, 29, 883–889. [Google Scholar] [CrossRef]
- Rech, T.H.; Vieira, S.R.R.; Nagel, F.; Brauner, J.S.; Scalco, R. Serum Neuron-Specific Enolase as Early Predictor of Outcome after in-Hospital Cardiac Arrest: A Cohort Study. Crit. Care 2006, 10, R133. [Google Scholar] [CrossRef]
- Shepheard, S.R.; Chataway, T.; Schultz, D.W.; Rush, R.A.; Rogers, M.-L. The Extracellular Domain of Neurotrophin Receptor P75 as a Candidate Biomarker for Amyotrophic Lateral Sclerosis. PLoS ONE 2014, 9, e87398. [Google Scholar] [CrossRef]
- Lowry, K.; Murray, S.; McLean, C.; Talman, P.; Mathers, S.; Lopes, E.; Cheema, S. A Potential Role for the P75 Low-Affinity Neurotrophin Receptor in Spinal Motor Neuron Degeneration in Murine and Human Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2001, 2, 127–134. [Google Scholar] [CrossRef]
- Ritala, J.F.; Lyne, S.B.; Sajanti, A.; Girard, R.; Koskimäki, J. Towards a Comprehensive Understanding of P75 Neurotrophin Receptor Functions and Interactions in the Brain. Neural Regen. Res. 2022, 17, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Shepheard, S.; Jin, J.; Hu, F.; Zhao, X.; Xue, L.; Xiang, L.; Qi, H.; Qu, Q.; Guo, F.; et al. Urinary Extracellular Domain of Neurotrophin Receptor P75 as a Biomarker for Amyotrophic Lateral Sclerosis in a Chinese Cohort. Sci. Rep. 2017, 7, 5127. [Google Scholar] [CrossRef] [PubMed]
- Jourdi, G.; Fleury, S.; Boukhatem, I.; Lordkipanidzé, M. Soluble P75 Neurotrophic Receptor as a Reliable Biomarker in Neurodegenerative Diseases: What Is the Evidence? Neural Regen. Res. 2024, 19, 536. [Google Scholar] [CrossRef]
- Schreiber, S.; Spotorno, N.; Schreiber, F.; Acosta-Cabronero, J.; Kaufmann, J.; Machts, J.; Debska-Vielhaber, G.; Garz, C.; Bittner, D.; Hensiek, N.; et al. Significance of CSF NfL and Tau in ALS. J. Neurol. 2018, 265, 2633–2645. [Google Scholar] [CrossRef] [PubMed]
- Zucchi, E.; Bonetto, V.; Sorarù, G.; Martinelli, I.; Parchi, P.; Liguori, R.; Mandrioli, J. Neurofilaments in Motor Neuron Disorders: Towards Promising Diagnostic and Prognostic Biomarkers. Mol. Neurodegener. 2020, 15, 58. [Google Scholar] [CrossRef]
- Zou, K.; Abdullah, M.; Michikawa, M. Current Biomarkers for Alzheimer’s Disease: From CSF to Blood. J. Pers. Med. 2020, 10, 85. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Miller, C.C.J. Neurofilaments and Neurological Disease. Bioessays 2003, 25, 346–355. [Google Scholar] [CrossRef]
- Menke, R.A.L.; Gray, E.; Lu, C.; Kuhle, J.; Talbot, K.; Malaspina, A.; Turner, M.R. CSF Neurofilament Light Chain Reflects Corticospinal Tract Degeneration in ALS. Ann. Clin. Transl. Neurol. 2015, 2, 748–755. [Google Scholar] [CrossRef]
- Shepherd, C.E.; McCann, H.; Thiel, E.; Halliday, G.M. Neurofilament-Immunoreactive Neurons in Alzheimer’s Disease and Dementia with Lewy Bodies. Neurobiol. Dis. 2002, 9, 249–257. [Google Scholar] [CrossRef]
- Zetterberg, H.; Jacobsson, J.; Rosengren, L.; Blennow, K.; Andersen, P.M. Cerebrospinal Fluid Neurofilament Light Levels in Amyotrophic Lateral Sclerosis: Impact of SOD1 Genotype. Eur. J. Neurol. 2007, 14, 1329–1333. [Google Scholar] [CrossRef]
- Shi, J.; Qin, X.; Chang, X.; Wang, H.; Guo, J.; Zhang, W. Neurofilament Markers in Serum and Cerebrospinal Fluid of Patients with Amyotrophic Lateral Sclerosis. J. Cell. Mol. Med. 2022, 26, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.; Teunissen, C.E.; Lehmann, S.; Otto, M.; Piehl, F.; Ziemssen, T.; Bittner, S.; Sormani, M.P.; Gattringer, T.; Abu-Rumeileh, S.; et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.-Y.; Lv, G.-P.; Gao, L.-N.; Lu, Y.; Guo, J.; Zang, D.-W. Neurofilament Subunit L Levels in the Cerebrospinal Fluid and Serum of Patients with Amyotrophic Lateral Sclerosis. Neurodegener. Dis. 2018, 18, 165–172. [Google Scholar] [CrossRef]
- Verde, F.; Steinacker, P.; Weishaupt, J.H.; Kassubek, J.; Oeckl, P.; Halbgebauer, S.; Tumani, H.; von Arnim, C.A.F.; Dorst, J.; Feneberg, E.; et al. Neurofilament Light Chain in Serum for the Diagnosis of Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2019, 90, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Simonini, C.; Zucchi, E.; Bedin, R.; Martinelli, I.; Gianferrari, G.; Fini, N.; Sorarù, G.; Liguori, R.; Vacchiano, V.; Mandrioli, J. CSF Heavy Neurofilament May Discriminate and Predict Motor Neuron Diseases with Upper Motor Neuron Involvement. Biomedicines 2021, 9, 1623. [Google Scholar] [CrossRef]
- Boylan, K.B.; Glass, J.D.; Crook, J.E.; Yang, C.; Thomas, C.S.; Desaro, P.; Johnston, A.; Overstreet, K.; Kelly, C.; Polak, M.; et al. Phosphorylated Neurofilament Heavy Subunit (pNF-H) in Peripheral Blood and CSF as a Potential Prognostic Biomarker in Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2013, 84, 467–472. [Google Scholar] [CrossRef]
- De Schaepdryver, M.; Goossens, J.; De Meyer, S.; Jeromin, A.; Masrori, P.; Brix, B.; Claeys, K.G.; Schaeverbeke, J.; Adamczuk, K.; Vandenberghe, R.; et al. Serum Neurofilament Heavy Chains as Early Marker of Motor Neuron Degeneration. Ann. Clin. Transl. Neurol. 2019, 6, 1971–1979. [Google Scholar] [CrossRef]
- Falzone, Y.M.; Domi, T.; Agosta, F.; Pozzi, L.; Schito, P.; Fazio, R.; Del Carro, U.; Barbieri, A.; Comola, M.; Leocani, L.; et al. Serum Phosphorylated Neurofilament Heavy-Chain Levels Reflect Phenotypic Heterogeneity and Are an Independent Predictor of Survival in Motor Neuron Disease. J. Neurol. 2020, 267, 2272–2280. [Google Scholar] [CrossRef]
- Zhou, Y.-N.; Chen, Y.-H.; Dong, S.-Q.; Yang, W.-B.; Qian, T.; Liu, X.-N.; Cheng, Q.; Wang, J.-C.; Chen, X.-J. Role of Blood Neurofilaments in the Prognosis of Amyotrophic Lateral Sclerosis: A Meta-Analysis. Front. Neurol. 2021, 12, 712245. [Google Scholar] [CrossRef]
- Thouvenot, E.; Demattei, C.; Lehmann, S.; Maceski-Maleska, A.; Hirtz, C.; Juntas-Morales, R.; Pageot, N.; Esselin, F.; Alphandéry, S.; Vincent, T.; et al. Serum Neurofilament Light Chain at Time of Diagnosis Is an Independent Prognostic Factor of Survival in Amyotrophic Lateral Sclerosis. Eur. J. Neurol. 2020, 27, 251–257. [Google Scholar] [CrossRef]
- Vacchiano, V.; Mastrangelo, A.; Zenesini, C.; Masullo, M.; Quadalti, C.; Avoni, P.; Polischi, B.; Cherici, A.; Capellari, S.; Salvi, F.; et al. Plasma and CSF Neurofilament Light Chain in Amyotrophic Lateral Sclerosis: A Cross-Sectional and Longitudinal Study. Front. Aging Neurosci. 2021, 13, 753242. [Google Scholar] [CrossRef]
- Lu, C.-H.; Macdonald-Wallis, C.; Gray, E.; Pearce, N.; Petzold, A.; Norgren, N.; Giovannoni, G.; Fratta, P.; Sidle, K.; Fish, M.; et al. Neurofilament Light Chain. Neurology 2015, 84, 2247–2257. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; Andersen, P.M.; Lombardi, V.; Malaspina, A. Neurofilament Light: A Candidate Biomarker of Presymptomatic Amyotrophic Lateral Sclerosis and Phenoconversion. Ann. Neurol. 2018, 84, 130–139. [Google Scholar] [CrossRef]
- Brodovitch, A.; Boucraut, J.; Delmont, E.; Parlanti, A.; Grapperon, A.-M.; Attarian, S.; Verschueren, A. Combination of Serum and CSF Neurofilament-Light and Neuroinflammatory Biomarkers to Evaluate ALS. Sci. Rep. 2021, 11, 703. [Google Scholar] [CrossRef]
- Behzadi, A.; Pujol-Calderón, F.; Tjust, A.E.; Wuolikainen, A.; Höglund, K.; Forsberg, K.; Portelius, E.; Blennow, K.; Zetterberg, H.; Andersen, P.M. Neurofilaments Can Differentiate ALS Subgroups and ALS from Common Diagnostic Mimics. Sci. Rep. 2021, 11, 22128. [Google Scholar] [CrossRef] [PubMed]
- Rosengren, L.E.; Karlsson, J.-E.; Karlsson, J.-O.; Persson, L.I.; Wikkelsø, C. Patients with Amyotrophic Lateral Sclerosis and Other Neurodegenerative Diseases Have Increased Levels of Neurofilament Protein in CSF. J. Neurochem. 1996, 67, 2013–2018. [Google Scholar] [CrossRef]
- Brettschneider, J.; Petzold, A.; Süßmuth, S.D.; Ludolph, A.C.; Tumani, H. Axonal Damage Markers in Cerebrospinal Fluid Are Increased in ALS. Neurology 2006, 66, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.F.; Bs, L.M.D.; Heckman, M.G.; Bs, N.N.D.; ScM, J.W.; Miller, T.M.; Pastor, P.; Trojanowski, J.Q.; Grossman, M.; Berry, J.D.; et al. Phosphorylated Neurofilament Heavy Chain: A Biomarker of Survival for C9ORF72-Associated Amyotrophic Lateral Sclerosis. Ann. Neurol. 2017, 82, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Tortelli, R.; Ruggieri, M.; Cortese, R.; D’Errico, E.; Capozzo, R.; Leo, A.; Mastrapasqua, M.; Zoccolella, S.; Leante, R.; Livrea, P.; et al. Elevated Cerebrospinal Fluid Neurofilament Light Levels in Patients with Amyotrophic Lateral Sclerosis: A Possible Marker of Disease Severity and Progression. Eur. J. Neurol. 2012, 19, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Gaiani, A.; Martinelli, I.; Bello, L.; Querin, G.; Puthenparampil, M.; Ruggero, S.; Toffanin, E.; Cagnin, A.; Briani, C.; Pegoraro, E.; et al. Diagnostic and Prognostic Biomarkers in Amyotrophic Lateral Sclerosis: Neurofilament Light Chain Levels in Definite Subtypes of Disease. JAMA Neurol. 2017, 74, 525–532. [Google Scholar] [CrossRef]
- Zecca, C.; Dell’abate, M.T.; Pasculli, G.; Capozzo, R.; Barone, R.; Arima, S.; Pollice, A.; Brescia, V.; Tortelli, R.; Logroscino, G. Role of Plasma Phosphorylated Neurofilament Heavy Chain (pNfH) in Amyotrophic Lateral Sclerosis. J. Cell. Mol. Med. 2022, 26, 3608–3615. [Google Scholar] [CrossRef] [PubMed]
- Steinacker, P.; Hendrich, C.; Sperfeld, A.-D.; Jesse, S.; Lehnert, S.; Pabst, A.; von Arnim, C.A.F.; Mottaghy, F.M.; Uttner, I.; Tumani, H.; et al. Concentrations of Beta-Amyloid Precursor Protein Processing Products in Cerebrospinal Fluid of Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration. J. Neural Transm. 2009, 116, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Gille, B.; De Schaepdryver, M.; Dedeene, L.; Goossens, J.; Claeys, K.G.; Bosch, L.V.D.; Tournoy, J.; Van Damme, P.; Poesen, K. Inflammatory Markers in Cerebrospinal Fluid: Independent Prognostic Biomarkers in Amyotrophic Lateral Sclerosis? J. Neurol. Neurosurg. Psychiatry 2019, 90, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Illán-Gala, I.; Alcolea, D.; Montal, V.; Dols-Icardo, O.; Muñoz, L.; de Luna, N.; Turón-Sans, J.; Cortés-Vicente, E.; Sánchez-Saudinós, M.B.; Subirana, A.; et al. CSF sAPPβ, YKL-40, and NfL along the ALS-FTD Spectrum. Neurology 2018, 91, e1619–e1628. [Google Scholar] [CrossRef]
- Strong, M.J.; Donison, N.S.; Volkening, K. Alterations in Tau Metabolism in ALS and ALS-FTSD. Front. Neurol. 2020, 11, 598907. [Google Scholar] [CrossRef]
- Süssmuth, S.D.; Reiber, H.; Tumani, H. Tau Protein in Cerebrospinal Fluid (CSF): A Blood–CSF Barrier Related Evaluation in Patients with Various Neurological Diseases. Neurosci. Lett. 2001, 300, 95–98. [Google Scholar] [CrossRef]
- Jimenez-Jimenez, F.J.; Hernanz, A.; Medina-Acebron, S.; de Bustos, F.; Zurdo, J.M.; Alonso, H.; Puertas, I.; Barcenilla, B.; Sayed, Y.; Cabrera-Valdivia, F. Tau Protein Concentrations in Cerebrospinal Fluid of Patients with Amyotrophic Lateral Sclerosis. Acta Neurol. Scand. 2005, 111, 114–117. [Google Scholar] [CrossRef]
- Ballatore, C.; Lee, V.M.-Y.; Trojanowski, J.Q. Tau-Mediated Neurodegeneration in Alzheimer’s Disease and Related Disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef]
- Virgilio, E.; Vecchio, D.; Crespi, I.; Serino, R.; Cantello, R.; Dianzani, U.; Comi, C. Cerebrospinal Tau Levels as a Predictor of Early Disability in Multiple Sclerosis. Mult. Scler. Relat. Disord. 2021, 56, 103231. [Google Scholar] [CrossRef]
- Didonna, A. Tau at the Interface between Neurodegeneration and Neuroinflammation. Genes. Immun. 2020, 21, 288–300. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Kopeikina, K.J.; Koffie, R.M.; de Calignon, A.; Hyman, B.T. Are Tangles as Toxic as They Look? J. Mol. Neurosci. 2011, 45, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Scarafino, A.; D’errico, E.; Introna, A.; Fraddosio, A.; Distaso, E.; Tempesta, I.; Morea, A.; Mastronardi, A.; Leante, R.; Ruggieri, M.; et al. Diagnostic and Prognostic Power of CSF Tau in Amyotrophic Lateral Sclerosis. J. Neurol. 2018, 265, 2353–2362. [Google Scholar] [CrossRef] [PubMed]
- Bourbouli, M.; Rentzos, M.; Bougea, A.; Zouvelou, V.; Constantinides, V.C.; Zaganas, I.; Evdokimidis, I.; Kapaki, E.; Paraskevas, G.P. Cerebrospinal Fluid TAR DNA-Binding Protein 43 Combined with Tau Proteins as a Candidate Biomarker for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Spectrum Disorders. Dement. Geriatr. Cogn. Disord. 2017, 44, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Wilke, C.; Deuschle, C.; Rattay, T.W.; Maetzler, W.; Synofzik, M. Total Tau Is Increased, but Phosphorylated Tau Not Decreased, in Cerebrospinal Fluid in Amyotrophic Lateral Sclerosis. Neurobiol. Aging 2015, 36, 1072–1074. [Google Scholar] [CrossRef]
- Grossman, M.; Elman, L.; McCluskey, L.; McMillan, C.T.; Boller, A.; Powers, J.; Rascovsky, K.; Hu, W.; Shaw, L.; Irwin, D.J.; et al. Phosphorylated Tau as a Candidate Biomarker for Amyotrophic Lateral Sclerosis. JAMA Neurol. 2014, 71, 442–448. [Google Scholar] [CrossRef]
- Agnello, L.; Colletti, T.; Sasso, B.L.; Vidali, M.; Spataro, R.; Gambino, C.M.; Giglio, R.V.; Piccoli, T.; Bivona, G.; La Bella, V.; et al. Tau Protein as a Diagnostic and Prognostic Biomarker in Amyotrophic Lateral Sclerosis. Eur. J. Neurol. 2021, 28, 1868–1875. [Google Scholar] [CrossRef]
- Cousins, K.A.Q.; Shaw, L.M.; Shellikeri, S.; Dratch, L.; Rosario, L.; Elman, L.B.; Quinn, C.; Amado, D.A.; Wolk, D.A.; Tropea, T.F.; et al. Elevated Plasma Phosphorylated Tau 181 in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2022, 92, 807–818. [Google Scholar] [CrossRef]
- Hu, W.T.; Watts, K.; Grossman, M.; Glass, J.; Lah, J.J.; Hales, C.; Shelnutt, M.; Van Deerlin, V.; Trojanowski, J.Q.; Levey, A.I. Reduced CSF P-Tau181 to Tau Ratio Is a Biomarker for FTLD-TDP. Neurology 2013, 81, 1945–1952. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.-F.; Wang, Q.; Krueger, B.J.; et al. Exome Sequencing in Amyotrophic Lateral Sclerosis Identifies Risk Genes and Pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef]
- Gao, L.; Pan, X.; Zhang, J.H.; Xia, Y. Glial Cells: An Important Switch for the Vascular Function of the Central Nervous System. Front. Cell. Neurosci. 2023, 17, 1166770. [Google Scholar] [CrossRef]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef]
- Beers, D.R.; Appel, S.H. Immune Dysregulation in Amyotrophic Lateral Sclerosis: Mechanisms and Emerging Therapies. Lancet Neurol. 2019, 18, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Frakes, A.E.; Braun, L.; Ferraiuolo, L.; Guttridge, D.C.; Kaspar, B.K. Additive Amelioration of ALS by Co-targeting Independent Pathogenic Mechanisms. Ann. Clin. Transl. Neurol. 2017, 4, 76–86. [Google Scholar] [CrossRef]
- Benninger, F.; Glat, M.J.; Offen, D.; Steiner, I. Glial Fibrillary Acidic Protein as a Marker of Astrocytic Activation in the Cerebrospinal Fluid of Patients with Amyotrophic Lateral Sclerosis. J. Clin. Neurosci. 2016, 26, 75–78. [Google Scholar] [CrossRef]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.J.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of Widespread Cerebral Microglial Activation in Amyotrophic Lateral Sclerosis: An [11C](R)-PK11195 Positron Emission Tomography Study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The Dual Role of Microglia in ALS: Mechanisms and Therapeutic Approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Hyun, H.-W.; Min, S.-J.; Kim, J.-E. CDK5 Inhibitors Prevent Astroglial Apoptosis and Reactive Astrogliosis by Regulating PKA and DRP1 Phosphorylations in the Rat Hippocampus. Neurosci. Res. 2017, 119, 24–37. [Google Scholar] [CrossRef]
- Middeldorp, J.; Hol, E.M. GFAP in Health and Disease. Prog. Neurobiol. 2011, 93, 421–443. [Google Scholar] [CrossRef] [PubMed]
- Foerch, C.; Niessner, M.; Back, T.; Bauerle, M.; De Marchis, G.M.; Ferbert, A.; Grehl, H.; Hamann, G.F.; Jacobs, A.; Kastrup, A.; et al. Diagnostic Accuracy of Plasma Glial Fibrillary Acidic Protein for Differentiating Intracerebral Hemorrhage and Cerebral Ischemia in Patients with Symptoms of Acute Stroke. Clin. Chem. 2012, 58, 237–245. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, K.K.W. Glial Fibrillary Acidic Protein: From Intermediate Filament Assembly and Gliosis to Neurobiomarker. Trends Neurosci. 2015, 38, 364–374. [Google Scholar] [CrossRef]
- Oeckl, P.; Weydt, P.; Steinacker, P.; Anderl-Straub, S.; Nordin, F.; Volk, A.E.; Diehl-Schmid, J.; Andersen, P.M.; Kornhuber, J.; Danek, A.; et al. Different Neuroinflammatory Profile in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Is Linked to the Clinical Phase. J. Neurol. Neurosurg. Psychiatry 2019, 90, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Verde, F.; Milone, I.; Maranzano, A.; Colombo, E.; Torre, S.; Solca, F.; Doretti, A.; Gentile, F.; Manini, A.; Bonetti, R.; et al. Serum Levels of Glial Fibrillary Acidic Protein in Patients with Amyotrophic Lateral Sclerosis. Ann. Clin. Transl. Neurol. 2022, 10, 118–129. [Google Scholar] [CrossRef]
- Kleindienst, A.; Hesse, F.; Bullock, M.R.; Buchfelder, M. The Neurotrophic Protein S100B: Value as a Marker of Brain Damage and Possible Therapeutic Implications. Prog. Brain Res. 2007, 161, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Sen, J.; Belli, A. S100B in Neuropathologic States: The CRP of the Brain? J. Neurosci. Res. 2007, 85, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Donato, R. Intracellular and Extracellular Roles of S100 Proteins. Microsc. Res. Tech. 2003, 60, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, D.S.; Lenz, G.; Karl, J.; Gonçalves, C.A.; Rodnight, R. Extracellular S100B Protein Modulates ERK in Astrocyte Cultures. Neuroreport 2000, 11, 807–809. [Google Scholar] [CrossRef]
- Rothermundt, M.; Peters, M.; Prehn, J.H.M.; Arolt, V. S100B in Brain Damage and Neurodegeneration. Microsc. Res. Tech. 2003, 60, 614–632. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Kuja-Panula, J.; Sorci, G.; Agneletti, A.L.; Donato, R.; Rauvala, H. Coregulation of Neurite Outgrowth and Cell Survival by Amphoterin and S100 Proteins through Receptor for Advanced Glycation End Products (RAGE) Activation. J. Biol. Chem. 2000, 275, 40096–40105. [Google Scholar] [CrossRef]
- de Souza, D.F.; Wartchow, K.; Hansen, F.; Lunardi, P.; Guerra, M.C.; Nardin, P.; Gonçalves, C.-A. Interleukin-6-Induced S100B Secretion Is Inhibited by Haloperidol and Risperidone. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 43, 14–22. [Google Scholar] [CrossRef]
- Parpura, V.; Verkhratsky, A. Homeostatic Function of Astrocytes: Calcium and Sodium Signalling. Transl. Neurosci. 2012, 3, 334–344. [Google Scholar] [CrossRef]
- Rezaei, O.; Pakdaman, H.; Gharehgozli, K.; Simani, L.; Vahedian-Azimi, A.; Asaadi, S.; Sahraei, Z.; Hajiesmaeili, M. S100 B: A New Concept in Neurocritical Care. Iran. J. Neurol. 2017, 16, 83–89. [Google Scholar] [PubMed Central]
- Yang, Y.H.; Nam, M.S.; Yang, E.S. Rapid Prenatal Diagnosis of Trisomy 21 by Real-Time Quantitative Polymerase Chain Reaction with Amplification of Small Tandem Repeats and S100B in Chromosome 21. Yonsei Med. J. 2005, 46, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.C. Pathophysiology of Status Epilepticus. Neurosci. Lett. 2018, 667, 84–91. [Google Scholar] [CrossRef]
- Süssmuth, S.D.; Sperfeld, A.D.; Hinz, A.; Brettschneider, J.; Endruhn, S.; Ludolph, A.C.; Tumani, H. CSF Glial Markers Correlate with Survival in Amyotrophic Lateral Sclerosis. Neurology 2010, 74, 982–987. [Google Scholar] [CrossRef] [PubMed]
- Steinacker, P.; Huss, A.; Mayer, B.; Grehl, T.; Grosskreutz, J.; Borck, G.; Kuhle, J.; Lulé, D.; Meyer, T.; Oeckl, P.; et al. Diagnostic and Prognostic Significance of Neurofilament Light Chain NF-L, but Not Progranulin and S100B, in the Course of Amyotrophic Lateral Sclerosis: Data from the German MND-Net. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 112–119. [Google Scholar] [CrossRef]
- Michetti, F.; Clementi, M.E.; Di Liddo, R.; Valeriani, F.; Ria, F.; Rende, M.; Di Sante, G.; Spica, V.R. The S100B Protein: A Multifaceted Pathogenic Factor More Than a Biomarker. Int. J. Mol. Sci. 2023, 24, 9605. [Google Scholar] [CrossRef]
- Mao, D.; Zheng, Y.; Xu, F.; Han, X.; Zhao, H. HMGB1 in Nervous System Diseases: A Common Biomarker and Potential Therapeutic Target. Front. Neurol. 2022, 13, 1029891. [Google Scholar] [CrossRef]
- Chen, R.; Kang, R.; Tang, D. The Mechanism of HMGB1 Secretion and Release. Exp. Mol. Med. 2022, 54, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Rosciszewski, G.; Cadena, V.; Auzmendi, J.; Cieri, M.B.; Lukin, J.; Rossi, A.R.; Murta, V.; Villarreal, A.; Reinés, A.; Gomes, F.C.A.; et al. Detrimental Effects of HMGB-1 Require Microglial-Astroglial Interaction: Implications for the Status Epilepticus -Induced Neuroinflammation. Front. Cell Neurosci. 2019, 13, 380. [Google Scholar] [CrossRef]
- Pedrazzi, M.; Patrone, M.; Passalacqua, M.; Ranzato, E.; Colamassaro, D.; Sparatore, B.; Pontremoli, S.; Melloni, E. Selective Proinflammatory Activation of Astrocytes by High-Mobility Group Box 1 Protein Signaling. J. Immunol. 2007, 179, 8525–8532. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Shaikh, M.F. Implication of HMGB1 Signaling Pathways in Amyotrophic Lateral Sclerosis (ALS): From Molecular Mechanisms to Pre-Clinical Results. Pharmacol. Res. 2020, 156, 104792. [Google Scholar] [CrossRef]
- Hwang, C.-S.; Liu, G.-T.; Chang, M.D.-T.; Liao, I.-L.; Chang, H.-T. Elevated Serum Autoantibody against High Mobility Group Box 1 as a Potent Surrogate Biomarker for Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2013, 58, 13–18. [Google Scholar] [CrossRef] [PubMed]
- van Maldeghem, I.; Nusman, C.M.; Visser, D.H. Soluble CD14 Subtype (sCD14-ST) as Biomarker in Neonatal Early-Onset Sepsis and Late-Onset Sepsis: A Systematic Review and Meta-Analysis. BMC Immunol. 2019, 20, 17. [Google Scholar] [CrossRef]
- Mussap, M.; Noto, A.; Fravega, M.; Fanos, V. Soluble CD14 Subtype Presepsin (sCD14-ST) and Lipopolysaccharide Binding Protein (LBP) in Neonatal Sepsis: New Clinical and Analytical Perspectives for Two Old Biomarkers. J. Matern. Fetal Neonatal Med. 2011, 24 (Suppl. 2), 12–14. [Google Scholar] [CrossRef]
- Beers, D.; Zhao, W.; Neal, D.; Thonhoff, J.; Thome, A.; Faridar, A.; Wen, S.; Wang, J.; Appel, S. Elevated Acute Phase Proteins Reflect Peripheral Inflammation and Disease Severity in Patients with Amyotrophic Lateral Sclerosis. Sci. Rep. 2020, 10, 15295. [Google Scholar] [CrossRef]
- Mabrey, F.L.; Morrell, E.D.; Bhatraju, P.K.M.; Sathe, N.A.; Sakr, S.S.; Sahi, S.K.B.; West, T.E.; Mikacenic, C.; Wurfel, M.M. Plasma Soluble CD14 Subtype Levels Are Associated With Clinical Outcomes in Critically Ill Subjects With Coronavirus Disease 2019. Crit. Care Explor. 2021, 3, e0591. [Google Scholar] [CrossRef]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Microglia and Aging: The Role of the TREM2–DAP12 and CX3CL1-CX3CR1 Axes. Int. J. Mol. Sci. 2018, 19, 318. [Google Scholar] [CrossRef]
- Zheng, H.; Cheng, B.; Li, Y.; Li, X.; Chen, X.; Zhang, Y. TREM2 in Alzheimer’s Disease: Microglial Survival and Energy Metabolism. Front. Aging Neurosci. 2018, 10, 395. [Google Scholar] [CrossRef]
- Guerreiro, R.J.; Lohmann, E.; Brás, J.M.; Gibbs, J.R.; Rohrer, J.D.; Gurunlian, N.; Dursun, B.; Bilgic, B.; Hanagasi, H.; Gurvit, H.; et al. Using Exome Sequencing to Reveal Mutations in TREM2 Presenting as a Frontotemporal Dementia-like Syndrome without Bone Involvement. JAMA Neurol. 2013, 70, 78–84. [Google Scholar] [CrossRef]
- Rayaprolu, S.; Mullen, B.; Baker, M.; Lynch, T.; Finger, E.; Seeley, W.W.; Hatanpaa, K.J.; Lomen-Hoerth, C.; Kertesz, A.; Bigio, E.H.; et al. TREM2 in Neurodegeneration: Evidence for Association of the p.R47H Variant with Frontotemporal Dementia and Parkinson’s Disease. Mol. Neurodegener. 2013, 8, 19. [Google Scholar] [CrossRef]
- Xie, M.; Zhao, S.; Bosco, D.B.; Nguyen, A.; Wu, L.-J. Microglial TREM2 in Amyotrophic Lateral Sclerosis. Dev. Neurobiol. 2022, 82, 125–137. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Green, C.; Altschuler, G.; Wei, W.; Bury, J.J.; Heath, P.R.; Wyles, M.; Gelsthorpe, C.; Highley, J.R.; Lorente-Pons, A.; et al. A Data-Driven Approach Links Microglia to Pathology and Prognosis in Amyotrophic Lateral Sclerosis. Acta Neuropathol. Commun. 2017, 5, 23. [Google Scholar] [CrossRef]
- Kleinberger, G.; Yamanishi, Y.; Suárez-Calvet, M.; Czirr, E.; Lohmann, E.; Cuyvers, E.; Struyfs, H.; Pettkus, N.; Wenninger-Weinzierl, A.; Mazaheri, F.; et al. TREM2 Mutations Implicated in Neurodegeneration Impair Cell Surface Transport and Phagocytosis. Sci. Transl. Med. 2014, 6, ra86–ra243. [Google Scholar] [CrossRef]
- Kanneganti, M.; Kamba, A.; Mizoguchi, E. Role of Chitotriosidase (Chitinase 1) under Normal and Disease Conditions. J. Ep. Biol. Pharmacol. 2012, 5, 1–9. [Google Scholar] [CrossRef]
- Llorens, F.; Thüne, K.; Tahir, W.; Kanata, E.; Diaz-Lucena, D.; Xanthopoulos, K.; Kovatsi, E.; Pleschka, C.; Garcia-Esparcia, P.; Schmitz, M.; et al. YKL-40 in the Brain and Cerebrospinal Fluid of Neurodegenerative Dementias. Mol. Neurodegener. 2017, 12, 83. [Google Scholar] [CrossRef]
- Masrori, P.; De Schaepdryver, M.; Floeter, M.K.; De Vocht, J.; Lamaire, N.; D’Hondt, A.; Traynor, B.; Poesen, K.; Van Damme, P. Prognostic Relationship of Neurofilaments, CHIT1, YKL-40 and MCP-1 in Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2022, 93, 681–682. [Google Scholar] [CrossRef]
- Sanfilippo, C.; Longo, A.; Lazzara, F.; Cambria, D.; Distefano, G.; Palumbo, M.; Cantarella, A.; Malaguarnera, L.; Di Rosa, M. CHI3L1 and CHI3L2 Overexpression in Motor Cortex and Spinal Cord of sALS Patients. Mol. Cell Neurosci. 2017, 85, 162–169. [Google Scholar] [CrossRef]
- Andrés-Benito, P.; Domínguez, R.; Colomina, M.J.; Llorens, F.; Povedano, M.; Ferrer, I. YKL40 in Sporadic Amyotrophic Lateral Sclerosis: Cerebrospinal Fluid Levels as a Prognosis Marker of Disease Progression. Aging 2018, 10, 2367–2382. [Google Scholar] [CrossRef]
- Thompson, A.G.; Gray, E.; Bampton, A.; Raciborska, D.; Talbot, K.; Turner, M.R. CSF Chitinase Proteins in Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1215–1220. [Google Scholar] [CrossRef]
- Thompson, A.G.; Gray, E.; Thézénas, M.-L.; Charles, P.D.; Evetts, S.; Hu, M.T.; Talbot, K.; Fischer, R.; Kessler, B.M.; Turner, M.R. Cerebrospinal Fluid Macrophage Biomarkers in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2018, 83, 258–268. [Google Scholar] [CrossRef]
- Abu-Rumeileh, S.; Vacchiano, V.; Zenesini, C.; Polischi, B.; de Pasqua, S.; Fileccia, E.; Mammana, A.; Di Stasi, V.; Capellari, S. Diagnostic-Prognostic Value and Electrophysiological Correlates of CSF Biomarkers of Neurodegeneration and Neuroinflammation in Amyotrophic Lateral Sclerosis. J. Neurol. 2020, 267, 1699–1708. [Google Scholar] [CrossRef]
- Varghese, A.M.; Sharma, A.; Mishra, P.; Vijayalakshmi, K.; Harsha, H.C.; Sathyaprabha, T.N.; Bharath, S.M.; Nalini, A.; Alladi, P.A.; Raju, T.R. Chitotriosidase-a Putative Biomarker for Sporadic Amyotrophic Lateral Sclerosis. Clin. Proteom. 2013, 10, 19. [Google Scholar] [CrossRef]
- Zlokovic, B.V. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef]
- Kakaroubas, N.; Brennan, S.; Keon, M.; Saksena, N.K. Pathomechanisms of Blood-Brain Barrier Disruption in ALS. Neurosci. J. 2019, 2019, 2537698. [Google Scholar] [CrossRef]
- Garbuzova-Davis, S.; Sanberg, P. Blood-CNS Barrier Impairment in ALS Patients versus an Animal Model. Front. Cell. Neurosci. 2014, 8, 21. [Google Scholar] [CrossRef]
- Winkler, E.A.; Sengillo, J.D.; Sullivan, J.S.; Henkel, J.S.; Appel, S.H.; Zlokovic, B.V. Blood-Spinal Cord Barrier Breakdown and Pericyte Reductions in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2013, 125, 111–120. [Google Scholar] [CrossRef]
- Musaeus, C.S.; Gleerup, H.S.; Høgh, P.; Waldemar, G.; Hasselbalch, S.G.; Simonsen, A.H. Cerebrospinal Fluid/Plasma Albumin Ratio as a Biomarker for Blood-Brain Barrier Impairment Across Neurodegenerative Dementias. J. Alzheimers Dis. 2020, 75, 429–436. [Google Scholar] [CrossRef]
- Fu, J.; Lai, X.; Wei, Q.; Chen, X.; Shang, H. Associations of Cerebrospinal Fluid Profiles with Severity and Mortality Risk of Amyotrophic Lateral Sclerosis. Front. Neurosci. 2024, 18, 1375892. [Google Scholar] [CrossRef]
- Li, J.-Y.; Cai, Z.-Y.; Sun, X.-H.; Shen, D.-C.; Yang, X.-Z.; Liu, M.-S.; Cui, L.-Y. Blood–Brain Barrier Dysfunction and Myelin Basic Protein in Survival of Amyotrophic Lateral Sclerosis with or without Frontotemporal Dementia. Neurol. Sci. 2022, 43, 3201–3210. [Google Scholar] [CrossRef]
- Klose, V.; Jesse, S.; Lewerenz, J.; Kassubek, J.; Dorst, J.; Rosenbohm, A.; Nagel, G.; Wernecke, D.; Roselli, F.; Tumani, H.; et al. Blood–CSF Barrier Integrity in Amyotrophic Lateral Sclerosis. Brain 2024, 14, e144. [Google Scholar] [CrossRef]
- Alarcan, H.; Vourc’h, P.; Berton, L.; Bretagne, I.B.-D.; Piver, E.; Andres, C.R.; Corcia, P.; Veyrat-Durebex, C.; Blasco, H. Implication of Central Nervous System Barrier Impairment in Amyotrophic Lateral Sclerosis: Gender-Related Difference in Patients. Int. J. Mol. Sci. 2023, 24, 11196. [Google Scholar] [CrossRef]
- Verde, F.; Ferrari, I.; Maranzano, A.; Ciusani, E.; Torre, S.; Milone, I.; Colombo, E.; Doretti, A.; Peverelli, S.; Ratti, A.; et al. Relationship between Cerebrospinal Fluid/Serum Albumin Quotient and Phenotype in Amyotrophic Lateral Sclerosis: A Retrospective Study on 328 Patients. Neurol. Sci. 2023, 44, 1679–1685. [Google Scholar] [CrossRef]
- Prell, T.; Vlad, B.; Gaur, N.; Stubendorff, B.; Grosskreutz, J. Blood–Brain Barrier Disruption Is Not Associated With Disease Aggressiveness in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2021, 15, 656456. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, M.J. Amyotrophic Lateral Sclerosis as a Synaptopathy. Neural Regen. Res. 2019, 14, 189. [Google Scholar] [CrossRef] [PubMed]
- Genç, B.; Jara, J.H.; Lagrimas, A.K.B.; Pytel, P.; Roos, R.P.; Mesulam, M.M.; Geula, C.; Bigio, E.H.; Özdinler, P.H. Apical Dendrite Degeneration, a Novel Cellular Pathology for Betz Cells in ALS. Sci. Rep. 2017, 7, 41765. [Google Scholar] [CrossRef]
- Wilson, D.M.; Cookson, M.R.; Bosch, L.V.D.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of Neurodegenerative Diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef]
- Nishimura, A.L.; Arias, N. Synaptopathy Mechanisms in ALS Caused by C9orf72 Repeat Expansion. Front. Cell. Neurosci. 2021, 15, 660693. [Google Scholar] [CrossRef]
- King, A.E.; Woodhouse, A.; Kirkcaldie, M.T.K.; Vickers, J.C. Excitotoxicity in ALS: Overstimulation, or Overreaction? Exp. Neurol. 2016, 275, 162–171. [Google Scholar] [CrossRef]
- Rosenblum, L.T.; Trotti, D. EAAT2 and the Molecular Signature of Amyotrophic Lateral Sclerosis. Adv. Neurobiol. 2017, 16, 117–136. [Google Scholar] [CrossRef]
- Rao, S.D.; Yin, H.Z.; Weiss, J.H. Disruption of Glial Glutamate Transport by Reactive Oxygen Species Produced in Motor Neurons. J. Neurosci. 2003, 23, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J.; Forrest, V.; Ince, P.G.; Richardson, J.P.; Wastell, H.J. CSF and Plasma Amino Acid Levels in Motor Neuron Disease: Elevation of CSF Glutamate in a Subset of Patients. Neurodegeneration 1995, 4, 209–216. [Google Scholar] [CrossRef]
- Karr, J.; Vagin, V.; Chen, K.; Ganesan, S.; Olenkina, O.; Gvozdev, V.; Featherstone, D.E. Regulation of Glutamate Receptor Subunit Availability by microRNAs. J. Cell Biol. 2009, 185, 685–697. [Google Scholar] [CrossRef]
- Fiszman, M.L.; Ricart, K.C.; Latini, A.; Rodríguez, G.; Sica, R.E.P. In Vitro Neurotoxic Properties and Excitatory Aminoacids Concentration in the Cerebrospinal Fluid of Amyotrophic Lateral Sclerosis Patients. Relationship with the Degree of Certainty of Disease Diagnoses. Acta Neurol. Scand. 2010, 121, 120–126. [Google Scholar] [CrossRef]
- Hemerková, P.; Vališ, M. Role of Oxidative Stress in the Pathogenesis of Amyotrophic Lateral Sclerosis: Antioxidant Metalloenzymes and Therapeutic Strategies. Biomolecules 2021, 11, 437. [Google Scholar] [CrossRef]
- Spreux-Varoquaux, O.; Bensimon, G.; Lacomblez, L.; Salachas, F.; Pradat, P.F.; Le Forestier, N.; Marouan, A.; Dib, M.; Meininger, V. Glutamate Levels in Cerebrospinal Fluid in Amyotrophic Lateral Sclerosis: A Reappraisal Using a New HPLC Method with Coulometric Detection in a Large Cohort of Patients. J. Neurol. Sci. 2002, 193, 73–78. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective Loss of Glial Glutamate Transporter GLT-1 in Amyotrophic Lateral Sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef]
- Ferrarese, C.; Pecora, N.; Frigo, M.; Appollobnio, I.; Frattola, L. Assessment of Reliability and Biological Significance of Glutamate Levels in Cerebrospinal Fluid. Ann. Neurol. 1993, 33, 316–319. [Google Scholar] [CrossRef]
- Kumar, A.; Bala, L.; Kalita, J.; Misra, U.; Singh, R.; Khetrapal, C.; Babu, G.N. Metabolomic Analysis of Serum by (1) H NMR Spectroscopy in Amyotrophic Lateral Sclerosis. Clin. Chim. Acta 2010, 411, 563–567. [Google Scholar] [CrossRef]
- Jia, R.; Chen, Q.; Zhou, Q.; Zhang, R.; Jin, J.; Hu, F.; Liu, X.; Qin, X.; Kang, L.; Zhao, S.; et al. Characteristics of Serum Metabolites in Sporadic Amyotrophic Lateral Sclerosis Patients Based on Gas Chromatography-Mass Spectrometry. Sci. Rep. 2021, 11, 20786. [Google Scholar] [CrossRef]
- Mouchard, M.-L.; Bes, S.; Mignon, M.; Meynial-Denis, D. Fasting Up-Regulates Muscle Glutamine Synthetase While It down-Regulates Liver Glutamine Synthetase in Male Rats during Aging. Eur. E-J. Clin. Nutr. Metab. 2008, 3, e309–e315. [Google Scholar] [CrossRef]
- Dulewicz, M.; Kulczyńska-Przybik, A.; Mroczko, B. Neurogranin and VILIP-1 as Molecular Indicators of Neurodegeneration in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2020, 21, 8335. [Google Scholar] [CrossRef] [PubMed]
- Díez-Guerra, F.J. Neurogranin, a Link between Calcium/Calmodulin and Protein Kinase C Signaling in Synaptic Plasticity. IUBMB Life 2010, 62, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Kvartsberg, H.; Duits, F.H.; Ingelsson, M.; Andreasen, N.; Öhrfelt, A.; Andersson, K.; Brinkmalm, G.; Lannfelt, L.; Minthon, L.; Hansson, O.; et al. Cerebrospinal Fluid Levels of the Synaptic Protein Neurogranin Correlates with Cognitive Decline in Prodromal Alzheimer’s Disease. Alzheimers Dement. 2015, 11, 1180–1190. [Google Scholar] [CrossRef]
- Brosseron, F.; Kleemann, K.; Kolbe, C.; Santarelli, F.; Castro-Gomez, S.; Tacik, P.; Latz, E.; Jessen, F.; Heneka, M.T. Interrelations of Alzheimer’s Disease Candidate Biomarkers Neurogranin, Fatty Acid-Binding Protein 3 and Ferritin to Neurodegeneration and Neuroinflammation. J. Neurochem. 2021, 157, 2210–2224. [Google Scholar] [CrossRef]
- Braunewell, K.H. The Visinin-like Proteins VILIP-1 and VILIP-3 in Alzheimer’s Disease—Old Wine in New Bottles. Front. Mol. Neurosci. 2012, 5, 22224. [Google Scholar] [CrossRef]
- de Wilde, M.C.; Overk, C.R.; Sijben, J.W.; Masliah, E. Meta-Analysis of Synaptic Pathology in Alzheimer’s Disease Reveals Selective Molecular Vesicular Machinery Vulnerability. Alzheimer’s Dement. 2016, 12, 633–644. [Google Scholar] [CrossRef]
- Zhao, C.; Braunewell, K.-H. Expression of the Neuronal Calcium Sensor VILIP-1 in the Rat Hippocampus. Neuroscience 2008, 153, 1202–1212. [Google Scholar] [CrossRef]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical Perspective on Oxidative Stress in Sporadic Amyotrophic Lateral Sclerosis. Free Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S.G. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention. Oxidative Med. Cell. Longev. 2020, 2020, e5021694. [Google Scholar] [CrossRef]
- Motataianu, A.; Serban, G.; Barcutean, L.; Balasa, R. Oxidative Stress in Amyotrophic Lateral Sclerosis: Synergy of Genetic and Environmental Factors. Int. J. Mol. Sci. 2022, 23, 9339. [Google Scholar] [CrossRef]
- González-Mingot, C.; Miana-Mena, F.J.; Iñarrea, P.J.; Iñiguez, C.; Capablo, J.L.; Osta, R.; Gil-Sánchez, A.; Brieva, L.; Larrodé, P. Mitochondrial Aconitase Enzymatic Activity: A Potential Long-Term Survival Biomarker in the Blood of ALS Patients. J. Clin. Med. 2023, 12, 3560. [Google Scholar] [CrossRef] [PubMed]
- Barja, G. Updating the Mitochondrial Free Radical Theory of Aging: An Integrated View, Key Aspects, and Confounding Concepts. Antioxid. Redox Signal. 2013, 19, 1420–1445. [Google Scholar] [CrossRef] [PubMed]
- Matuz-Mares, D.; González-Andrade, M.; Araiza-Villanueva, M.G.; Vilchis-Landeros, M.M.; Vázquez-Meza, H. Mitochondrial Calcium: Effects of Its Imbalance in Disease. Antioxidants 2022, 11, 801. [Google Scholar] [CrossRef]
- Mehta, A.R.; Walters, R.; Waldron, F.M.; Pal, S.; Selvaraj, B.T.; Macleod, M.R.; Hardingham, G.E.; Chandran, S.; Gregory, J.M. Targeting Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Brain Commun. 2019, 1, fcz009. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Heiman-Patterson, T.; Wiedau-Pazos, M.; Liu, S.; Zhang, J.; Apple, S. Edaravone Efficacy in Amyotrophic Lateral Sclerosis with Reduced Forced Vital Capacity: Post-Hoc Analysis of Study 19 (MCI186-19) [Clinical Trial NCT01492686]. PLoS ONE 2022, 17, e0258614. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, M.; Brown, R.H.; Matson, W.; Smart, R.; Hayden, D.; O’Donnell, H.; Flint Beal, M.; Cudkowicz, M. Increased Oxidative Damage to DNA in ALS Patients. Free Radic. Biol. Med. 2000, 29, 652–658. [Google Scholar] [CrossRef]
- Abu-Qare, A.W.; Abou-Donia, M.B. Biomarkers of Apoptosis: Release of Cytochrome c, Activation of Caspase-3, Induction of 8-Hydroxy-2’-Deoxyguanosine, Increased 3-Nitrotyrosine, and Alteration of P53 Gene. J. Toxicol. Env. Health B Crit. Rev. 2001, 4, 313–332. [Google Scholar] [CrossRef]
- Milne, G.L.; Sanchez, S.C.; Musiek, E.S.; Morrow, J.D. Quantification of F2-Isoprostanes as a Biomarker of Oxidative Stress. Nat. Protoc. 2007, 2, 221–226. [Google Scholar] [CrossRef]
- Mitsumoto, H.; Santella, R.M.; Liu, X.; Bogdanov, M.; Zipprich, J.; Wu, H.-C.; Mahata, J.; Kilty, M.; Bednarz, K.; Bell, D.; et al. Oxidative Stress Biomarkers in Sporadic ALS. Amyotroph. Lateral Scler. 2008, 9, 177–183. [Google Scholar] [CrossRef]
- Csala, M.; Kardon, T.; Legeza, B.; Lizák, B.; Mandl, J.; Margittai, É.; Puskás, F.; Száraz, P.; Szelényi, P.; Bánhegyi, G. On the Role of 4-Hydroxynonenal in Health and Disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Dalleau, S.; Baradat, M.; Guéraud, F.; Huc, L. Cell Death and Diseases Related to Oxidative Stress:4-Hydroxynonenal (HNE) in the Balance. Cell Death Differ. 2013, 20, 1615–1630. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.G.; Henry, Y.K.; Mattson, M.P.; Appel, S.H. Presence of 4-Hydroxynonenal in Cerebrospinal Fluid of Patients with Sporadic Amyotrophic Lateral Sclerosis. Ann. Neurol. 1998, 44, 696–699. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.P.; Henry, Y.K.; Henkel, J.S.; Smith, R.G.; Appel, S.H. Increased Lipid Peroxidation in Sera of ALS Patients. Neurology 2004, 62, 1758–1765. [Google Scholar] [CrossRef] [PubMed]
- Han, H.J.; Shin, H.Y.; Choi, Y.-C.; Kim, S.M.; Kim, S.W. Serum Uric Acid Level Predicts the Progression of Amyotrophic Lateral Sclerosis Following Treatment with Edaravone. Redox Rep 2022, 27, 79–84. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Keizman, D.; Ish-Shalom, M.; Berliner, S.; Maimon, N.; Vered, Y.; Artamonov, I.; Tsehori, J.; Nefussy, B.; Drory, V. Low Uric Acid Levels in Serum of Patients with ALS: Further Evidence for Oxidative Stress? J. Neurol. Sci. 2009, 285, 95–99. [Google Scholar] [CrossRef]
- Yu, Z.F.; Bruce-Keller, A.J.; Goodman, Y.; Mattson, M.P. Uric Acid Protects Neurons against Excitotoxic and Metabolic Insults in Cell Culture, and against Focal Ischemic Brain Injury in Vivo. J. Neurosci. Res. 1998, 53, 613–625. [Google Scholar] [CrossRef]
- Gao, X.; Chen, H.; Choi, H.K.; Curhan, G.; Schwarzschild, M.A.; Ascherio, A. Diet, Urate, and Parkinson’s Disease Risk in Men. Am. J. Epidemiol. 2008, 167, 831–838. [Google Scholar] [CrossRef]
- Paganoni, S.; Zhang, M.; Zárate, A.Q.; Jaffa, M.; Yu, H.; Cudkowicz, M.E.; Wills, A.-M. Uric Acid Levels Predict Survival in Men with Amyotrophic Lateral Sclerosis. J. Neurol. 2012, 259, 1923–1928. [Google Scholar] [CrossRef]
- Nagase, M.; Yamamoto, Y.; Miyazaki, Y.; Yoshino, H. Increased Oxidative Stress in Patients with Amyotrophic Lateral Sclerosis and the Effect of Edaravone Administration. Redox Rep. 2016, 21, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Hirayama, T.; Takazawa, T.; Kawabe, K.; Iwasaki, Y. Relationships between Disease Progression and Serum Levels of Lipid, Urate, Creatinine and Ferritin in Japanese Patients with Amyotrophic Lateral Sclerosis: A Cross-Sectional Study. Intern. Med. 2012, 51, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Baek, S.; Park, J.-S.; Piao, L.; Oh, K.-W.; Kim, S.H. Prognostic Role of Serum Levels of Uric Acid in Amyotrophic Lateral Sclerosis. J. Clin. Neurol. 2015, 11, 376–382. [Google Scholar] [CrossRef]
- Neubauer, K.; Kempinski, R.; Matusiewicz, M.; Bednarz-Misa, I.; Krzystek-Korpacka, M. Nonenzymatic Serum Antioxidant Capacity in IBD and Its Association with the Severity of Bowel Inflammation and Corticosteroids Treatment. Medicina 2019, 55, 88. [Google Scholar] [CrossRef] [PubMed]
- Iazzolino, B.; Grassano, M.; Moglia, C.; Canosa, A.; Manera, U.; Vasta, R.; Cabras, S.; Callegaro, S.; Matteoni, E.; Di Pede, F.; et al. High serum uric acid levels are protective against cognitive impairment in amyotrophic lateral sclerosis. J. Neurol. 2024, 271, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Zoccolella, S.; Simone, I.L.; Capozzo, R.; Tortelli, R.; Leo, A.; D’Errico, E.; Logroscino, G. An Exploratory Study of Serum Urate Levels in Patients with Amyotrophic Lateral Sclerosis. J. Neurol. 2011, 258, 238–243. [Google Scholar] [CrossRef]
- Chang, S.-H.; Tian, X.-B.; Wang, J.; Liu, M.-Q.; Huang, C.-N.; Qi, Y.; Zhang, L.-J.; Gao, C.-L.; Zhang, D.-Q.; Sun, L.-S.; et al. Increased Cerebrospinal Fluid Uric Acid Levels in Guillain–Barré Syndrome. Front. Neurol. 2020, 11, 589928. [Google Scholar] [CrossRef]
- Freischmidt, A.; Müller, K.; Ludolph, A.C.; Weishaupt, J.H. Systemic Dysregulation of TDP-43 Binding microRNAs in Amyotrophic Lateral Sclerosis. Acta Neuropathol. Commun. 2013, 1, 42. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Non-Coding Region of C9ORF72 Causes Chromosome 9p-Linked Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Matamala, J.M.; Arias-Carrasco, R.; Sanchez, C.; Uhrig, M.; Bargsted, L.; Matus, S.; Maracaja-Coutinho, V.; Abarzua, S.; van Zundert, B.; Verdugo, R.; et al. Genome-Wide Circulating microRNA Expression Profiling Reveals Potential Biomarkers for Amyotrophic Lateral Sclerosis. Neurobiol. Aging 2018, 64, 123–138. [Google Scholar] [CrossRef]
- Loffreda, A.; Nizzardo, M.; Arosio, A.; Ruepp, M.-D.; Calogero, R.A.; Volinia, S.; Galasso, M.; Bendotti, C.; Ferrarese, C.; Lunetta, C.; et al. miR-129-5p: A Key Factor and Therapeutic Target in Amyotrophic Lateral Sclerosis. Prog. Neurobiol. 2020, 190, 101803. [Google Scholar] [CrossRef] [PubMed]
- Haramati, S.; Chapnik, E.; Sztainberg, Y.; Eilam, R.; Zwang, R.; Gershoni, N.; McGlinn, E.; Heiser, P.W.; Wills, A.-M.; Wirguin, I.; et al. miRNA Malfunction Causes Spinal Motor Neuron Disease. Proc. Natl. Acad. Sci. USA 2010, 107, 13111–13116. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Croce, C.M. Causes and Consequences of microRNA Dysregulation. Cancer J. 2012, 18, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, D.; Comi, G.P.; Bresolin, N.; Corti, S. MicroRNAs as Regulators of Cell Death Mechanisms in Amyotrophic Lateral Sclerosis. J. Cell. Mol. Med. 2019, 23, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Read, D.E.; Deepti, A.; Cawley, K.; Gupta, A.; Oommen, D.; Verfaillie, T.; Matus, S.; Smith, M.A.; Mott, J.L.; et al. Perk-Dependent Repression of miR-106b-25 Cluster Is Required for ER Stress-Induced Apoptosis. Cell Death Dis. 2012, 3, e333. [Google Scholar] [CrossRef]
- De Felice, B.; Annunziata, A.; Fiorentino, G.; Borra, M.; Biffali, E.; Coppola, C.; Cotrufo, R.; Brettschneider, J.; Giordana, M.L.; Dalmay, T.; et al. miR-338-3p Is over-Expressed in Blood, CFS, Serum and Spinal Cord from Sporadic Amyotrophic Lateral Sclerosis Patients. Neurogenetics 2014, 15, 243–253. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Waller, R.; Goodall, E.F.; Milo, M.; Cooper-Knock, J.; Da Costa, M.; Hobson, E.; Kazoka, M.; Wollff, H.; Heath, P.R.; Shaw, P.J.; et al. Serum miRNAs miR-206, 143-3p and 374b-5p as Potential Biomarkers for Amyotrophic Lateral Sclerosis (ALS). Neurobiol. Aging 2017, 55, 123–131. [Google Scholar] [CrossRef]
- Umu, S.U.; Langseth, H.; Bucher-Johannessen, C.; Fromm, B.; Keller, A.; Meese, E.; Lauritzen, M.; Leithaug, M.; Lyle, R.; Rounge, T.B. A Comprehensive Profile of Circulating RNAs in Human Serum. bioRxiv 2017, 15, 242–250. [Google Scholar] [CrossRef]
- Toivonen, J.M.; Manzano, R.; Oliván, S.; Zaragoza, P.; García-Redondo, A.; Osta, R. MicroRNA-206: A Potential Circulating Biomarker Candidate for Amyotrophic Lateral Sclerosis. PLoS ONE 2014, 9, e89065. [Google Scholar] [CrossRef]
- Magen, I.; Yacovzada, N.S.; Yanowski, E.; Coenen-Stass, A.; Grosskreutz, J.; Lu, C.-H.; Greensmith, L.; Malaspina, A.; Fratta, P.; Hornstein, E. Circulating miR-181 Is a Prognostic Biomarker for Amyotrophic Lateral Sclerosis. Nat. Neurosci. 2021, 24, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Freischmidt, A.; Müller, K.; Zondler, L.; Weydt, P.; Mayer, B.; von Arnim, C.A.; Hübers, A.; Dorst, J.; Otto, M.; Holzmann, K.; et al. Serum microRNAs in Sporadic Amyotrophic Lateral Sclerosis. Neurobiol. Aging 2015, 36, 2660.e15–2660.e20. [Google Scholar] [CrossRef] [PubMed]
- Mandolesi, G.; De Vito, F.; Musella, A.; Gentile, A.; Bullitta, S.; Fresegna, D.; Sepman, H.; Di Sanza, C.; Haji, N.; Mori, F.; et al. miR-142-3p Is a Key Regulator of IL-1β-Dependent Synaptopathy in Neuroinflammation. J. Neurosci. 2017, 37, 546–561. [Google Scholar] [CrossRef]
- Shefner, J.M.; Musaro, A.; Ngo, S.T.; Lunetta, C.; Steyn, F.J.; Robitaille, R.; De Carvalho, M.; Rutkove, S.; Ludolph, A.C.; Dupuis, L. Skeletal Muscle in Amyotrophic Lateral Sclerosis. Brain 2023, 146, 4425–4436. [Google Scholar] [CrossRef] [PubMed]
- Freischmidt, A.; Müller, K.; Zondler, L.; Weydt, P.; Volk, A.E.; Božič, A.L.; Walter, M.; Bonin, M.; Mayer, B.; von Arnim, C.A.F.; et al. Serum microRNAs in Patients with Genetic Amyotrophic Lateral Sclerosis and Pre-Manifest Mutation Carriers. Brain 2014, 137 Pt 11, 2938–2950. [Google Scholar] [CrossRef]
- McCombe, P.A.; Henderson, R.D. Effects of Gender in Amyotrophic Lateral Sclerosis. Gend. Med. 2010, 7, 557–570. [Google Scholar] [CrossRef]
- Sheinerman, K.S.; Toledo, J.B.; Tsivinsky, V.G.; Irwin, D.; Grossman, M.; Weintraub, D.; Hurtig, H.I.; Chen-Plotkin, A.; Wolk, D.A.; McCluskey, L.F.; et al. Circulating Brain-Enriched MicroRNAs as Novel Biomarkers for Detection and Differentiation of Neurodegenerative Diseases. Alzheimer’s Res. Ther. 2017, 9, 89. [Google Scholar] [CrossRef]
- Liguori, M.; Nuzziello, N.; Introna, A.; Consiglio, A.; Licciulli, F.; D’errico, E.; Scarafino, A.; Distaso, E.; Simone, I.L. Dysregulation of MicroRNAs and Target Genes Networks in Peripheral Blood of Patients With Sporadic Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2018, 11, 288. [Google Scholar] [CrossRef]
- Tasca, E.; Pegoraro, V.; Merico, A.; Angelini, C. Circulating microRNAs as Biomarkers of Muscle Differentiation and Atrophy in ALS. Clin. Neuropathol. 2016, 35, 22–30. [Google Scholar] [CrossRef]
- Kumar, P.; Dezso, Z.; MacKenzie, C.; Oestreicher, J.; Agoulnik, S.; Byrne, M.; Bernier, F.; Yanagimachi, M.; Aoshima, K.; Oda, Y. Circulating miRNA Biomarkers for Alzheimer’s Disease. PLoS ONE 2013, 8, e69807. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, S.; Zhang, J.; Lou, L.; Liu, W.; Gao, C.; Miao, L.; Sun, F.; Chen, W.; Cao, X.; et al. MicroRNA-142-3p Promotes Renal Cell Carcinoma Progression by Targeting RhoBTB3 to Regulate HIF-1 Signaling and GGT/GSH Pathways. Sci. Rep. 2023, 13, 5935. [Google Scholar] [CrossRef]
- Burns, T.C.; Li, M.D.; Mehta, S.; Awad, A.J.; Morgan, A.A. Mouse Models Rarely Mimic the Transcriptome of Human Neurodegenerative Diseases: A Systematic Bioinformatics-Based Critique of Preclinical Models. Eur. J. Pharmacol. 2015, 759, 101–117. [Google Scholar] [CrossRef]
- García-Segura, L.; Pérez-Andrade, M.; Miranda-Ríos, J. The Emerging Role of MicroRNAs in the Regulation of Gene Expression by Nutrients. J. Nutr. Nutr. 2013, 6, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Siddiqui, S.; Gabriely, G.; Lanser, A.J.; Dake, B.; Murugaiyan, G.; Doykan, C.E.; Wu, P.M.; Gali, R.R.; Iyer, L.K.; et al. Modulating Inflammatory Monocytes with a Unique microRNA Gene Signature Ameliorates Murine ALS. J. Clin. Investig. 2012, 122, 3063–3087. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.A.; Southam, K.A.; Blizzard, C.A.; King, A.E.; Dickson, T.C. Axonal Degeneration, Distal Collateral Branching and Neuromuscular Junction Architecture Alterations Occur Prior to Symptom Onset in the SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. J. Chem. Neuroanat. 2016, 76 Pt A, 35–47. [Google Scholar] [CrossRef]
- Pun, S.; Santos, A.F.; Saxena, S.; Xu, L.; Caroni, P. Selective Vulnerability and Pruning of Phasic Motoneuron Axons in Motoneuron Disease Alleviated by CNTF. Nat. Neurosci. 2006, 9, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Moghadam-Kia, S.; Oddis, C.V.; Aggarwal, R. Approach to Asymptomatic Creatine Kinase Elevation. CCJM 2016, 83, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Boyer, J.G.; Ferrier, A.; Kothary, R. More than a Bystander: The Contributions of Intrinsic Skeletal Muscle Defects in Motor Neuron Diseases. Front. Physiol. 2013, 4, 356. [Google Scholar] [CrossRef]
- Dupuis, L.; de Aguilar, J.-L.G.; Echaniz-Laguna, A.; Eschbach, J.; Rene, F.; Oudart, H.; Halter, B.; Huze, C.; Schaeffer, L.; Bouillaud, F.; et al. Muscle Mitochondrial Uncoupling Dismantles Neuromuscular Junction and Triggers Distal Degeneration of Motor Neurons. PLoS ONE 2009, 4, e5390. [Google Scholar] [CrossRef]
- Manzano, R.; Toivonen, J.M.; Oliván, S.; Calvo, A.C.; Moreno-Igoa, M.; Muñoz, M.J.; Zaragoza, P.; García-Redondo, A.; Osta, R. Altered Expression of Myogenic Regulatory Factors in the Mouse Model of Amyotrophic Lateral Sclerosis. Neurodegener. Dis. 2011, 8, 386–396. [Google Scholar] [CrossRef]
- Ma, G.; Wang, Y.; Li, Y.; Cui, L.; Zhao, Y.; Zhao, B.; Li, K. MiR-206, a Key Modulator of Skeletal Muscle Development and Disease. Int. J. Biol. Sci. 2015, 11, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Sun, X.; Xu, D.; Xiong, Y.; Zuo, B. MicroRNA, miR-374b, Directly Targets Myf6 and Negatively Regulates C2C12 Myoblasts Differentiation. Biochem. Biophys. Res. Commun. 2015, 467, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Joilin, G.; Leigh, P.N.; Newbury, S.F.; Hafezparast, M. An Overview of MicroRNAs as Biomarkers of ALS. Front. Neurol. 2019, 10, 186. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, V.; Merico, A.; Angelini, C. MyomiRNAs Dysregulation in ALS Rehabilitation. Brain Sci. 2019, 9, 8. [Google Scholar] [CrossRef]
- Joyce, N.C.; Oskarsson, B.; Jin, L.-W. Muscle Biopsy Evaluation in Neuromuscular Disorders. Phys. Med. Rehabil. Clin. North. Am. 2012, 23, 609–631. [Google Scholar] [CrossRef]
- Shang, H.-F.; Chen, X.-P.; Wei, Q.-Q.; Ou, R.-W.; Hou, Y.-B.; Zhang, L.-Y.; Yuan, X.-Q.; Yao, Y.-Q.; Jia, D.-S.; Zhang, Q.; et al. Creatine Kinase in the Diagnosis and Prognostic Prediction of Amyotrophic Lateral Sclerosis: A Retrospective Case-Control Study. Neural Regen. Res. 2021, 16, 591. [Google Scholar] [CrossRef]
- Awano, H.; Matsumoto, M.; Nagai, M.; Shirakawa, T.; Maruyama, N.; Iijima, K.; Nabeshima, Y.-I.; Matsuo, M. Diagnostic and Clinical Significance of the Titin Fragment in Urine of Duchenne Muscular Dystrophy Patients. Clin. Chim. Acta 2018, 476, 111–116. [Google Scholar] [CrossRef]
- Al-Sarraj, S.; King, A.; Cleveland, M.; Pradat, P.-F.; Corse, A.; Rothstein, J.D.; Leigh, P.N.; Abila, B.; Bates, S.; Wurthner, J.; et al. Mitochondrial Abnormalities and Low Grade Inflammation Are Present in the Skeletal Muscle of a Minority of Patients with Amyotrophic Lateral Sclerosis; an Observational Myopathology Study. Acta Neuropathol. Commun. 2014, 2, 165. [Google Scholar] [CrossRef]
- Ceccanti, M.; Pozzilli, V.; Cambieri, C.; Libonati, L.; Onesti, E.; Frasca, V.; Fiorini, I.; Petrucci, A.; Garibaldi, M.; Palma, E.; et al. Creatine Kinase and Progression Rate in Amyotrophic Lateral Sclerosis. Cells 2020, 9, 1174. [Google Scholar] [CrossRef]
- Tai, H.; Cui, L.; Liu, M.; Guan, Y.; Li, X.; Shen, D.; Zhang, K.; Liu, S.; Wu, S.; Ding, Q.; et al. Creatine Kinase Level and Its Relationship with Quantitative Electromyographic Characteristics in Amyotrophic Lateral Sclerosis. Clin. Neurophysiol. 2018, 129, 926–930. [Google Scholar] [CrossRef]
- Lang, F.; Aravamudhan, S.; Nolte, H.; Tuerk, C.; Hölper, S.; Müller, S.; Günther, S.; Blaauw, B.; Braun, T.; Krüger, M. Dynamic Changes in the Mouse Skeletal Muscle Proteome during Denervation-Induced Atrophy. Dis. Models Mech. 2017, 10, 881–896. [Google Scholar] [CrossRef]
- Kontrogianni-Konstantopoulos, A.; Ackermann, M.A.; Bowman, A.L.; Yap, S.V.; Bloch, R.J. Muscle Giants: Molecular Scaffolds in Sarcomerogenesis. Physiol. Rev. 2009, 89, 1217–1267. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, N.; Asai, T.; Abe, C.; Inada, A.; Kawauchi, T.; Miyashita, K.; Maeda, M.; Matsuo, M.; Nabeshima, Y.-I. Establishment of a Highly Sensitive Sandwich ELISA for the N-Terminal Fragment of Titin in Urine. Sci. Rep. 2016, 6, 39375. [Google Scholar] [CrossRef] [PubMed]
- Tanihata, J.; Minamisawa, S. Urinary Titin Is Not an Early Biomarker of Skeletal Muscle Atrophy Induced by Muscle Denervation in Mice. PLoS ONE 2023, 18, e0289185. [Google Scholar] [CrossRef]
- Yamada, S.; Hashizume, A.; Hijikata, Y.; Ito, D.; Kishimoto, Y.; Iida, M.; Koike, H.; Hirakawa, A.; Katsuno, M. Ratio of Urinary N-Terminal Titin Fragment to Urinary Creatinine Is a Novel Biomarker for Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1072–1079. [Google Scholar] [CrossRef]
- Meng, L.; Bian, A.; Jordan, S.; Wolff, A.; Shefner, J.M.; Andrews, J. Profile of Medical Care Costs in Patients with Amyotrophic Lateral Sclerosis in the Medicare Programme and under Commercial Insurance. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 134–142. [Google Scholar] [CrossRef]
- Rolland, Y.; Vellas, B. CHAPTER 73-Sarcopenia. In Brocklehurst’s Textbook of Geriatric Medicine and Gerontology, 7th ed.; Fillit, H.M., Rockwood, K., Woodhouse, K., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2010; pp. 587–593. [Google Scholar] [CrossRef]
- Al-Zaidy, S.A.; Sahenk, Z.; Rodino-Klapac, L.R.; Kaspar, B.; Mendell, J.R. Follistatin Gene Therapy Improves Ambulation in Becker Muscular Dystrophy. J. Neuromuscul. Dis. 2015, 2, 185–192. [Google Scholar] [CrossRef]
- Morrison, B.M.; Lachey, J.L.; Warsing, L.C.; Ting, B.L.; Pullen, A.E.; Underwood, K.W.; Kumar, R.; Sako, D.; Grinberg, A.; Wong, V.; et al. A Soluble Activin Type IIB Receptor Improves Function in a Mouse Model of Amyotrophic Lateral Sclerosis. Exp. Neurol. 2009, 217, 258–268. [Google Scholar] [CrossRef]
- Li, J.; Fredericks, M.; Cannell, M.; Wang, K.; Sako, D.; Maguire, M.C.; Grenha, R.; Liharska, K.; Krishnan, L.; Bloom, T.; et al. ActRIIB:ALK4-Fc Alleviates Muscle Dysfunction and Comorbidities in Murine Models of Neuromuscular Disorders. J. Clin. Investig. 2021, 131, e138634. [Google Scholar] [CrossRef]
- Duong, T.; Darras, B.; Morrow, J.; Muntoni, F.; Servais, L.; Rabbia, M.; Gerber, M.; Kletzl, H.; Gaki, E.; Fletcher, S.; et al. P227 MANATEE: GYM329 (RO7204239) in Combination with Risdiplam Treatment in Patients with Spinal Muscular Atrophy (SMA). Neuromuscul. Disord. 2023, 33, S92. [Google Scholar] [CrossRef]
- Donini, L.; Tanel, R.; Zuccarino, R.; Basso, M. Protein Biomarkers for the Diagnosis and Prognosis of Amyotrophic Lateral Sclerosis. Neurosci. Res. 2023, 197, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liem, R.K.H. α-Internexin and Peripherin: Expression, Assembly, Functions, and Roles in Disease. Methods Enzym. 2016, 568, 477–507. [Google Scholar] [CrossRef]
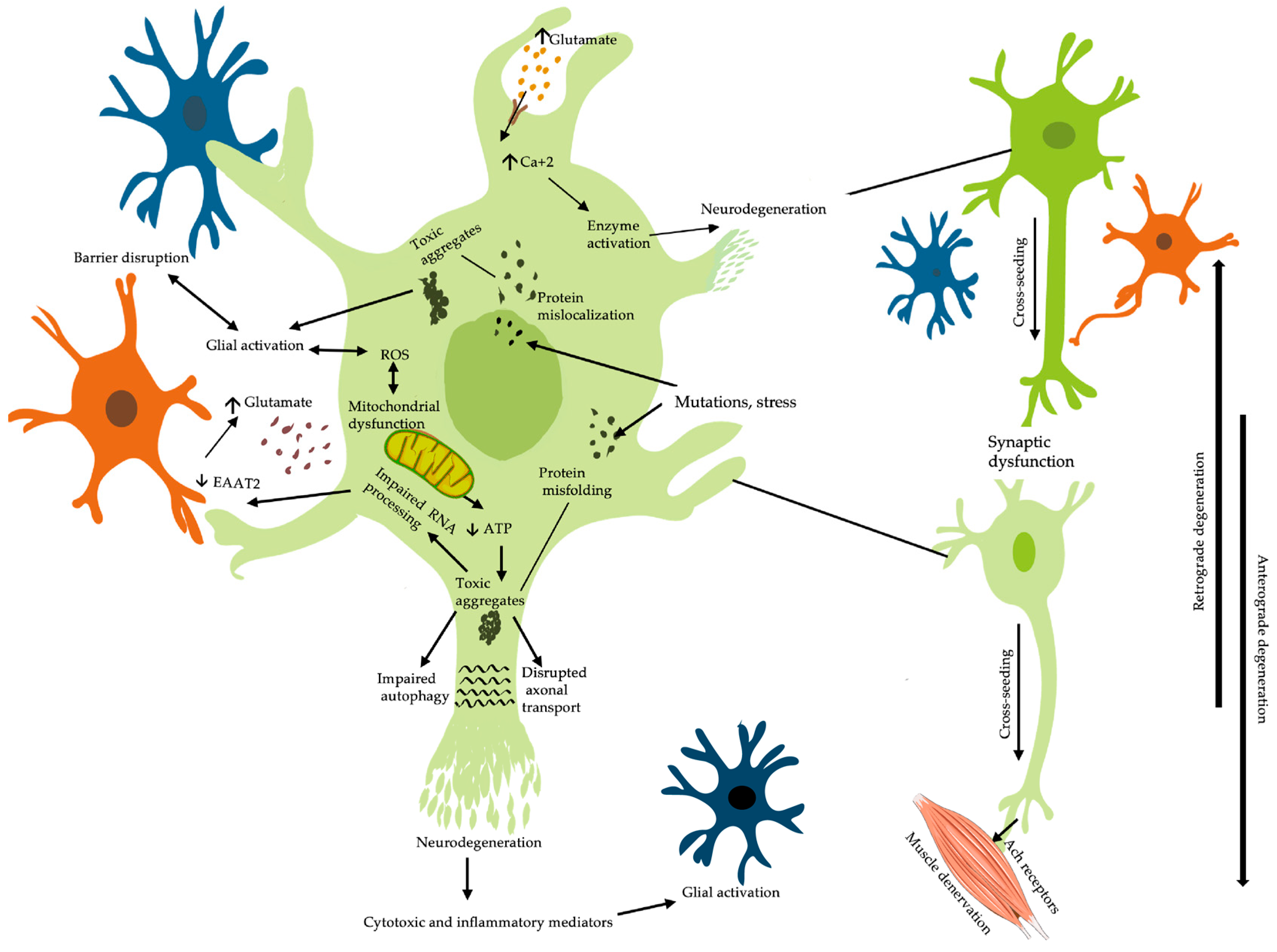
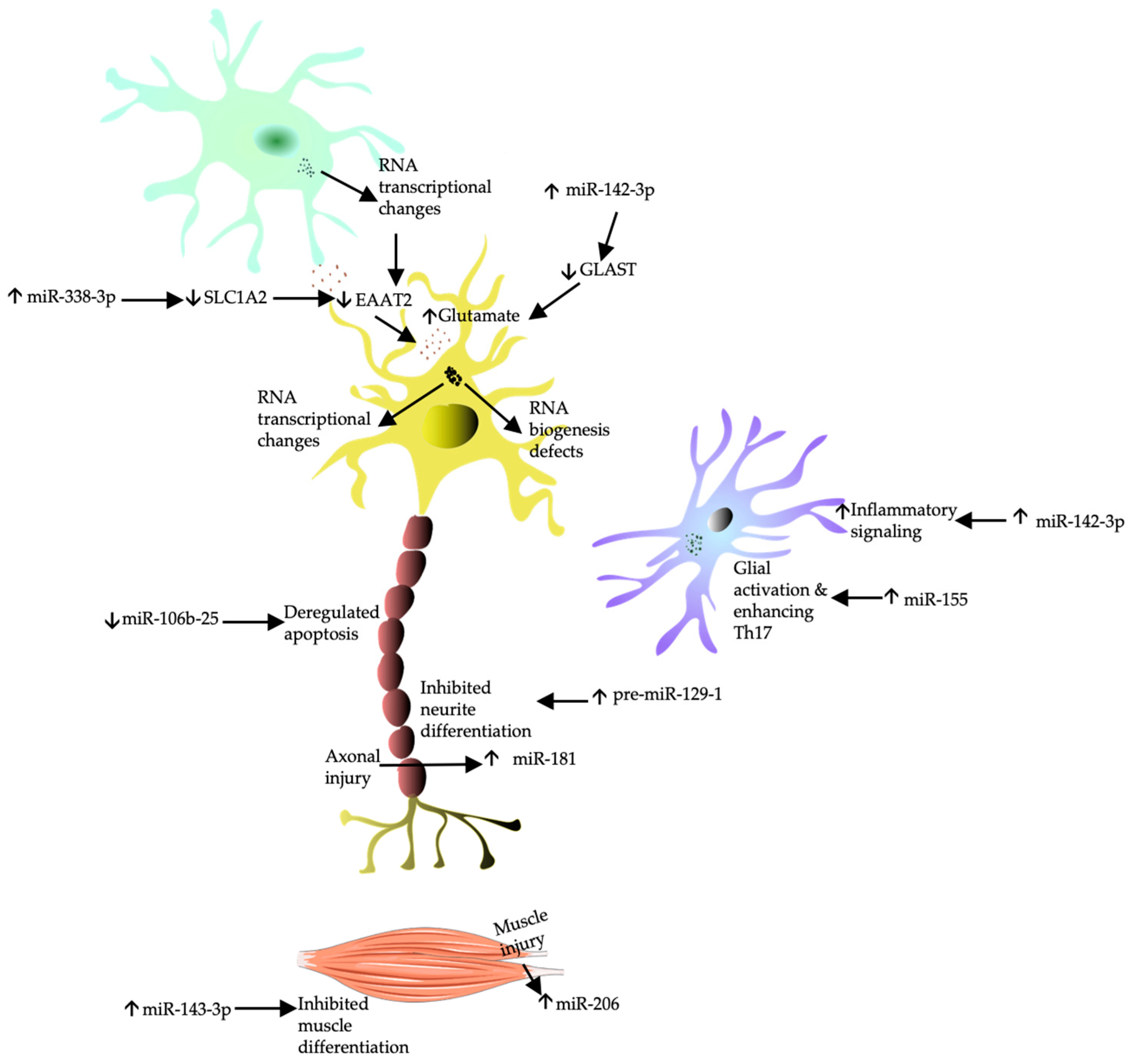


{kind=link}
{kind=link}
{kind=link}
| Pathophysiology | Biomarker | Detection Technique | Clinical Findings |
|---|---|---|---|
| Proteinopathy | ELISA [57,58,59,63] | Assists diagnosis vs. controls [57,58,59,60] Prognostic [57,59,61] | |
| TDP-43 | Simoa [60,61] | ||
| Mass spectrometry [62] | |||
| SOD1 | ELISA [78,81] LC-MS [82] | Assists diagnosis vs. controls [82] Prognostic [83] | |
| DPRs | MSM [18,91,92] Simoa [93] | Diagnostic of mutation [18,91,92,93] | |
| Neuroinflammation | GFAP | ELISA [163] Simoa [164] | Assists diagnosis vs. controls [156,162] Prognostic [164] |
| S100B | ELISA [176,177] | Prognostic [176,177]. | |
| HMGB1 ab | ELISA [184] | Assists diagnosis vs. controls [184] | |
| sCD14 | ELISA [187] | Assists diagnosis vs. controls [187] Prognostic [176,187] | |
| TREM2 | ELISA [194,195] | Assists diagnosis vs. controls [194,195] Prognostic [193,195] | |
| CHIT1 & YKL-40 | ELISA [200,201] LC/MS [202] | Assists diagnosis vs. controls [136,201,203] Assists diagnosis vs. mimics [135,201,203] Prognostic [135,136,198,200,202] | |
| BBB disruption | Q-Alb | Immunoturbidimetric assay [219] | Prognostic [218] |
| Oxidative stress | 8OH2′dG IsoP | ELISA [249,252] Liquid chromatography [252] | Assists diagnosis vs. controls [249,252] Prognostic [249,252] |
| HNE | ELISA [255,256] Liquid chromatography [255,256] | Assists diagnosis vs. controls [255,256] | |
| UA | Liquid chromatography [269] | Assists diagnosis vs. controls [259,263,264,265] Prognostic [257,259,264,265] | |
| Synaptopathy | Glutamate | Liquid chromatography [226,228] Ion-exchange chromatography [224] Gas chromatography-MS [234] (1) H NMR spectroscopy [233] | Assists diagnosis vs controls [224,231] Prognostic [231,232] |
| Aberrant RNA processing | miR-181 | PCR [286] | Assists diagnosis vs. controls and prognostic [286] |
| miR-206 | PCR [280,282,291,305] | Assists diagnosis vs. controls [280,283,291,305] | |
| miR-338-3p | PCR [278] | Assists diagnosis vs. controls and prognostic [278]. | |
| [278].Neurodegeneration | NSE | ECLIA [98] | Assists diagnosis vs. controls and mimics [98]. |
| p75ECD | ELISA [102,105] | Assists diagnosis vs. controls [102,105] Prognostic [102,105] | |
| NFs | ELISA [111,114,115,116,117,118,119,120,121,134,135,136] Simoa [61,117,123,124] ELICA [125,126] Multiplex method [126] MSM [130] | Assists diagnosis vs. controls [112,113,115,122,124,125,128,129,131,132,133]. Assists diagnosis vs. mimics [114,118,124,128,129,130,131,132]. Prognostic [61,111,114,115,116,117,119,121,123,124,125,128,129,132,136] | |
| Tau | ELISA [8,78,107,144,145,146] Luminex [147], Simoa [94] CLIA [148] | Prognostic [8,94,144,147] | |
| Muscle changes | CK | Enzymatic method [307] | Prognostic [312] |
| N-titin | ELISA [318] | Assists diagnosis vs. controls and prognostic [318] | |
| myostatin/follistatin | ELISA [291] | Assists diagnosis vs. controls [291] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshehri, R.S.; Abuzinadah, A.R.; Alrawaili, M.S.; Alotaibi, M.K.; Alsufyani, H.A.; Alshanketi, R.M.; AlShareef, A.A. A Review of Biomarkers of Amyotrophic Lateral Sclerosis: A Pathophysiologic Approach. Int. J. Mol. Sci. 2024, 25, 10900. https://doi.org/10.3390/ijms252010900
Alshehri RS, Abuzinadah AR, Alrawaili MS, Alotaibi MK, Alsufyani HA, Alshanketi RM, AlShareef AA. A Review of Biomarkers of Amyotrophic Lateral Sclerosis: A Pathophysiologic Approach. International Journal of Molecular Sciences. 2024; 25(20):10900. https://doi.org/10.3390/ijms252010900
Chicago/Turabian StyleAlshehri, Rawiah S., Ahmad R. Abuzinadah, Moafaq S. Alrawaili, Muteb K. Alotaibi, Hadeel A. Alsufyani, Rajaa M. Alshanketi, and Aysha A. AlShareef. 2024. "A Review of Biomarkers of Amyotrophic Lateral Sclerosis: A Pathophysiologic Approach" International Journal of Molecular Sciences 25, no. 20: 10900. https://doi.org/10.3390/ijms252010900