
Lymphocytes Change Their Phenotype and Function in Systemic Lupus Erythematosus and Lupus Nephritis

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Β Lymphocytes Related Abnormalities
2.1. Dysregulated Function of B Lymphocytes
2.1.1. Loss of Self-Tolerance
2.1.2. Intrafollicular and Extrafollicular B Lymphocyte Activation
2.1.3. Cytokine Production
2.1.4. Autoantibodies Production
2.2. Changes in B Cell Phenotype Related to SLE
2.2.1. Expression of IgD and CD27 Molecules
2.2.2. Age Associated B Cells
2.2.3. B Regulatory Cells
3. T Lymphocytes Related Abnormalities
3.1. CD4 Lymphocytes
3.1.1. T Helper 1 Lymphocytes
3.1.2. T Helper 2 Lymphocytes
3.1.3. Regulatory T Lymphocytes
Interactions of Regulatory T Cells with Other T Lymphocyte Subtypes
The Role of FoxP3 Isoforms in the Function of Regulatory T Cells
3.1.4. T Helper 22 Lymphocytes
3.1.5. T Follicular Helper Lymphocytes
3.1.6. T Helper 9 Lymphocytes
3.1.7. T Helper 17 Lymphocytes
3.2. CD8+ T Lymphocytes
3.3. γδT Cells
3.4. Double Negative T Lymphocytes
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCs | Age Associated B cells |
| ANA | Antinuclear antibodies |
| AP-1 | Activator protein-1 |
| APCs | Antigen-presenting cells |
| APRIL | A proliferative-inducing ligand |
| ASC | Antibody-secreting cells |
| asN | Activated naïve B cells |
| BAFF-Rs | B-cell activating factor receptors |
| BCMA | B cell maturation antigen |
| BCRs | B-cell receptors |
| BLys | B lymphocyte stimulator |
| Bregs | B regulatory cells |
| CCR6 | C-C-motif chemokine receptor 6 |
| CD | Clusters of differentiation |
| CM | Central memory |
| CNS | Central Nervous System |
| cTfh | Circulating Tfh |
| CTLA-4 | Cytotoxic T-lymphocyte associated protein 4 |
| CXCR5 | C-X-C chemokine receptor type 5 |
| DCs | Dendritic cells |
| DN | Double negative |
| DN T cells | Double-Negative T cells |
| EF | Extrafollicular |
| GARP | Glycoprotein A Repetitions Predominant |
| GCs | Germinal centers |
| GITR | Glucocorticoid-induced TNFR-related protein |
| GWAS | Genome-wide association studies |
| IC | Immune-complex |
| ICOS | Inducible costimulator |
| IFNs | Interferons |
| IgD | Immunoglobulin D |
| ILs | Interleukins |
| IRF4 | Interferon Regulatory Factor 4 |
| LAP | Latency-associated peptide |
| LDGs | Low-density granulocytes |
| MHC | Major histocompatibility complex |
| mTOR | Mammalian Target of Rapamycin |
| NETs | Neutrophil extracellular traps |
| NFAT | Nuclear factor of activated T-cells |
| NGS | Next generation sequencing |
| NK | Natural killer |
| NLE | Neonatal lupus erythematosus |
| PD-1 | Programmed death-1 |
| pDC | Plasmacytoid DC |
| PLC-γ2 | Phospholipase C-γ2 |
| PRRs | Pattern recognition receptors |
| ROCK | Rho-associated coiled-coil domain protein kinase |
| SFB | Segmented filamentous bacteria |
| SLAMF4 | Signaling lymphocytic activation molecule family member 4 |
| SLE | Systemic lupus erythematosus |
| SLEDAI | Systemic Lupus Erythematosus Disease Activity Index |
| TACI | Transmembrane activator, calcium modulator and cyclophilin ligand interactor |
| Tfh | T follicular helper |
| Tfr | Follicular regulatory T cells |
| TGF-β | Transforming growth factor-b |
| Th | T helper lymphocytes |
| Th1 | T helper 1 cells |
| Th17 | T helper 17 lymphocytes |
| TLRs | Toll-like receptors |
| TNF | Tumor necrosis factor |
| Tregs | Regulatory T cells |
References
- Yap, D.Y.H.; Chan, T.M. B Cell Abnormalities in Systemic Lupus Erythematosus and Lupus Nephritis—Role in Pathogenesis and Effect of Immunosuppressive Treatments. Int. J. Mol. Sci. 2019, 20, 6231. [Google Scholar] [CrossRef] [PubMed]
- Fanouriakis, A.; Tziolos, N.; Bertsias, G.; Boumpas, D.T. Update οn the diagnosis and management of systemic lupus erythematosus. Ann. Rheum. Dis. 2020, 80, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Kaul, A.; Gordon, C.; Crow, M.K.; Touma, Z.; Urowitz, M.B.; van Vollenhoven, R.; Ruiz-Irastorza, G.; Hughes, G. Systemic lupus erythematosus. Nat. Rev. Dis. Primers 2016, 2, 16039. [Google Scholar] [CrossRef] [PubMed]
- Tselios, K.; Gladman, D.; Touma, Z.; Su, J.; Anderson, N.; Urowitz, M. Disease course patterns in systemic lupus erythematosus. Lupus 2018, 28, 114–122. [Google Scholar] [CrossRef]
- Anders, H.J.; Saxena, R.; Zhao, M.H.; Parodis, I.; Salmon, J.E.; Mohan, C. Lupus nephritis. Nat. Rev. Dis. Primers 2020, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Almaani, S.; Meara, A.; Rovin, B.H. Update on Lupus Nephritis. Clin. J. Am. Soc. Nephrol. 2017, 12, 825–835. [Google Scholar] [CrossRef]
- Parikh, S.V.; Almaani, S.; Brodsky, S.; Rovin, B.H. Update on Lupus Nephritis: Core Curriculum 2020. Am. J. Kidney Dis. 2020, 76, 265–281. [Google Scholar] [CrossRef]
- Tsokos, G.C. Systemic lupus erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef]
- Frangou, E.; Vassilopoulos, D.; Boletis, J.; Boumpas, D.T. An emerging role of neutrophils and NETosis in chronic inflammation and fibrosis in systemic lupus erythematosus (SLE) and ANCA-associated vasculitides (AAV): Implications for the pathogenesis and treatment. Autoimmun. Rev. 2019, 18, 751–760. [Google Scholar] [CrossRef]
- Weidenbusch, M.; Kulkarni, O.P.; Anders, H.-J. The innate immune system in human systemic lupus erythematosus. Clin. Sci. 2017, 131, 625–634. [Google Scholar] [CrossRef]
- Muñoz, L.E.; Lauber, K.; Schiller, M.; Manfredi, A.A.; Herrmann, M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat. Rev. Rheumatol. 2010, 6, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.R.; Pisitkun, P.; Voynova, E.; Deane, J.A.; Scott, B.L.; Caspi, R.R.; Bolland, S. Dual signaling by innate and adaptive immune receptors is required for TLR7-induced B-cell–mediated autoimmunity. Proc. Natl. Acad. Sci. USA 2012, 109, 16276–16281. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Lu, M.-P.; Wang, J.-H.; Xu, M.; Yang, S.-R. Immunological pathogenesis and treatment of systemic lupus erythematosus. World J. Pediatr. 2019, 16, 19–30. [Google Scholar] [CrossRef]
- Courtney, P.A.; Crockard, A.D.; Williamson, K.; Irvine, A.E.; Kennedy, R.J.; Bell, A.L. Increased apoptotic peripheral blood neutrophils in systemic lupus erythematosus: Relations with disease activity, antibodies to double stranded DNA, and neutropenia. Ann. Rheum. Dis. 1999, 58, 309–314. [Google Scholar] [CrossRef]
- Donnelly, S.; Roake, W.; Brown, S.; Young, P.; Naik, H.; Wordsworth, P.; Isenberg, D.A.; Reid, K.B.M.; Eggleton, P. Impaired recognition of apoptotic neutrophils by the C1q/calreticulin and CD91 pathway in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 1543–1556. [Google Scholar] [CrossRef]
- Alves, C.M.O.S.; Marzocchi-Machado, C.M.; Louzada-Junior, P.; Azzolini, A.E.C.S.; Polizello, A.C.M.; de Carvalho, I.F.; Lucisano-Valim, Y.M. Superoxide anion production by neutrophils is associated with prevalent clinical manifestations in systemic lupus erythematosus. Clin. Rheumatol. 2007, 27, 701–708. [Google Scholar] [CrossRef]
- Banchereau, R.; Hong, S.; Cantarel, B.; Baldwin, N.; Baisch, J.; Edens, M.; Cepika, A.-M.; Acs, P.; Turner, J.; Anguiano, E.; et al. Personalized Immunomonitoring Uncovers Molecular Networks that Stratify Lupus Patients. Cell 2016, 165, 551–565. [Google Scholar] [CrossRef]
- Coit, P.; Yalavarthi, S.; Ognenovski, M.; Zhao, W.; Hasni, S.; Wren, J.D.; Kaplan, M.J.; Sawalha, A.H. Epigenome profiling reveals significant DNA demethylation of interferon signature genes in lupus neutrophils. J. Autoimmun. 2015, 58, 59–66. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef]
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting Neutrophils Induce Endothelial Damage, Infiltrate Tissues, and Expose Immunostimulatory Molecules in Systemic Lupus Erythematosus. J. Immunol. 2011, 187, 538–552. [Google Scholar] [CrossRef] [PubMed]
- Leffler, J.; Martin, M.; Gullstrand, B.; Tydén, H.; Lood, C.; Truedsson, L.; Bengtsson, A.A.; Blom, A.M. Neutrophil Extracellular Traps That Are Not Degraded in Systemic Lupus Erythematosus Activate Complement Exacerbating the Disease. J. Immunol. 2012, 188, 3522–3531. [Google Scholar] [CrossRef] [PubMed]
- Truedsson, L.; Bengtsson, A.A.; Sturfelt, G. Complement deficiencies and systemic lupus erythematosus. Autoimmunity 2007, 40, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Dall’Era, M.C.; Cardarelli, P.M.; Preston, B.T.; Witte, A.; Davis, J.C., Jr. Type I interferon correlates with serological and clinical manifestations of SLE. Ann. Rheum. Dis. 2005, 64, 1692–1697. [Google Scholar] [CrossRef]
- Song, G.G.; Kim, J.-H.; Seo, Y.H.; Choi, S.J.; Ji, J.D.; Lee, Y.H. Associations between interleukin 1 polymorphisms and susceptibility to systemic lupus erythematosus: A meta-analysis. Hum. Immunol. 2013, 75, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Onda, H.; Tanigawa, A.; Ohshima, S.; Fujiwara, H.; Mima, T.; Katada, Y.; Deguchi, H.; Suemura, M.; Miyake, T.; et al. Isolation and Expression Profiling of Genes Upregulated in the Peripheral Blood Cells of Systemic Lupus Erythematosus Patients. DNA Res. 2005, 12, 429–439. [Google Scholar] [CrossRef]
- Jørgensen, T.N.; Roper, E.; Thurman, J.M.; Marrack, P.; Kotzin, B.L. Type I interferon signaling is involved in the spontaneous development of lupus-like disease in B6.Nba2 and (B6.Nba2 × NZW)F1 mice. Genes Immun. 2007, 8, 653–662. [Google Scholar] [CrossRef]
- Bruera, S.; Chavula, T.; Madan, R.; Agarwal, S.K. Targeting type I interferons in systemic lupus erythematous. Front. Pharmacol. 2023, 13, 1046687. [Google Scholar] [CrossRef]
- Canny, S.P.; Jackson, S.W. B cells in systemic lupus erythematosus: From disease mechanisms to targeted therapies. Rheum. Dis. Clin. N. Am. 2021, 47, 395–413. [Google Scholar] [CrossRef]
- Tenbrock, K.; Rauen, T. T cell dysregulation in SLE. Clin. Immunol. 2022, 239, 109031. [Google Scholar] [CrossRef]
- Frangou, E.A.; Bertsias, G.K.; Boumpas, D.T. Gene expression and regulation in systemic lupus erythematosus. Eur. J. Clin. Investig. 2013, 43, 1084–1096. [Google Scholar] [CrossRef]
- Holborow, E.J.; Weir, D.M.; Johnson, G.D. A Serum Factor in Lupus Erythematosus with Affinity for Tissue Nuclei. BMJ 1957, 2, 732–734. [Google Scholar] [CrossRef] [PubMed]
- Chan, O.T.; Hannum, L.G.; Haberman, A.M.; Madaio, M.P.; Shlomchik, M.J. A Novel Mouse with B Cells but Lacking Serum Antibody Reveals an Antibody-independent Role for B Cells in Murine Lupus. J. Exp. Med. 1999, 189, 1639–1648. [Google Scholar] [CrossRef]
- Shlomchik, M.J.; Chan, O.I.M.; Madaio, M.P. The central and multiple roles of B cells in lupus pathogenesis. Immunol. Rev. 1999, 169, 107–121. [Google Scholar] [CrossRef]
- Suzuki, N.; Sakane, T. Induction of excessive B cell proliferation and differentiation by an in vitro stimulus in culture in human systemic lupus erythematosus. J. Clin. Investig. 1989, 83, 937–944. [Google Scholar] [CrossRef]
- Dörner, T.; Giesecke, C.; E Lipsky, P. Mechanisms of B cell autoimmunity in SLE. Arthritis Res. Ther. 2011, 13, 243. [Google Scholar] [CrossRef]
- Karrar, S.; Graham, D.S.C. Review: Abnormal B Cell Development in Systemic Lupus Erythematosus: What the Genetics Tell Us. Arthritis Rheumatol. 2018, 70, 496–507. [Google Scholar] [CrossRef]
- Nemazee, D. Mechanisms of central tolerance for B cells. Nat. Rev. Immunol. 2017, 17, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Iwata, S.; Tanaka, Y. B-cell subsets, signaling and their roles in secretion of autoantibodies. Lupus 2016, 25, 850–856. [Google Scholar] [CrossRef]
- Kang, N.; Liu, X.; You, X.; Sun, W.; Haneef, K.; Sun, X.; Liu, W. Aberrant B-Cell Activation in Systemic Lupus Erythematosus. Kidney Dis. 2022, 8, 437–445. [Google Scholar] [CrossRef]
- Kitaura, Y.; Jang, I.K.; Wang, Y.; Han, Y.-C.; Inazu, T.; Cadera, E.J.; Schlissel, M.; Hardy, R.R.; Gu, H. Control of the B Cell-Intrinsic Tolerance Programs by Ubiquitin Ligases Cbl and Cbl-b. Immunity 2007, 26, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Juang, Y.-T.; Chowdhury, B.; Magilavy, A.; Fisher, C.U.; Nguyen, H.; Nambiar, M.P.; Kyttaris, V.; Weinstein, A.; Bahjat, R.; et al. Differential Expression and Molecular Associations of Syk in Systemic Lupus Erythematosus T Cells. J. Immunol. 2008, 181, 8145–8152. [Google Scholar] [CrossRef]
- Maleknia, S.; Salehi, Z.; Tabar, V.R.; Sharifi-Zarchi, A.; Kavousi, K. An integrative Bayesian network approach to highlight key drivers in systemic lupus erythematosus. Arthritis Res. Ther. 2020, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Xu, H.; Zhang, C.; Hong, X.; Liu, D.; Tang, D.; Xiong, Z.; Dai, Y. Immune cell and TCR/BCR repertoire profiling in systemic lupus erythematosus patients by single-cell sequencing. Aging 2021, 13, 24432–24448. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-F.; Chang, C.-B.; Hsu, J.-M.; Lu, M.-C.; Lai, N.-S.; Li, C.; Tung, C.-H. Hydroxychloroquine inhibits CD154 expression in CD4+ T lymphocytes of systemic lupus erythematosus through NFAT, but not STAT5, signaling. Arthritis Res. Ther. 2017, 19, 183. [Google Scholar] [CrossRef]
- Fujii, Y.; Fujii, K.; Iwata, S.; Suzuki, K.; Azuma, T.; Saito, K.; Tanaka, Y. Abnormal intracellular distribution of NFAT1 in T lymphocytes from patients with systemic lupus erythematosus and characteristic clinical features. Clin. Immunol. 2006, 119, 297–306. [Google Scholar] [CrossRef]
- Han, Y.; Zeng, F.; Tan, G.; Yang, C.; Tang, H.; Luo, Y.; Feng, J.; Xiong, H.; Guo, Q. Hydrogen Sulfide Inhibits Abnormal Proliferation of Lymphocytes via AKT/GSK3β Signal Pathway in Systemic Lupus Erythematosus Patients. Cell. Physiol. Biochem. 2013, 31, 795–804. [Google Scholar] [CrossRef]
- Tang, H.; Tan, G.; Guo, Q.; Pang, R.; Zeng, F. Abnormal activation of the Akt-GSK3beta signaling pathway in peripheral blood T cells from patients with systemic lupus erythematosus. Cell Cycle 2009, 8, 2789–2793. [Google Scholar] [CrossRef]
- Ji, Y.R.; Yang, Z.X.; Han, Z.-B.; Meng, L.; Liang, L.; Feng, X.M.; Yang, S.G.; Chi, Y.; Chen, D.D.; Wang, Y.W.; et al. Mesenchymal Stem Cells Support Proliferation and Terminal Differentiation of B Cells. Cell. Physiol. Biochem. 2012, 30, 1526–1537. [Google Scholar] [CrossRef]
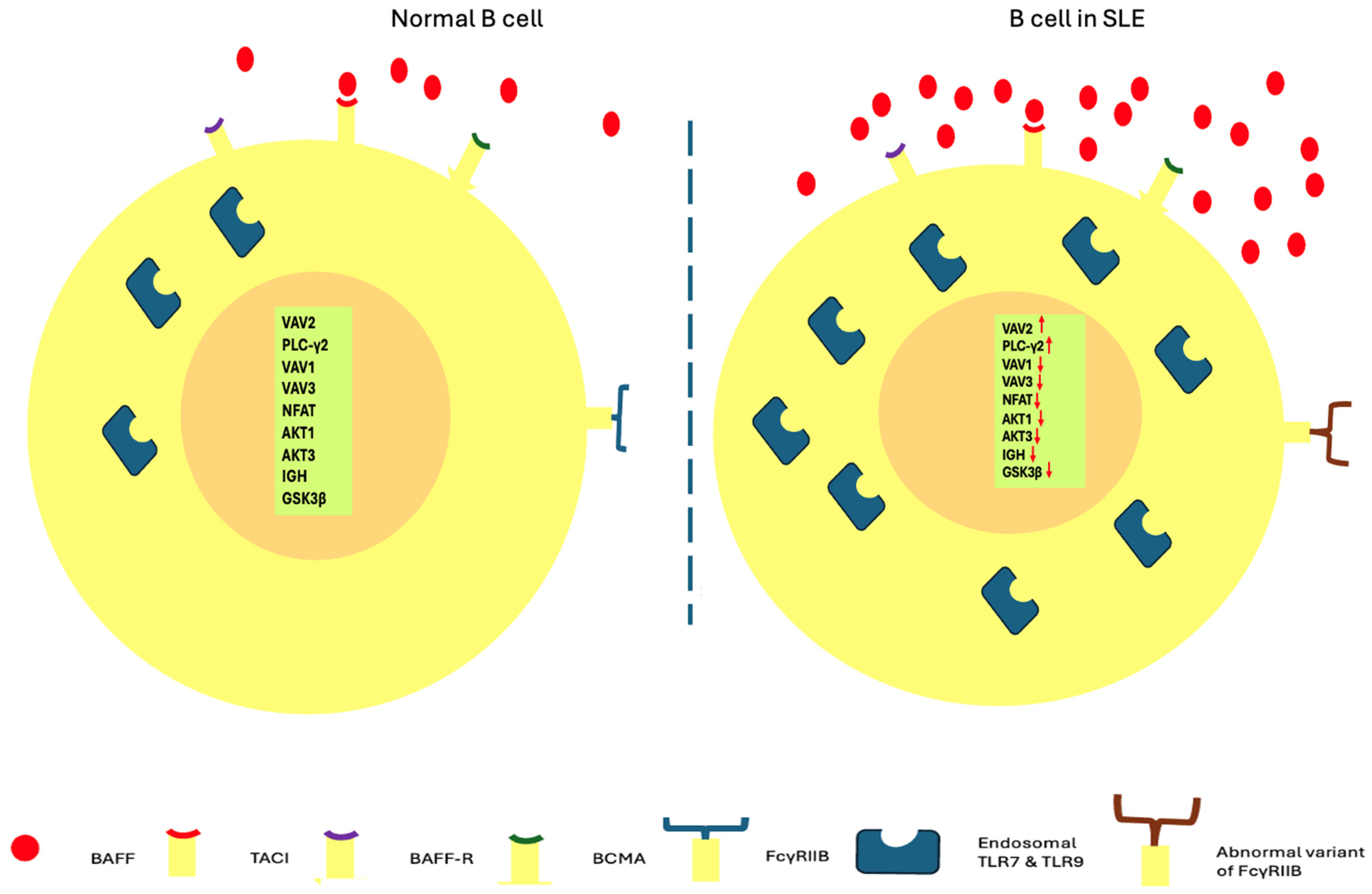
- Taher, T.E.; Parikh, K.; Flores-Borja, F.; Mletzko, S.; Isenberg, D.A.; Peppelenbosch, M.P.; Mageed, R.A. Protein phosphorylation and kinome profiling reveal altered regulation of multiple signaling pathways in B lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum. 2010, 62, 2412–2423. [Google Scholar] [CrossRef]
- Rawlings, D.J.; Schwartz, M.A.; Jackson, S.W.; Meyer-Bahlburg, A. Integration of B cell responses through Toll-like receptors and antigen receptors. Nat. Rev. Immunol. 2012, 12, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, S.; Greidinger, E.L. Endosomal Toll-like receptors in autoimmunity: Mechanisms for clinical diversity. Therapy 2009, 6, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.R.; Shupe, J.; Nickerson, K.; Kashgarian, M.; Flavell, R.A.; Shlomchik, M.J. Toll-like Receptor 7 and TLR9 Dictate Autoantibody Specificity and Have Opposing Inflammatory and Regulatory Roles in a Murine Model of Lupus. Immunity 2006, 25, 417–428. [Google Scholar] [CrossRef]
- Berland, R.; Fernandez, L.; Kari, E.; Han, J.-H.; Lomakin, I.; Akira, S.; Wortis, H.H.; Kearney, J.F.; Ucci, A.A.; Imanishi-Kari, T. Toll-like Receptor 7-Dependent Loss of B Cell Tolerance in Pathogenic Autoantibody Knockin Mice. Immunity 2006, 25, 429–440. [Google Scholar] [CrossRef]
- Lartigue, A.; Courville, P.; Auquit, I.; François, A.; Arnoult, C.; Tron, F.; Gilbert, D.; Musette, P. Role of TLR9 in Anti-Nucleosome and Anti-DNA Antibody Production in lpr Mutation-Induced Murine Lupus. J. Immunol. 2006, 177, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Tilstra, J.S.; John, S.; Gordon, R.A.; Leibler, C.; Kashgarian, M.; Bastacky, S.; Nickerson, K.M.; Shlomchik, M.J. B cell–intrinsic TLR9 expression is protective in murine lupus. J. Clin. Investig. 2020, 130, 3172–3187. [Google Scholar] [CrossRef]
- Jackson, S.W.; Scharping, N.E.; Kolhatkar, N.S.; Khim, S.; Schwartz, M.A.; Li, Q.-Z.; Hudkins, K.L.; Alpers, C.E.; Liggitt, D.; Rawlings, D.J. Opposing Impact of B Cell–Intrinsic TLR7 and TLR9 Signals on Autoantibody Repertoire and Systemic Inflammation. J. Immunol. 2014, 192, 4525–4532. [Google Scholar] [CrossRef]
- Hwang, S.-H.; Lee, H.; Yamamoto, M.; Jones, L.A.; Dayalan, J.; Hopkins, R.; Zhou, X.J.; Yarovinsky, F.; Connolly, J.E.; de Lafaille, M.A.C.; et al. B Cell TLR7 Expression Drives Anti-RNA Autoantibody Production and Exacerbates Disease in Systemic Lupus Erythematosus–Prone Mice. J. Immunol. 2012, 189, 5786–5796. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, K.M.; Christensen, S.R.; Shupe, J.; Kashgarian, M.; Kim, D.; Elkon, K.; Shlomchik, M.J. TLR9 Regulates TLR7- and MyD88-Dependent Autoantibody Production and Disease in a Murine Model of Lupus. J. Immunol. 2010, 184, 1840–1848. [Google Scholar] [CrossRef]
- Julià, A.; López-Longo, F.J.; Venegas, J.J.P.; Bonàs-Guarch, S.; Olivé, À.; Andreu, J.L.; Aguirre-Zamorano, M.; Vela, P.; Nolla, J.M.; de la Fuente, J.L.M.; et al. Genome-wide association study meta-analysis identifies five new loci for systemic lupus erythematosus. Arthritis Res. Ther. 2018, 20, 100. [Google Scholar] [CrossRef]
- Vaughn, S.E.; Foley, C.; Lu, X.; Patel, Z.H.; Zoller, E.E.; Magnusen, A.F.; Williams, A.H.; Ziegler, J.T.; Comeau, M.E.; Marion, M.C.; et al. Lupus risk variants in the PXK locus alter B-cell receptor internalization. Front. Genet. 2015, 5, 450. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zhao, J.; Sakurai, D.; Kaufman, K.M.; Edberg, J.C.; Kimberly, R.P.; Kamen, D.L.; Gilkeson, G.S.; Jacob, C.O.; Scofield, R.H.; et al. MicroRNA-3148 Modulates Allelic Expression of Toll-Like Receptor 7 Variant Associated with Systemic Lupus Erythematosus. PLOS Genet. 2013, 9, e1003336. [Google Scholar] [CrossRef]
- Sun, C.; E Molineros, J.; Looger, L.L.; Zhou, X.-J.; Kim, K.; Okada, Y.; Ma, J.; Qi, Y.-Y.; Kim-Howard, X.; Motghare, P.; et al. High-density genotyping of immune-related loci identifies new SLE risk variants in individuals with Asian ancestry. Nat. Genet. 2016, 48, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Adrianto, I.; Wen, F.; Templeton, A.; Wiley, G.; King, J.B.; Lessard, C.J.; Bates, J.S.; Hu, Y.; Kelly, J.A.; Kaufman, K.M.; et al. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat. Genet. 2011, 43, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Groom, J.R.; Fletcher, C.A.; Walters, S.N.; Grey, S.T.; Watt, S.V.; Sweet, M.J.; Smyth, M.J.; Mackay, C.R.; Mackay, F. BAFF and MyD88 signals promote a lupuslike disease independent of T cells. J. Exp. Med. 2007, 204, 1959–1971. [Google Scholar] [CrossRef]
- Du, S.W.; Jacobs, H.M.; Arkatkar, T.; Rawlings, D.J.; Jackson, S.W. Integrated B Cell, Toll-like, and BAFF Receptor Signals Promote Autoantibody Production by Transitional B Cells. J. Immunol. 2018, 201, 3258–3268. [Google Scholar] [CrossRef]
- Parodis, I.; Stockfelt, M.; Sjöwall, C. B Cell Therapy in Systemic Lupus Erythematosus: From Rationale to Clinical Practice. Front. Med. 2020, 7, 316. [Google Scholar] [CrossRef]
- Müller-Winkler, J.; Mitter, R.; Rappe, J.C.; Vanes, L.; Schweighoffer, E.; Mohammadi, H.; Wack, A.; Tybulewicz, V.L. Critical requirement for BCR, BAFF, and BAFFR in memory B cell survival. J. Exp. Med. 2020, 218, e20191393. [Google Scholar] [CrossRef]
- Gross, J.A.; Johnston, J.; Mudri, S.; Enselman, R.; Dillon, S.R.; Madden, K.; Xu, W.; Parrish-Novak, J.; Foster, D.; Lofton-Day, C.; et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature 2000, 404, 995–999. [Google Scholar] [CrossRef]
- Mackay, F.; Woodcock, S.A.; Lawton, P.; Ambrose, C.; Baetscher, M.; Schneider, P.; Tschopp, J.; Browning, J.L. Mice Transgenic for Baff Develop Lymphocytic Disorders along with Autoimmune Manifestations. J. Exp. Med. 1999, 190, 1697–1710. [Google Scholar] [CrossRef]
- Gavin, A.L.; Duong, B.; Skog, P.; Aït-Azzouzene, D.; Greaves, D.R.; Scott, M.L.; Nemazee, D. ΔBAFF, a Splice Isoform of BAFF, Opposes Full-Length BAFF Activity In Vivo in Transgenic Mouse Models. J. Immunol. 2005, 175, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Arkatkar, T.; Jacobs, H.M.; Du, S.W.; Li, Q.-Z.; Hudkins, K.L.; Alpers, C.E.; Rawlings, D.J.; Jackson, S.W. TACI deletion protects against progressive murine lupus nephritis induced by BAFF overexpression. Kidney Int. 2018, 94, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.; Stohl, W.; Chatham, W.; McCune, W.J.; Chevrier, M.; Ryel, J.; Recta, V.; Zhong, J.; Freimuth, W. Association of plasma B lymphocyte stimulator levels and disease activity in systemic lupus erythematosus. Arthritis Rheum. 2008, 58, 2453–2459. [Google Scholar] [CrossRef]
- Salazar-Camarena, D.C.; Ortiz-Lazareno, P.C.; Cruz, A.; Oregon-Romero, E.; Machado-Contreras, J.R.; Muñoz-Valle, J.F.; Orozco-López, M.; Marín-Rosales, M.; A Palafox-Sánchez, C. Association of BAFF, APRIL serum levels, BAFF-R, TACI and BCMA expression on peripheral B-cell subsets with clinical manifestations in systemic lupus erythematosus. Lupus 2015, 25, 582–592. [Google Scholar] [CrossRef]
- Blank, M.C.; Stefanescu, R.N.; Masuda, E.; Marti, F.; King, P.D.; Redecha, P.B.; Wurzburger, R.J.; Peterson, M.G.; Tanaka, S.; Pricop, L. Decreased transcription of the human FCGR2B gene mediated by the -343 G/C promoter polymorphism and association with systemic lupus erythematosus. Hum. Genet. 2005, 117, 220–227. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fc-receptors as regulators of immunity. Adv. Immunol. 2007, 96, 179–204. [Google Scholar]
- Jacobson, B.A.; Panka, D.J.; Nguyen, K.-A.; Erikson, J.; Abbas, A.K.; Marshak-Rothstein, A. Anatomy of autoantibody production: Dominant localization of antibody-producing cells to T cell zones in fas-deficient mice. Immunity 1995, 3, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Nutt, S.L.; Hodgkin, P.D.; Tarlinton, D.M.; Corcoran, L.M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 2015, 15, 160–171. [Google Scholar] [CrossRef] [PubMed]
- William, J.; Euler, C.; Christensen, S.; Shlomchik, M.J. Evolution of Autoantibody Responses via Somatic Hypermutation Outside of Germinal Centers. Science 2002, 297, 2066–2070. [Google Scholar] [CrossRef]
- Tipton, C.M.; Fucile, C.F.; Darce, J.; Chida, A.; Ichikawa, T.; Gregoretti, I.; Schieferl, S.; Hom, J.; Jenks, S.; Feldman, R.J.; et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat. Immunol. 2015, 16, 755–765. [Google Scholar] [CrossRef]
- Kitani, A.; Hara, M.; Hirose, T.; Harigai, M.; Suzuki, K.; Kawakami, M.; Awaguchi, Y.; Hidak, T.; Kawagoe, A.M.; Nakamura, H. Autostimulatory effects of IL-6 on excessive B cell differentiation in patients with systemic lupus erythematosus: Analysis of IL-6 production and IL-6R expression. Clin. Exp. Immunol. 1992, 88, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Ryffel, B.; Car, B.D.; Gunn, H.; Roman, D.; Hiestand, P.; Mihatsch, M.J. Interleukin-6 exacerbates glomerulonephritis in (NZB x NZW)F1 mice. Am. J. Pathol. 1994, 144, 927–937. [Google Scholar] [PubMed]
- Liang, B.; Gardner, D.B.; Griswold, D.E.; Bugelski, P.J.; Song, X.Y.R. Anti-interleukin-6 monoclonal antibody inhibits autoimmune responses in a murine model of systemic lupus erythematosus. Immunology 2006, 119, 296–305. [Google Scholar] [CrossRef]
- Arkatkar, T.; Du, S.W.; Jacobs, H.M.; Dam, E.M.; Hou, B.; Buckner, J.H.; Rawlings, D.J.; Jackson, S.W. B cell–derived IL-6 initiates spontaneous germinal center formation during systemic autoimmunity. J. Exp. Med. 2017, 214, 3207–3217. [Google Scholar] [CrossRef] [PubMed]
- E Munroe, M.; Lu, R.; Zhao, Y.D.; A Fife, D.; Robertson, J.M.; Guthridge, J.M.; Niewold, T.B.; Tsokos, G.C.; Keith, M.P.; Harley, J.B.; et al. Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann. Rheum. Dis. 2016, 75, 2014–2021. [Google Scholar] [CrossRef]
- Ettinger, R.; Kuchen, S.; E Lipsky, P. Interleukin 21 as a target of intervention in autoimmune disease. Ann. Rheum. Dis. 2008, 67, iii83–iii86. [Google Scholar] [CrossRef]
- Vinuesa, C.G.; Cook, M.C.; Angelucci, C.; Athanasopoulos, V.; Rui, L.; Hill, K.M.; Yu, D.; Domaschenz, H.; Whittle, B.; Lambe, T.; et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature 2005, 435, 452–458. [Google Scholar] [CrossRef]
- Ettinger, R.; Kuchen, S.; Lipsky, P.E. The role of IL-21 in regulating B-cell function in health and disease. Immunol. Rev. 2008, 223, 60–86. [Google Scholar] [CrossRef]
- Bubier, J.A.; Sproule, T.J.; Foreman, O.; Spolski, R.; Shaffer, D.J.; Morse, H.C.; Leonard, W.J.; Roopenian, D.C. A critical role for IL-21 receptor signaling in the pathogenesis of systemic lupus erythematosus in BXSB-Yaa mice. Proc. Natl. Acad. Sci. USA 2009, 106, 1518–1523. [Google Scholar] [CrossRef]
- Webb, R.; Merrill, J.T.; Kelly, J.A.; Sestak, A.; Kaufman, K.M.; Langefeld, C.D.; Ziegler, J.; Kimberly, R.P.; Edberg, J.C.; Ramsey-Goldman, R.; et al. A polymorphism within IL21R confers risk for systemic lupus erythematosus. Arthritis Rheum. 2009, 60, 2402–2407. [Google Scholar] [CrossRef]
- Avery, D.T.; Deenick, E.K.; Ma, C.S.; Suryani, S.; Simpson, N.; Chew, G.Y.; Chan, T.D.; Palendira, U.; Bustamante, J.; Boisson-Dupuis, S.; et al. B cell–intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J. Exp. Med. 2010, 207, 155–171. [Google Scholar] [CrossRef]
- Abbasifard, M.; Kamiab, Z.; Hasani, M.; Rahnama, A.; Saeed-Askari, P.; Khorramdelazad, H. Assessing the expression of immunosuppressive cytokines in the newly diagnosed systemic lupus Erythematosus patients: A focus on B cells. BMC Immunol. 2020, 21, 1–12. [Google Scholar] [CrossRef]
- Fillatreau, S.; Sweenie, C.H.; McGeachy, M.J.; Gray, D.; Anderton, S.M. B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 2002, 3, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Gabryšová, L.; Howes, A.; Saraiva, M.; O’Garra, A. The regulation of IL-10 expression. Curr. Top. Microbiol. Immunol. 2014, 380, 157–190. [Google Scholar]
- Dambuza, I.M.; Cheng-Rong, Y.; Choi, J.K.; Yu, C.-R.; Wang, R.; Mattapallil, M.J.; Wingfield, P.T.; Caspi, R.R.; Egwuagu, C.E. IL-12p35 induces expansion of IL-10 and IL-35-expressing regulatory B cells and ameliorates autoimmune disease. Nat. Commun. 2017, 8, 719. [Google Scholar] [CrossRef]
- Qiu, F.; Song, L.; Yang, N.; Li, X. Glucocorticoid downregulates expression of IL-12 family cytokines in systemic lupus erythematosus patients. Lupus 2013, 22, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Wong, C.K.; Dong, J.; Chu, M.; Jiao, D.; Kam, N.W.; Lam, C.W.K.; Tam, L.S. Remission of systemic lupus erythematosus disease activity with regulatory cytokine interleukin (IL)-35 in Murphy Roths Large (MRL)/lpr mice. Clin. Exp. Immunol. 2015, 181, 253–266. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Liu, M.; Liu, B. Interleukin-35 as a New Biomarker of Renal Involvement in Lupus Nephritis Patients. Tohoku J. Exp. Med. 2018, 244, 263–270. [Google Scholar] [CrossRef]
- Keegan, A.D.; Leonard, W.J.; Zhu, J. Recent advances in understanding the role of IL-4 signaling. Fac. Rev. 2021, 10, 71. [Google Scholar] [CrossRef]
- Sugimoto, K.; Morimoto, S.; Kaneko, H.; Nozawa, K.; Tokano, Y.; Takasaki, Y.; Hashimoto, H. Decreased IL-4 Producing CD4 + T Cells in Patients with Active Systemic Lupus Erythematosus-relation to IL-12R Expression. Autoimmunity 2002, 35, 381–387. [Google Scholar] [CrossRef]
- Wong, C.K.; Ho, C.Y.; Li, E.; Lam, C. Elevation of proinflammatory cytokine (IL-18, IL-17, IL-12) and Th2 cytokine (IL-4) concentrations in patients with systemic lupus erythematosus. Lupus 2000, 9, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Flores, S.; Hernández-Molina, G.; Enríquez, A.B.; Faz-Muñoz, D.; Esquivel, Y.; Pacheco-Molina, C.; Furuzawa-Carballeda, J. Cytokines and Effector/Regulatory Cells Characterization in the Physiopathology of Cutaneous Lupus Erythematous: A Cross-Sectional Study. Mediat. Inflamm. 2016, 2016, 7074829. [Google Scholar] [CrossRef]
- Dong, C.; Fu, T.; Ji, J.; Li, Z.; Gu, Z. The role of interleukin-4 in rheumatic diseases. Clin. Exp. Pharmacol. Physiol. 2018, 45, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Saxena, V.; Zang, S.; Li, L.; Finkelman, F.D.; Witte, D.P.; Jacob, C.O. Differential Contribution of IL-4 and STAT6 vs STAT4 to the Development of Lupus Nephritis. J. Immunol. 2003, 170, 4818–4825. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Z.; Kang, Z.; Zhou, X.J.; Liu, S.; Guo, S.; Jin, Q.; Li, T.; Zhou, L.; Wu, X.; et al. Interleukin 4-driven reversal of self-reactive B cell anergy contributes to the pathogenesis of systemic lupus erythematosus. Ann. Rheum. Dis. 2023, 82, 1444–1454. [Google Scholar] [CrossRef] [PubMed]
- Izmirly, P.M.; Rivera, T.L.; Buyon, J.P. Neonatal Lupus Syndromes. Rheum. Dis. Clin. N. Am. 2007, 33, 267–285. [Google Scholar] [CrossRef]
- Arbuckle, M.R.; McClain, M.T.; Rubertone, M.V.; Scofield, R.H.; Dennis, G.J.; James, J.A.; Harley, J.B. Development of Autoantibodies before the Clinical Onset of Systemic Lupus Erythematosus. N. Engl. J. Med. 2003, 349, 1526–1533. [Google Scholar] [CrossRef]
- Hedberg, A.; Fismen, S.; Fenton, K.A.; Mortensen, E.S.; Rekvig, O.P. Deposition of chromatin-IgG complexes in skin of nephritic MRL-lpr/lpr mice is associated with increased local matrix metalloprotease activities. Exp. Dermatol. 2010, 19, e265–e274. [Google Scholar] [CrossRef]
- Lou, H.; Ling, G.S.; Cao, X. Autoantibodies in systemic lupus erythematosus: From immunopathology to therapeutic target. J. Autoimmun. 2022, 132, 102861. [Google Scholar] [CrossRef]
- Deocharan, B.; Qing, X.; Lichauco, J.; Putterman, C. Alpha-actinin is a cross-reactive renal target for pathogenic anti-DNA antibodies. J. Immunol. 2002, 168, 3072–3078. [Google Scholar] [CrossRef]
- Ehrenstein, M.R.; Katz, D.R.; Griffiths, M.H.; Papadaki, L.; Winkler, T.H.; Kalden, J.R.; Isenberg, D.A. Human IgG anti-DNA antibodies deposit in kidneys and induce proteinuria in SCID mice. Kidney Int. 1995, 48, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Menke, J.; Hsu, M.-Y.; Byrne, K.T.; Lucas, J.A.; Rabacal, W.A.; Croker, B.P.; Zong, X.-H.; Stanley, E.R.; Kelley, V.R. Sunlight Triggers Cutaneous Lupus through a CSF-1-Dependent Mechanism in MRL-Fas lpr Mice. J. Immunol. 2008, 181, 7367–7379. [Google Scholar] [CrossRef]
- Selmi, C.; Barin, J.G.; Rose, N.R. Current trends in autoimmunity and the nervous system. J. Autoimmun. 2016, 75, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Chan, O.; Shlomchik, M.J. A New Role for B Cells in Systemic Autoimmunity: B Cells Promote Spontaneous T Cell Activation in MRL-lpr/lprMice. J. Immunol. 1998, 160, 51–59. [Google Scholar] [CrossRef]
- Shlomchik, M.J.; Luo, W.; Weisel, F. Linking signaling and selection in the germinal center. Immunol. Rev. 2019, 288, 49–63. [Google Scholar] [CrossRef]
- Cancro, M.P. Age-Associated B Cells. Annu. Rev. Immunol. 2020, 38, 315–340. [Google Scholar] [CrossRef]
- Wang, Q.; Feng, D.; Jia, S.; Lu, Q.; Zhao, M. B-Cell Receptor Repertoire: Recent Advances in Autoimmune Diseases. Clin. Rev. Allergy Immunol. 2024, 66, 1–23. [Google Scholar] [CrossRef]
- de Rie, M.A.; Schumacher, T.N.; van Schijndel, G.M.; van Lier, R.A.; Miedema, F. Regulatory role of CD19 molecules in B-cell activation and differentiation. Cell Immunol. 1989, 118, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Grimsholm, O. CD27 on human memory B cells-more than just a surface marker. Clin. Exp. Immunol. 2023, 213, 164–172. [Google Scholar] [CrossRef]
- Klein, U.; Rajewsky, K.; Küppers, R. Human immunoglobulin (Ig)M+IgD+ peripheral blood B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes: CD27 as a general marker for somatically mutated (memory) B cells. J. Exp. Med. 1998, 188, 1679–1689. [Google Scholar] [CrossRef]
- Gutzeit, C.; Chen, K.; Cerutti, A. The enigmatic function of IgD: Some answers at last. Eur. J. Immunol. 2018, 48, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- Sachinidis, A.; Xanthopoulos, K.; Garyfallos, A. Age-Associated B Cells (ABCs) in the Prognosis, Diagnosis and Therapy of Systemic Lupus Erythematosus (SLE). Mediterr. J. Rheumatol. 2020, 31, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Moysidou, E.; Lioulios, G.; Xochelli, A.; Nikolaidou, V.; Christodoulou, M.; Mitsoglou, Z.; Stai, S.; Fylaktou, A.; Papagianni, A.; Stangou, M. Different Types of Chronic Inflammation Engender Distinctive Immunosenescent Profiles in Affected Patients. Int. J. Mol. Sci. 2022, 23, 14688. [Google Scholar] [CrossRef]
- Sachinidis, A.; Trachana, M.; Taparkou, A.; Gavriilidis, G.; Verginis, P.; Psomopoulos, F.; Adamichou, C.; Boumpas, D.; Garyfallos, A. Investigating the Role of T-bet+ B Cells (ABCs/DN) in the Immunopathogenesis of Systemic Lupus Erythematosus. Mediterr. J. Rheumatol. 2023, 34, 117–120. [Google Scholar] [CrossRef]
- Moysidou, E.; Lioulios, G.; Christodoulou, M.; Xochelli, A.; Stai, S.; Iosifidou, M.; Iosifidou, A.; Briza, S.; Briza, D.I.; Fylaktou, A.; et al. Increase in Double Negative B Lymphocytes in Patients with Systemic Lupus Erythematosus in Remission and Their Correlation with Early Differentiated T Lymphocyte Subpopulations. Curr. Issues Mol. Biol. 2023, 45, 6667–6681. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.K.Y.; Gong, L.; Kwong, D.L.W.; Lee, V.H.F.; Lee, A.W.M.; Guan, X.Y.; Kam, N.W.; Dai, W. Functions of double-negative B cells in autoimmune diseases, infections, and cancers. EMBO Mol. Med. 2023, 15, e17341. [Google Scholar] [CrossRef]
- Beckers, L.; Somers, V.; Fraussen, J. IgD-CD27- double negative (DN) B cells: Origins and functions in health and disease. Immunol. Lett. 2023, 255, 67–76. [Google Scholar] [CrossRef]
- Li, Y.; Li, Z.; Hu, F. Double-negative (DN) B cells: An under-recognized effector memory B cell subset in autoimmunity. Clin. Exp. Immunol. 2021, 205, 119–127. [Google Scholar] [CrossRef]
- Szelinski, F.; Lino, A.C.; Dörner, T. B cells in systemic lupus erythematosus. Curr. Opin. Rheumatol. 2022, 34, 125–132. [Google Scholar] [CrossRef]
- Mouat, I.C.; Goldberg, E.; Horwitz, M.S. Age-associated B cells in autoimmune diseases. Cell. Mol. Life Sci. 2022, 79, 1–11. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, H.; Liu, S.; Xia, F.; Kang, Z.; Zhang, Y.; Liu, Y.; Xiao, H.; Chen, L.; Huang, C.; et al. Excessive CD11c+Tbet+ B cells promote aberrant TFH differentiation and affinity-based germinal center selection in lupus. Proc. Natl. Acad. Sci. USA 2019, 116, 18550–18560. [Google Scholar] [CrossRef] [PubMed]
- Moneta, G.M.; Bracaglia, C.; Caiello, I.; Farroni, C.; Pires Marafon, D.; Carlomagno, R.; Hiraki, L.; Vivarelli, M.; Gianviti, A.; Carbogno, S.; et al. Persistently active interferon-γ pathway and expansion of T-bet+ B cells in a subset of patients with childhood-onset systemic lupus erythematosus. Eur. J. Immunol. 2023, 53, e2250319. [Google Scholar] [CrossRef] [PubMed]
- Ruan, P.; Wang, S.; Yang, M.; Wu, H. The ABC-associated immunosenescence and lifestyle interventions in autoimmune disease. Rheumatol. Immunol. Res. 2022, 3, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Rubtsova, K.; Rubtsov, A.V.; Thurman, J.M.; Mennona, J.M.; Kappler, J.W.; Marrack, P. B cells expressing the transcription factor T-bet drive lupus-like autoimmunity. J. Clin. Investig. 2017, 127, 1392–1404. [Google Scholar] [CrossRef]
- Rauch, E.; Amendt, T.; Krol, A.L.; Lang, F.B.; Linse, V.; Hohmann, M.; Keim, A.-C.; Kreutzer, S.; Kawengian, K.; Buchholz, M.; et al. T-bet+ B cells are activated by and control endogenous retroviruses through TLR-dependent mechanisms. Nat. Commun. 2024, 15, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Rubtsov, A.V.; Marrack, P.; Rubtsova, K. T-bet expressing B cells—Novel target for autoimmune therapies? Cell Immunol. 2017, 321, 35–39. [Google Scholar] [CrossRef]
- Yasaka, K.; Yamazaki, T.; Sato, H.; Shirai, T.; Cho, M.; Ishida, K.; Ito, K.; Tanaka, T.; Ogasawara, K.; Harigae, H.; et al. Phospholipase D4 as a signature of toll-like receptor 7 or 9 signaling is expressed on blastic T-bet + B cells in systemic lupus erythematosus. Arthritis Res. Ther. 2023, 25, 1–13. [Google Scholar] [CrossRef]
- Wang, S.; Wang, J.; Kumar, V.; Karnell, J.L.; Naiman, B.; Gross, P.S.; Rahman, S.; Zerrouki, K.; Hanna, R.; Morehouse, C.; et al. IL-21 drives expansion and plasma cell differentiation of autoreactive CD11chiT-bet+ B cells in SLE. Nat. Commun. 2018, 9, 1758. [Google Scholar] [CrossRef]
- Jenks, S.A.; Cashman, K.S.; Zumaquero, E.; Marigorta, U.M.; Patel, A.V.; Wang, X.; Tomar, D.; Woodruff, M.C.; Simon, Z.; Bugrovsky, R.; et al. Distinct Effector B Cells Induced by Unregulated Toll-like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Immunity 2018, 49, 725–739.e6. [Google Scholar] [CrossRef]
- Mizoguchi, A.; Bhan, A.K. A Case for Regulatory B Cells. J. Immunol. 2006, 176, 705–710. [Google Scholar] [CrossRef]
- Yang, M.; Rui, K.; Wang, S.; Lu, L. Regulatory B cells in autoimmune diseases. Cell. Mol. Immunol. 2013, 10, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Yanaba, K.; Bouaziz, J.-D.; Fujimoto, M.; Tedder, T.F. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J. Clin. Investig. 2008, 118, 3420–3430. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Baba, A.; Yokota, T.; Nishikawa, H.; Ohkawa, Y.; Kayama, H.; Kallies, A.; Nutt, S.L.; Sakaguchi, S.; Takeda, K.; et al. Interleukin-10-Producing Plasmablasts Exert Regulatory Function in Autoimmune Inflammation. Immunity 2014, 41, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-X.; Yu, C.-R.; Dambuza, I.M.; Mahdi, R.M.; Dolinska, M.B.; Sergeev, Y.V.; Wingfield, P.T.; Kim, S.-H.; E Egwuagu, C. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat. Med. 2014, 20, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Vadasz, Z.; Peri, R.; Eiza, N.; Slobodin, G.; Balbir-Gurman, A.; Toubi, E. The Expansion of CD25highIL-10highFoxP3high B Regulatory Cells Is in Association with SLE Disease Activity. J. Immunol. Res. 2015, 2015, 254245. [Google Scholar] [CrossRef]
- Yang, X.; Yang, J.; Chu, Y.; Xue, Y.; Xuan, D.; Zheng, S.; Zou, H. T Follicular Helper Cells and Regulatory B Cells Dynamics in Systemic Lupus Erythematosus. PLoS ONE 2014, 9, e88441. [Google Scholar] [CrossRef]
- Kashipaz, M.R.A.; Huggins, M.L.; Lanyon, P.; Robins, A.; Powell, R.J.; Todd, I. Assessment of Be1 and Be2 cells in systemic lupus erythematosus indicates elevated interleukin-10 producing CD5+ B cells. Lupus 2003, 12, 356–363. [Google Scholar] [CrossRef]
- Díaz-Alderete, A.; Crispin, J.C.; Vargas-Rojas, M.I.; Alcocer-Varela, J. IL-10 production in B cells is confined to CD154+ cells in patients with systemic lupus erythematosus. J. Autoimmun. 2004, 23, 379–383. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, P.; Ma, L.; Shan, Y.; Jiang, Z.; Wang, J.; Jiang, Y. Increased Interleukin 21 and Follicular Helper T-like Cells and Reduced Interleukin 10+ B cells in Patients with New-onset Systemic Lupus Erythematosus. J. Rheumatol. 2014, 41, 1781–1792. [Google Scholar] [CrossRef]
- Blair, P.A.; Noreña, L.Y.; Flores-Borja, F.; Rawlings, D.J.; Isenberg, D.A.; Ehrenstein, M.R.; Mauri, C. CD19+CD24hiCD38hi B Cells Exhibit Regulatory Capacity in Healthy Individuals but Are Functionally Impaired in Systemic Lupus Erythematosus Patients. Immunity 2010, 32, 129–140. [Google Scholar] [CrossRef]
- Heinemann, K.; Wilde, B.; Hoerning, A.; Tebbe, B.; Kribben, A.; Witzke, O.; Dolff, S. Decreased IL-10+ regulatory B cells (Bregs) in lupus nephritis patients. Scand. J. Rheumatol. 2016, 45, 312–316. [Google Scholar] [CrossRef]
- Watanabe, R.; Ishiura, N.; Nakashima, H.; Kuwano, Y.; Okochi, H.; Tamaki, K.; Sato, S.; Tedder, T.F.; Fujimoto, M. Regulatory B Cells (B10 Cells) Have a Suppressive Role in Murine Lupus: CD19 and B10 Cell Deficiency Exacerbates Systemic Autoimmunity. J. Immunol. 2010, 184, 4801–4809. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Z.; Xiao, H.; Liu, X.; Zhu, G.; Yu, D.; Han, G.; Chen, G.; Hou, C.; Ma, N.; et al. Foxd3 suppresses interleukin-10 expression in B cells. Immunology 2017, 150, 478–488. [Google Scholar] [CrossRef]
- Menon, M.; Blair, P.A.; Isenberg, D.A.; Mauri, C. A Regulatory Feedback between Plasmacytoid Dendritic Cells and Regulatory B Cells Is Aberrant in Systemic Lupus Erythematosus. Immunity 2016, 44, 683–697. [Google Scholar] [CrossRef]
- Wang, X.; Wei, Y.; Xiao, H.; Liu, X.; Zhang, Y.; Han, G.; Chen, G.; Hou, C.; Zhang, L.; Ma, N.; et al. Pre-existing CD19-independent GL7 − Breg cells are expanded during inflammation and in mice with lupus-like disease. Mol. Immunol. 2016, 71, 54–63. [Google Scholar] [CrossRef] [PubMed]
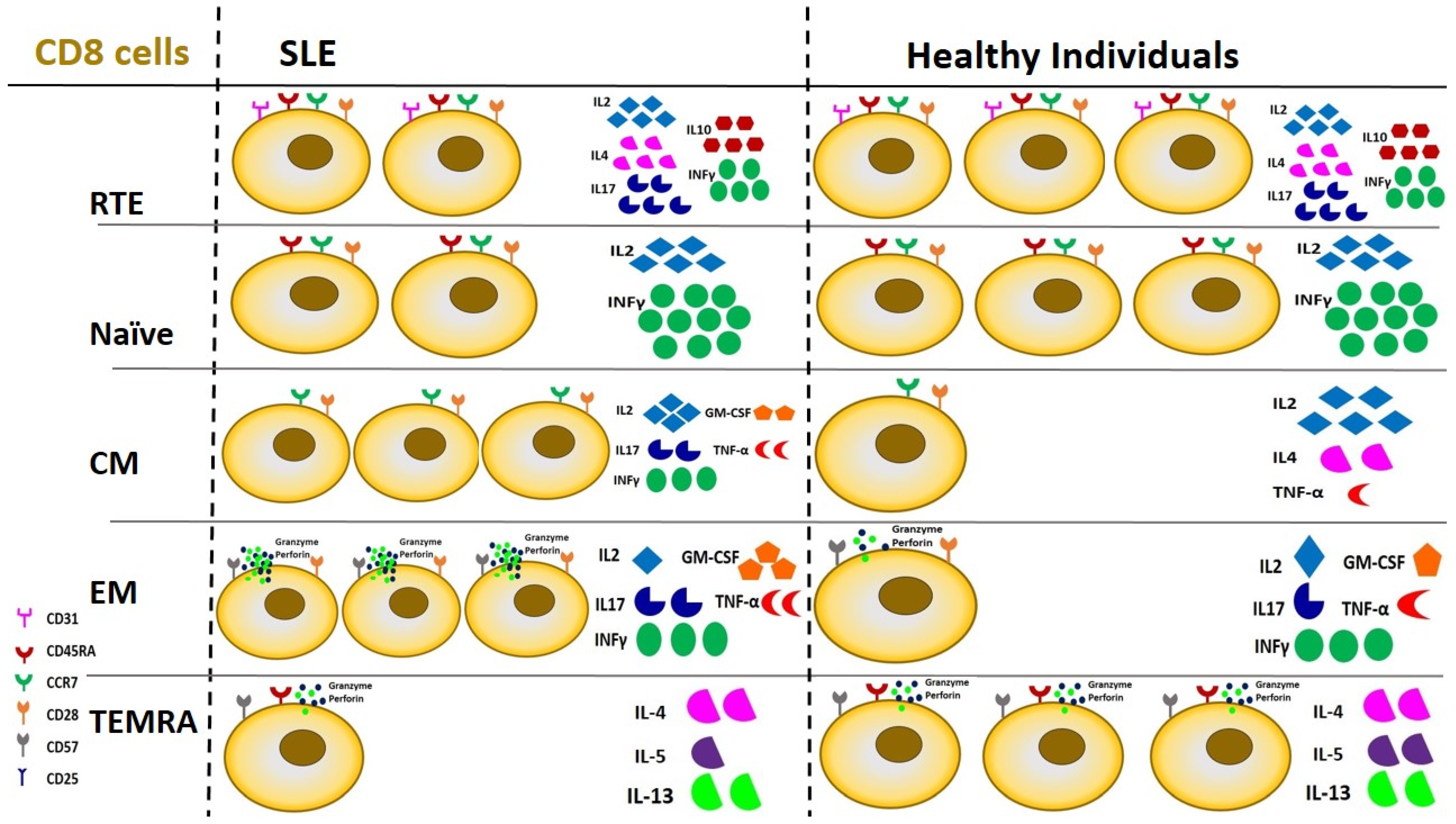
- Katsuyama, T.; Tsokos, G.C.; Moulton, V.R. Aberrant T Cell Signaling and Subsets in Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 1088. [Google Scholar] [CrossRef]
- Lioulios, G.; Mitsoglou, Z.; Fylaktou, A.; Xochelli, A.; Christodoulou, M.; Stai, S.; Moysidou, E.; Konstantouli, A.; Nikolaidou, V.; Papagianni, A.; et al. Exhausted but Not Senescent T Lymphocytes Predominate in Lupus Nephritis Patients. Int. J. Mol. Sci. 2022, 23, 13928. [Google Scholar] [CrossRef]
- Liu, L.; Takeda, K.; Akkoyunlu, M. Disease Stage-Specific Pathogenicity of CD138 (Syndecan 1)-Expressing T Cells in Systemic Lupus Erythematosus. Front. Immunol. 2020, 11, 1569. [Google Scholar] [CrossRef]
- Lee, K.; Park, J.; Tanno, H.; Georgiou, G.; Diamond, B.; Kim, S.J. Peripheral T cell activation, not thymic selection, expands the T follicular helper repertoire in a lupus-prone murine model. Proc. Natl. Acad. Sci. USA 2023, 120, e2309780120. [Google Scholar] [CrossRef]
- Cuda, C.M.; Li, S.; Liang, S.; Yin, Y.; Potula, H.H.S.K.; Xu, Z.; Sengupta, M.; Chen, Y.; Butfiloski, E.; Baker, H.; et al. Pre-B Cell Leukemia Homeobox 1 Is Associated with Lupus Susceptibility in Mice and Humans. J. Immunol. 2012, 188, 604–614. [Google Scholar] [CrossRef]
- Li, W.; Deng, C.; Yang, H.; Wang, G. The Regulatory T Cell in Active Systemic Lupus Erythematosus Patients: A Systemic Review and Meta-Analysis. Front. Immunol. 2019, 10, 159. [Google Scholar] [CrossRef]
- Guo, C.; Liu, Q.; Zong, D.; Zhang, W.; Zuo, Z.; Yu, Q.; Sha, Q.; Zhu, L.; Gao, X.; Fang, J.; et al. Single-cell transcriptome profiling and chromatin accessibility reveal an exhausted regulatory CD4+ T cell subset in systemic lupus erythematosus. Cell Rep. 2022, 41. [Google Scholar] [CrossRef]
- Jin, X.; Chen, J.; Wu, J.; Lu, Y.; Li, B.; Fu, W.; Wang, W.; Cui, D. Aberrant Expansion of Follicular Helper T Cell Subsets in Patients with Systemic Lupus Erythematosus. Front. Immunol. 2022, 13, 928359. [Google Scholar] [CrossRef]
- Zhang, X.; Lindwall, E.; Gauthier, C.; Lyman, J.; Spencer, N.; Alarakhia, A.; Fraser, A.; Ing, S.; Chen, M.; Webb-Detiege, T.; et al. Circulating CXCR5+CD4+helper T cells in systemic lupus erythematosus patients share phenotypic properties with germinal center follicular helper T cells and promote antibody production. Lupus 2015, 24, 909–917. [Google Scholar] [CrossRef]
- Isgro, J.; Gupta, S.; Jacek, E.; Pavri, T.; Duculan, R.; Kim, M.; Kirou, K.A.; Salmon, J.E.; Pernis, A.B. Enhanced Rho-Associated Protein Kinase Activation in Patients With Systemic Lupus Erythematosus. Arthritis Rheum. 2013, 65, 1592–1602. [Google Scholar] [CrossRef]
- Chen, Y.; Tian, B. IFN-γ promotes the development of systemic lupus erythematosus through the IFNGR1/2-PSTAT1-TBX21 signaling axis. Am. J. Transl. Res. 2022, 14, 6874–6888. [Google Scholar]
- Schneider, L.; da Silva, A.C.C.; Junior, L.C.W.; Alegretti, A.P.; dos Santos, A.S.P.; Santos, M.; Sassi, R.; Heemann, B.; Pfaffenseller, B.; Brenol, J.C.T.; et al. Vitamin D levels and cytokine profiles in patients with systemic lupus erythematosus. Lupus 2015, 24, 1191–1197. [Google Scholar] [CrossRef]
- Chen, W.; Li, W.; Zhang, Z.; Tang, X.; Wu, S.; Yao, G.; Li, K.; Wang, D.; Xu, Y.; Feng, R.; et al. Lipocalin-2 Exacerbates Lupus Nephritis by Promoting Th1 Cell Differentiation. J. Am. Soc. Nephrol. 2020, 31, 2263–2277. [Google Scholar] [CrossRef]
- Zumaquero, E.; Stone, S.L.; Scharer, C.D.; Jenks, S.A.; Nellore, A.; Mousseau, B.; Rosal-Vela, A.; Botta, D.; Bradley, J.E.; Wojciechowski, W.; et al. IFNγ induces epigenetic programming of human T-bethi B cells and promotes TLR7/8 and IL-21 induced differentiation. Elife 2019, 8, e41641. [Google Scholar] [CrossRef]
- Xiang, S.; Zhang, J.; Zhang, M.; Qian, S.; Wang, R.; Wang, Y.; Xiang, Y.; Ding, X. Imbalance of helper T cell type 1, helper T cell type 2 and associated cytokines in patients with systemic lupus erythematosus: A meta-analysis. Front. Pharmacol. 2022, 13, 988512. [Google Scholar] [CrossRef]
- Lit, L.C.W.; Wong, C.K.; Li, E.K.M.; Tam, L.S.; Lam, C.W.K.; Lo, Y.M.D. Elevated gene expression of Th1/Th2 associated transcription factors is correlated with disease activity in patients with systemic lupus erythematosus. J. Rheumatol. 2007, 34, 89–96. [Google Scholar]
- Wang, H.; Li, C.; Ren, G.; Yang, C.; Sun, J.; Zhao, L.; Sun, W.; Ju, J.; Xu, D. Updated insight into the role of Th2-associated immunity in systemic lupus erythematosus. Autoimmun. Rev. 2023, 22, 103213. [Google Scholar] [CrossRef] [PubMed]
- Meloun, A.; León, B. Sensing of protease activity as a triggering mechanism of Th2 cell immunity and allergic disease. Front. Allergy 2023, 4, 1265049. [Google Scholar] [CrossRef] [PubMed]
- Mowen, K.A.; Glimcher, L.H. Signaling pathways in Th2 development. Immunol. Rev. 2004, 202, 203–222. [Google Scholar] [CrossRef]
- Han, M.; Ma, J.; Ouyang, S.; Wang, Y.; Zheng, T.; Lu, P.; Zheng, Z.; Zhao, W.; Li, H.; Wu, Y.; et al. The kinase p38α functions in dendritic cells to regulate Th2-cell differentiation and allergic inflammation. Cell. Mol. Immunol. 2022, 19, 805–819. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Ramachandran, P. Immunology of human fibrosis. Nat. Immunol. 2023, 24, 1423–1433. [Google Scholar] [CrossRef]
- Yuan, S.; Zeng, Y.; Li, J.; Wang, C.; Li, W.; He, Z.; Ye, J.; Li, F.; Chen, Y.; Lin, X.; et al. Phenotypical changes and clinical significance of CD4+/CD8+ T cells in SLE. Lupus Sci. Med. 2022, 9, e000660. [Google Scholar] [CrossRef]
- Wangriatisak, K.; Kochayoo, P.; Thawornpan, P.; Leepiyasakulchai, C.; Suangtamai, T.; Ngamjanyaporn, P.; Khowawisetsut, L.; Khaenam, P.; Pisitkun, P.; Chootong, P. CD4+T-cell cooperation promoted pathogenic function of activated naïve B cells of patients with SLE. Lupus Sci. Med. 2022, 9, e000739. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, F.V.; Ferreira, V.; Mesquita, D.; Andrade, L.E.C. CD4 T lymphocyte subsets display heterogeneous susceptibility to apoptosis induced by serum from patients with systemic lupus erythematosus. Hortic. Bras. 2023, 63, 1–8. [Google Scholar] [CrossRef]
- Li, H.; Boulougoura, A.; Endo, Y.; Tsokos, G.C. Abnormalities of T cells in systemic lupus erythematosus: New insights in pathogenesis and therapeutic strategies. J. Autoimmun. 2022, 132, 102870. [Google Scholar] [CrossRef]
- Chen, P.-M.; Katsuyama, E.; Satyam, A.; Li, H.; Rubio, J.; Jung, S.; Andrzejewski, S.; Becherer, J.D.; Tsokos, M.G.; Abdi, R.; et al. CD38 reduces mitochondrial fitness and cytotoxic T cell response against viral infection in lupus patients by suppressing mitophagy. Sci. Adv. 2022, 8, eabo4271. [Google Scholar] [CrossRef]
- Lin, C.R.; Wei, T.Y.W.; Tsai, H.Y.; Wu, Y.T.; Wu, P.Y.; Chen, S.T. Glycosylation-dependent interaction between CD69 and S100A8/S100A9 complex is required for regulatory T-cell differentiation. FASEB J. 2015, 29, 5006–5017. [Google Scholar] [CrossRef]
- Cabral, J.; Hanley, S.A.; Gerlach, J.Q.; O’Leary, N.; Cunningham, S.; Ritter, T.; Ceredig, R.; Joshi, L.; Griffin, M.D. Distinctive Surface Glycosylation Patterns Associated With Mouse and Human CD4+ Regulatory T Cells and Their Suppressive Function. Front. Immunol. 2017, 8, 987. [Google Scholar] [CrossRef]
- Suen, J.L.; Li, H.T.; Jong, Y.J.; Chiang, B.L.; Yen, J.H. Altered homeostasis of CD4+ FoxP3+ regulatory T-cell subpopulations in systemic lupus erythematosus. Immunology 2009, 127, 196–205. [Google Scholar] [CrossRef]
- Piantoni, S.; Regola, F.; Zanola, A.; Andreoli, L.; Dall’ara, F.; Tincani, A.; Airo’, P. Effector T-cells are expanded in systemic lupus erythematosus patients with high disease activity and damage indexes. Lupus 2018, 27, 143–149. [Google Scholar] [CrossRef]
- Ip, W.K.E.; Hoshi, N.; Shouval, D.S.; Snapper, S.; Medzhitov, R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 2017, 356, 513–519. [Google Scholar] [CrossRef]
- Houssiau, F.; Lefebvre, C.; Berghe, M.V.; Lambert, M.; Devogelaer, J.-P.; Renauld, J.-C. Serum interleukin 10 titers in systemic lupus erythematosus reflect disease activity. Lupus 1995, 4, 393–395. [Google Scholar] [CrossRef]
- Fischer, J.; Dirks, J.; Klaussner, J.; Haase, G.; Holl-Wieden, A.; Hofmann, C.; Hackenberg, S.; Girschick, H.; Morbach, H. Effect of Clonally Expanded PD-1highCXCR5–CD4+ Peripheral T Helper Cells on B Cell Differentiation in the Joints of Patients With Antinuclear Antibody–Positive Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2021, 74, 150–162. [Google Scholar] [CrossRef]
- Llorente, L.; Zou, W.; Levy, Y.; Richaud-Patin, Y.; Wijdenes, J.; Alcocer-Varela, J.; Morel-Fourrier, B.; Brouet, J.C.; Alarcon-Segovia, D.; Galanaud, P.; et al. Role of interleukin 10 in the B lymphocyte hyperactivity and autoantibody production of human systemic lupus erythematosus. J. Exp. Med. 1995, 181, 839–844. [Google Scholar] [CrossRef]
- Fujimura, K.; Oyamada, A.; Iwamoto, Y.; Yoshikai, Y.; Yamada, H. CD4 T cell-intrinsic IL-2 signaling differentially affects Th1 and Th17 development. J. Leukoc. Biol. 2013, 94, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Ou, Q.; Power, R.; Griffin, M.D. Revisiting regulatory T cells as modulators of innate immune response and inflammatory diseases. Front. Immunol. 2023, 14, 1287465. [Google Scholar] [CrossRef] [PubMed]
- Hansmann, L.; Schmidl, C.; Kett, J.; Steger, L.; Andreesen, R.; Hoffmann, P.; Rehli, M.; Edinger, M. Dominant Th2 Differentiation of Human Regulatory T Cells upon Loss of FOXP3 Expression. J. Immunol. 2012, 188, 1275–1282. [Google Scholar] [CrossRef]
- Kiernan, C.H.; Asmawidjaja, P.S.; Fahy, N.; Witte-Bouma, J.; Wolvius, E.B.; Brama, P.A.J.; Lubberts, E.; Farrell, E. Allogeneic Chondrogenic Mesenchymal Stromal Cells Alter Helper T Cell Subsets in CD4+ Memory T Cells. Tissue Eng. Part A 2020, 26, 490–502. [Google Scholar] [CrossRef]
- Yao, X.; Wang, Q.; Chen, C.; Zeng, P.; Hou, L.; Zhou, J.; Huang, Y.; Ma, W. Wu-Teng-Gao External Treatment Improves Th17/Treg Balance in Rheumatoid Arthritis. Evid. Based Complement. Altern. Med. 2022, 2022, 5105545. [Google Scholar]
- Lohr, J.; Knoechel, B.; Caretto, D.; Abbas, A.K. Balance of Th1 and Th17 effector and peripheral regulatory T cells. Microbes Infect. 2009, 11, 589–593. [Google Scholar] [CrossRef]
- Wang, C.; Yang, J.; Xie, L.; Saimaier, K.; Zhuang, W.; Han, M.; Liu, G.; Lv, J.; Shi, G.; Li, N.; et al. Methyl Butyrate Alleviates Experimental Autoimmune Encephalomyelitis and Regulates the Balance of Effector T Cells and Regulatory T Cells. Inflammation 2021, 45, 977–991. [Google Scholar] [CrossRef]
- Lopez-Ocasio, M.; Buszko, M.; Blain, M.; Wang, K.; Shevach, E.M. T Follicular Regulatory Cell Suppression of T Follicular Helper Cell Function Is Context-Dependent in vitro. Front. Immunol. 2020, 11, 637. [Google Scholar] [CrossRef]
- Qiu, H.; Wu, H.; Chan, V.; Lau, C.-S.; Lu, Q. Transcriptional and epigenetic regulation of follicular T-helper cells and their role in autoimmunity. Autoimmunity 2017, 50, 71–81. [Google Scholar] [CrossRef]
- Szabó, K.; Jámbor, I.; Pázmándi, K.; Nagy, N.; Papp, G.; Tarr, T. Altered Circulating Follicular T Helper Cell Subsets and Follicular T Regulatory Cells Are Indicators of a Derailed B Cell Response in Lupus, Which Could Be Modified by Targeting IL-21R. Int. J. Mol. Sci. 2022, 23, 12209. [Google Scholar] [CrossRef]
- Weinstein, K.N.; Domeier, P.P.; Ziegler, S.F. A Splice of Life: The Discovery, Function, and Clinical Implications of FOXP3 Isoforms in Autoimmune Disease. Int. Immunol. 2024, dxae049. [Google Scholar] [CrossRef] [PubMed]
- Seitz, C.; Joly, A.-L.; Fang, F.; Frith, K.; Gray, P.; Andersson, J. The FOXP3 full-length isoform controls the lineage-stability of CD4+FOXP3+ regulatory T cells. Clin. Immunol. 2022, 237, 108957. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, Q.; Yang, S.; Chen, S.; Fu, Y.; Spath, S.; Domeier, P.; Hagin, D.; Anover-Sombke, S.; Haouili, M.; et al. FOXP3 exon 2 controls Treg stability and autoimmunity. Sci. Immunol. 2022, 7, eabo5407. [Google Scholar] [CrossRef]
- Phillips, R. FOXP3 splice variant is associated with autoimmune disease. Nat. Rev. Rheumatol. 2022, 18, 493. [Google Scholar] [CrossRef]
- Suzuki, K.; Setoyama, Y.; Yoshimoto, K.; Tsuzaka, K.; Abe, T.; Takeuchi, T. Decreased mRNA Expression of Two FOXP3 Isoforms in Peripheral Blood Mononuclear Cells from Patients with Rheumatoid Arthritis and Systemic Lupus Erythematosus. Int. J. Immunopathol. Pharmacol. 2011, 24, 7–14. [Google Scholar] [CrossRef]
- Jakiela, B.; Iwaniec, T.; Plutecka, H.; Celinska-Lowenhoff, M.; Dziedzina, S.; Musial, J. Signs of impaired immunoregulation and enhanced effector T-cell responses in the primary antiphospholipid syndrome. Lupus 2015, 25, 389–398. [Google Scholar] [CrossRef]
- Wallace, C.H.; Wu, B.X.; Salem, M.; Ansa-Addo, E.A.; Metelli, A.; Sun, S.; Gilkeson, G.; Shlomchik, M.J.; Liu, B.; Li, Z. B lymphocytes confer immune tolerance via cell surface GARP-TGF-β complex. JCI Insight 2018, 3, e99863. [Google Scholar] [CrossRef]
- Zhou, A.X.; Kozhaya, L.; Fujii, H.; Unutmaz, D. GARP–TGF-β Complexes Negatively Regulate Regulatory T Cell Development and Maintenance of Peripheral CD4+ T Cells In Vivo. J. Immunol. 2013, 190, 5057–5064. [Google Scholar] [CrossRef]
- Zimmer, N.; Trzeciak, E.R.; Graefen, B.; Satoh, K.; Tuettenberg, A. GARP as a Therapeutic Target for the Modulation of Regulatory T Cells in Cancer and Autoimmunity. Front. Immunol. 2022, 13, 928450. [Google Scholar] [CrossRef]
- Azizi, G.; Yazdani, R.; Mirshafiey, A. Th22 cells in autoimmunity: A review of current knowledge. Eur. Ann. Allergy Clin. Immunol. 2015, 47, 108–117. [Google Scholar]
- Doulabi, H.; Masoumi, E.; Rastin, M.; Azarnaminy, A.F.; Esmaeili, S.-A.; Mahmoudi, M. The role of Th22 cells, from tissue repair to cancer progression. Cytokine 2022, 149, 155749. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, D.; Dumoutier, L.; Constantinescu, S.; Kruijer, W.; Schuringa, J.J.; Renauld, J.-C. Interleukin-22 (IL-22) Activates the JAK/STAT, ERK, JNK, and p38 MAP Kinase Pathways in a Rat Hepatoma Cell Line. J. Biol. Chem. 2002, 277, 33676–33682. [Google Scholar] [CrossRef]
- Mitra, A.; Raychaudhuri, S.K. IL-22 induced cell proliferation is regulated by PI3K/Akt/mTOR signaling cascade. Cytokine 2012, 60, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Jiang, Y.; Ma, H.; Wu, J.; Jiang, Z.; Zhao, L. Elevated levels of CCR6+ T helper 22 cells correlate with skin and renal impairment in systemic lupus erythematosus. Sci. Rep. 2017, 7, 12962. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Yang, G.; Xiao, F.; Xie, J.; Wang, S.; Lu, L.; Cui, D. Role of Th22 Cells in the Pathogenesis of Autoimmune Diseases. Front. Immunol. 2021, 12, 688066. [Google Scholar] [CrossRef]
- Ye, Z.; Zhao, L.; Gao, Q.; Jiang, Y.; Jiang, Z.; Chu, C.Q. Analysis of IL-22 and Th22 Cells by Flow Cytometry in Systemic Lupus Erythematosus. Methods Mol. Biol. 2020, 2108, 29–42. [Google Scholar]
- Yang, J.; Yang, X.; Wang, L.; Li, M. B cells control lupus autoimmunity by inhibiting Th17 and promoting Th22 cells. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Yang, X.-Y.; Wang, H.-Y.; Zhao, X.-Y.; Wang, L.-J.; Lv, Q.-H.; Wang, Q.-Q. Th22, but not Th17 Might be a Good Index to Predict the Tissue Involvement of Systemic Lupus Erythematosus. J. Clin. Immunol. 2013, 33, 767–774. [Google Scholar] [CrossRef]
- Kim, S.J.; Lee, K.; Diamond, B. Follicular Helper T Cells in Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 1793. [Google Scholar] [CrossRef] [PubMed]
- Morita, R.; Schmitt, N.; Bentebibel, S.-E.; Ranganathan, R.; Bourdery, L.; Zurawski, G.; Foucat, E.; Dullaers, M.; Oh, S.; Sabzghabaei, N.; et al. Human Blood CXCR5+CD4+ T Cells Are Counterparts of T Follicular Cells and Contain Specific Subsets that Differentially Support Antibody Secretion. Immunity 2011, 34, 108–121. [Google Scholar] [CrossRef]
- Walker, L.S.K. The link between circulating follicular helper T cells and autoimmunity. Nat. Rev. Immunol. 2022, 22, 567–575. [Google Scholar] [CrossRef]
- Choi, J.Y.; Ho, J.H.E.; Pasoto, S.G.; Bunin, V.; Kim, S.T.; Carrasco, S.; Borba, E.F.; Gonçalves, C.R.; Costa, P.R.; Kallas, E.G.; et al. Circulating follicular helper-like T cells in systemic lupus erythematosus: Association with disease activity. Arthritis Rheumatol. 2015, 67, 988–999. [Google Scholar] [CrossRef]
- Xu, H.; Liu, J.; Cui, X.; Zuo, Y.; Zhang, Z.; Li, Y.; Tao, R.; Li, Y.; Pang, J. Increased frequency of circulating follicular helper T cells in lupus patients is associated with autoantibody production in a CD40L-dependent manner. Cell. Immunol. 2015, 295, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Mao, M.; Xu, S.; Lin, L.; Dong, D.; Xue, M.; He, S.; Cai, G. Impact of Corticosteroids on the Proportions of Circulating Tfh Cell Subsets in Patients With Systemic Lupus Erythematous. Front. Med. 2022, 9, 949334. [Google Scholar] [CrossRef]
- Liarski, V.M.; Kaverina, N.; Chang, A.; Brandt, D.; Yanez, D.; Talasnik, L.; Carlesso, G.; Herbst, R.; Utset, T.O.; Labno, C.; et al. Cell Distance Mapping Identifies Functional T Follicular Helper Cells in Inflamed Human Renal Tissue. Sci. Transl. Med. 2014, 6, 230ra46. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wang, Z.; Chang, C.; Lu, L.; Lau, C.S.; Lu, Q. Th9 cells and IL-9 in autoimmune disorders: Pathogenesis and therapeutic potentials. Hum. Immunol. 2017, 78, 120–128. [Google Scholar] [CrossRef]
- Leng, R.-X.; Pan, H.-F.; Ye, D.-Q.; Xu, Y. Potential roles of IL-9 in the pathogenesis of systemic lupus erythematosus. Am. J. Clin. Exp. Immunol. 2012, 1, 28–32. [Google Scholar]
- Yang, J.; Li, Q.; Yang, X.; Li, M. Interleukin-9 Is Associated with Elevated Anti-Double-Stranded DNA Antibodies in Lupus-Prone Mice. Mol. Med. 2015, 21, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Paredes, J.L.; Fernandez-Ruiz, R.; Niewold, T.B. T Cells in Systemic Lupus Erythematosus. Rheum. Dis. Clin. N. Am. 2021, 47, 379–393. [Google Scholar] [CrossRef]
- Duan, J.; Kasper, D.L. Regulation of T cells by gut commensal microbiota. Curr. Opin. Rheumatol. 2011, 23, 372–376. [Google Scholar] [CrossRef]
- Silverman, G.J.; Azzouz, D.F.; Alekseyenko, A.V. Systemic Lupus Erythematosus and dysbiosis in the microbiome: Cause or effect or both? Curr. Opin. Immunol. 2019, 61, 80–85. [Google Scholar] [CrossRef]
- Brockmann, L.; Tran, A.; Huang, Y.; Edwards, M.; Ronda, C.; Wang, H.H.; Ivanov, I.I. Intestinal microbiota-specific Th17 cells possess regulatory properties and suppress effector T cells via c-MAF and IL-10. Immunity 2023, 56, 2719–2735.e7. [Google Scholar] [CrossRef]
- Zhang, L.; Qing, P.; Yang, H.; Wu, Y.; Liu, Y.; Luo, Y. Gut Microbiome and Metabolites in Systemic Lupus Erythematosus: Link, Mechanisms and Intervention. Front. Immunol. 2021, 12, 686501. [Google Scholar] [CrossRef] [PubMed]
- López, P.; de Paz, B.; Rodríguez-Carrio, J.; Hevia, A.; Sánchez, B.; Margolles, A.; Suárez, A. Th17 responses and natural IgM antibodies are related to gut microbiota composition in systemic lupus erythematosus patients. Sci. Rep. 2016, 6, 24072. [Google Scholar] [CrossRef] [PubMed]
- Kosiewicz, M.M.; Dryden, G.W.; Chhabra, A.; Alard, P. Relationship between gut microbiota and development of T cell associated disease. FEBS Lett. 2014, 588, 4195–4206. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Guo, P.; Zhang, J.; He, T.; Kim, S.W.; Zhang, G.; Ma, X. Nutrients Mediate Intestinal Bacteria-Mucosal Immune Crosstalk. Front. Immunol. 2018, 9, 5. [Google Scholar] [CrossRef]
- Romagnani, S.; Maggi, E.; Liotta, F.; Cosmi, L.; Annunziato, F. Properties and Origin of Human Th17 Cells. Mol. Immunol. 2009, 47, 3–7. [Google Scholar] [CrossRef]
- Valmori, D.; Raffin, C.; Raimbaud, I.; Ayyoub, M. Human RORγt+ TH17 Cells Preferentially Differentiate from Naive FOXP3+Treg in the Presence of Lineage-Specific Polarizing Factors. Proc. Natl. Acad. Sci. USA 2010, 107, 19402–19407. [Google Scholar] [CrossRef]
- Pang, B.; Zhen, Y.; Hu, C.; Ma, Z.; Lin, S.; Yi, H. Myeloid-derived suppressor cells shift Th17/Treg ratio and promote systemic lupus erythematosus progression through arginase-1/miR-322-5p/TGF-β pathway. Clin. Sci. 2020, 134, 2209–2222. [Google Scholar] [CrossRef]
- Zhu, M.; Mo, H.; Li, D.; Luo, X.; Zhang, L. Th17/Treg imbalance induced by increased incidence of atherosclerosis in patients with systemic lupus erythematosus (SLE). Clin. Rheumatol. 2013, 32, 1045–1052. [Google Scholar]
- Huang, J.; Li, X.; Zhu, Q.; Wang, M.; Xie, Z.; Zhao, T. Imbalance of Th17 cells, Treg cells and associated cytokines in patients with systemic lupus erythematosus: A meta-analysis. Front. Immunol. 2024, 15, 1425847. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-Y.; Chiu, W.-C.; Huang, Y.-L.; Su, Y.-J. Identify differential inflammatory cellular and serology pathways between children and adult patients in the lupus registry. Medicine 2022, 101, e29916. [Google Scholar] [CrossRef]
- Michels-van Amelsfort, J.M.; Walter, G.J.; Taams, L.S. CD4+CD25+ Regulatory T Cells in Systemic Sclerosis and Other Rheumatic Diseases. Expert Rev. Clin. Immunol. 2011, 7, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Kis-Toth, K.; Comte, D.; Karampetsou, M.P.; Kyttaris, V.C.; Kannan, L.; Terhorst, C.; Tsokos, G.C. Selective Loss of Signaling Lymphocytic Activation Molecule Family Member 4-Positive CD8+ T Cells Contributes to the Decreased Cytotoxic Cell Activity in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2016, 68, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rodríguez, N.; Apostolidis, S.A.; Penaloza-MacMaster, P.; Martín Villa, J.M.; Barouch, D.H.; Tsokos, G.C.; Crispín, J.C. Programmed cell death 1 and Helios distinguish TCR-αβ+ double-negative (CD4-CD8-) T cells that derive from self-reactive CD8 T cells. J. Immunol. 2015, 194, 4207–4214. [Google Scholar] [CrossRef]
- Kopetschke, K.; Klocke, J.; Grießbach, A.-S.; Humrich, J.Y.; Biesen, R.; Dragun, D.; Burmester, G.-R.; Enghard, P.; Riemekasten, G. The cellular signature of urinary immune cells in Lupus nephritis: New insights into potential biomarkers. Arthritis Res. Ther. 2015, 17, 1–9. [Google Scholar] [CrossRef]
- Sun, L.; Su, Y.; Jiao, A.; Wang, X.; Zhang, B. T cells in health and disease. Signal Transduct. Target. Ther. 2023, 8, 235. [Google Scholar] [CrossRef]
- Caccamo, N.; Battistini, L.; Bonneville, M.; Poccia, F.; Fournié, J.J.; Meraviglia, S.; Borsellino, G.; Kroczek, R.A.; La Mendola, C.; Scotet, E. CXCR5 identifies a subset of Vgamma9Vdelta2 T cells which secrete IL-4 and IL-10 and help B cells for antibody production. J. Immunol. 2006, 177, 5290–5295. [Google Scholar] [CrossRef] [PubMed]
- Rampoldi, F.; Ullrich, L.; Prinz, I. Revisiting the Interaction of γδ T-Cells and B-Cells. Cells 2020, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kang, N.; Zhou, J.; Guo, Y.; Zhang, X.; Cui, L.; Ba, D.; He, W. Downregulation of CD94/NKG2A inhibitory receptor on decreased γδ T cells in patients with systemic lupus erythematosus. Scand. J. Immunol. 2012, 76, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Robak, E.; Niewiadomska, H.; Robak, T.; Bartkowiak, J.; Błoński, J.Z.; Woźniacka, A.; Pomorski, L.; Sysa-Jędrzejowska, A. Lymphocyctes Tgammadelta in clinically normal skin and peripheral blood of patients with systemic lupus erythematosus and their correlation with disease activity. Mediat. Inflamm. 2001, 10, 179–189. [Google Scholar] [CrossRef]
- Paul, S.; Singh, A.K.; Shilpi null Lal, G. Phenotypic and functional plasticity of gamma-delta (γδ) T cells in inflammation and tolerance. Int. Rev. Immunol. 2014, 33, 537–558. [Google Scholar] [CrossRef]
- Wu, M.; Yang, J.; Li, X.; Chen, J. The Role of γδ T Cells in Systemic Lupus Erythematosus. J. Immunol. Res. 2016, 2016, 2932531. [Google Scholar] [CrossRef]
- Zhang, J.; Wencker, M.; Marliac, Q.; Berton, A.; Hasan, U.; Schneider, R.; Laubreton, D.; Cherrier, D.E.; Mathieu, A.L.; Rey, A.; et al. Zeb1 Represses TCR Signaling, Promotes the Proliferation of T Cell Progenitors and Is Essential for NK1.1+ T Cell Development. Cell. Mol. Immunol. 2021, 18, 2140–2152. [Google Scholar] [CrossRef]
- Li, H.; Adamopoulos, I.E.; Moulton, V.R.; Stillman, I.E.; Herbert, Z.; Moon, J.J.; Sharabi, A.; Krishfield, S.; Tsokos, M.G.; Tsokos, G.C. Systemic lupus erythematosus favors the generation of IL-17 producing double negative T cells. Nat. Commun. 2020, 11, 2859. [Google Scholar] [CrossRef] [PubMed]
- Crispín, J.C.; Oukka, M.; Bayliss, G.; Cohen, R.A.; Van Beek, C.A.; Stillman, I.E.; Kyttaris, V.C.; Juang, Y.T.; Tsokos, G.C. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J. Immunol. 2008, 181, 8761–8766. [Google Scholar] [CrossRef]
- Dowdell, K.C.; Niemela, J.E.; Price, S.; Davis, J.; Hornung, R.L.; Oliveira, J.B.; Puck, J.M.; Jaffe, E.S.; Pittaluga, S.; Cohen, J.I.; et al. Somatic FAS mutations are common in patients with genetically undefined autoimmune lymphoproliferative syndrome. Blood 2010, 115, 5164–5169. [Google Scholar] [CrossRef]
- Alunno, A.; Bistoni, O.; Bartoloni Bocci, E.; Caterbi, S.; Bigerna, B.; Pucciarini, A.; Tabarrini, A.; Mannucci, R.; Beghelli, D.; Falini, B.; et al. IL-17-producing double-negative T cells are expanded in the peripheral blood, infiltrate the salivary gland and are partially resistant to corticosteroid therapy in patients with Sjögren’s syndrome. Reumatismo 2013, 65, 192–198. [Google Scholar] [CrossRef]
- Russell, T.B.; Kurre, P. Double-negative T cells are non–ALPS-specific markers of immune dysregulation found in patients with aplastic anemia. Blood 2010, 116, 5072–5073. [Google Scholar] [CrossRef]
- Wiener, A.; Schippers, A.; Wagner, N.; Tacke, F.; Ostendorf, T.; Honke, N.; Tenbrock, K.; Ohl, K. CXCR5 is critically involved in progression of lupus through regulation of B cell and double-negative T cell trafficking. Clin. Exp. Immunol. 2016, 185, 22–32. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotypic Alterations of B Lymphocytes in SLE | |
|---|---|
| Upregulation | Downregulation |
| Switched Memory (CD19+IgD-CD27+) | Naïve (CD19+IgD+CD27-) |
| Double Negative (CD19+IgD-CD27-) | Non-Switched Memory (CD19+IgD+CD27+) |
| Age Associated B cells (CD19+CD21-CD11c+Tbet+) | |
| B regulatory cells (secreting IL10, IL5) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moysidou, E.; Christodoulou, M.; Lioulios, G.; Stai, S.; Karamitsos, T.; Dimitroulas, T.; Fylaktou, A.; Stangou, M. Lymphocytes Change Their Phenotype and Function in Systemic Lupus Erythematosus and Lupus Nephritis. Int. J. Mol. Sci. 2024, 25, 10905. https://doi.org/10.3390/ijms252010905
Moysidou E, Christodoulou M, Lioulios G, Stai S, Karamitsos T, Dimitroulas T, Fylaktou A, Stangou M. Lymphocytes Change Their Phenotype and Function in Systemic Lupus Erythematosus and Lupus Nephritis. International Journal of Molecular Sciences. 2024; 25(20):10905. https://doi.org/10.3390/ijms252010905
Chicago/Turabian StyleMoysidou, Eleni, Michalis Christodoulou, Georgios Lioulios, Stamatia Stai, Theodoros Karamitsos, Theodoros Dimitroulas, Asimina Fylaktou, and Maria Stangou. 2024. "Lymphocytes Change Their Phenotype and Function in Systemic Lupus Erythematosus and Lupus Nephritis" International Journal of Molecular Sciences 25, no. 20: 10905. https://doi.org/10.3390/ijms252010905