
Single-Cell Transcriptomics Reveals Cellular Heterogeneity and Drivers in Serrated Pathway-Driven Colorectal Cancer Progression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
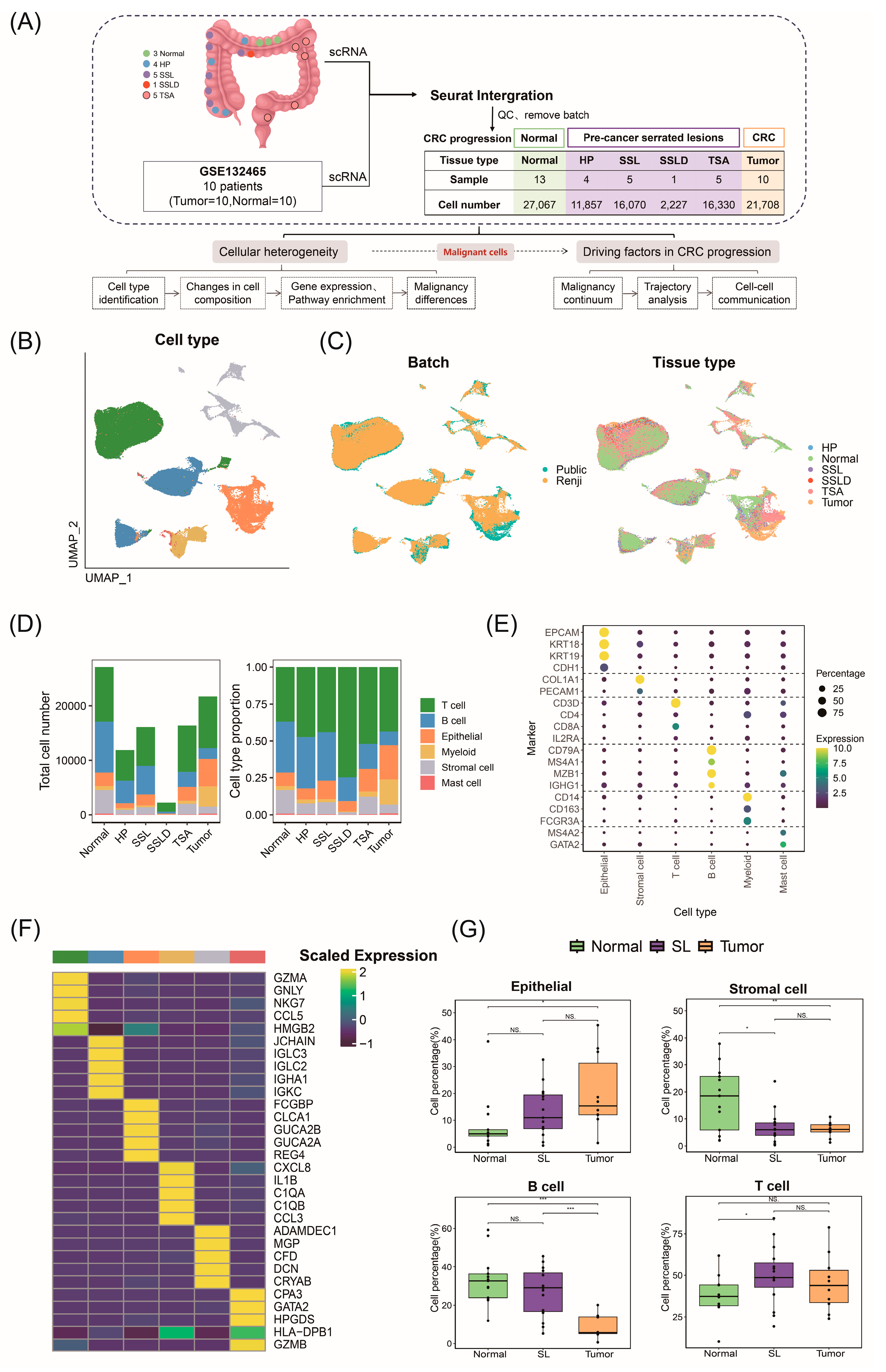
2.1. ScRNA-Seq Atlas and Cellular Composition of Stepwise Progression of CRC
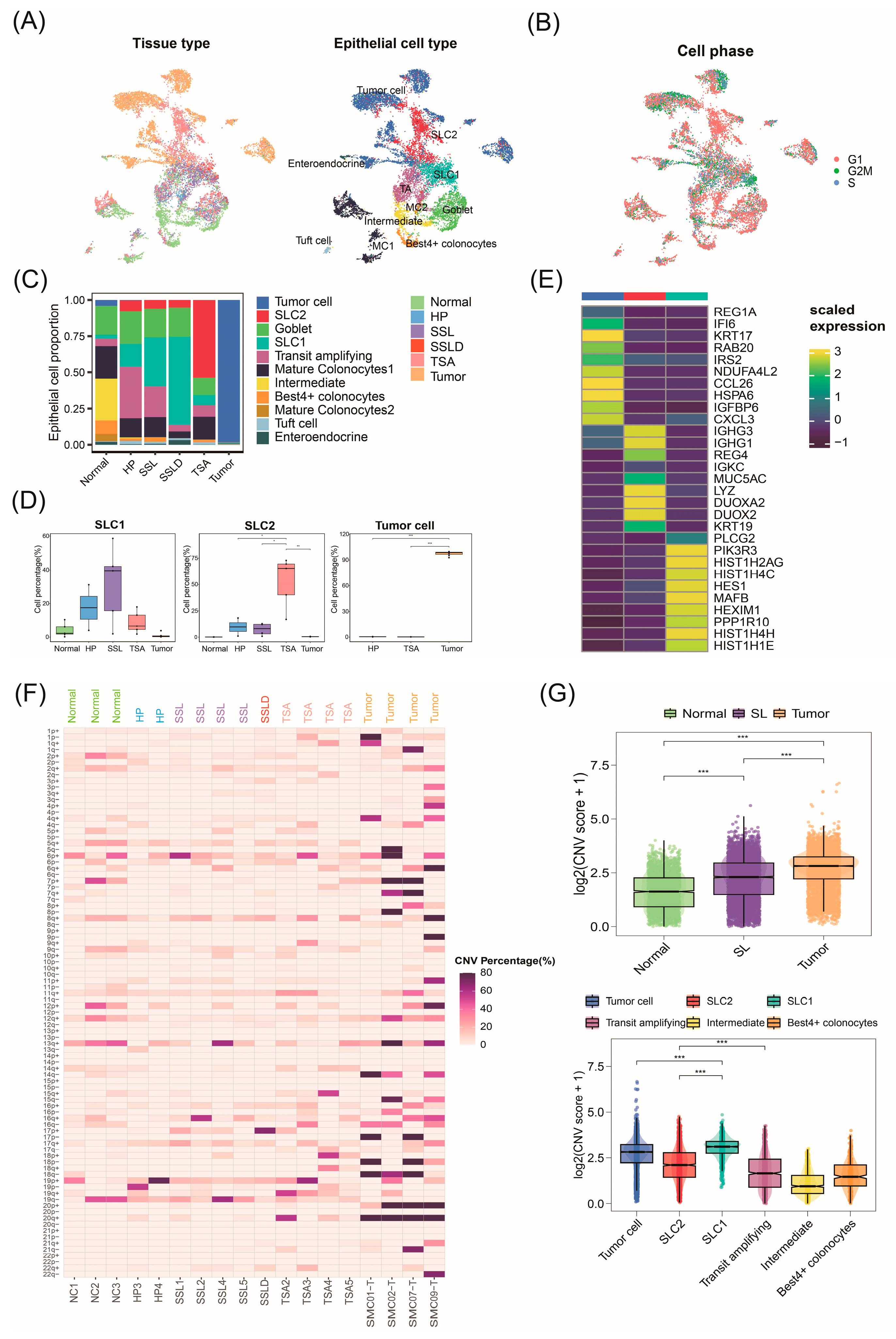
2.2. Invasive Malignant Epithelial Cell Subtypes Associated with CRC Progression
2.3. Malignant Origins of CRC Identified by Copy Number Variation Analysis
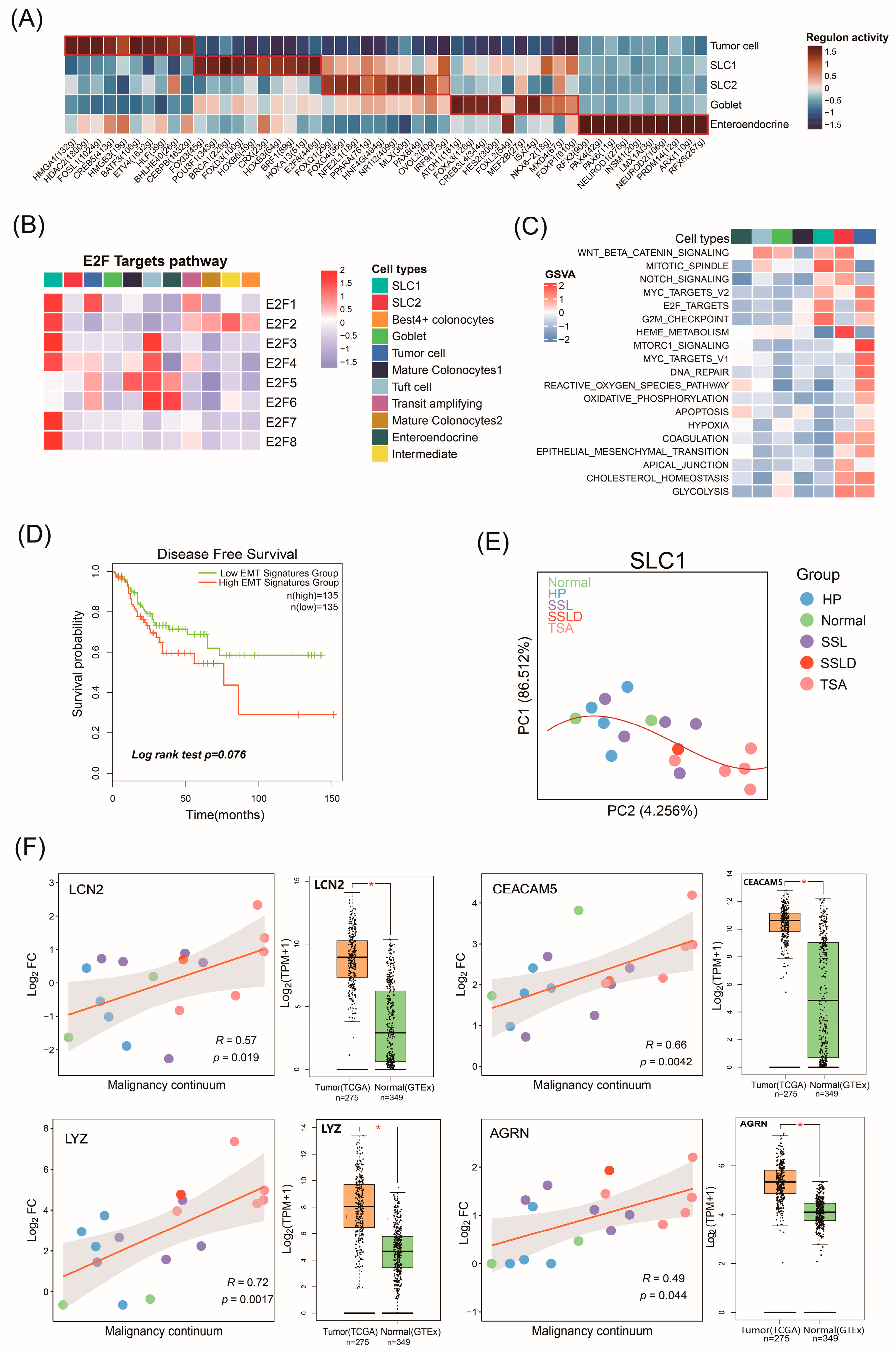
2.4. The Biological Functions of Malignant Cell Subtypes Involved in CRC Progression
2.5. Potential Malignant Continuum Formed by SLC1
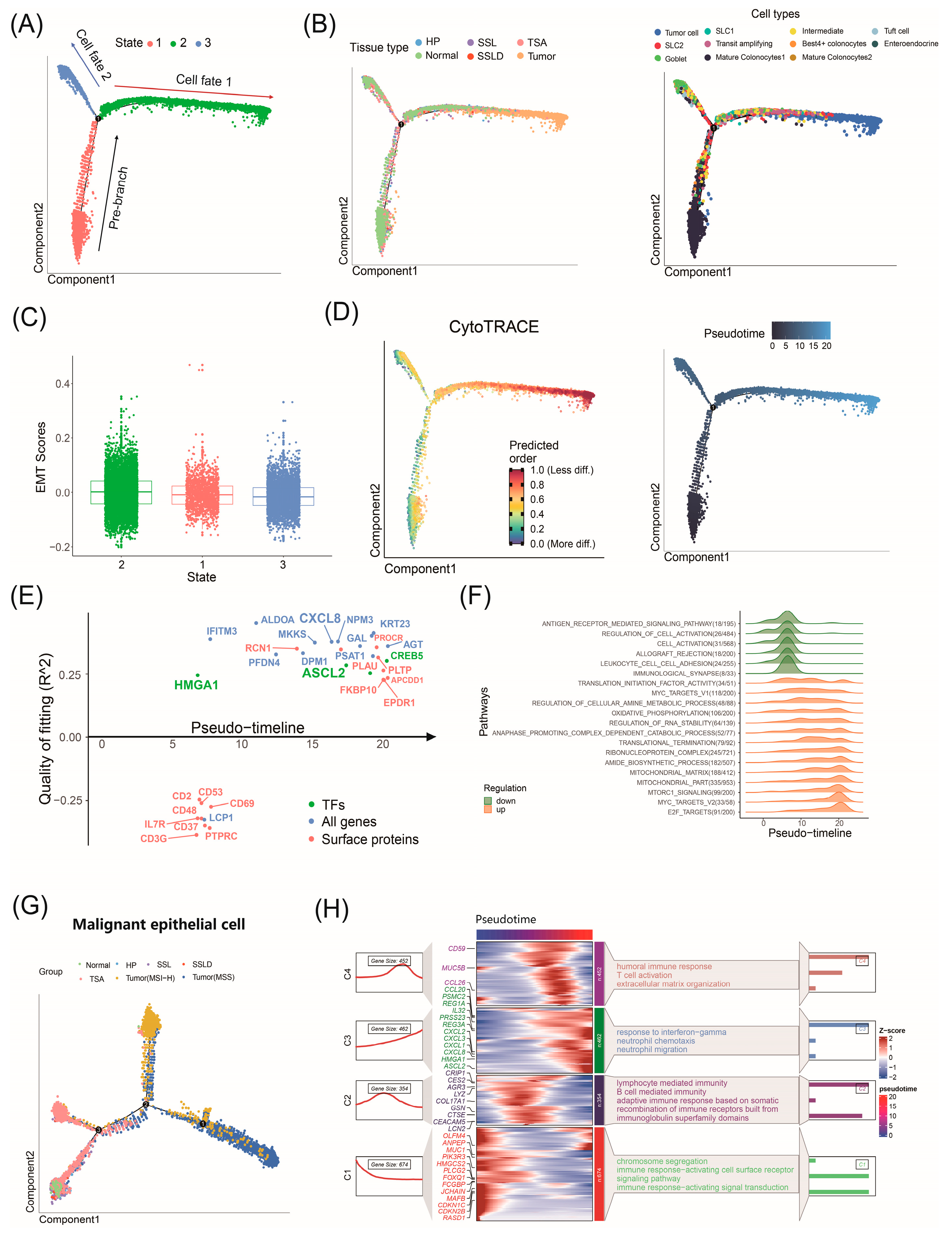
2.6. Factors Driving CRC Progression Identified by Trajectory Analysis
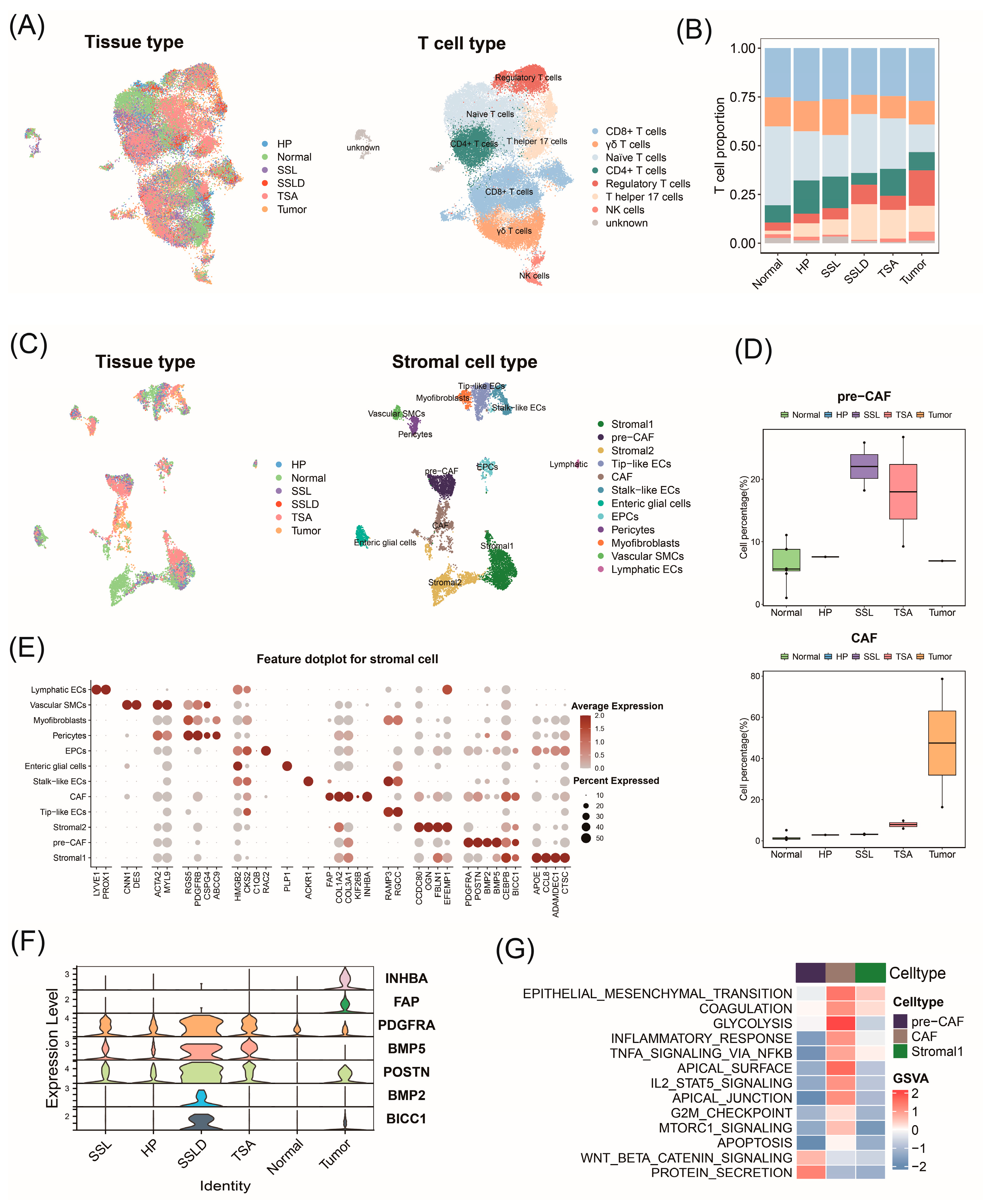
2.7. Transcriptomic Landscape Alterations in Stromal and Immune Cells throughout Progression of CRC
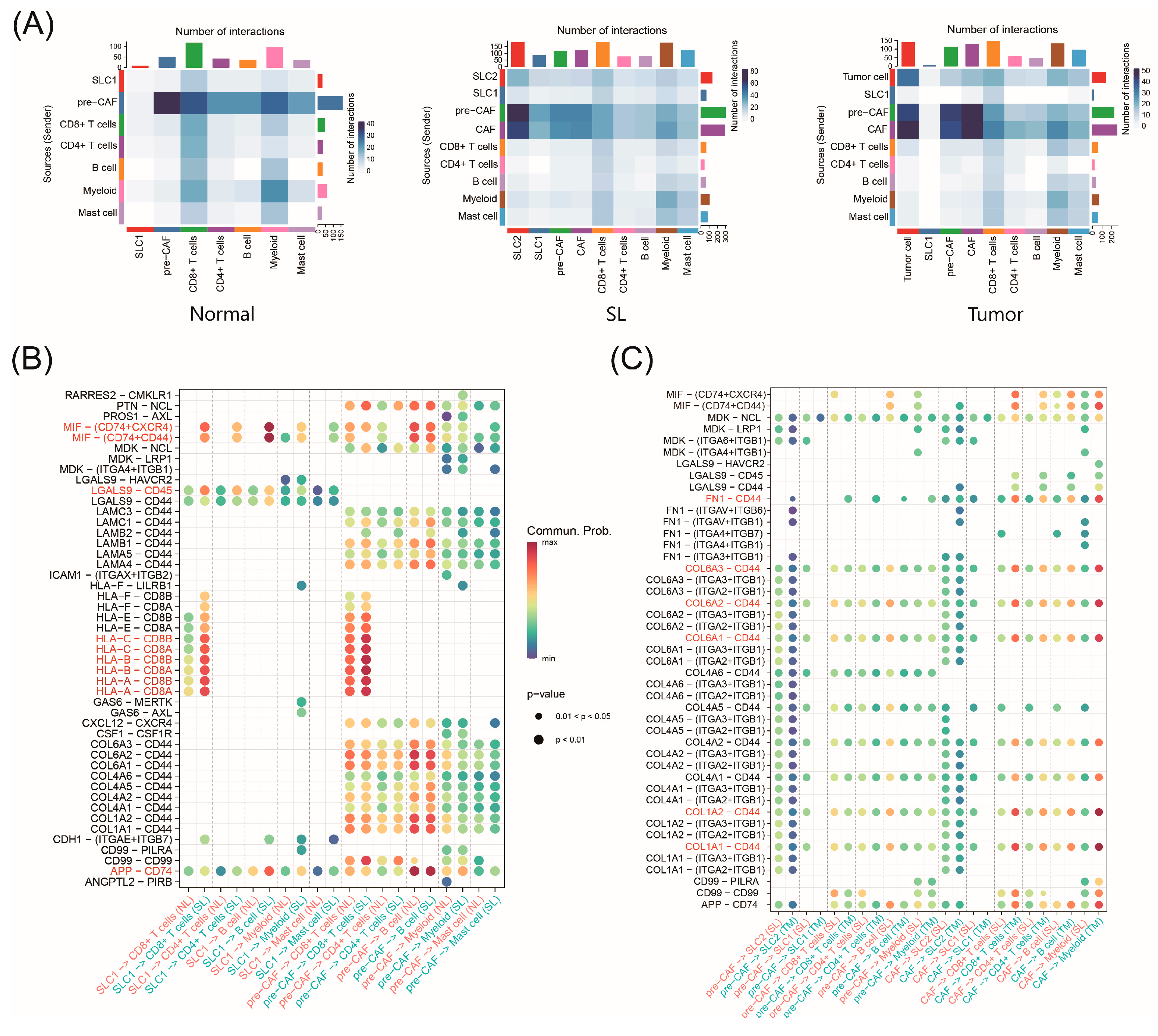
2.8. Cell–Cell Interactions during CRC Progression
3. Discussion
4. Materials and Methods
Dataset Preparation and Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.; Hemann, M.T. Drugs, Bugs, and Cancer: Fusobacterium nucleatum Promotes Chemoresistance in Colorectal Cancer. Cell 2017, 170, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Pita-Fernández, S.; Alhayek-Aí, M.; González-Martín, C.; López-Calviño, B.; Seoane-Pillado, T.; Pértega-Díaz, S. Intensive follow-up strategies improve outcomes in nonmetastatic colorectal cancer patients after curative surgery: A systematic review and meta-analysis. Ann. Oncol. 2015, 26, 644–656. [Google Scholar] [CrossRef]
- Fodde, R.; Smits, R.; Clevers, H. APC, Signal transduction and genetic instability in colorectal cancer. Nat. Rev. Cancer 2001, 1, 55–67. [Google Scholar] [CrossRef]
- Ijspeert, J.E.G.; Vermeulen, L.; Meijer, G.A.; Dekker, E. Serrated neoplasia—Role in colorectal carcinogenesis and clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 401–409. [Google Scholar] [CrossRef]
- Ahadi, M.; Sokolova, A.; Brown, I.; Chou, A.; Gill, A.J. The 2019 World Health Organization Classification of appendiceal, colorectal and anal canal tumours: An update and critical assessment. Pathology 2021, 53, 454–461. [Google Scholar] [CrossRef]
- Leggett, B.; Whitehall, V. Role of the Serrated Pathway in Colorectal Cancer Pathogenesis. Gastroenterology 2010, 138, 2088–2100. [Google Scholar] [CrossRef] [PubMed]
- Bramsen, J.B.; Rasmussen, M.H.; Ongen, H.; Mattesen, T.B.; Ørntoft, M.-B.W.; Árnadóttir, S.S.; Sandoval, J.; Laguna, T.; Vang, S.; Øster, B.; et al. Molecular-Subtype-Specific Biomarkers Improve Prediction of Prognosis in Colorectal Cancer. Cell Rep. 2017, 19, 1268–1280. [Google Scholar] [CrossRef]
- Peng, X.L.; Moffitt, R.A.; Torphy, R.J.; Volmar, K.E.; Yeh, J.J. De novo compartment deconvolution and weight estimation of tumor samples using DECODER. Nat. Commun. 2019, 10, 4729. [Google Scholar] [CrossRef]
- Di, Z.; Zhou, S.; Xu, G.; Ren, L.; Li, C.; Ding, Z.; Huang, K.; Liang, L.; Yuan, Y. Single-cell and WGCNA uncover a prognostic model and potential oncogenes in colorectal cancer. Biol. Proced. Online 2022, 24, 13. [Google Scholar] [CrossRef]
- Hudson, T.J.; Anderson, W.; Aretz, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; Gerhard, D.S.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-J.; Lu, X.-F.; Chen, H.; Wang, X.-Y.; Cheng, W.; Zhang, Q.-W.; Chen, J.-N.; Wang, X.-Y.; Jin, J.-Z.; Yan, F.-R.; et al. Single-cell Transcriptomics Reveals Early Molecular and Immune Alterations Underlying the Serrated Neoplasia Pathway Toward Colorectal Cancer. Cell Mol. Gastroenterol. Hepatol. 2023, 15, 393–424. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, H.; Chu, Y.; Song, Y.; Ding, L.; Zhu, B.; Zhai, W.; Wang, X.; Kuang, Y.; Ren, F.; et al. CREPT is required for murine stem cell maintenance during intestinal regeneration. Nat. Commun. 2021, 12, 270. [Google Scholar] [CrossRef] [PubMed]
- Natasja, L.d.V.; van Vincent, U.; Marieke, E.I.; Abdelaal, T.; Van Der Breggen, R.; Sarasqueta, A.F.; Mahfouz, A.; Peeters, K.C.M.J.; Höllt, T.; Lelieveldt, B.P.F.; et al. High-dimensional cytometric analysis of colorectal cancer reveals novel mediators of antitumour immunity. Gut 2020, 69, 691–703. [Google Scholar] [CrossRef]
- Xu, Y.; Wei, Z.; Feng, M.; Zhu, D.; Mei, S.; Wu, Z.; Feng, Q.; Chang, W.; Ji, M.; Liu, C.; et al. Tumor-infiltrated activated B cells suppress liver metastasis of colorectal cancers. Cell Rep. 2022, 40, 111295. [Google Scholar] [CrossRef]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, X.; Luo, X.; Wang, J.; Hu, J.; Feng, Y. PIK3R3 inhibits cell senescence through p53/p21 signaling. Cell Death Dis. 2020, 11, 798. [Google Scholar] [CrossRef]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019, 35, 559–572.e7. [Google Scholar] [CrossRef]
- Astrosini, C.; Roeefzaad, C.; Dai, Y.; Dieckgraefe, B.K.; Jöns, T.; Kemmner, W. REG1A expression is a prognostic marker in colorectal cancer and associated with peritoneal carcinomatosis. Int. J. Cancer 2008, 123, 409–413. [Google Scholar] [CrossRef]
- Kent, L.N.; Leone, G. The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 2019, 19, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Pizzolato, G.; Moparthi, L.; Söderholm, S.; Cantù, C.; Koch, S. The oncogenic transcription factor FOXQ1 is a differential regulator of Wnt target genes. J. Cell Sci. 2022, 135, jcs260082. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Feng, J.; Chen, W.; Wang, W.; Wu, X.; Zhang, J.; Lai, M. Lipocalin2 suppresses metastasis of colorectal cancer by attenuating NF-κB-dependent activation of snail and epithelial mesenchymal transition. Mol. Cancer 2016, 15, 77. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukarasu, P.; Sukumar, S.; Sathaiah, M.; Mahan, M.; Pragatheeshwar, K.D.; Pingpank, J.F.; Zeh, H.; Bartels, C.J.; Lee, K.K.; Bartlett, D.L. C-stage in Colon Cancer: Implications of Carcinoembryonic Antigen Biomarker in Staging, Prognosis, and Management. J. Natl. Cancer Inst. 2011, 103, 689–697. [Google Scholar] [CrossRef]
- Rickelt, S.; Condon, C.; Mana, M.; Whittaker, C.; Pfirschke, C.; Roper, J.; Patil, D.T.; Brown, I.; Mattia, A.R.; Zukerberg, L.; et al. Agrin in the Muscularis Mucosa Serves as a Biomarker Distinguishing Hyperplastic Polyps from Sessile Serrated Lesions. Clin. Cancer Res. 2020, 26, 1277–1287. [Google Scholar] [CrossRef]
- van der Flier, L.G.; van Gijn, M.E.; Hatzis, P.; Kujala, P.; Haegebarth, A.; Stange, D.E.; Begthel, H.; Born, M.v.D.; Guryev, V.; Oving, I.; et al. Transcription Factor Achaete Scute-Like 2 Controls Intestinal Stem Cell Fate. Cell 2009, 136, 903–912. [Google Scholar] [CrossRef]
- Pennel, K.A.; Quinn, J.A.; Nixon, C.; Inthagard, J.; van Wyk, H.C.; Chang, D.; Rebus, S.; GPOL Group; Hay, J.; Maka, N.N.; et al. CXCL8 expression is associated with advanced stage, right sidedness, and distinct histological features of colorectal cancer. J. Pathol. Clin. Res. 2022, 8, 509–520. [Google Scholar] [CrossRef]
- Koliaraki, V.; Pallangyo, C.K.; Greten, F.R.; Greten, F.R.; Kollias, G. Mesenchymal Cells in Colon Cancer. Gastroenterology 2017, 152, 964–979. [Google Scholar] [CrossRef]
- Karagiannis, G.S.; Poutahidis, T.; Erdman, S.E.; Kirsch, R.; Riddell, R.H.; Diamandis, E.P. Cancer-Associated Fibroblasts Drive the Progression of Metastasis through both Paracrine and Mechanical Pressure on Cancer Tissue. Mol. Cancer Res. 2012, 10, 1403–1418. [Google Scholar] [CrossRef]
- Puré, E.; Blomberg, R. Pro-tumorigenic roles of fibroblast activation protein in cancer: Back to the basics. Oncogene 2018, 37, 4343–4357. [Google Scholar] [CrossRef]
- Fang, S.; Dai, Y.; Mei, Y.; Yang, M.; Hu, L.; Yang, H.; Guan, X.; Li, J. Clinical significance and biological role of cancer-derived Type I collagen in lung and esophageal cancers. Thorac. Cancer 2019, 10, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.; Nelson, C.M.; Nandadasa, S.; Aramaki-Hattori, N.; Lindner, D.J.; Alban, T.; Inagaki, J.; Ohtsuki, T.; Oohashi, T.; Apte, S.S.; et al. Stromal Versican Regulates Tumor Growth by Promoting Angiogenesis. Sci. Rep. 2017, 7, 17225. [Google Scholar] [CrossRef] [PubMed]
- Vanuytsel, T.; Senger, S.; Fasano, A.; Shea-Donohue, T. Major signaling pathways in intestinal stem cells. Biochim. Biophys. Acta 2013, 1830, 2410–2426. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Chen, L.; Chen, G.; Smaldini, P.; Bongers, G.; Catalan-Dibene, J.; Furtado, G.C.; Lira, S.A. Interleukin 1 beta and Matrix Metallopeptidase 3 Contributeto Development of Epidermal Growth Factor Receptor–Dependent Serrated Polyps in Mouse Cecum. Gastroenterology 2019, 157, 1572–1583.e8. [Google Scholar] [CrossRef]
- Li, H.; Fan, X.; Houghton, J. Tumor microenvironment: The role of the tumor stroma in cancer. J. Cell Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef]
- Swartz, M.A.; Iida, N.; Roberts, E.W.; Sangaletti, S.; Wong, M.H.; Yull, F.E.; Coussens, L.M.; Declerck, Y.A. Tumor Microenvironment Complexity: Emerging Roles in Cancer Therapy. Cancer Res. 2012, 72, 2473–2480. [Google Scholar] [CrossRef]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Lin, D.; Fan, W.; Zhang, R.; Zhao, E.; Li, P.; Zhou, W.; Peng, J.; Li, L. Molecular subtype identification and prognosis stratification by a metabolism-related gene expression signature in colorectal cancer. J. Transl. Med. 2021, 19, 279. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Seeber, A.; Battaglin, F.; Zimmer, K.; Kocher, F.; Baca, Y.; Xiu, J.; Spizzo, G.; Novotny-Diermayr, V.; Rieder, D.; Puccini, A.; et al. Comprehensive Analysis of R-Spondin Fusions and RNF43 Mutations Implicate Novel Therapeutic Options in Colorectal Cancer. Clin. Cancer Res. 2022, 28, 1863–1870. [Google Scholar] [CrossRef] [PubMed]
- Utsumi, T.; Yamada, Y.; Diaz-Meco, M.T.; Moscat, J.; Nakanishi, Y. Sessile serrated lesions with dysplasia: Is it possible to nip them in the bud? J. Gastroenterol. 2023, 58, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Bettington, M.; Walker, N.; Clouston, A.; Brown, I.; Leggett, B.; Whitehall, V. The serrated pathway to colorectal carcinoma: Current concepts and challenges. Histopathology 2013, 62, 367–386. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Zhang, Y.; Chen, X.; Sheng, Q.; Yang, J.; Zhu, Y.; Wang, Y.; Yan, F.; Fang, J. Single-Cell Transcriptomics Reveals Cellular Heterogeneity and Drivers in Serrated Pathway-Driven Colorectal Cancer Progression. Int. J. Mol. Sci. 2024, 25, 10944. https://doi.org/10.3390/ijms252010944
Wang J, Zhang Y, Chen X, Sheng Q, Yang J, Zhu Y, Wang Y, Yan F, Fang J. Single-Cell Transcriptomics Reveals Cellular Heterogeneity and Drivers in Serrated Pathway-Driven Colorectal Cancer Progression. International Journal of Molecular Sciences. 2024; 25(20):10944. https://doi.org/10.3390/ijms252010944
Chicago/Turabian StyleWang, Jiahui, Yu Zhang, Xinyi Chen, Qi Sheng, Junluo Yang, Yuyao Zhu, Yuhang Wang, Fangrong Yan, and Jingya Fang. 2024. "Single-Cell Transcriptomics Reveals Cellular Heterogeneity and Drivers in Serrated Pathway-Driven Colorectal Cancer Progression" International Journal of Molecular Sciences 25, no. 20: 10944. https://doi.org/10.3390/ijms252010944