

Molecular Landscape of Bladder Cancer: Key Genes, Transcription Factors, and Drug Interactions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
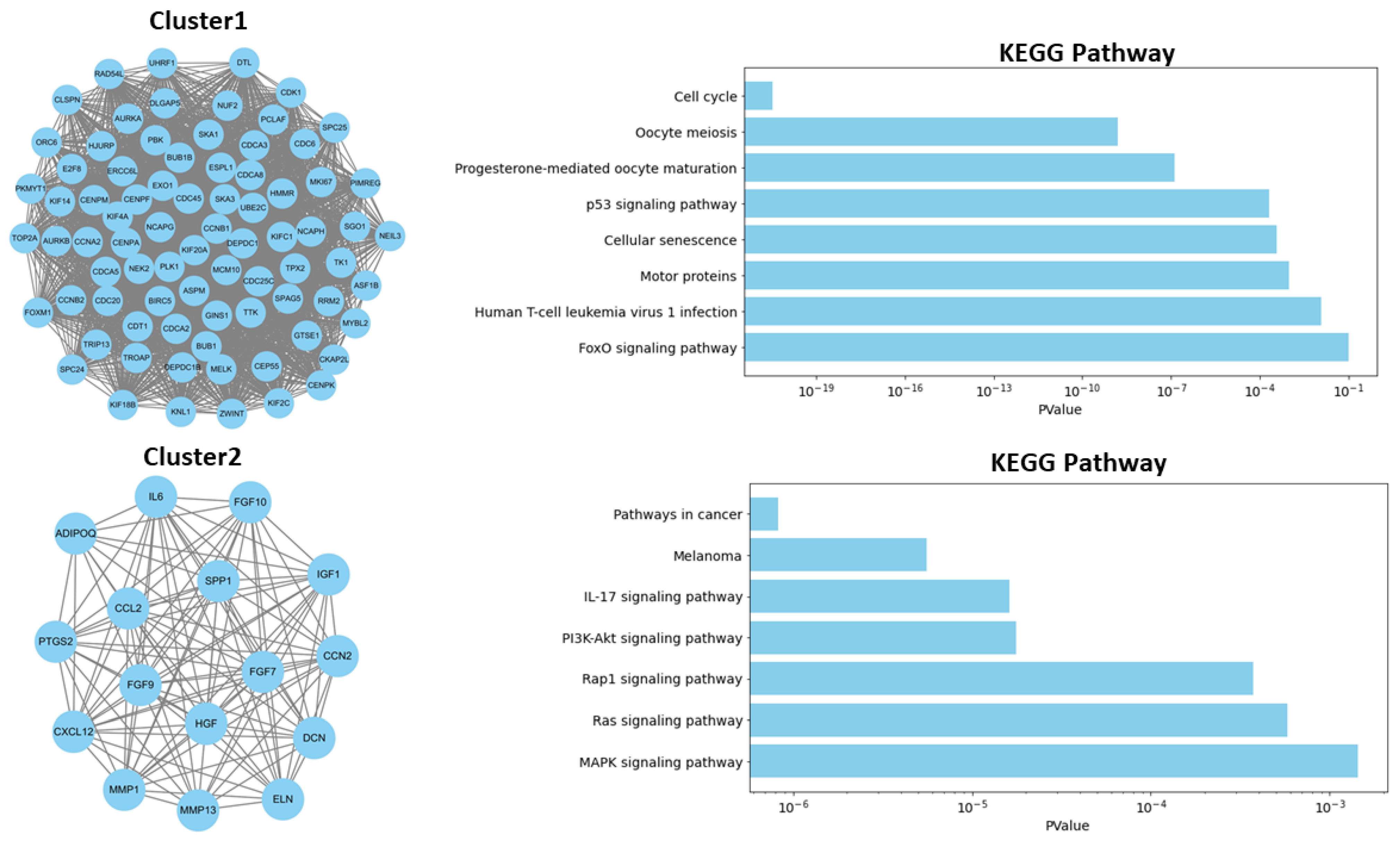
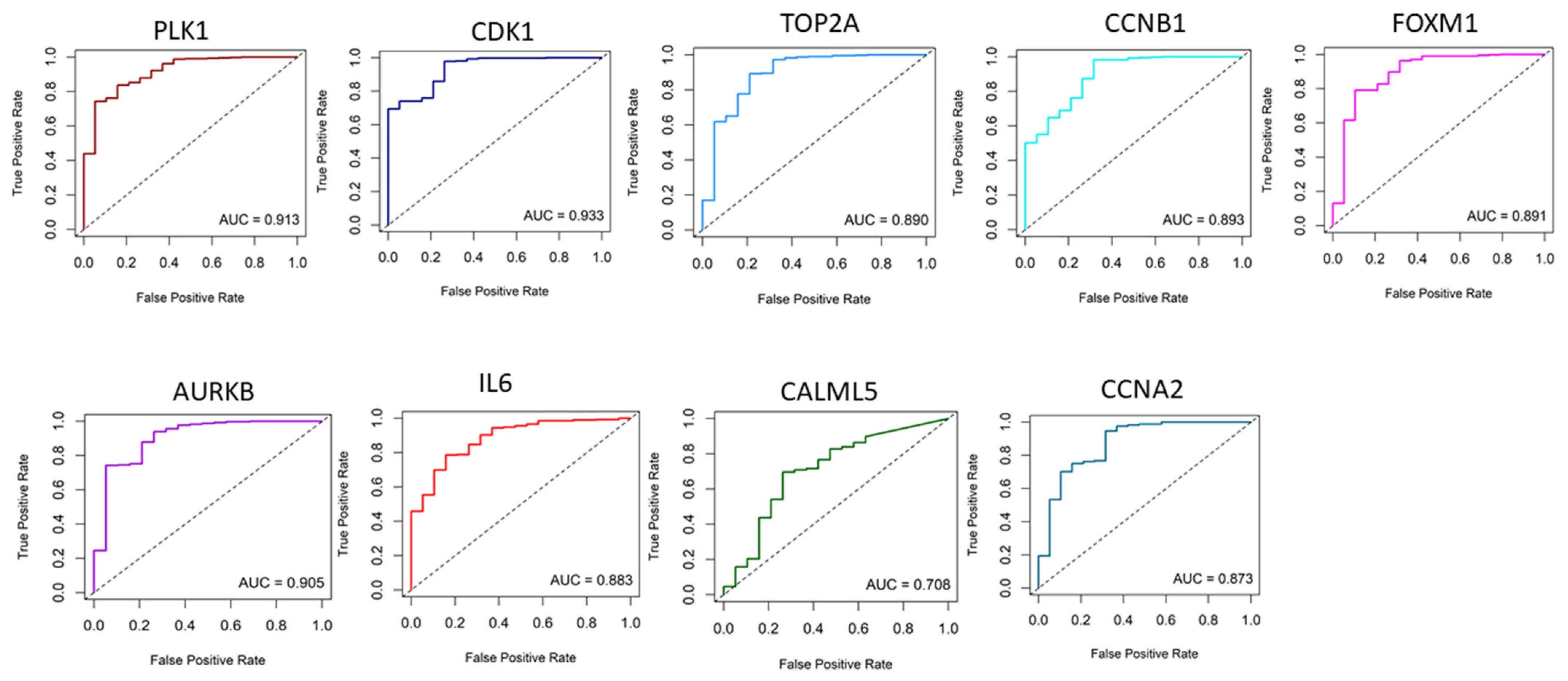
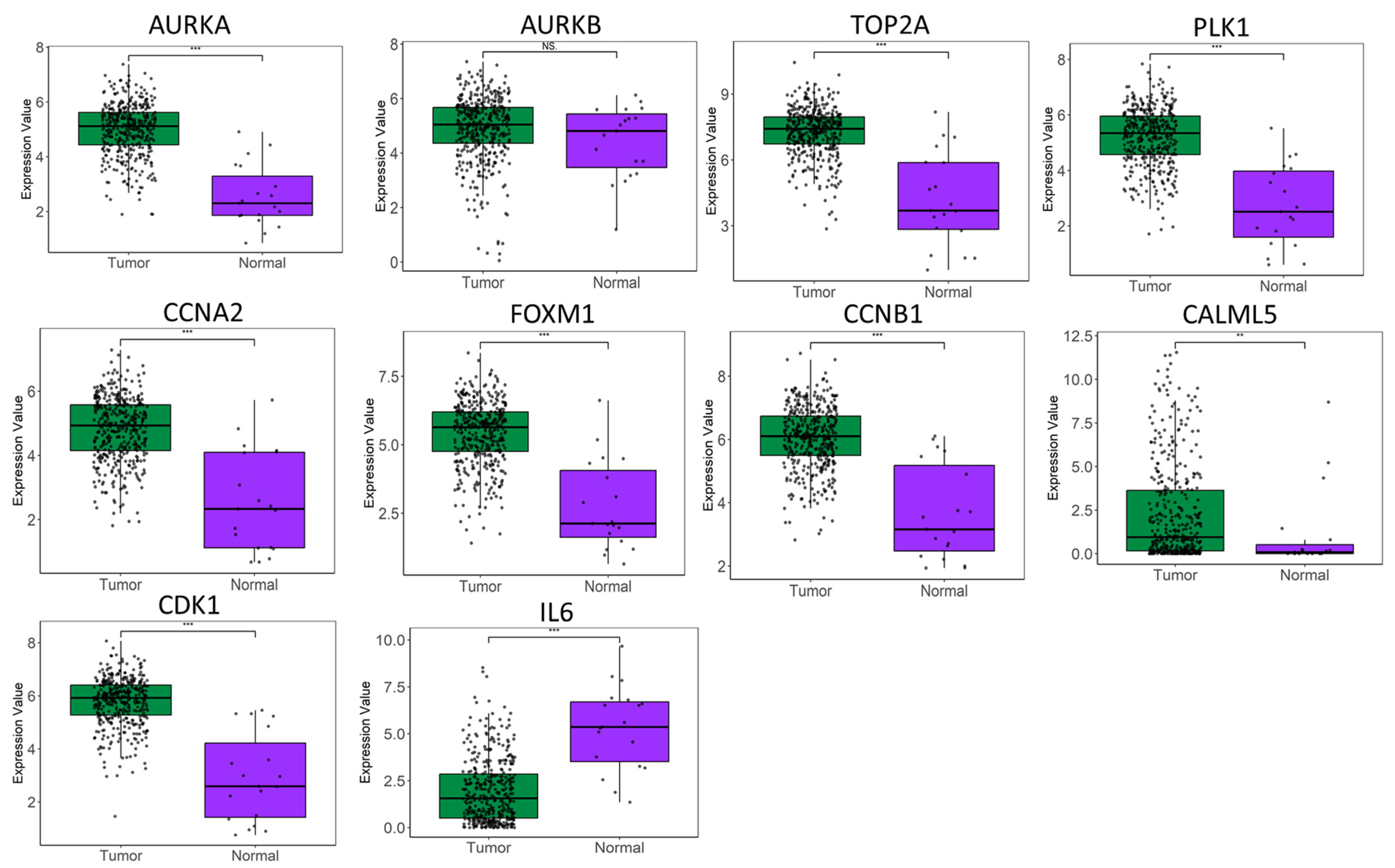
2.1. Identification of Key Regulators or Hub Genes
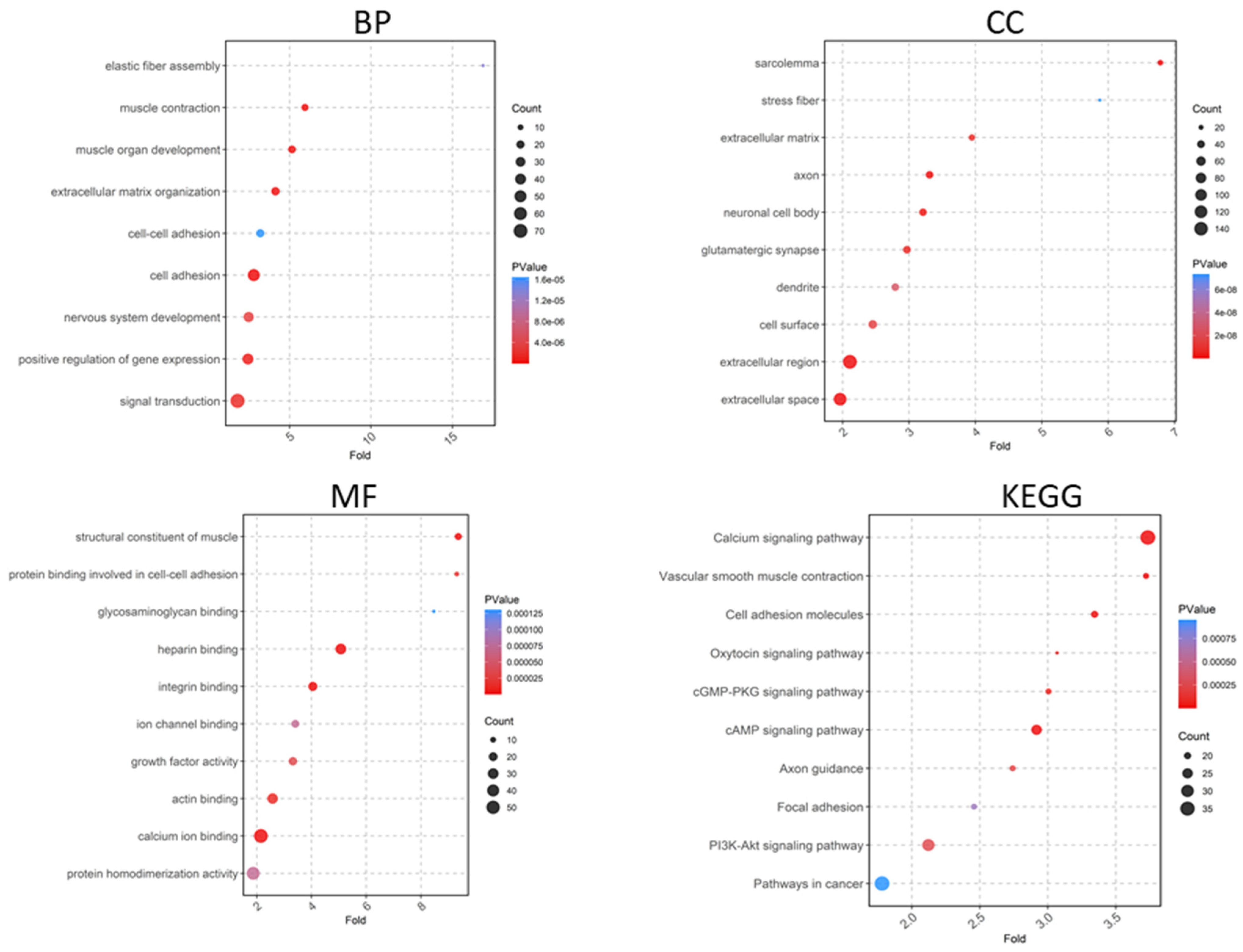
2.2. Functional Enrichment Analysis of DEGs
2.3. Relationship of Hub Genes and Disease-Related Genes
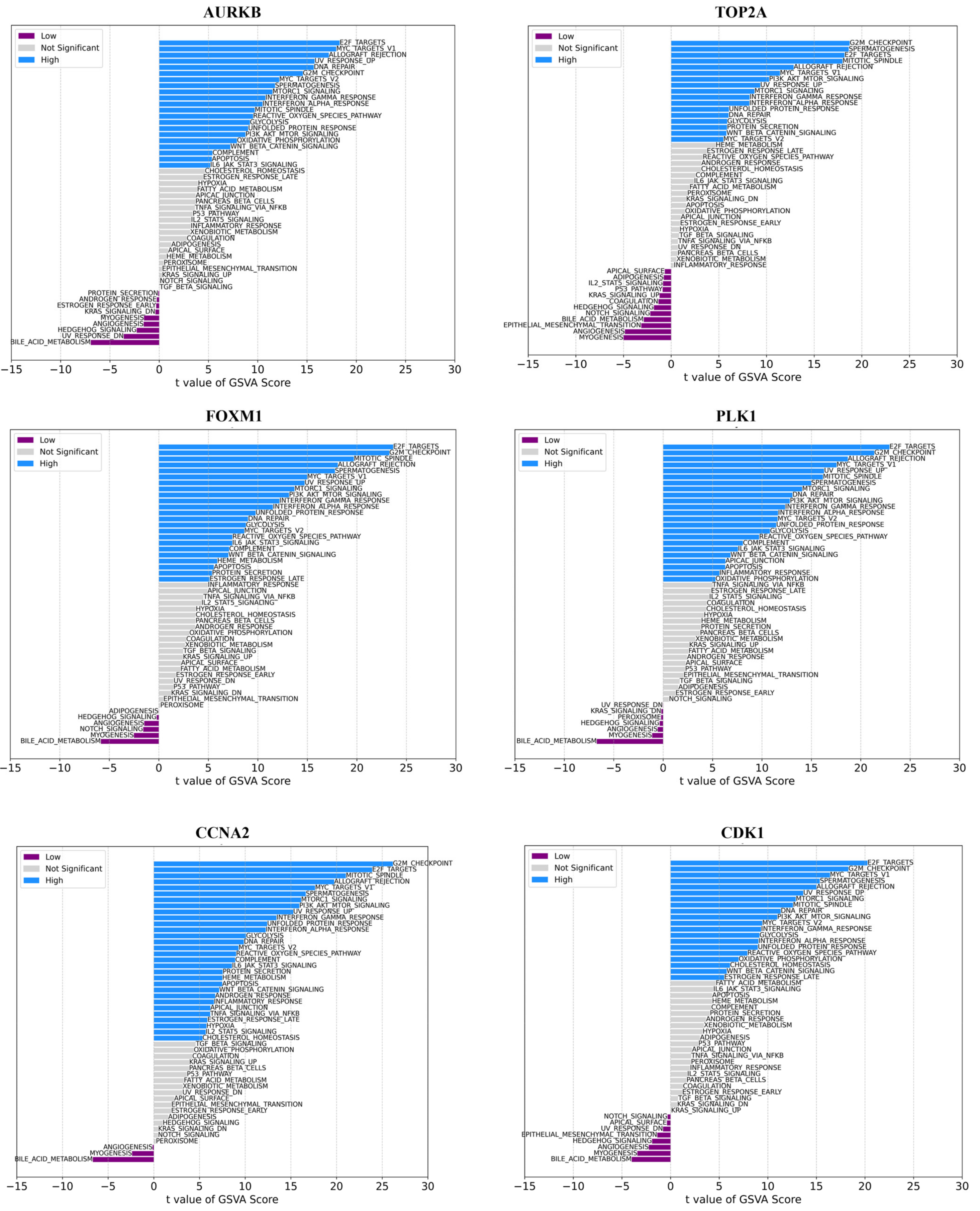
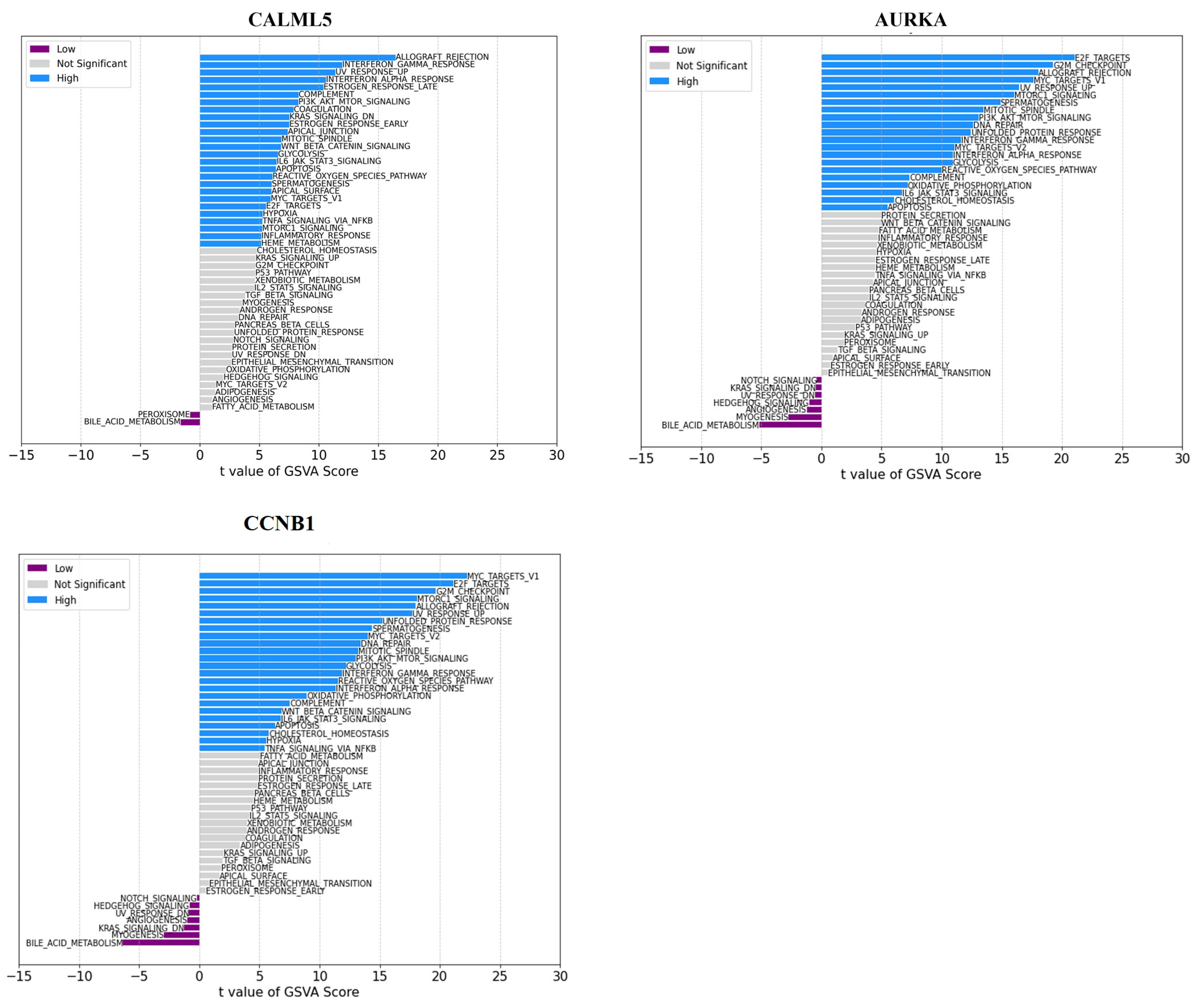
2.4. Gene Set Variation Analysis (GSVA) Analyses
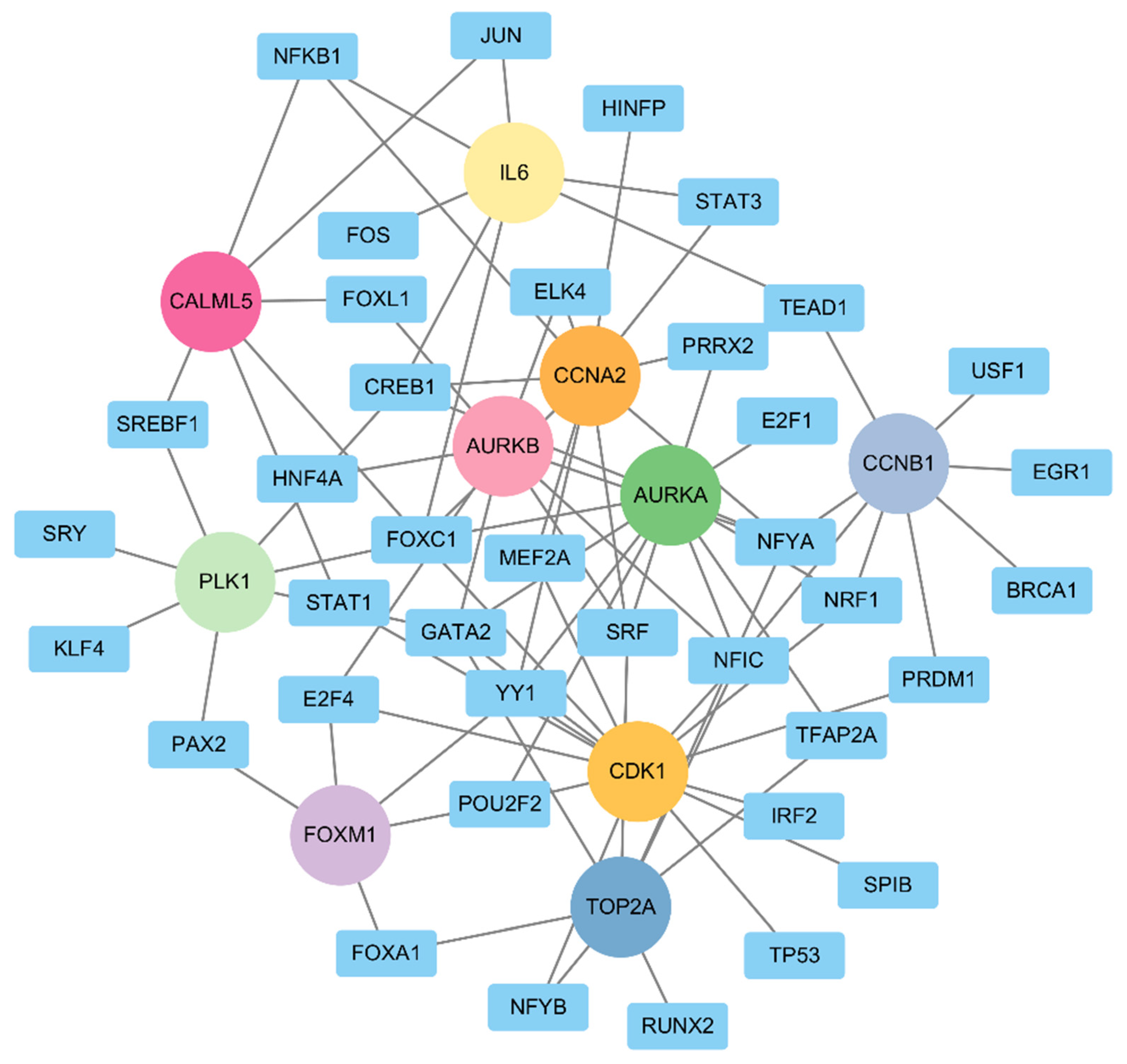
2.5. TF–Gene Regulatory Network Analyses
2.6. Potential Drug Identification
3. Discussion
4. Material and Methods
4.1. Dataset
4.2. Functional Enrichment Analysis of the DEGs
4.3. Key Regulators (KRs)
4.4. Molecular Complex Detection (MCODE)
4.5. Receiver Operating Characteristic (ROC) Curve Analyses
4.6. Gene Set Variation Analysis (GSVA)
4.7. Gene–Transcriptional Factor (TF) Regulatory Network
4.8. Drug–Gene Interactions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Richters, A.; Aben, K.K.H.; Kiemeney, L.A.L.M. The Global Burden of Urinary Bladder Cancer: An Update. World J. Urol. 2020, 38, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, D.S.; Shipley, W.U.; Feldman, A.S. Bladder Cancer. Lancet 2009, 374, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Xu, P. Global Colorectal Cancer Burden in 2020 and Projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef] [PubMed]
- Hensley, P.J.; Duan, Z.; Bree, K.; Sood, A.; Zhao, H.; Lobo, N.; Contieri, R.; Campbell, M.T.; Guo, C.C.; Navai, N.; et al. Competing Mortality Risk from Second Primary Malignancy in Bladder Cancer Patients Following Radical Cystectomy: Implications for Survivorship. Urol. Oncol. Semin. Orig. Investig. 2023, 41, 108.e11–108.e17. [Google Scholar] [CrossRef]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Padala, S.A.; Barsouk, A. Epidemiology of Bladder Cancer. Med. Sci. 2020, 8, 15. [Google Scholar] [CrossRef]
- Freedman, N.D. Association Between Smoking and Risk of Bladder Cancer Among Men and Women. JAMA 2011, 306, 737. [Google Scholar] [CrossRef]
- Ebrahimi, H.; Amini, E.; Pishgar, F.; Moghaddam, S.S.; Nabavizadeh, B.; Rostamabadi, Y.; Aminorroaya, A.; Fitzmaurice, C.; Farzadfar, F.; Nowroozi, M.R.; et al. Global, Regional and National Burden of Bladder Cancer, 1990 to 2016: Results from the GBD Study 2016. J. Urol. 2019, 201, 893–901. [Google Scholar] [CrossRef]
- Tran, L.; Xiao, J.-F.; Agarwal, N.; Duex, J.E.; Theodorescu, D. Advances in Bladder Cancer Biology and Therapy. Nat. Rev. Cancer 2021, 21, 104–121. [Google Scholar] [CrossRef]
- Lindskrog, S.V.; Prip, F.; Lamy, P.; Taber, A.; Groeneveld, C.S.; Birkenkamp-Demtröder, K.; Jensen, J.B.; Strandgaard, T.; Nordentoft, I.; Christensen, E.; et al. An Integrated Multi-Omics Analysis Identifies Prognostic Molecular Subtypes of Non-Muscle-Invasive Bladder Cancer. Nat. Commun. 2021, 12, 2301. [Google Scholar] [CrossRef]
- Batista, R.; Vinagre, N.; Meireles, S.; Vinagre, J.; Prazeres, H.; Leão, R.; Máximo, V.; Soares, P. Biomarkers for Bladder Cancer Diagnosis and Surveillance: A Comprehensive Review. Diagnostics 2020, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Nedjadi, T.; Benabdelkamal, H.; Albarakati, N.; Masood, A.; Al-Sayyad, A.; Alfadda, A.A.; Alanazi, I.O.; Al-Ammari, A.; Al-Maghrabi, J. Circulating Proteomic Signature for Detection of Biomarkers in Bladder Cancer Patients. Sci. Rep. 2020, 10, 10999. [Google Scholar] [CrossRef] [PubMed]
- Grębowski, R.; Saluk, J.; Bijak, M.; Szemraj, J.; Wigner-Jeziorska, P. The Role of SOD2 and NOS2 Genes in the Molecular Aspect of Bladder Cancer Pathophysiology. Sci. Rep. 2023, 13, 14491. [Google Scholar] [CrossRef] [PubMed]
- Chin, F.-W.; Chan, S.-C.; Veerakumarasivam, A. Homeobox Gene Expression Dysregulation as Potential Diagnostic and Prognostic Biomarkers in Bladder Cancer. Diagnostics 2023, 13, 2641. [Google Scholar] [CrossRef] [PubMed]
- Aine, M.; Eriksson, P.; Liedberg, F.; Sjödahl, G.; Höglund, M. Biological Determinants of Bladder Cancer Gene Expression Subtypes. Sci. Rep. 2015, 5, 10957. [Google Scholar] [CrossRef]
- Quan, J.; Zhang, W.; Yu, C.; Bai, Y.; Cui, J.; Lv, J.; Zhang, Q. Bioinformatic Identification of Prognostic Indicators in Bladder Cancer. Biomark. Med. 2020, 14, 1243–1254. [Google Scholar] [CrossRef]
- Chatterjee, D.; Mou, S.I.; Sultana, T.; Hosen, M.I.; Faruk, M.O. Identification and Validation of Prognostic Signature Genes of Bladder Cancer by Integrating Methylation and Transcriptomic Analysis. Sci. Rep. 2024, 14, 368. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An Immeasurable Source of Knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Su, S.; Chhabra, G.; Singh, C.K.; Ndiaye, M.A.; Ahmad, N. PLK1 Inhibition-Based Combination Therapies for Cancer Management. Transl. Oncol. 2022, 16, 101332. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Q.; Wang, X. PLK1, A Potential Target for Cancer Therapy. Transl. Oncol. 2017, 10, 22–32. [Google Scholar] [CrossRef]
- Li, L.; Jiang, P.; Hu, W.; Zou, F.; Li, M.; Rao, T.; Ruan, Y.; Yu, W.; Ning, J.; Cheng, F. AURKB Promotes Bladder Cancer Progression by Deregulating the P53 DNA Damage Response Pathway via MAD2L2. J. Transl. Med. 2024, 22, 295. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-X.; Wu, H.-Y.; Yang, Y.; Ma, M.-M.; Zhang, Y.-W.; Huang, H.-Z.; Li, S.-H.; Pan, S.-L.; Tang, J.; Peng, J.-H. CCNB1 Is Involved in Bladder Cancer Pathogenesis and Silencing CCNB1 Decelerates Tumor Growth and Improves Prognosis of Bladder Cancer. Exp. Ther. Med. 2023, 26, 382. [Google Scholar] [CrossRef] [PubMed]
- Pinto Marques, H.; Gomes da Silva, S.; De Martin, E.; Agopian, V.G.; Martins, P.N. Emerging Biomarkers in HCC Patients: Current Status. Int. J. Surg. 2020, 82, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Liu, A.; Dai, L.; Yu, X.; Zhang, Z.; Xiong, Q.; Yang, J.; Liu, F.; Xu, J.; Xue, Y.; et al. Prognostic Value of TOP2A in Bladder Urothelial Carcinoma and Potential Molecular Mechanisms. BMC Cancer 2019, 19, 604. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Liu, P.; Wen, W.; Grubbs, C.J.; Townsend, R.R.; Malone, J.P.; Lubet, R.A.; You, M. Cross-Species Comparison of Orthologous Gene Expression in Human Bladder Cancer and Carcinogen-Induced Rodent Models. Am. J. Transl. Res. 2010, 3, 8–27. [Google Scholar]
- Lee, S.-J.; Lee, E.-J.; Kim, S.-K.; Jeong, P.; Cho, Y.-H.; Yun, S.J.; Kim, S.; Kim, G.-Y.; Choi, Y.H.; Cha, E.-J.; et al. Identification of Pro-Inflammatory Cytokines Associated with Muscle Invasive Bladder Cancer; The Roles of IL-5, IL-20, and IL-28A. PLoS ONE 2012, 7, e40267. [Google Scholar] [CrossRef]
- Sathe, A.; Nawroth, R. Targeting the PI3K/AKT/MTOR Pathway in Bladder Cancer. In Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2018; pp. 335–350. [Google Scholar]
- Wu, G.; Weng, W.; Xia, P.; Yan, S.; Zhong, C.; Xie, L.; Xie, Y.; Fan, G. Wnt Signalling Pathway in Bladder Cancer. Cell. Signal. 2021, 79, 109886. [Google Scholar] [CrossRef]
- Huang, Y.; Li, G.-M. DNA Mismatch Repair in the Context of Chromatin. Cell Biosci. 2020, 10, 10. [Google Scholar] [CrossRef]
- Pal, A.; Greenblatt, H.M.; Levy, Y. Prerecognition Diffusion Mechanism of Human DNA Mismatch Repair Proteins along DNA: Msh2-Msh3 versus Msh2-Msh6. Biochemistry 2020, 59, 4822–4832. [Google Scholar] [CrossRef]
- Viswanath, K.K.; Kuo, S.-Y.; Tu, C.-W.; Hsu, Y.-H.; Huang, Y.-W.; Hu, C.-C. The Role of Plant Transcription Factors in the Fight against Plant Viruses. Int. J. Mol. Sci. 2023, 24, 8433. [Google Scholar] [CrossRef]
- Essebier, A.; Lamprecht, M.; Piper, M.; Bodén, M. Bioinformatics Approaches to Predict Target Genes from Transcription Factor Binding Data. Methods 2017, 131, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-T.; Xu, T.; Iqbal, M.; Hsieh, T.-C.; Luo, Z.; Liang, G.; Farnham, P.J.; Rhie, S.K.; Goldkorn, A. FOXC1 Binds Enhancers and Promotes Cisplatin Resistance in Bladder Cancer. Cancers 2022, 14, 1717. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Lin, A.; Chen, J.; Zhang, X.; Liu, H.; Li, H.; Hu, Y.; Zhang, X.; Zhang, J.; Qiu, L.; et al. CREB1 Directly Activates the Transcription of Ribonucleotide Reductase Small Subunit M2 and Promotes the Aggressiveness of Human Colorectal Cancer. Oncotarget 2016, 7, 78055–78068. [Google Scholar] [CrossRef] [PubMed]
- LaPlant, K.D.; Louzon, P.D. Pazopanib: An Oral Multitargeted Tyrosine Kinase Inhibitor for Use in Renal Cell Carcinoma. Ann. Pharmacother. 2010, 44, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, J.; Quispe, C.; Patra, J.K.; Singh, Y.D.; Panda, M.K.; Das, G.; Adetunji, C.O.; Michael, O.S.; Sytar, O.; Polito, L.; et al. Paclitaxel: Application in Modern Oncology and Nanomedicine-Based Cancer Therapy. Oxid. Med. Cell Longev. 2021, 2021, 3687700. [Google Scholar] [CrossRef]
- Di Bello, F.; Siech, C.; Jannello, L.M.I.; de Angelis, M.; Rodriguez Peñaranda, N.; Tian, Z.; Goyal, J.A.; Baudo, A.; Collà Ruvolo, C.; Califano, G.; et al. Contemporary Survival in Metastatic Bladder Cancer Patients: A Population-based Study. Int. J. Cancer 2024, 155, 1762–1768. [Google Scholar] [CrossRef]
- Peyrottes, A.; Ouzaid, I.; Califano, G.; Hermieu, J.-F.; Xylinas, E. Neoadjuvant Immunotherapy for Muscle-Invasive Bladder Cancer. Medicina 2021, 57, 769. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Taverna, F.; Goveia, J.; Karakach, T.K.; Khan, S.; Rohlenova, K.; Treps, L.; Subramanian, A.; Schoonjans, L.; Dewerchin, M.; Eelen, G.; et al. BIOMEX: An Interactive Workflow for (Single Cell) Omics Data Interpretation and Visualization. Nucleic Acids Res. 2020, 48, W385–W394. [Google Scholar] [CrossRef]
- Huang, D.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A Novel Biological Module-Centric Algorithm to Functionally Analyze Large Gene Lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene Ontology Analysis for RNA-Seq: Accounting for Selection Bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING Database in 2023: Protein–Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. CytoHubba: Identifying Hub Objects and Sub-Networks from Complex Interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W. An Automated Method for Finding Molecular Complexes in Large Protein Interaction Networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef]
- Kwon, Y.; Han, K.; Suh, Y.J.; Jung, I. Stability Selection for LASSO with Weights Based on AUC. Sci. Rep. 2023, 13, 5207. [Google Scholar] [CrossRef]
- Danishuddin; Haque, M.A.; Malik, M.Z.; Arya, R.; Singh, P.; Lee, J.-S.; Kim, J.-J.; Lee, K.-W.; Jung, T.-S. Unveiling the Mechanisms Underlying the Immunotherapeutic Potential of Gene–MiRNA and Drugs in Head and Neck Cancer. Pharmaceuticals 2024, 17, 921. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Rauluseviciute, I.; Riudavets-Puig, R.; Blanc-Mathieu, R.; Castro-Mondragon, J.A.; Ferenc, K.; Kumar, V.; Lemma, R.B.; Lucas, J.; Chèneby, J.; Baranasic, D.; et al. JASPAR 2024: 20th Anniversary of the Open-Access Database of Transcription Factor Binding Profiles. Nucleic Acids Res. 2024, 52, D174–D182. [Google Scholar] [CrossRef]
- Cannon, M.; Stevenson, J.; Stahl, K.; Basu, R.; Coffman, A.; Kiwala, S.; McMichael, J.F.; Kuzma, K.; Morrissey, D.; Cotto, K.; et al. DGIdb 5.0: Rebuilding the Drug–Gene Interaction Database for Precision Medicine and Drug Discovery Platforms. Nucleic Acids Res. 2024, 52, D1227–D1235. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing 2023. Available online: https://cran.r-project.org/doc/manuals/r-release/fullrefman.pdf (accessed on 10 July 2024).



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danishuddin; Haque, M.A.; Khan, S.; Kim, J.-J.; Ahmad, K. Molecular Landscape of Bladder Cancer: Key Genes, Transcription Factors, and Drug Interactions. Int. J. Mol. Sci. 2024, 25, 10997. https://doi.org/10.3390/ijms252010997
Danishuddin, Haque MA, Khan S, Kim J-J, Ahmad K. Molecular Landscape of Bladder Cancer: Key Genes, Transcription Factors, and Drug Interactions. International Journal of Molecular Sciences. 2024; 25(20):10997. https://doi.org/10.3390/ijms252010997
Chicago/Turabian StyleDanishuddin, Md Azizul Haque, Shawez Khan, Jong-Joo Kim, and Khurshid Ahmad. 2024. "Molecular Landscape of Bladder Cancer: Key Genes, Transcription Factors, and Drug Interactions" International Journal of Molecular Sciences 25, no. 20: 10997. https://doi.org/10.3390/ijms252010997
APA StyleDanishuddin, Haque, M. A., Khan, S., Kim, J.-J., & Ahmad, K. (2024). Molecular Landscape of Bladder Cancer: Key Genes, Transcription Factors, and Drug Interactions. International Journal of Molecular Sciences, 25(20), 10997. https://doi.org/10.3390/ijms252010997