
Lipoprotein (a) as a Cardiovascular Risk Factor in Controversial Clinical Scenarios: A Narrative Review

 , , ,
, , ,  , , ,
, , ,  , ,
, ,
Abstract
:1. Introduction

{kind=link}
{kind=link}
| Scientific Society | Screening Indications/Recommendations | Cut-Off Values |
|---|---|---|
| ACC/AHA—Guideline 2019 [5] | - A relative indication for its measurement is family history of premature ASCVD. - Classed as a “risk-enhancing factor” in patients 40 to 75 years old, without diabetes mellitus but with 10-year ASCVD risk–7.5 to 19.9% would favor initiation of statin therapy. | >50 mg/dL (125 nmol/L) |
| ESC/EAS—Guideline 2019 [14] | - Measure in all patients at least once in their adult lifetime to identify those who may have a very high lifetime risk of atherosclerotic cardiovascular disease (ASCVD), similar to those with heterozygous familial hypercholesterolemia (FH). | >180 mg/dL (430 nmol/L) [very high risk] - Normal value not specified |
| National Lipid Association (NLA)—Scientific Statement 2021 [15] | Reasonable to refine ASCVD risk assessment in adults with: - first-degree relatives with premature ASCVD (<55 years of age in men and <65 years of age in women); - a personal history of premature ASCVD; and - primary severe hypercholesterolemia or suspected FH. | >50 mg/dL (125 nmol/L) |
| Canadian Cardiovascular Society (CCS)—Guideline 2021 [16] | Recommend measuring Lp(a) level once in a person’s lifetime as a part of the initial lipid screening. | >50 mg/dL (125 nmol/L) |
| European Atherosclerosis Society (EAS)—Consensus Statement 2022 [17] | Lp(a) should be measured at least once in adults to identify those with high cardiovascular risk. | >50 mg/dL (125 nmol/L) |
2. Results
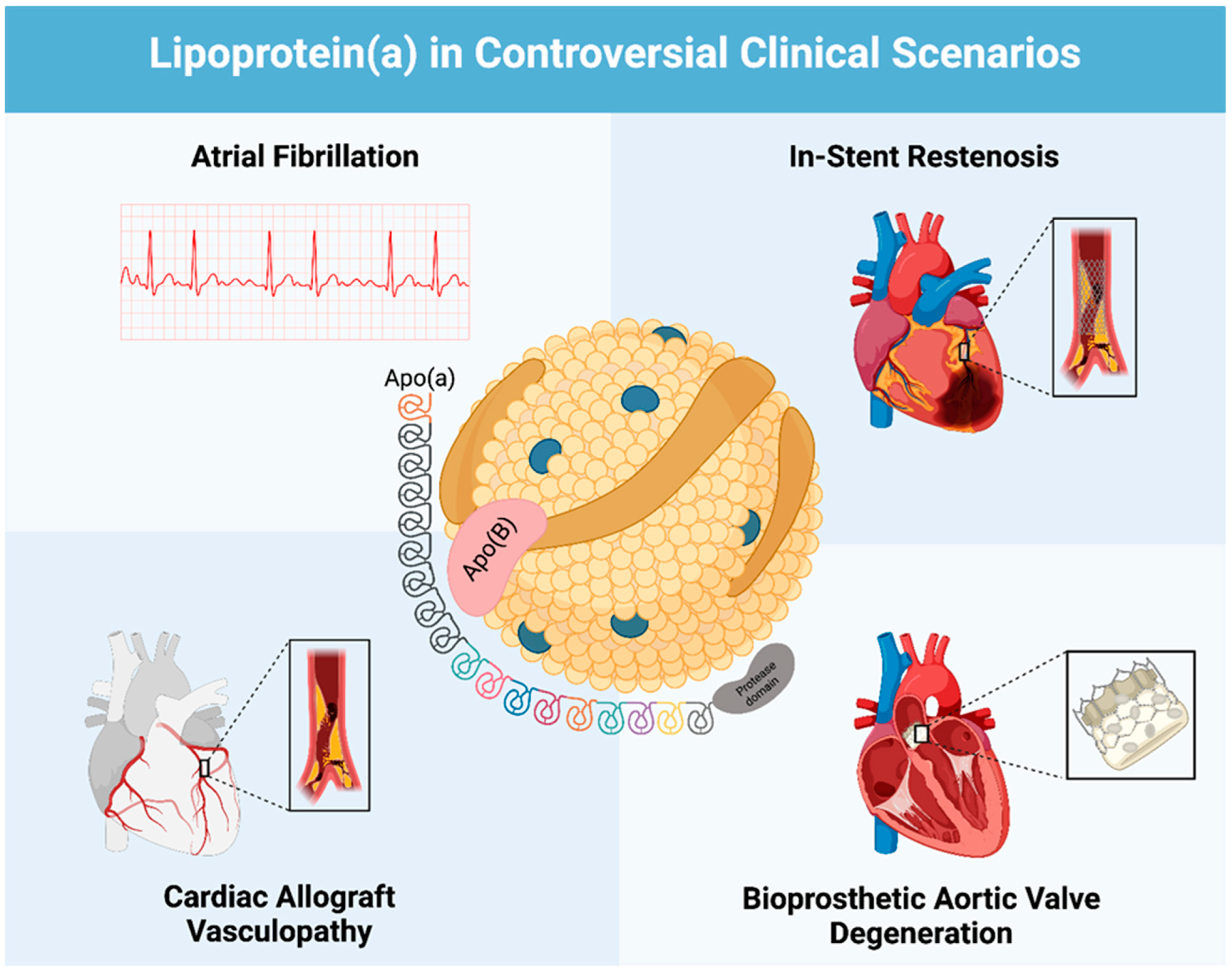
2.1. Lp(a) and Atrial Fibrillation
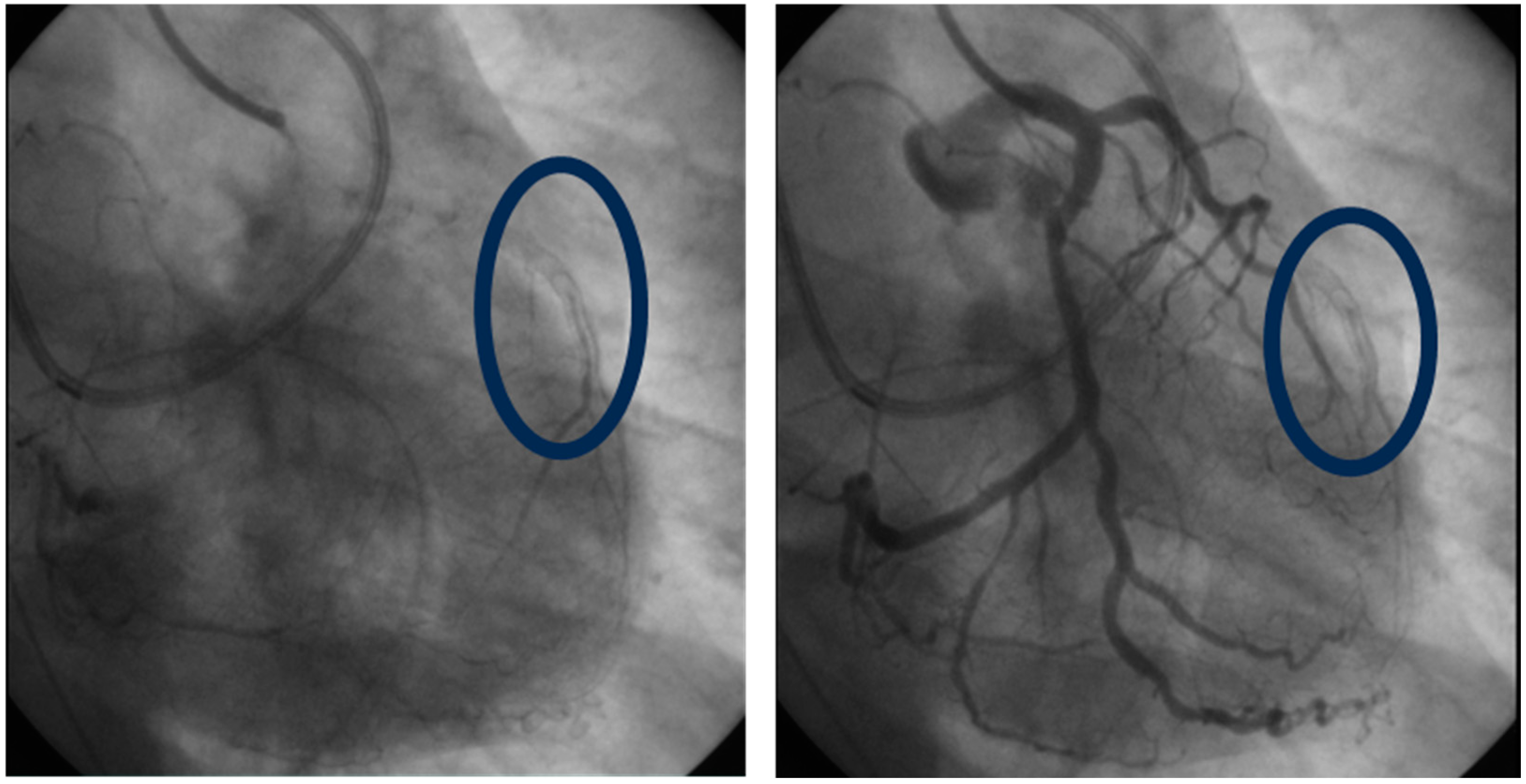
2.2. Lp(a) and In-Stent Restenosis
2.3. Lp(a) and Cardiac Allograft Vasculopathy
2.4. Lp(a) and Bioprosthetic Aortic Valve Degeneration
3. Discussion
3.1. Lipoprotein (a) Structure and Function
3.2. Lp(a) lowering therapies
3.3. Lp(a) and Atrial Fibrillation
3.4. Lp(a) and In-Stent Restenosis
3.5. Lp(a) and Cardiac Allograft Vasculopathy
3.6. Lp(a) and Bioprosthetic Aortic Valve Degeneration
4. Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; De Ferranti, S.; Després, J.-P.; Fullerton, H.J.; et al. Executive Summary: Heart Disease and Stroke Statistics—2016 Update: A Report From the American Heart Association. Circulation 2016, 133, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Kostner, K.M.; Kostner, G.M. Lipoprotein (a): A historical appraisal. J. Lipid Res. 2017, 58, 1–14. [Google Scholar] [CrossRef]
- Kamstrup, P.R. Lipoprotein(a) and ischemic heart disease—A causal association? A review. Atherosclerosis 2010, 211, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Noureen, A.; Kronenberg, F.; Utermann, G. Structure, function, and genetics of lipoprotein (a). J. Lipid Res. 2016, 57, 1339–1359. [Google Scholar] [CrossRef]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the primary prevention of cardiovascular disease: A report of the american college of cardiology/American heart association task force on clinical practice guidelines. Circulation 2019, 140, e596–e646. [Google Scholar] [CrossRef]
- Björnson, E.; Adiels, M.; Taskinen, M.-R.; Burgess, S.; Chapman, M.J.; Packard, C.J.; Borén, J. Lipoprotein(a) Is Markedly More Atherogenic Than LDL. J. Am. Coll. Cardiol. 2024, 83, 385–395. [Google Scholar] [CrossRef]
- Zawacki, A.W.; Dodge, A.; Woo, K.M.; Ralphe, J.C.; Peterson, A.L. In pediatric familial hypercholesterolemia, lipoprotein(a) is more predictive than LDL-C for early onset of cardiovascular disease in family members. J. Clin. Lipidol. 2018, 12, 1445–1451. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Nicholls, S.J.; Langsted, A.; Ray, K.K.; Tybjærg-Hansen, A. Advances in lipid-lowering therapy through gene-silencing technologies. Nat. Rev. Cardiol. 2018, 15, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Tao, J.-Y.; Cai, D.-P.; He, Y.-M. Interaction of lipoprotein(a) with low-density lipoprotein cholesterol on first incident acute myocardial infarction. Clin. Chim. Acta 2020, 501, 1–5. [Google Scholar] [CrossRef]
- Langsted, A.; Nordestgaard, B.G. Lipoprotein(a): Is it more, less or equal to LDL as a causal factor for cardiovascular disease and mortality? Curr. Opin. Infect. Dis. 2020, 31, 125–131. [Google Scholar] [CrossRef]
- Smolders, B.; Lemmens, R.; Thijs, V. Lipoprotein (a) and Stroke. Stroke 2007, 38, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, Z.; Ji, R.; Wang, Y.; Zhang, Y.; Yu, K. Relationship between carotid atherosclerosis and lipoprotein (a) in patients with acute ischemic stroke. Front. Neurol. 2024, 15, 1383771. [Google Scholar] [CrossRef] [PubMed]
- Fujino, M.; Nicholls, S.J. Lipoprotein(a): Cardiovascular risk and emerging therapies. Expert Rev. Cardiovasc. Ther. 2023, 21, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Koschinsky, M.L.; Bajaj, A.; Boffa, M.B.; Dixon, D.L.; Ferdinand, K.C.; Gidding, S.S.; Gill, E.A.; Jacobson, T.A.; Michos, E.D.; Safarova, M.S.; et al. A focused update to the 2019 NLA scientific statement on use of lipoprotein(a) in clinical practice. J. Clin. Lipidol. 2024, 18, e308–e319. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.J.; Grégoire, J.; Pearson, G.J.; Barry, A.R.; Couture, P.; Dawes, M.; Francis, G.A.; Genest, J., Jr.; Grover, S.; Gupta, M.; et al. 2016 Canadian Cardiovascular Society Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in the Adult. Can. J. Cardiol. 2016, 32, 1263–1282. [Google Scholar] [CrossRef]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; A Ference, B.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: A European Atherosclerosis Society consensus statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef]
- Tao, J.; Yang, X.; Qiu, Q.; Gao, F.; Chen, W.; Hu, L.; Xu, Y.; Yi, Y.; Hu, H.; Jiang, L. Low lipoprotein(a) concentration is associated with atrial fibrillation: A large retrospective cohort study. Lipids Health Dis. 2022, 21, 119, Erratum in Lipids Health Dis. 2023, 22, 36. [Google Scholar] [CrossRef]
- Garg, P.K.; Guan, W.; Karger, A.B.; Steffen, B.T.; O’neal, W.; Heckbert, S.R.; Michos, E.D.; Tsai, M.Y. Lp(a) (Lipoprotein [a]) and Risk for Incident Atrial Fibrillation. Circ. Arrhythmia Electrophysiol. 2020, 13, e008401. [Google Scholar] [CrossRef]
- Xia, J.; Guo, C.; Liu, K.; Xie, Y.; Cao, H.; Peng, W.; Sun, Y.; Liu, X.; Li, B.; Zhang, L. Association of Lipoprotein (a) variants with risk of cardiovascular disease: A Mendelian randomization study. Lipids Health Dis. 2021, 20, 1–11. [Google Scholar] [CrossRef]
- Mohammadi-Shemirani, P.; Chong, M.; Narula, S.; Perrot, N.; Conen, D.; Roberts, J.D.; Thériault, S.; Bossé, Y.; Lanktree, M.B.; Pigeyr, M.; et al. Elevated Lipoprotein(a) and Risk of Atrial Fibrillation: An Observational and Mendelian Randomization Study. J. Am. Coll. Cardiol. 2022, 79, 1579–1590. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Baars, D.P.; Desai, R.; Singh, D.; Pinto-Sietsma, S.-J. Association Between Lipoprotein (a) and Risk of Atrial Fibrillation: A Systematic Review and Meta-analysis of Mendelian Randomization Studies. Curr. Probl. Cardiol. 2024, 49, 102024. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Qin, D.; Yang, L.; Lin, Y.; Zhai, L.; Zhang, Y.; Yang, G.; Wang, K.; Tong, D.; Li, X.; et al. Causal effects of plasma lipids on the risk of atrial fibrillation: A multivariable mendelian randomization study. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Aronis, K.N.; Zhao, D.; Hoogeveen, R.C.; Alonso, A.; Ballantyne, C.M.; Guallar, E.; Jones, S.R.; Martin, S.S.; Nazarian, S.; Steffen, B.T.; et al. Associations of Lipoprotein(a) Levels With Incident Atrial Fibrillation and Ischemic Stroke: The ARIC (Athero-sclerosis Risk in Communities) Study. J. Am. Heart Assoc. 2017, 6, e007372. [Google Scholar] [CrossRef] [PubMed]
- Mora, S.; Akinkuolie, A.O.; Sandhu, R.K.; Conen, D.; Albert, C.M. Paradoxical Association of Lipoprotein Measures With Incident Atrial Fibrillation. Circ. Arrhythmia Electrophysiol. 2014, 7, 612–619. [Google Scholar] [CrossRef]
- Wehinger, A.; Kastrati, A.; Elezi, S.; Baum, H.; Braun, S.; Neumann, F.-J.; Schömig, A. Lipoprotein(a) and coronary thrombosis and restenosis after stent placement. J. Am. Coll. Cardiol. 1999, 33, 1005–1012. [Google Scholar] [CrossRef]
- Ribichini, F.; Steffenino, G.; Dellavalle, A.; Vado, A.; Ferrero, V.; Camilla, T.; Giubergia, S.; Uslenghi, E. Plasma Lipoprotein(a) Is Not a Predictor for Restenosis After Elective High-Pressure Coronary Stenting. Circulation 1998, 98, 1172–1177. [Google Scholar] [CrossRef]
- Khosravi, A.; Pourmoghaddas, M.; Ziaie, F.; Enteshari, A.; Khaledifa, A.; Bahonar, A. Does Lipoprotein (a) Level Have a Predictive Value in Restenosis after Coronary Stenting? Int. J. Prev. Med. 2011, 2, 158–163. [Google Scholar]
- Qin, S.-Y.; Liu, J.; Jiang, H.-X.; Hu, B.-L.; Zhou, Y.; Olkkonen, V.M. Association between baseline lipoprotein (a) levels and restenosis after coronary stenting: Meta-analysis of 9 cohort studies. Atherosclerosis 2013, 227, 360–366. [Google Scholar] [CrossRef]
- Kamitani, T.; Taniguchi, T.; Miyai, N.; Kawasaki, T.; Kawasaki, S.; Sugihara, H. Association Between Plasma Lipoprotein(a) Concentration and Restenosis After Stent Implantation. Circ. J. 2005, 69, 644–649. [Google Scholar] [CrossRef]
- Yuan, X.; Han, Y.; Hu, X.; Jiang, M.; Feng, H.; Fang, Y.; Liu, M.; Chen, Y.; Gao, L. Lipoprotein (a) is related to In-Stent neo-atherosclerosis incidence rate and plaque vulnerability: Optical Coherence Tomography Study. Int. J. Cardiovasc. Imaging 2022, 39, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.K.; Farina, J.M.; Awad, K.; Ali, N.B.; Pereyra, M.; Scalia, I.G.; Abbas, M.T.; Allam, M.N.; A Kamel, M.; A Abu Rmilah, A.; et al. Lipoprotein(a) and long-term in-stent restenosis after percutaneous coronary intervention. Eur. J. Prev. Cardiol. 2024. [CrossRef]
- Lee, F.; Nair, V.; Chih, S. Cardiac allograft vasculopathy: Insights on pathogenesis and therapy. Clin. Transplant. 2020, 34, e13794. [Google Scholar] [CrossRef] [PubMed]
- Barbir, M.; Kushwaha, S.; Hunt, B.; Macken, A.; Yacoub, M.; Thompson, G.; Robinson, D.; Mitchell, A. Lipoprotein(a) and accelerated coronary artery disease in cardiac transplant recipients. Lancet 1992, 340, 1500–1502. [Google Scholar] [CrossRef]
- Chang, G.; DeNofrio, D.; Desai, S.; Kelley, M.P.; Rader, D.J.; Acker, M.A.; Loh, E. Lipoprotein(a) levels and heart transplantation atherosclerosis. Am. Heart J. 1998, 136, 329–334. [Google Scholar] [CrossRef]
- Farina, J.M.; Chao, C.-J.; Pereyra, M.; Roarke, M.; Said, E.F.; Barry, T.; Alsidawi, S.; Sell-Dottin, K.; Sweeney, J.P.; Fortuin, D.F.; et al. Role of lipoprotein(a) concentrations in bioprosthetic aortic valve degeneration. Heart 2023, 110, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Botezatu, S.B.; Tzolos, E.; Kaiser, Y.; Cartlidge, T.R.G.; Kwiecinski, J.; Barton, A.K.; Yu, X.; Williams, M.C.; van Beek, E.J.R.; White, A.; et al. Serum lipoprotein(a) and bioprosthetic aortic valve degeneration. Eur. Heart J.-Cardiovasc. Imaging 2023, 24, 759–767. [Google Scholar] [CrossRef]
- Lipoprotein(a)—Structure, Epidemiology, Function and Diagnostics of a Cardiovascular Risk Marker. Available online: https://benthamopenarchives.com/abstract.php?ArticleCode=TOCCHEMJ-1-79 (accessed on 31 August 2024).
- Marcovina, S.M.; Albers, J.J. Lipoprotein (a) measurements for clinical application. J. Lipid Res. 2016, 57, 526–537. [Google Scholar] [CrossRef]
- von Zychlinski, A.; Williams, M.; McCormick, S.; Kleffmann, T. Absolute quantification of apolipoproteins and associated proteins on human plasma lipoproteins. J. Proteom. 2014, 106, 181–190. [Google Scholar] [CrossRef]
- McLean, J.W.; Tomlinson, J.E.; Kuang, W.-J.; Eaton, D.L.; Chen, E.Y.; Fless, G.M.; Scanu, A.M.; Lawn, R.M. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature 1987, 330, 132–137. [Google Scholar] [CrossRef]
- Marcovina, S.M.; Albers, J.J.; Gabel, B.; Koschinsky, M.L.; Gaur, V.P. Effect of the number of apolipoprotein(a) kringle 4 domains on immunochemical measurements of lipoprotein(a). Clin. Chem. 1995, 41, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Scanu, A.M.; Edelstein, C. Learning about the structure and biology of human lipoprotein [a] through dissection by enzymes of the elastase family: Facts and speculations. J. Lipid Res. 1997, 38, 2193–2206. [Google Scholar] [CrossRef] [PubMed]
- Cai, A.; Li, L.; Zhang, Y.; Mo, Y.; Mai, W.; Zhou, Y. Lipoprotein(a): A Promising Marker for Residual Cardiovascular Risk Assessment. Dis. Markers 2013, 35, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, T.A. Lipoprotein(a), Cardiovascular Disease, and Contemporary Management. Mayo Clin. Proc. 2013, 88, 1294–1311. [Google Scholar] [CrossRef]
- Clarke, R.; Peden, J.F.; Hopewell, J.C.; Kyriakou, T.; Goel, A.; Heath, S.; Parish, S.; Barlera, S.; Franzosi, M.G.; Rust, S.; et al. Genetic Variants Associated with Lp(a) Lipoprotein Level and Coronary Disease. N. Engl. J. Med. 2009, 361, 2518–2528. [Google Scholar] [CrossRef]
- Deo, R.C.; Wilson, J.G.; Xing, C.; Lawson, K.; Kao, W.H.L.; Reich, D.; Tandon, A.; Akylbekova, E.; Patterson, N.; Mosley, T.H.; et al. Single-Nucleotide Polymorphisms in LPA Explain Most of the Ancestry-Specific Variation in Lp(a) Levels in African Americans. PLoS ONE 2011, 6, e14581. [Google Scholar] [CrossRef]
- Lim, E.T.; Würtz, P.; Havulinna, A.S.; Palta, P.; Tukiainen, T.; Rehnström, K.; Esko, T.; Mägi, R.; Inouye, M.; Lappalainen, T.; et al. Distribution and Medical Impact of Loss-of-Function Variants in the Finnish Founder Population. PLOS Genet. 2014, 10, e10044942014. [Google Scholar] [CrossRef]
- Koschinsky, M.L.; Marcovina, S.M. Structure-function relationships in apolipoprotein(a): Insights into lipoprotein(a) assembly and pathogenicity. Curr. Opin. Infect. Dis. 2004, 15, 167–174. [Google Scholar] [CrossRef]
- Yano, Y.; Shimokawa, K.; Okada, Y.; Noma, A. Immunolocalization of Lipoprotein(a) in Wounded Tissues. J. Histochem. Cytochem. 1997, 45, 559–568. [Google Scholar] [CrossRef]
- Lippi, G.; Guidi, G. Lipoprotein(a): From ancestral benefit to modern pathogen? QJM Int. J. Med. 2000, 93, 75–84. [Google Scholar] [CrossRef]
- Cui, K.; Wang, H.-Y.; Yin, D.; Zhu, C.; Song, W.; Wang, H.; Jia, L.; Zhang, D.; Song, C.; Feng, L.; et al. Benefit and Risk of Prolonged Dual Antiplatelet Therapy After Percutaneous Coronary Intervention With Drug-Eluting Stents in Patients With Elevated Lipoprotein(a) Concentrations. Front. Cardiovasc. Med. 2021, 8, 807925. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, H.S.; Trainor, P.; Carlisle, S.; Tsai, M.Y.; Criqui, M.H.; DeFilippis, A.; Tsimikas, S. Aspirin and Cardiovascular Risk in Individuals With Elevated Lipoprotein(a): The Multi-Ethnic Study of Atherosclerosis. J. Am. Heart Assoc. 2024, 13, e033562. [Google Scholar] [CrossRef] [PubMed]
- Skålén, K.; Gustafsson, M.; Rydberg, E.K.; Hultén, L.M.; Wiklund, O.; Innerarity, T.L.; Borén, J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 2002, 417, 750–754. [Google Scholar] [CrossRef]
- Wolf, D.; Gerhardt, T.; Winkels, H.; Michel, N.A.; Pramod, A.B.; Ghosheh, Y.; Brunel, S.; Buscher, K.; Miller, J.; McArdle, S.; et al. Pathogenic Autoimmunity in Atherosclerosis Evolves From Initially Protective Apolipoprotein B 100 –Reactive CD4 + T-Regulatory Cells. Circulation 2020, 142, 1279–1293. [Google Scholar] [CrossRef] [PubMed]
- Malick, W.A.; Goonewardena, S.N.; Koenig, W.; Rosenson, R.S. Clinical Trial Design for Lipoprotein(a)-Lowering Therapies. J. Am. Coll. Cardiol. 2023, 81, 1633–1645. [Google Scholar] [CrossRef]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.-C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.-J. Therapeutic siRNA: State of the art. Signal Transduct. Target. Ther. 2020, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- O’donoghue, M.L.; Rosenson, R.S.; Gencer, B.; López, J.A.G.; Lepor, N.E.; Baum, S.J.; Stout, E.; Gaudet, D.; Knusel, B.; Kuder, J.F.; et al. Small Interfering RNA to Reduce Lipoprotein(a) in Cardiovascular Disease. N. Engl. J. Med. 2022, 387, 1855–1864. [Google Scholar] [CrossRef]
- Staerk, L.; Sherer, J.A.; Ko, D.; Benjamin, E.J.; Helm, R.H. Atrial Fibrillation. Circ. Res. 2017, 120, 1501–1517. [Google Scholar] [CrossRef]
- Brundel, B.J.; Henning, R.H.; Kampinga, H.H.; Van Gelder, I.C. Molecular mechanisms of remodeling in human atrial fibrillation. Cardiovasc. Res. 2002, 54, 315–324. [Google Scholar] [CrossRef]
- Boffa, M.B.; Koschinsky, M.L. Oxidized phospholipids as a unifying theory for lipoprotein(a) and cardiovascular disease. Nat. Rev. Cardiol. 2019, 16, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Bouchareb, R.; Mahmut, A.; Nsaibia, M.J.; Boulanger, M.-C.; Dahou, A.; Lépine, J.-L.; Laflamme, M.-H.; Hadji, F.; Couture, C.; Trahan, S.; et al. Autotaxin Derived From Lipoprotein(a) and Valve Interstitial Cells Promotes Inflammation and Mineralization of the Aortic Valve. Circulation 2015, 132, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.L.; Lane, D.A.; Banach, M.; Mastej, M.; Kasperczyk, S.; Jóźwiak, J.J.; Lip, G.Y.; Al-Shaer, B.; Andrusewicz, W.; Andrzejczuk-Rosa, M.; et al. Lipid levels, atrial fibrillation and the impact of age: Results from the LIPIDOGRAM2015 study. Atherosclerosis 2020, 312, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Wennberg, A.; Gigante, B.; Walldius, G.; Hammar, N.; Modig, K. Lipid levels in midlife and risk of atrial fibrillation over 3 decades—Experience from the Swedish AMORIS cohort: A cohort study. PLOS Med. 2022, 19, e1004044. [Google Scholar] [CrossRef] [PubMed]
- Enkhmaa, B.; Anuurad, E.; Berglund, L. Lipoprotein (a): Impact by ethnicity and environmental and medical conditions. J. Lipid Res. 2016, 57, 1111–1125. [Google Scholar] [CrossRef]
- Giustino, G.; Colombo, A.; Camaj, A.; Yasumura, K.; Mehran, R.; Stone, G.W.; Kini, A.; Sharma, S.K. Coronary In-Stent Restenosis. J. Am. Coll. Cardiol. 2022, 80, 348–372. [Google Scholar] [CrossRef]
- Nakamura, D.; Dohi, T.; Ishihara, T.; Kikuchi, A.; Mori, N.; Yokoi, K.; Shiraki, T.; Mizote, I.; Mano, T.; Higuchi, Y.; et al. Predictors and outcomes of neoatherosclerosis in patients with in-stent restenosis. EuroIntervention 2021, 17, 489–496. [Google Scholar] [CrossRef]
- Akiyama, T.; Ozaki, K.; Takano, T.; Yoneyama, S.; Kubota, N.; Okubo, T.; Ikegami, R.; Hoyano, M.; Yanagawa, T.; Inomata, T. Efficacy of Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitor Treatment for Repeated In-stent Restenosis in a Coronary Artery. Intern. Med. 2023, 62, 3361–3365. [Google Scholar] [CrossRef]
- Verbeek, R.; Hoogeveen, R.M.; Langsted, A.; A Stiekema, L.C.; Verweij, S.L.; Hovingh, G.K.; Wareham, N.J.; Khaw, K.-T.; Boekholdt, S.M.; Nordestgaard, B.G.; et al. Cardiovascular disease risk associated with elevated lipoprotein(a) attenuates at low low-density lipoprotein cholesterol levels in a primary prevention setting. Eur. Heart J. 2018, 39, 2589–2596. [Google Scholar] [CrossRef]
- González-Quijano, M.; Grande-Trillo, A.; Esteve-Ruiz, I.; Aranda-Dios, A.; Sobrino-Márquez, J.M.; Rangel-Sousa, D. Elevated Lipoprotein A Levels and Development of Moderate or Severe Cardiac Allograft Vasculopathy in Patients with Heart Transplants. Transplant. Proc. 2023, 55, 2295–2298. [Google Scholar] [CrossRef]
- Lindman, B.R.; Clavel, M.-A.; Mathieu, P.; Iung, B.; Lancellotti, P.; Otto, C.M.; Pibarot, P. Calcific aortic stenosis. Nat. Rev. Dis. Prim. 2016, 2, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Capoulade, R.; Chan, K.L.; Yeang, C.; Mathieu, P.; Bossé, Y.; Dumesnil, J.G.; Tam, J.W.; Teo, K.K.; Mahmut, A.; Yang, X.; et al. Oxidized Phospholipids, Lipoprotein(a), and Progression of Calcific Aortic Valve Stenosis. J. Am. Coll. Cardiol. 2015, 66, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Freeman, R.V.; Otto, C.M. Spectrum of Calcific Aortic Valve Disease. Circulation 2005, 111, 3316–3326. [Google Scholar] [CrossRef] [PubMed]
- Kostyunin, A.E.; Yuzhalin, A.E.; Rezvova, M.A.; Ovcharenko, E.A.; Glushkova, T.V.; Kutikhin, A.G. Degeneration of Bioprosthetic Heart Valves: Update 2020. J. Am. Heart Assoc. 2020, 9, e018506. [Google Scholar] [CrossRef]
- Côté, N.; Pibarot, P.; Clavel, M.-A. Incidence, risk factors, clinical impact, and management of bioprosthesis structural valve degeneration. Curr. Opin. Cardiol. 2017, 32, 123–129. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdalla, H.M.; Mahmoud, A.K.; Khedr, A.E.; Farina, J.M.; Scalia, I.G.; Abbas, M.T.; Awad, K.A.; Baba Ali, N.; Bismee, N.N.; Attaripour Esfahani, S.; et al. Lipoprotein (a) as a Cardiovascular Risk Factor in Controversial Clinical Scenarios: A Narrative Review. Int. J. Mol. Sci. 2024, 25, 11029. https://doi.org/10.3390/ijms252011029
Abdalla HM, Mahmoud AK, Khedr AE, Farina JM, Scalia IG, Abbas MT, Awad KA, Baba Ali N, Bismee NN, Attaripour Esfahani S, et al. Lipoprotein (a) as a Cardiovascular Risk Factor in Controversial Clinical Scenarios: A Narrative Review. International Journal of Molecular Sciences. 2024; 25(20):11029. https://doi.org/10.3390/ijms252011029
Chicago/Turabian StyleAbdalla, Hesham M., Ahmed K. Mahmoud, Ahmed E. Khedr, Juan M. Farina, Isabel G. Scalia, Mohammed Tiseer Abbas, Kamal A. Awad, Nima Baba Ali, Nadera N. Bismee, Sogol Attaripour Esfahani, and et al. 2024. "Lipoprotein (a) as a Cardiovascular Risk Factor in Controversial Clinical Scenarios: A Narrative Review" International Journal of Molecular Sciences 25, no. 20: 11029. https://doi.org/10.3390/ijms252011029
APA StyleAbdalla, H. M., Mahmoud, A. K., Khedr, A. E., Farina, J. M., Scalia, I. G., Abbas, M. T., Awad, K. A., Baba Ali, N., Bismee, N. N., Attaripour Esfahani, S., Javadi, N., Pereyra, M., Alsidawi, S., Lester, S. J., Ayoub, C., & Arsanjani, R. (2024). Lipoprotein (a) as a Cardiovascular Risk Factor in Controversial Clinical Scenarios: A Narrative Review. International Journal of Molecular Sciences, 25(20), 11029. https://doi.org/10.3390/ijms252011029