
Regulation of Mitochondrial and Peroxisomal Metabolism in Female Obesity and Type 2 Diabetes
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Estrogen Influence in Liver Metabolic Dysfunction, Obesity, and T2D
3. PPAR Involvement in Obesity through β-Oxidation of Fatty Acids
3.1. Biogenesis and PPARs Function
3.2. Fatty Acid β-Oxidation and MAFLD Generation
3.3. E2 Regulation of PPARs
4. Cholesterol Synthesis Regulation
4.1. Implication of Peroxisomes–Mitochondria in Cholesterol Synthesis
4.2. Estrogen Regulation of Cholesterol Synthesis
5. Bile Acids Metabolism
5.1. Bile Acids Synthesis and Signaling Function
5.2. Estrogen Regulation of Bile Acid Metabolism
5.3. Effect of Dysfunctional Peroxisomes–Mitochondria on Bile Acid Metabolism
6. Lipoprotein Metabolism
6.1. Key Regulators of Lipoprotein Metabolism in Health and Disease
6.2. Estrogen’s Protective Effects on Lipoprotein Failure Metabolism Diseases
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Casanova, A.; Wevers, A.; Navarro-Ledesma, S.; Pruimboom, L. Mitochondria: It Is All about Energy. Front. Physiol. 2023, 14, 1114231. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Reumann, S.; Hu, J. Peroxisome Biogenesis and Function. Arab. Book 2009, 7, e0123. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandusse, S.; Denis, S.; Faust, P.L.; Wanders, R.J. Bile Acids: The Role of Peroxisomes. J. Lipid Res. 2009, 50, 2139–2147. [Google Scholar] [CrossRef]
- Islinger, M.; Voelkl, A.; Fahimi, H.D.; Schrader, M. The Peroxisome: An Update on Mysteries 2.0. Histochem. Cell Biol. 2018, 150, 443–471. [Google Scholar] [CrossRef]
- Tanaka, H.; Okazaki, T.; Aoyama, S.; Yokota, M.; Koike, M.; Okada, Y.; Fujiki, Y.; Gotoh, Y. Peroxisomes Control Mitochondrial Dynamics and the Mitochondrion-Dependent Apoptosis Pathway. J. Cell Sci. 2019, 132, jcs224766. [Google Scholar] [CrossRef]
- Kim, J.-A.; Wei, Y.; Sowers, J.R. Role of Mitochondrial Dysfunction in Insulin Resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Kleiboeker, B.; Lodhi, I.J. Peroxisomal Regulation of Energy Homeostasis: Effect on Obesity and Related Metabolic Disorders. Mol. Metab. 2022, 65, 101577. [Google Scholar] [CrossRef]
- Russo, M.P.; Grande-Ratti, M.F.; Burgos, M.A.; Molaro, A.A.; Bonella, M.B. Prevalencia de Diabetes, Características Epidemiológicas y Complicaciones Vasculares. Arch. Cardiol. Mex. 2023, 93, 030–036. [Google Scholar] [CrossRef] [PubMed]
- GBD 2015 Obesity Collaborators. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef]
- Schrader, M.; Fahimi, H.D. Peroxisomes and Oxidative Stress. Biochim. Biophys. Acta Mol. Cell Res. 2006, 1763, 1755–1766. [Google Scholar] [CrossRef]
- Palmisano, B.T.; Zhu, L.; Stafford, J.M. Role of Estrogens in the Regulation of Liver Lipid Metabolism. In Sex and Gender Factors Affecting Metabolic Homeostasis, Diabetes and Obesity; Springer: Berlin/Heidelberg, Germany, 2017; Volume 1043, pp. 227–256. [Google Scholar] [CrossRef]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular Matrix as a Driver of Progressive Fibrosis. J. Clin. Investig. 2018, 128, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Mizobuchi, Y.; Yasuda, M.; Shiba, M.; Ma, Y.-R.; Horie, T.; Liu, F.; Ito, S. Inhibitory Effect of Oestradiol on Activation of Rat Hepatic Stellate Cells In Vivo and In Vitro. Gut 1999, 44, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Al Mahtab, M.; Ghosh, J.; Bhatia, S.; Nagral, A.; Bangar, M.; Menezes, S.; Butt, N.; Manchanayake, J.H.; Singh, S.P. Gender Differences in Nonalcoholic Fatty Liver Disease. Euroasian J. Hepato-Gastroenterol. 2022, 12, S19–S25. [Google Scholar] [CrossRef]
- Simpson, E.; Jones, M.; Misso, M.; Hewitt, K.; Hill, R.; Maffei, L.; Carani, C.; Boon, W.C. Estrogen, a Fundamental Player in Energy Homeostasis. J. Steroid Biochem. Mol. Biol. 2005, 95, 3–8. [Google Scholar] [CrossRef]
- Coelingh Bennink, H.J.T. Are All Estrogens the Same? Maturitas 2004, 47, 269–275. [Google Scholar] [CrossRef]
- Grodin, J.M.; Siiteri, P.K.; Macdonald, P.C. Source of Estrogen Production in Postmenopausal Women. J. Clin. Endocrinol. Metab. 1973, 36, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Shen, Y.; Li, R. Estrogen Synthesis and Signaling Pathways during Aging: From Periphery to Brain. Trends Mol. Med. 2013, 19, 197–209. [Google Scholar] [CrossRef]
- Romero-Aleshire, M.J.; Diamond-Stanic, M.K.; Hasty, A.H.; Hoyer, P.B.; Brooks, H.L. Loss of Ovarian Function in the VCD Mouse-Model of Menopause Leads to Insulin Resistance and a Rapid Progression into the Metabolic Syndrome. Am. J. Physiol. Integr. Comp. Physiol. 2009, 297, R587–R592. [Google Scholar] [CrossRef] [PubMed]
- Ntikoudi, A.; Spyrou, A.; Evangelou, E.; Dokoutsidou, E.; Mastorakos, G. The Effect of Menopausal Status, Insulin Resistance and Body Mass Index on the Prevalence of Non-Alcoholic Fatty Liver Disease. Healthcare 2024, 12, 1081. [Google Scholar] [CrossRef]
- Barros, R.P.A.; Gustafsson, J.Å. Estrogen Receptors and the Metabolic Network. Cell Metab. 2011, 14, 289–299. [Google Scholar] [CrossRef]
- Van Sinderen, M.; Steinberg, G.; Jorgensen, S.B.; Honeyman, J.; Chow, J.D.; Simpson, E.R.; Jones, M.E.; Boon, W.C. Sexual Dimorphism in the Glucose Homeostasis Phenotype of the Aromatase Knockout (ArKO) Mice. J. Steroid Biochem. Mol. Biol. 2017, 170, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Nakajima, M.; Yokoi, T. Cytochrome P450-Mediated Metabolism of Estrogens and Its Regulation in Human. Cancer Lett. 2005, 227, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 12808. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Chen, G.G.; Liu, Y.; Su, X.; Hu, B.; Leung, B.C.S.; Wang, Y.; Ho, R.L.K.; Yang, S.; Lu, G.; et al. Cytochrome P4501A2 Metabolizes 17β-Estradiol to Suppress Hepatocellular Carcinoma. PLoS ONE 2016, 11, e0153863. [Google Scholar] [CrossRef] [PubMed]
- Konopka, A.R.; Asante, A.; Lanza, I.R.; Robinson, M.M.; Johnson, M.L.; Man, C.D.; Cobelli, C.; Amols, M.H.; Irving, B.A.; Nair, K. Defects in Mitochondrial Efficiency and H2O2 Emissions in Obese Women Are Restored to a Lean Phenotype with Aerobic Exercise Training. Diabetes 2015, 64, 2104–2115. [Google Scholar] [CrossRef]
- Smith, A.J.; Phipps, W.R.; Thomas, W.; Schmitz, K.H.; Kurzer, M.S. The Effects of Aerobic Exercise on Estrogen Metabolism in Healthy Premenopausal Women. Cancer Epidemiol. Biomark. Prev. 2013, 22, 756–764. [Google Scholar] [CrossRef]
- Liu, J.; Lu, W.; Shi, B.; Klein, S.; Su, X. Peroxisomal Regulation of Redox Homeostasis and Adipocyte Metabolism. Redox Biol. 2019, 24, 101167. [Google Scholar] [CrossRef]
- Demarquoy, F.J.; Le Borgne, O. Crosstalk between Mitochondria and Peroxisomes. World J. Biol. Chem. 2015, 6, 301–309. [Google Scholar] [CrossRef]
- Lismont, C.; Nordgren, M.; Van Veldhoven, P.P.; Fransen, M. Redox Interplay between Mitochondria and Peroxisomes. Front. Cell Dev. Biol. 2015, 3, 35. [Google Scholar] [CrossRef]
- Schrader, M.; Costello, J.L.; Godinho, L.F.; Azadi, A.S.; Islinger, M. Proliferation and Fission of Peroxisomes—An Update. Biochim. Biophys. Acta-Mol. Cell Res. 2016, 1863, 971–983. [Google Scholar] [CrossRef]
- Brown, L.; Baker, A. Peroxisome Biogenesis and the Role of Protein Import. J. Cell. Mol. Med. 2003, 7, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Agrawal, G.; Subramani, S. Peroxisome Assembly: Matrix and Membrane Protein Biogenesis. J. Cell Biol. 2011, 193, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.; Brocard, C. PEX11 Proteins Attract Mff and Human Fis1 to Coordinate Peroxisomal Fission. J. Cell Sci. 2012, 125, 3813–3826. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.K.; Hashimoto, T. Peroxisomal β-Oxidation and Peroxisome Proliferator-Activated Receptor α: An Adaptive Metabolic System. Annu. Rev. Nutr. 2001, 21, 193–230. [Google Scholar] [CrossRef]
- Dominy, J.E.; Puigserver, P. Mitochondrial Biogenesis through Activation of Nuclear Signaling Proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a015008. [Google Scholar] [CrossRef]
- Alfonso-Prieto, M.; Biarnés, X.; Vidossich, P.; Rovira, C. The Molecular Mechanism of the Catalase Reaction. J. Am. Chem. Soc. 2009, 131, 11751–11761. [Google Scholar] [CrossRef]
- Okumoto, K.; Tamura, S.; Honsho, M.; Fujiki, Y. Peroxisome: Metabolic Functions and Biogenesis. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2020; Volume 1299. [Google Scholar]
- Talley, J.T.; Mohiuddin, S.S. Biochemistry, Fatty Acid Oxidation; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Engelen, M.; Kemp, S.; de Visser, M.; van Geel, B.M.; Wanders, R.J.; Aubourg, P.; Poll-The, B.T. X-Linked Adrenoleukodystrophy (X-ALD): Clinical Presentation and Guidelines for Diagnosis, Follow-up and Management. Orphanet J. Rare Dis. 2012, 7, 51. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Vreken, P.; Ferdinandusse, S.; Jansen, G.A.; Waterham, H.R.; van Roermund, C.W.T.; Van Grunsven, E.G. Peroxisomal Fatty Acid Alpha- and Beta-Oxidation in Humans: Enzymology, Peroxisomal Metabolite Transporters and Peroxisomal Diseases. Biochem. Soc. Trans. 2001, 29, 250–267. [Google Scholar] [CrossRef]
- Wanders, R.J.; Waterham, H.R. Peroxisomal Disorders: The Single Peroxisomal Enzyme Deficiencies. Biochim. Biophys. Acta-Mol. Cell Res. 2006, 1763, 1707–1720. [Google Scholar] [CrossRef]
- Moreno-Fernandez, M.E.; Giles, D.A.; Stankiewicz, T.E.; Sheridan, R.; Karns, R.; Cappelletti, M.; Lampe, K.; Mukherjee, R.; Sina, C.; Sallese, A.; et al. Peroxisomal β-Oxidation Regulates Whole Body Metabolism, Inflammatory Vigor, and Pathogenesis of Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2018, 3, e93626. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Vaz, F.M.; Waterham, H.R.; Ferdinandusse, S. Fatty Acid Oxidation in Peroxisomes: Enzymology, Metabolic Crosstalk with Other Organelles and Peroxisomal Disorders. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2020; Volume 1299. [Google Scholar]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular Mechanisms of Hepatic Lipid accumulation in Non-Alcoholic Fatty Liver Disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed]
- Dasarathy, S.; Yang, Y.; McCullough, A.J.; Marczewski, S.; Bennett, C.; Kalhan, S.C. Elevated Hepatic Fatty Acid Oxidation, High Plasma Fibroblast Growth Factor 21, and Fasting Bile Acids in Nonalcoholic Steatohepatitis. Eur. J. Gastroenterol. Hepatol. 2011, 23, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Sprecher, H.W.; Kolattukudy, P.E. Estrogen-induced Production of a Peroxisome Proliferator-Activated Receptor (PPAR) Ligand in a PPARγ-Expressing Tissue. J. Biol. Chem. 1998, 273, 30131–30138. [Google Scholar] [CrossRef]
- Yoon, M. The role of PPARα in Lipid Metabolism and Obesity: Focusing on the Effects of Estrogen on PPARα Actions. Pharmacol. Res. 2009, 60, 151–159. [Google Scholar] [CrossRef]
- Yoon, M. PPAR in Obesity: Sex Difference and Estrogen Involvement. PPAR Res. 2010, 2010, 584296. [Google Scholar] [CrossRef]
- Foryst-Ludwig, A.; Clemenz, M.; Hohmann, S.; Hartge, M.; Sprang, C.; Frost, N.; Krikov, M.; Bhanot, S.; Barros, R.; Morani, A.; et al. Metabolic Actions of Estrogen Receptor Beta (ERβ) Are Mediated by a Negative Cross-Talk with PPARγ. PLoS Genet. 2008, 4, e1000108. [Google Scholar] [CrossRef]
- Corona, J.C.; Duchen, M.R. PPARγ as a Therapeutic Target to Rescue Mitochondrial Function in Neurological Disease. Free Radic. Biol. Med. 2016, 100, 153–163. [Google Scholar] [CrossRef]
- Villena, J.A.; Kralli, A. ERRα: A Metabolic Function for the Oldest Orphan. Trends Endocrinol. Metab. 2008, 19, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-Y.; McDonnell, D.P. Molecular Pathways: The Metabolic Regulator Estrogen-Related Receptor α as a Therapeutic Target in Cancer. Clin. Cancer Res. 2012, 18, 6089–6095. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocr. Rev. 2013, 34, 309–338. [Google Scholar] [CrossRef]
- D’Eon, T.M.; Souza, S.C.; Aronovitz, M.; Obin, M.S.; Fried, S.K.; Greenberg, A.S. Estrogen Regulation of Adiposity and Fuel Partitioning: Evidence of Genomic and Non-Genomic Regulation of Lipogenic and Oxidative Pathways. J. Biol. Chem. 2005, 280, 35983–35991. [Google Scholar] [CrossRef] [PubMed]
- Olivier, L.M.; Krisans, S.K. Peroxisomal Protein Targeting and Identification of Peroxisomal Targeting Signals in Cholesterol Biosynthetic Enzymes. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2000, 1529, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Liscurn, L. Chapter 15 Cholesterol Biosynthesis. In New Comprehensive Biochemistry; Elsevier: Amsterdam, The Netherlands, 2002; Volume 36. [Google Scholar] [CrossRef]
- Murray, R.K.; Granner, D.K.; Mayes, P.A.; Rodwell, V.W. Harper’s Illustrated Biochemistry; McGraw-Hill: New York, NY, USA, 2003; ISBN 0071389016. [Google Scholar]
- Cerqueira, N.M.F.S.A.; Oliveira, E.F.; Gesto, D.S.; Santos-Martins, D.; Moreira, C.; Moorthy, H.N.; Ramos, M.J.; Fernandes, P.A. Cholesterol Biosynthesis: A Mechanistic Overview. Biochemistry 2016, 55, 5483–5506. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Regulation of the Mevalonate Pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Charles, K.N.; Shackelford, J.E.; Faust, P.L.; Fliesler, S.J.; Stangl, H.; Kovacs, W.J. Functional Peroxisomes Are Essential for Efficient Cholesterol Sensing and Synthesis. Front. Cell Dev. Biol. 2020, 8, 560266. [Google Scholar] [CrossRef]
- Botham, K.M.; Mayes, P.A. Chapter 26: Cholesterol Synthesis, Transport, & Excretion. In Harper’s Illustrated Biochemistry; McGraw-Hill: New York, NY, USA, 2015; p. 219. [Google Scholar]
- Wilds, A.L.; Djerassi, C. The Synthesis of Estradiol and 1-Methylestradiol from Cholesterol. J. Am. Chem. Soc. 1946, 68, 2125–2133. [Google Scholar] [CrossRef]
- Pallottini, V.; Martini, C.; Pascolini, A.; Cavallini, G.; Gori, Z.; Bergamini, E.; Incerpi, S.; Trentalance, A. 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Deregulation and Age-Related Hypercholesterolemia: A New Role for ROS. Mech. Ageing Dev. 2005, 126, 845–851. [Google Scholar] [CrossRef]
- Kovacs, W.J.; Shackelford, J.E.; Tape, K.N.; Richards, M.J.; Faust, P.L.; Fliesler, S.J.; Krisans, S.K. Disturbed Cholesterol Homeostasis in a Peroxisome-Deficient PEX2 Knockout Mouse Model. Mol. Cell. Biol. 2004, 24, 1–13. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative Stress, Mitochondrial Damage and Neurodegenerative Diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Su, W.; Chi, Y.; An, Y.A. Editorial: Lipid Droplets and Mitochondria in Metabolic Diseases. Front. Physiol. 2023, 14, 1266356. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, Z.; Shen, W.-J.; Azhar, S. Cellular Cholesterol Delivery, Intracellular Processing and Utilization for Biosynthesis of Steroid Hormones. Nutr. Metab. 2010, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Philipp, B.; Shapiro, D. Estrogen Regulation of Hepatic 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase and Acetyl-CoA Carboxylase in Xenopus Laevis. J. Biol. Chem. 1981, 256, 2922–2927. [Google Scholar] [CrossRef] [PubMed]
- Ness, G.C.; Chambers, C.M. Feedback and Hormonal Regulation of Hepatic 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase: The Concept of Cholesterol Buffering Capacity. Proc. Soc. Exp. Biol. Med. 2000, 224, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Trapani, L.; Pallottini, V. Age-Related Hypercholesterolemia and HMG-CoA Reductase Dysregulation: Sex Does Matter (A Gender Perspective). Curr. Gerontol. Geriatr. Res. 2010, 2010, 420139. [Google Scholar] [CrossRef]
- Trapani, L.; Violo, F.; Pallottini, V. Hypercholesterolemia and 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Regulation in Aged Female Rats. Exp. Gerontol. 2010, 45, 119–128. [Google Scholar] [CrossRef]
- Schulz, E.; Anter, E.; Zou, M.-H.; Keaney, J.J.F. Estradiol-Mediated Endothelial Nitric Oxide Synthase Association with Heat Shock Protein 90 Requires Adenosine Monophosphate-Dependent Protein Kinase. Circulation 2005, 111, 3473–3480. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile acids: Regulation of Synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef]
- Chiang, J.Y. Regulation of Bile Acid Synthesis: Pathways, Nuclear Receptors, and Mechanisms. J. Hepatol. 2004, 40, 539–551. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile Acid Metabolism and Signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar]
- Schroor, M.M.; Sennels, H.P.; Fahrenkrug, J.; Jørgensen, H.L.; Plat, J.; Mensink, R.P. Diurnal Variation of Markers for Cholesterol Synthesis, Cholesterol Absorption, and Bile Acid Synthesis: A Systematic Review and the Bispebjerg Study of Diurnal Variations. Nutrients 2019, 11, 1439. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Garruti, G.; Baccetto, R.L.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2017, 16, S4–S14. [Google Scholar] [CrossRef] [PubMed]
- García, G.C. Fisiopatología de La Colestasis. Med. Interna Mex. 2006, 22, 411–421. [Google Scholar]
- Chen, I.; Cassaro, S. Physiology, Bile Acids; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Sinal, C.J.; Tohkin, M.; Miyata, M.; Ward, J.M.; Lambert, G.; Gonzalez, F.J. Targeted Disruption of the Nuclear Receptor FXR/BAR Impairs Bile Acid and Lipid Homeostasis. Cell 2000, 102, 731–744. [Google Scholar] [CrossRef]
- Kim, I.; Ahn, S.-H.; Inagaki, T.; Choi, M.; Ito, S.; Guo, G.L.; Kliewer, S.A.; Gonzalez, F.J. Differential Regulation of Bile Acid Homeostasis by the Farnesoid X Receptor in Liver and Intestine. J. Lipid Res. 2007, 48, 2664–2672. [Google Scholar] [CrossRef]
- Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Ren, S.; Gil, G.; Dent, P. Bile Acids as Regulatory Molecules. J. Lipid Res. 2009, 50, 1509–1520. [Google Scholar] [CrossRef]
- Fang, Y.; Han, S.I.; Mitchell, C.; Gupta, S.; Studer, E.; Grant, S.; Hylemon, P.B.; Dent, P. Bile Acids Induce Mitochondrial ROS, Which Promote Activation of Receptor Tyrosine Kinases and Signaling Pathways in Rat Hepatocytes. Hepatology 2004, 40, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Duran-Sandoval, D.; Mautino, G.; Martin, G.; Percevault, F.; Barbier, O.; Fruchart, J.-C.; Kuipers, F.; Staels, B. Glucose Regulates the Expression of the Farnesoid X Receptor in Liver. Diabetes 2004, 53, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G Protein-Coupled Receptor Responsive to Bile Acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [PubMed]
- Duboc, H.; Taché, Y.; Hofmann, A.F. The Bile Acid TGR5 Membrane Receptor: From Basic Research to Clinical Application. Dig. Liver Dis. 2014, 46, 302–312. [Google Scholar] [CrossRef]
- Mumford, S.L.; Dasharathy, S.; Pollack, A.Z.; Schisterman, E.F. Variations in Lipid Levels According to Menstrual Cycle Phase: Clinical Implications. Clin. Lipidol. 2011, 6, 225–234. [Google Scholar] [CrossRef]
- Journe, F.; Durbecq, V.; Chaboteaux, C.; Rouas, G.; Laurent, G.; Nonclercq, D.; Sotiriou, C.; Body, J.-J.; Larsimont, D. Association between Farnesoid X Receptor Expression and Cell Proliferation in Estrogen Receptor-Positive Luminal-like Breast Cancer from Postmenopausal Patients. Breast Cancer Res. Treat. 2009, 115, 523–535. [Google Scholar] [CrossRef]
- Knebel, B.; Hartwig, S.; Haas, J.; Lehr, S.; Goeddeke, S.; Susanto, F.; Bohne, L.; Jacob, S.; Koellmer, C.; Nitzgen, U.; et al. Peroxisomes Compensate Hepatic Lipid Overflow in Mice with Fatty Liver. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2015, 1851, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Knebel, B.; Göddeke, S.; Hartwig, S.; Hörbelt, T.; Fahlbusch, P.; Al-Hasani, H.; Jacob, S.; Koellmer, C.; Nitzgen, U.; Schiller, M.; et al. Alteration of Liver Peroxisomal and Mitochondrial Functionality in the NZO Mouse Model of Metabolic Syndrome. Proteom.–Clin. Appl. 2018, 12, 1700028. [Google Scholar] [CrossRef]
- Mueller, A.M.; Kleemann, R.; Gart, E.; van Duyvenvoorde, W.; Verschuren, L.; Caspers, M.; Menke, A.; Krömmelbein, N.; Salic, K.; Burmeister, Y.; et al. Cholesterol Accumulation as a Driver of Hepatic Inflammation Under Translational Dietary Conditions Can Be Attenuated by a Multicomponent Medicine. Front. Endocrinol. 2021, 12, 601160. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liu, J.; Zhao, K.; Gao, L.; Zhao, J. Cholesterol-Induced Toxicity: An Integrated View of the Role of Cholesterol in Multiple Diseases. Cell Metab. 2021, 33, 1911–1925. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, A.; Canbay, A. Why Bile Acids Are So Important in Non-Alcoholic Fatty Liver Disease (NAFLD) Progression. Cells 2019, 8, 1358. [Google Scholar] [CrossRef]
- Abrigo, J.; Olguín, H.; Gutierrez, D.; Tacchi, F.; Arrese, M.; Cabrera, D.; Valero-Breton, M.; Elorza, A.A.; Simon, F.; Cabello-Verrugio, C. Bile Acids Induce Alterations in Mitochondrial Function in Skeletal Muscle Fibers. Antioxidants 2022, 11, 1706. [Google Scholar] [CrossRef]
- Gusdon, A.M.; Song, K.-X.; Qu, S. Nonalcoholic Fatty Liver Disease: Pathogenesis and Therapeutics from a Mitochondria-Centric Perspective. Oxidative Med. Cell. Longev. 2014, 2014, 637027. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative Stress, Cardiolipin and Mitochondrial Dysfunction in Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2014, 20, 14205–14218. [Google Scholar] [CrossRef]
- Dornas, W.; Schuppan, D. Mitochondrial Oxidative Injury: A Key Player in Nonalcoholic Fatty Liver Disease. Am. J. Physiol. Liver Physiol. 2020, 319, G400–G411. [Google Scholar] [CrossRef]
- Perez, M.J.; Briz, O. Bile-Acid-Induced Cell Injury and Protection. World J. Gastroenterol. 2009, 15, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Lake, A.D.; Novak, P.; Shipkova, P.; Aranibar, N.; Robertson, D.; Reily, M.D.; Lu, Z.; Lehman-McKeeman, L.D.; Cherrington, N.J. Decreased Hepatotoxic Bile Acid Composition and Altered Synthesis in Progressive Human Nonalcoholic Fatty Liver Disease. Toxicol. Appl. Pharmacol. 2013, 268, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Jiang, Z.; Zhang, L. Bile Acid Regulation: A Novel Therapeutic Strategy in Non-Alcoholic Fatty Liver Disease. Pharmacol. Ther. 2018, 190, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R. Introduction to Lipids and Lipoproteins. Endotext. 2021. Available online: https://www.ncbi.nlm.nih.gov/sites/books/NBK305896/ (accessed on 15 October 2024).
- Lent-Schochet, D.; Jialal, I. Biochemistry, Lipoprotein Metabolism. Adv. Lipid Res. 2020, 13, 1–89. [Google Scholar]
- Feingold, K.R. Lipid and Lipoprotein Metabolism. Endocrinol. Metab. Clin. N. Am. 2022, 51, 437–458. [Google Scholar] [CrossRef]
- Wang, H.H.; Garruti, G.; Liu, M.; Portincasa, P.; Wang, D.Q.-H. Cholesterol and Lipoprotein Metabolism and Atherosclerosis: Recent Advances in Reverse Cholesterol Transport. Ann. Hepatol. 2017, 16, S27–S42. [Google Scholar] [CrossRef]
- Wang, A.-B.; Liu, D.-P.; Liang, C.-C. Regulation of Human Apolipoprotein B Gene Expression at Multiple Levels. Exp. Cell Res. 2003, 290, 1–12. [Google Scholar] [CrossRef]
- Hussain, M.M.; Shi, J.; Dreizen, P. Microsomal Triglyceride Transfer Protein and Its Role in ApoB-Lipoprotein Assembly. J. Lipid Res. 2003, 44, 22–32. [Google Scholar] [CrossRef]
- Gugliucci, A. Triglyceride-Rich Lipoprotein Metabolism: Key Regulators of Their Flux. J. Clin. Med. 2023, 12, 4399. [Google Scholar] [CrossRef]
- Adiels, M.; Olofsson, S.-O.; Taskinen, M.-R.; Borén, J. Overproduction of Very Low–Density Lipoproteins Is the Hallmark of the Dyslipidemia in the Metabolic Syndrome. Arter. Thromb. Vasc. Biol. 2008, 28, 1225–1236. [Google Scholar] [CrossRef]
- Van Zwol, W.; Van De Sluis, B.; Ginsberg, H.N.; Kuivenhoven, J.A. VLDL Biogenesis and Secretion: It Takes a Village. Circ. Res. 2024, 134, 226–244. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liang, C.-P.; Westerterp, M.; Senokuchi, T.; Welch, C.L.; Wang, Q.; Matsumoto, M.; Accili, D.; Tall, A.R. Hepatic Insulin Signaling Regulates VLDL Secretion and Atherogenesis in Mice. J. Clin. Investig. 2009, 119, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, H.N.; Packard, C.J.; Chapman, M.J.; Borén, J.; Aguilar-Salinas, C.A.; Averna, M.; Ference, B.A.; Gaudet, D.; Hegele, R.A.; Kersten, S.; et al. Triglyceride-Rich Lipoproteins and Their Remnants: Metabolic Insights, Role in Atherosclerotic Cardiovascular Disease, and Emerging Therapeutic Strategies—A Consensus Statement from the European Atherosclerosis Society. Eur. Heart J. 2021, 42, 4791–4806. [Google Scholar] [CrossRef] [PubMed]
- Desmarchelier, C.; Borel, P.; Lairon, D.; Maraninchi, M.; Valéro, R. Effect of Nutrient and Micronutrient Intake on Chylomicron Production and Postprandial Lipemia. Nutrients 2019, 11, 1299. [Google Scholar] [CrossRef]
- Xiao, C.; Stahel, P.; Lewis, G.F. Regulation of Chylomicron Secretion: Focus on Post-Assembly Mechanisms. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 487–501. [Google Scholar] [CrossRef]
- Lin, M.C.; Gordon, D.; Wetterau, J.R. Microsomal Triglyceride Transfer Protein (MTP) Regulation in HepG2 Cells: Insulin Negatively Regulates MTP Gene Expression. J. Lipid Res. 1995, 36, 1073–1081. [Google Scholar] [CrossRef]
- Au, W.-S.; Kung, H.-F.; Lin, M.C. Regulation of Microsomal Triglyceride Transfer Protein Gene by Insulin in HepG2 Cells: Roles of MAPKerk and MAPKp38. Diabetes 2003, 52, 1073–1080. [Google Scholar] [CrossRef]
- Tomlinson, D.R. Mitogen-Activated Protein Kinases as Glucose Transducers for Diabetic Complications. Diabetologia 1999, 42, 1271–1281. [Google Scholar] [CrossRef]
- Huang, Y.; Li, X.; Wang, M.; Ning, H.; Li, Y.; Sun, C. Lipoprotein Lipase Links Vitamin D, Insulin Resistance, and Type 2 Diabetes: A Cross-Sectional Epidemiological Study. Cardiovasc. Diabetol. 2013, 12, 17. [Google Scholar] [CrossRef]
- Young, S.G.; Fong, L.G.; Beigneux, A.P.; Allan, C.M.; He, C.; Jiang, H.; Nakajima, K.; Meiyappan, M.; Birrane, G.; Ploug, M. GPIHBP1 and Lipoprotein Lipase, Partners in Plasma Triglyceride Metabolism. Cell Metab. 2019, 30, 51–65. [Google Scholar] [CrossRef]
- Kersten, S. Physiological Regulation of Lipoprotein Lipase. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2014, 1841, 919–933. [Google Scholar] [CrossRef] [PubMed]
- Panarotto, D.; Rémillard, P.; Bouffard, L.; Maheux, P. Insulin Resistance Affects the Regulation of Lipoprotein Lipase in the Postprandial Period and in an Adipose Tissue-Specific Manner. Eur. J. Clin. Investig. 2002, 32, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Waterham, H.R. Biochemistry of Mammalian Peroxisomes Revisited. Annu. Rev. Biochem. 2006, 75, 295–332. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J. Metabolic Functions of Peroxisomes in Health and Disease. Biochimie 2014, 98, 36–44. [Google Scholar] [CrossRef]
- Rasmiena, A.A.; Barlow, C.K.; Stefanovic, N.; Huynh, K.; Tan, R.; Sharma, A.; Tull, D.; de Haan, J.B.; Meikle, P.J. Plasmalogen Modulation Attenuates Atherosclerosis in ApoE- and ApoE/GPx1-Deficient Mice. Atherosclerosis 2015, 243, 598–608. [Google Scholar] [CrossRef]
- Szafran, H.; Smielak-Korombel, W. The Role of Estrogens in Hormonal Regulation of Lipid Metabolism in Women. Prz. Lek. 1998, 55, 266–270. [Google Scholar]
- Magkos, F.; Patterson, B.W.; Mohammed, B.S.; Klein, S.; Mittendorfer, B. Women Produce Fewer but Triglyceride-Richer Very Low-Density Lipoproteins than Men. J. Clin. Endocrinol. Metab. 2007, 92, 1311–1318. [Google Scholar] [CrossRef]
- Matthan, N.R.; Jalbert, S.M.; Barrett, P.H.R.; Dolnikowski, G.G.; Schaefer, E.J.; Lichtenstein, A.H. Gender-Specific Differences in the Kinetics of Nonfasting TRL, IDL, and LDL Apolipoprotein B-100 in Men and Premenopausal Women. Arter. Thromb. Vasc. Biol. 2008, 28, 1838–1843. [Google Scholar] [CrossRef]
- Côté, I.; Yasari, S.; Pighon, A.; Barsalani, R.; Rabasa-Lhoret, R.; Prud’Homme, D.; Lavoie, J.-M. Liver Fat Accumulation May Be Dissociated from Adiposity Gain in Ovariectomized Rats. Climacteric 2012, 15, 594–601. [Google Scholar] [CrossRef]
- de Oliveira, M.C.; Gilglioni, E.H.; de Boer, B.A.; Runge, J.H.; de Waart, D.R.; Salgueiro, C.L.; Ishii-Iwamoto, E.L.; Elferink, R.P.O.; Gaemers, I.C. Bile Acid Receptor Agonists INT747 and INT777 Decrease Oestrogen Deficiency-Related Postmenopausal Obesity and Hepatic Steatosis in Mice. Biochim. Biophys. Acta-Mol. Basis Dis. 2016, 1862, 2054–2062. [Google Scholar] [CrossRef]
- Villa, A.; Della Torre, S.; Stell, A.; Cook, J.; Brown, M.; Maggi, A. Tetradian Oscillation of Estrogen Receptor α Is Necessary to Prevent Liver Lipid Deposition. Proc. Natl. Acad. Sci. USA 2012, 109, 11806–11811. [Google Scholar] [CrossRef] [PubMed]
- Frias, J.P.; Macaraeg, G.B.; Ofrecio, J.; Yu, J.G.; Olefsky, J.M.; Kruszynska, Y.T. Decreased Susceptibility to Fatty Acid–Induced Peripheral Tissue Insulin Resistance in Women. Diabetes 2001, 50, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Brown, W.C.; Cai, Q.; Krust, A.; Chambon, P.; McGuinness, O.P.; Stafford, J.M. Estrogen Treatment after Ovariectomy Protects against Fatty Liver and May Improve Pathway-Selective Insulin Resistance. Diabetes 2013, 62, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Hevener, A.; Reichart, D.; Janez, A.; Olefsky, J. Female Rats Do Not Exhibit Free Fatty Acid-Induced Insulin Resistance. Diabetes 2002, 51, 1907–1912. [Google Scholar] [CrossRef]
- Di Croce, L.; Bruscalupi, G.; Trentalance, A. Independent Behavior of Rat Liver LDL Receptor and HMGCoA Reductase under Estrogen Treatment. Biochem. Biophys. Res. Commun. 1996, 224, 345–350. [Google Scholar] [CrossRef]
- De Marinis, E.; Martini, C.; Trentalance, A.; Pallottini, V. Sex Differences in Hepatic Regulation of Cholesterol Homeostasis. J. Endocrinol. 2008, 198, 635–643. [Google Scholar] [CrossRef]
- Fukata, Y.; Yu, X.; Imachi, H.; Nishiuchi, T.; Lyu, J.; Seo, K.; Takeuchi, A.; Iwama, H.; Masugata, H.; Hoshikawa, H.; et al. 17β-Estradiol Regulates Scavenger Receptor Class BI Gene Expression via Protein Kinase C in Vascular Endothelial Cells. Endocrine 2014, 46, 644–650. [Google Scholar] [CrossRef]
- Zhu, L.; Shi, J.; Luu, T.N.; Neuman, J.C.; Trefts, E.; Yu, S.; Palmisano, B.T.; Wasserman, D.H.; Linton, M.F.; Stafford, J.M. Hepatocyte Estrogen Receptor Alpha Mediates Estrogen Action to Promote Reverse Cholesterol Transport during Western-Type Diet Feeding. Mol. Metab. 2018, 8, 106–116. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogenic Control of Mitochondrial Function. Redox Biol. 2020, 31, 101435. [Google Scholar] [CrossRef]
- de Toda, I.M.; González-Sánchez, M.; Cerro, E.D.-D.; Valera, G.; Carracedo, J.; Guerra-Pérez, N. Sex Differences in Markers of Oxidation and Inflammation. Implications for Ageing. Mech. Ageing Dev. 2023, 211, 111797. [Google Scholar] [CrossRef]
- Ruiz-Romero, G.A.; Álvarez-Delgado, C. Effects of Estrogens in Mitochondria: An Approach to Type 2 Diabetes. AIMS Mol. Sci. 2024, 11, 72–98. [Google Scholar] [CrossRef]
- Li, A.; Zheng, N.; Ding, X. Mitochondrial Abnormalities: A Hub in Metabolic Syndrome-Related Cardiac Dysfunction Caused by Oxidative Stress. Heart Fail. Rev. 2022, 27, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
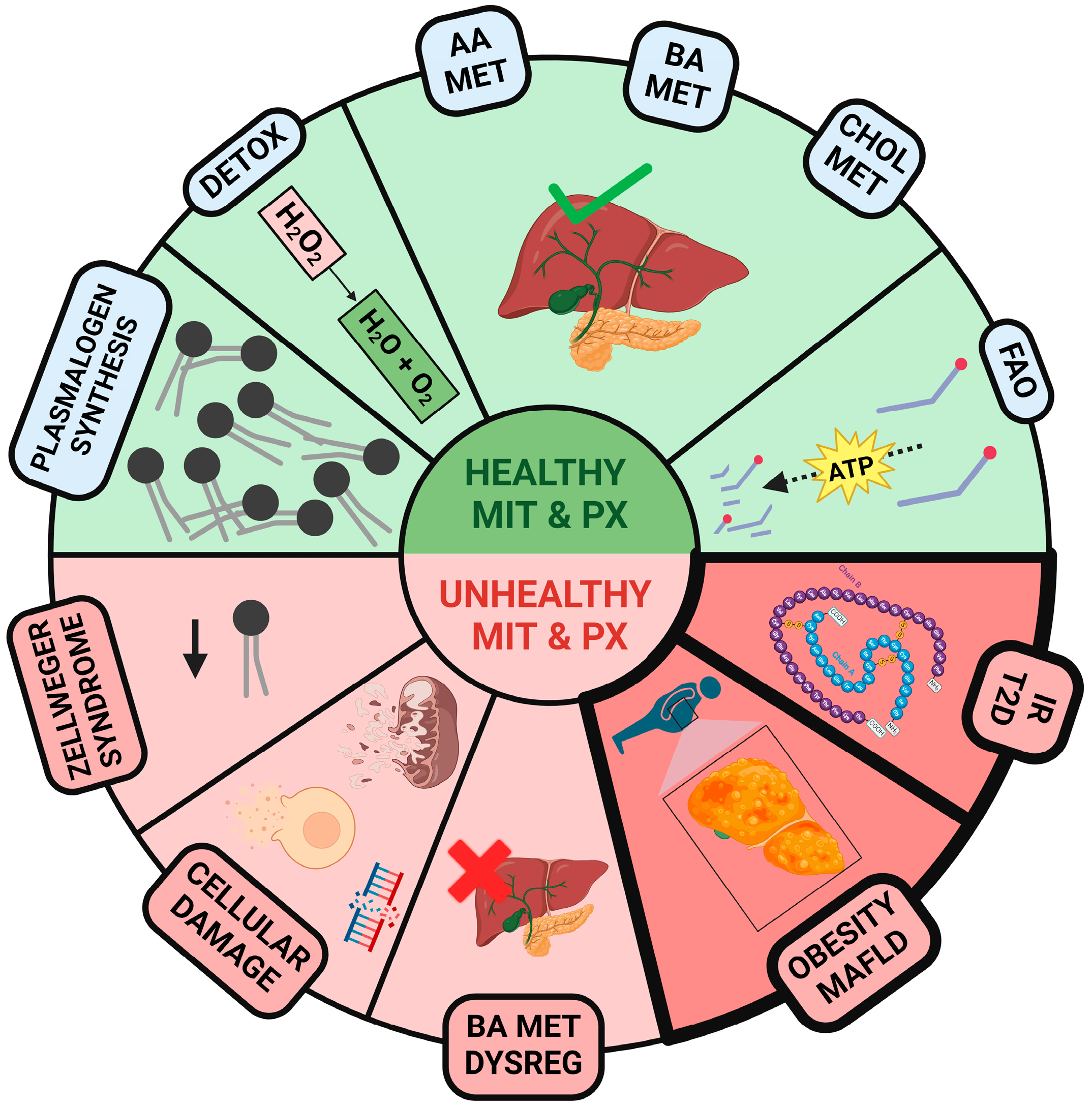
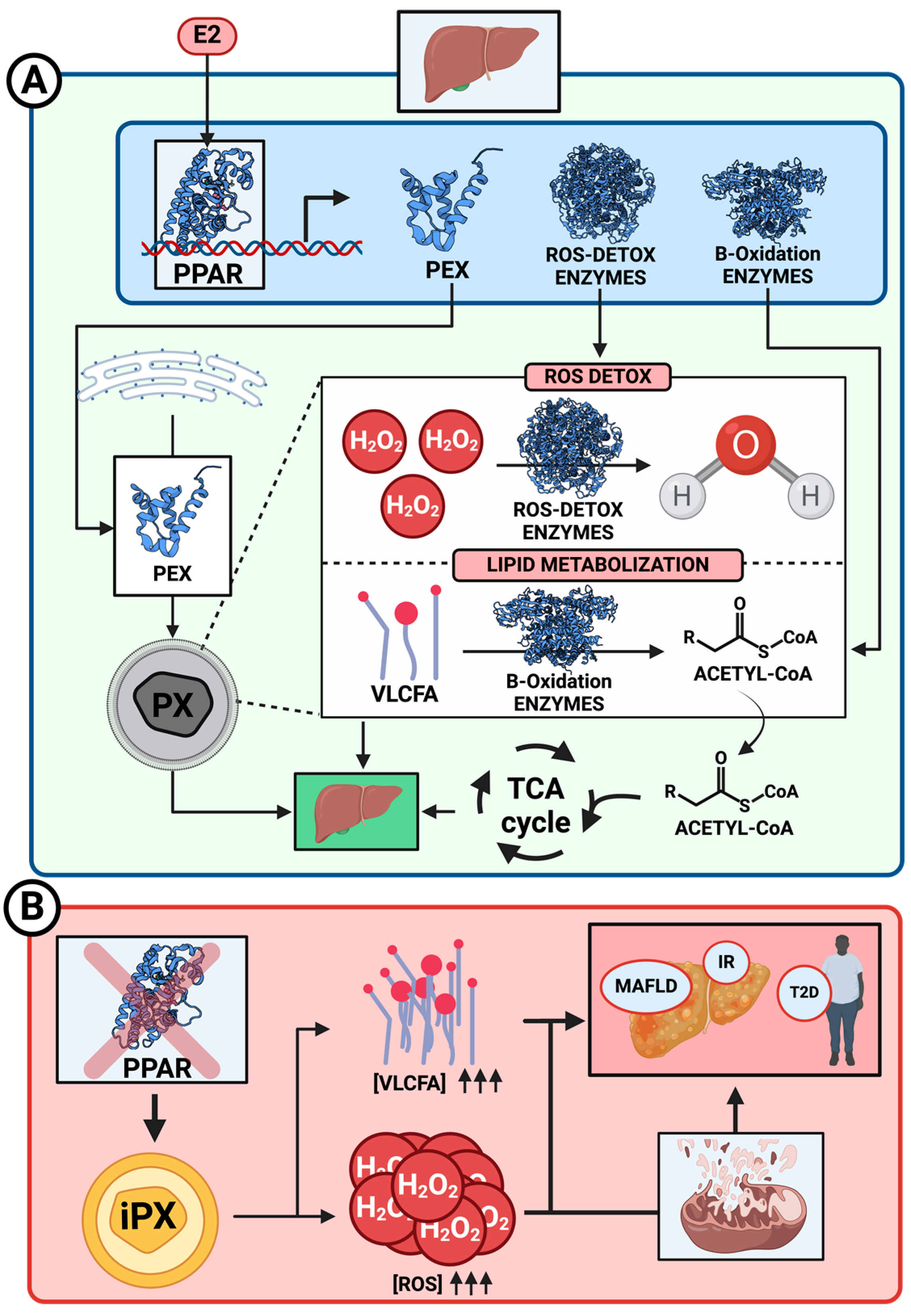
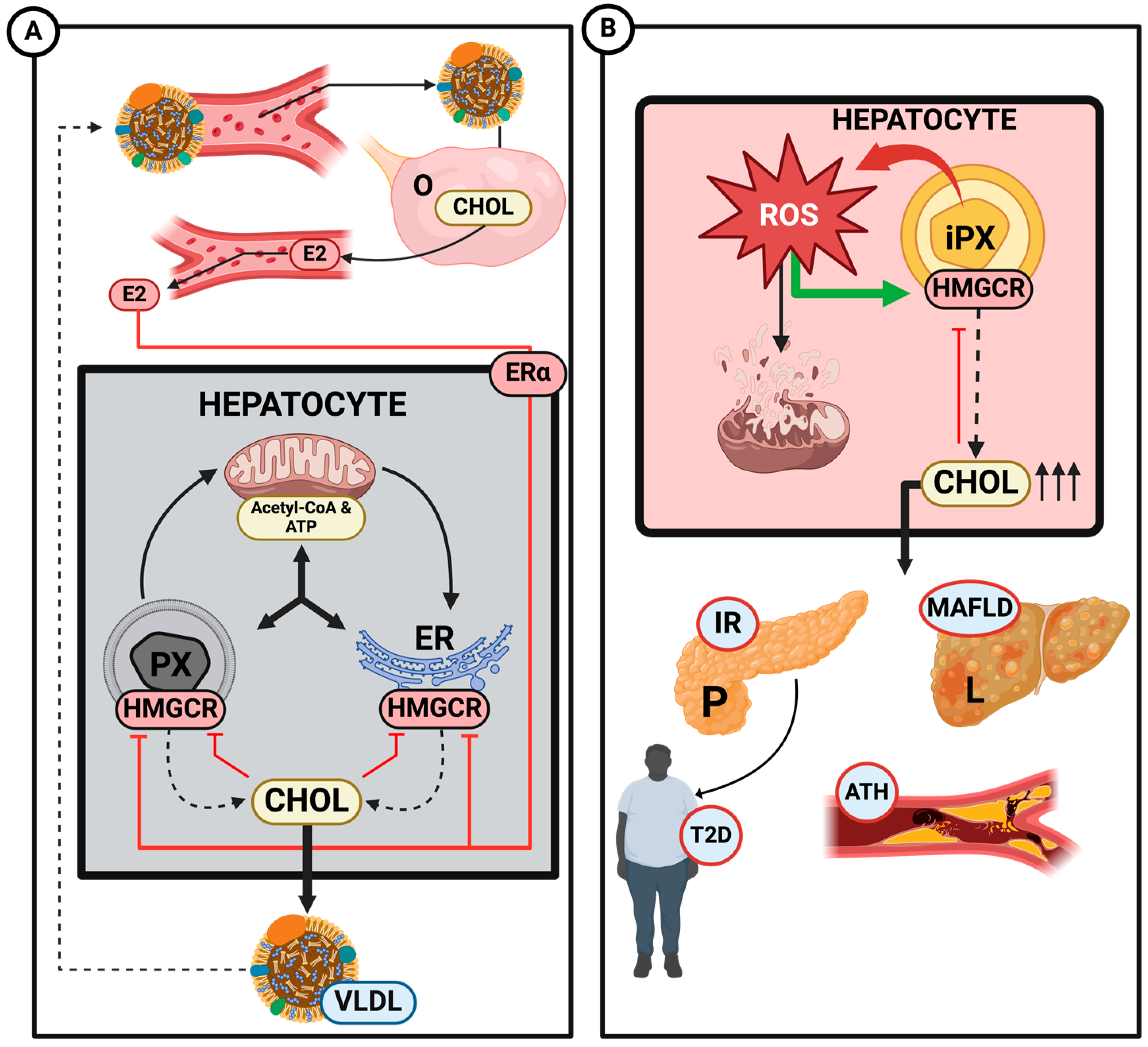
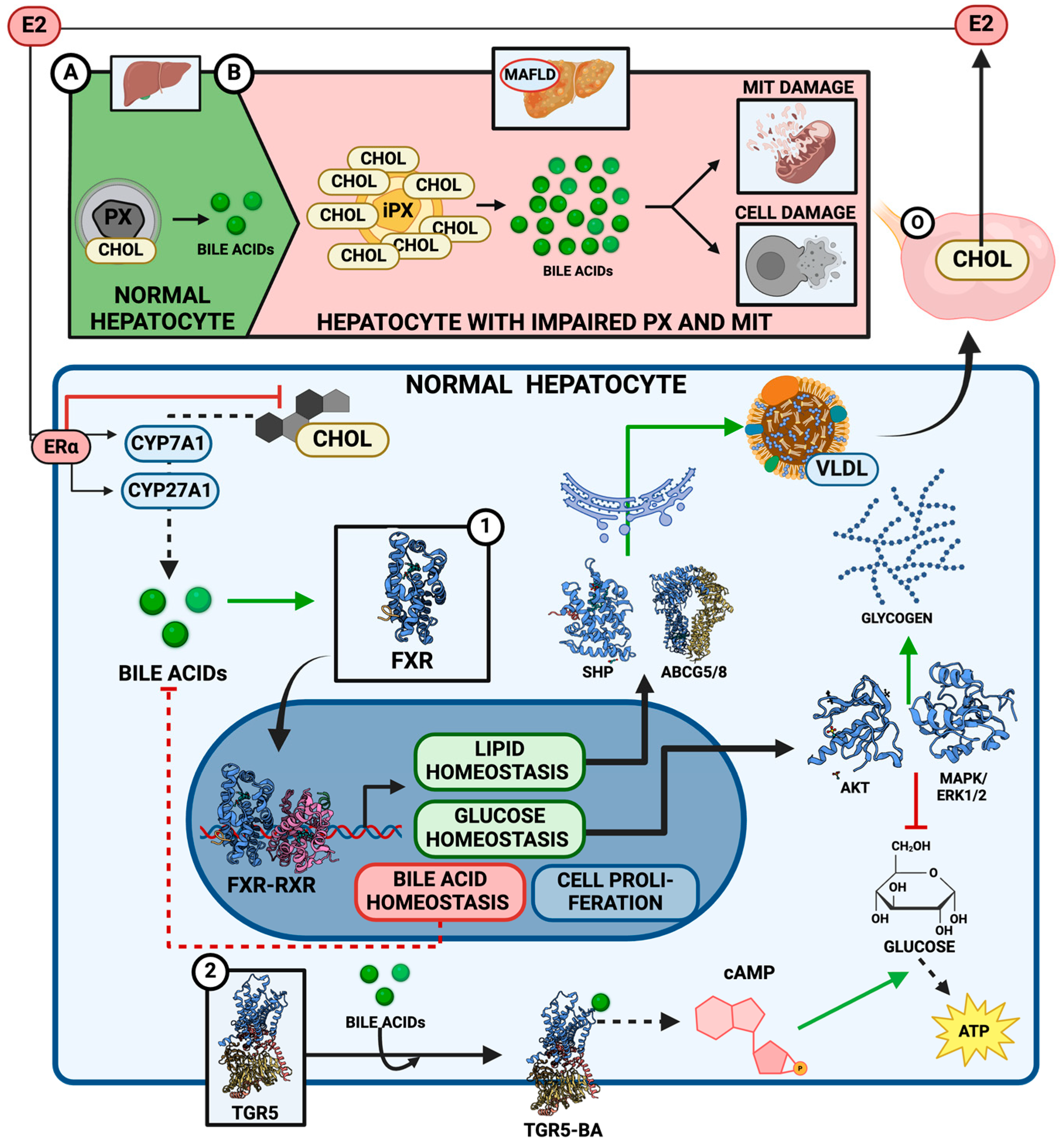

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antelo-Cea, D.A.; Martínez-Rojas, L.; Cabrerizo-Ibáñez, I.; Roudi Rashtabady, A.; Hernández-Alvarez, M.I. Regulation of Mitochondrial and Peroxisomal Metabolism in Female Obesity and Type 2 Diabetes. Int. J. Mol. Sci. 2024, 25, 11237. https://doi.org/10.3390/ijms252011237
Antelo-Cea DA, Martínez-Rojas L, Cabrerizo-Ibáñez I, Roudi Rashtabady A, Hernández-Alvarez MI. Regulation of Mitochondrial and Peroxisomal Metabolism in Female Obesity and Type 2 Diabetes. International Journal of Molecular Sciences. 2024; 25(20):11237. https://doi.org/10.3390/ijms252011237
Chicago/Turabian StyleAntelo-Cea, Damián A., Laura Martínez-Rojas, Izan Cabrerizo-Ibáñez, Ayda Roudi Rashtabady, and María Isabel Hernández-Alvarez. 2024. "Regulation of Mitochondrial and Peroxisomal Metabolism in Female Obesity and Type 2 Diabetes" International Journal of Molecular Sciences 25, no. 20: 11237. https://doi.org/10.3390/ijms252011237
APA StyleAntelo-Cea, D. A., Martínez-Rojas, L., Cabrerizo-Ibáñez, I., Roudi Rashtabady, A., & Hernández-Alvarez, M. I. (2024). Regulation of Mitochondrial and Peroxisomal Metabolism in Female Obesity and Type 2 Diabetes. International Journal of Molecular Sciences, 25(20), 11237. https://doi.org/10.3390/ijms252011237