
Co-Stimulation with TWEAK and TGF-β1 Induces Steroid-Insensitive TSLP and CCL5 Production in BEAS-2B Human Bronchial Epithelial Cells

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
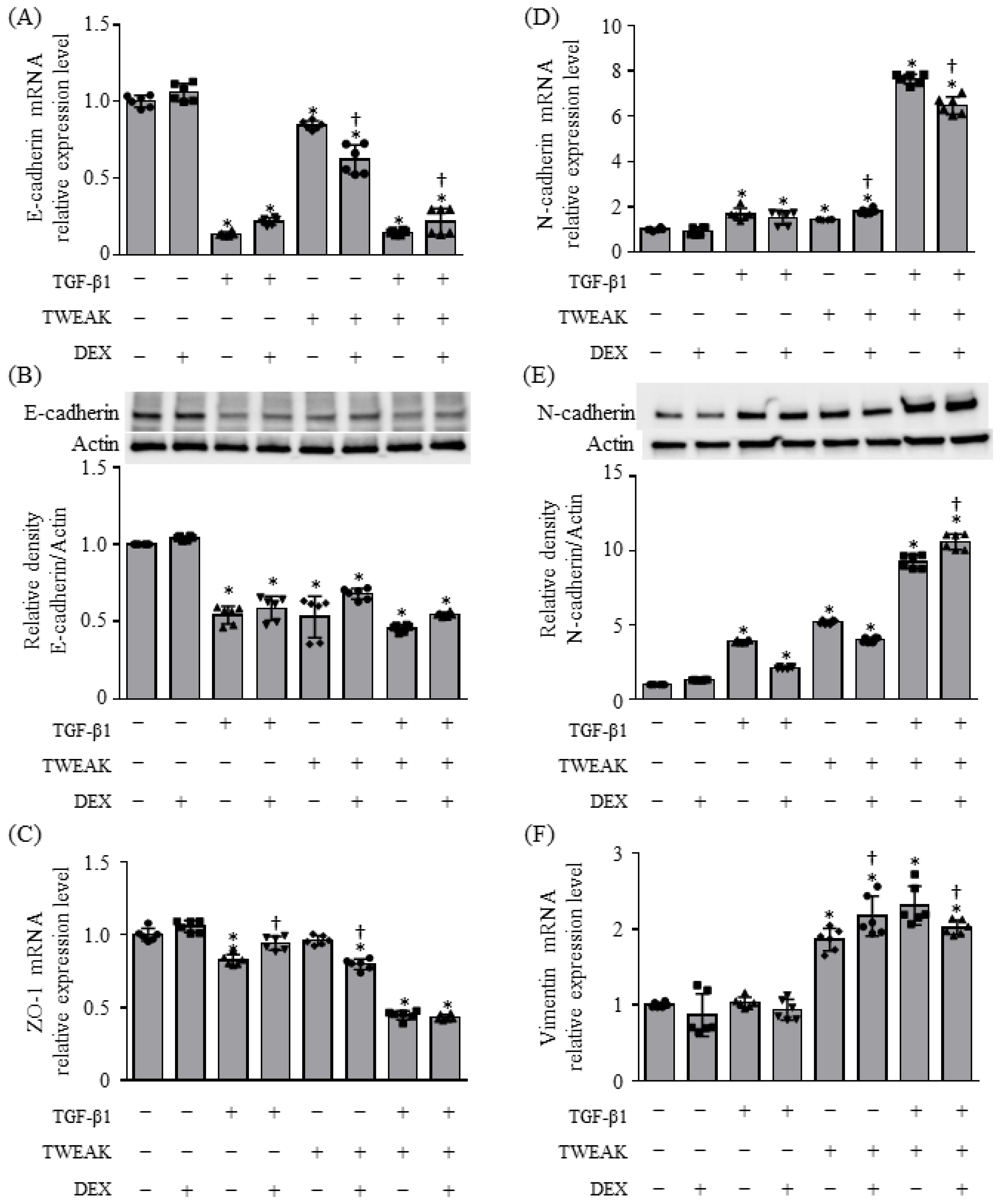
2.1. EMT and Cytokine Production Induced by Co-Stimulation with TWEAK and TGF-β1 Were Steroid Unresponsiveness
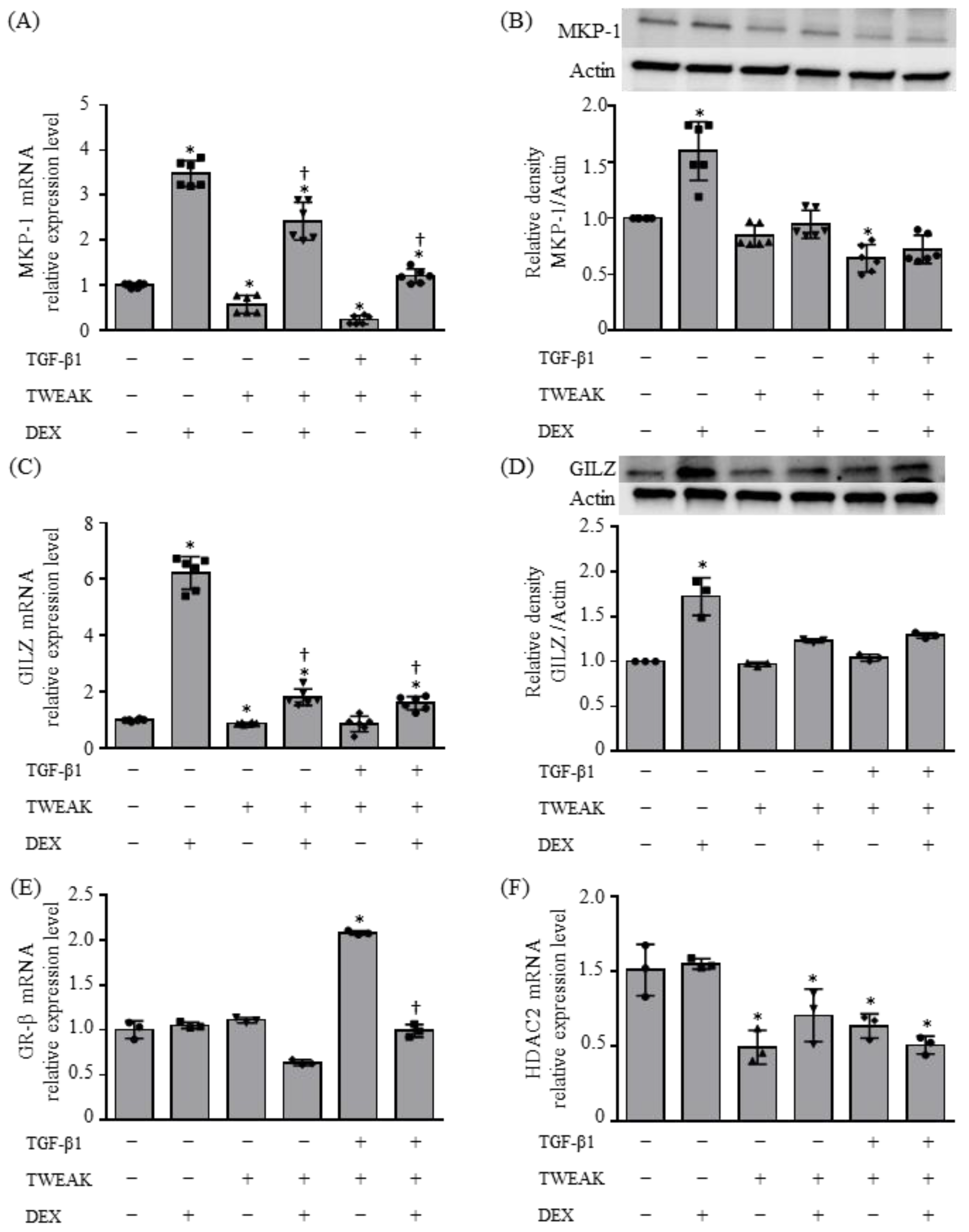
2.2. Molecular Mechanisms Resulting in Steroid Unresponsiveness, Induced by the Co-Stimulation of TWEAK and TGF-β1
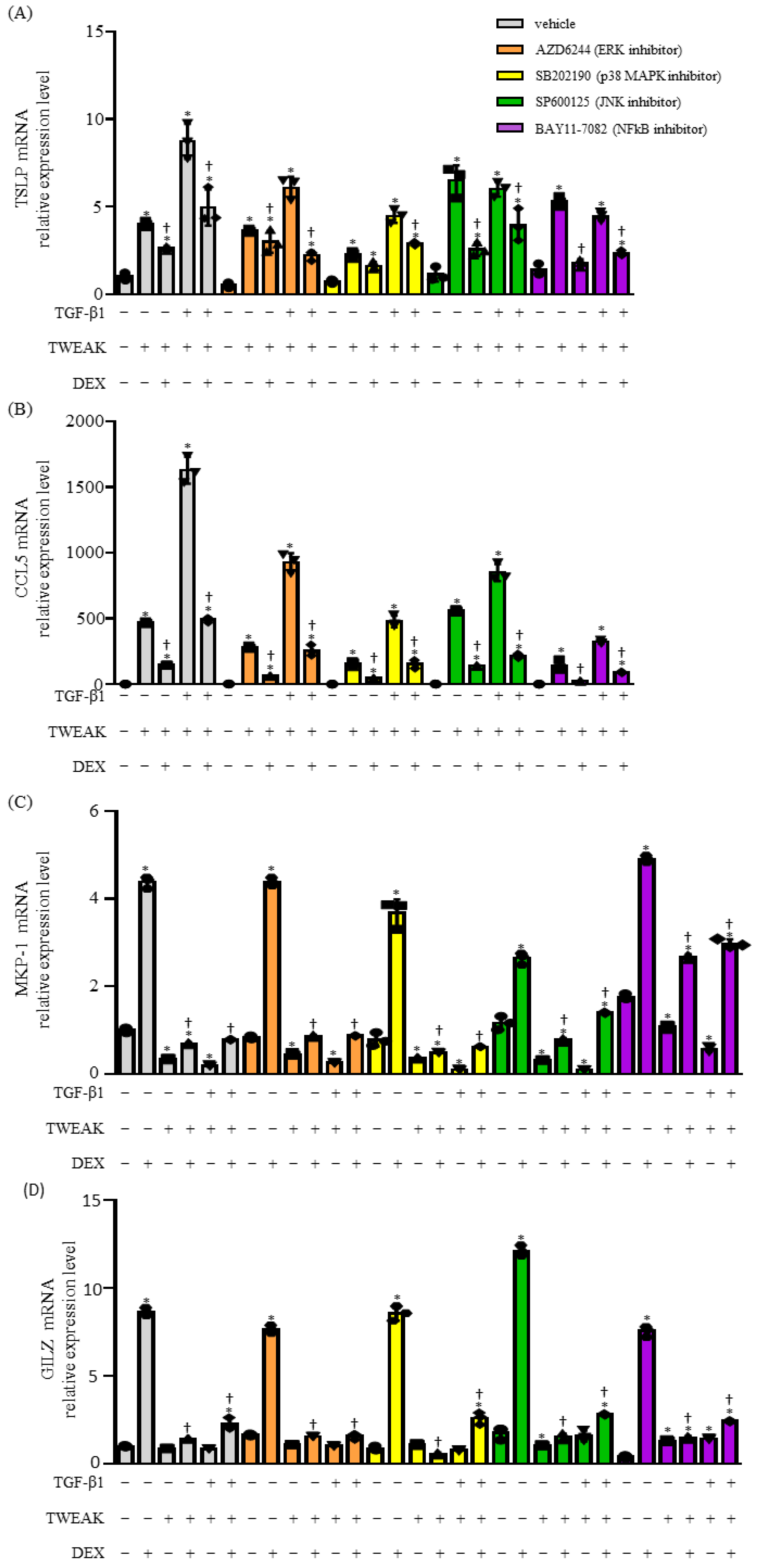
2.3. The Co-Stimulation of TWEAK- and TGF-β1-Induced Steroid Unresponsiveness and the Production of Cytokines Require the Mitogen-Activated Protein Kinase (MAPK) and Nuclear Factor Kappa Beta (NF-κB) Signaling Pathways
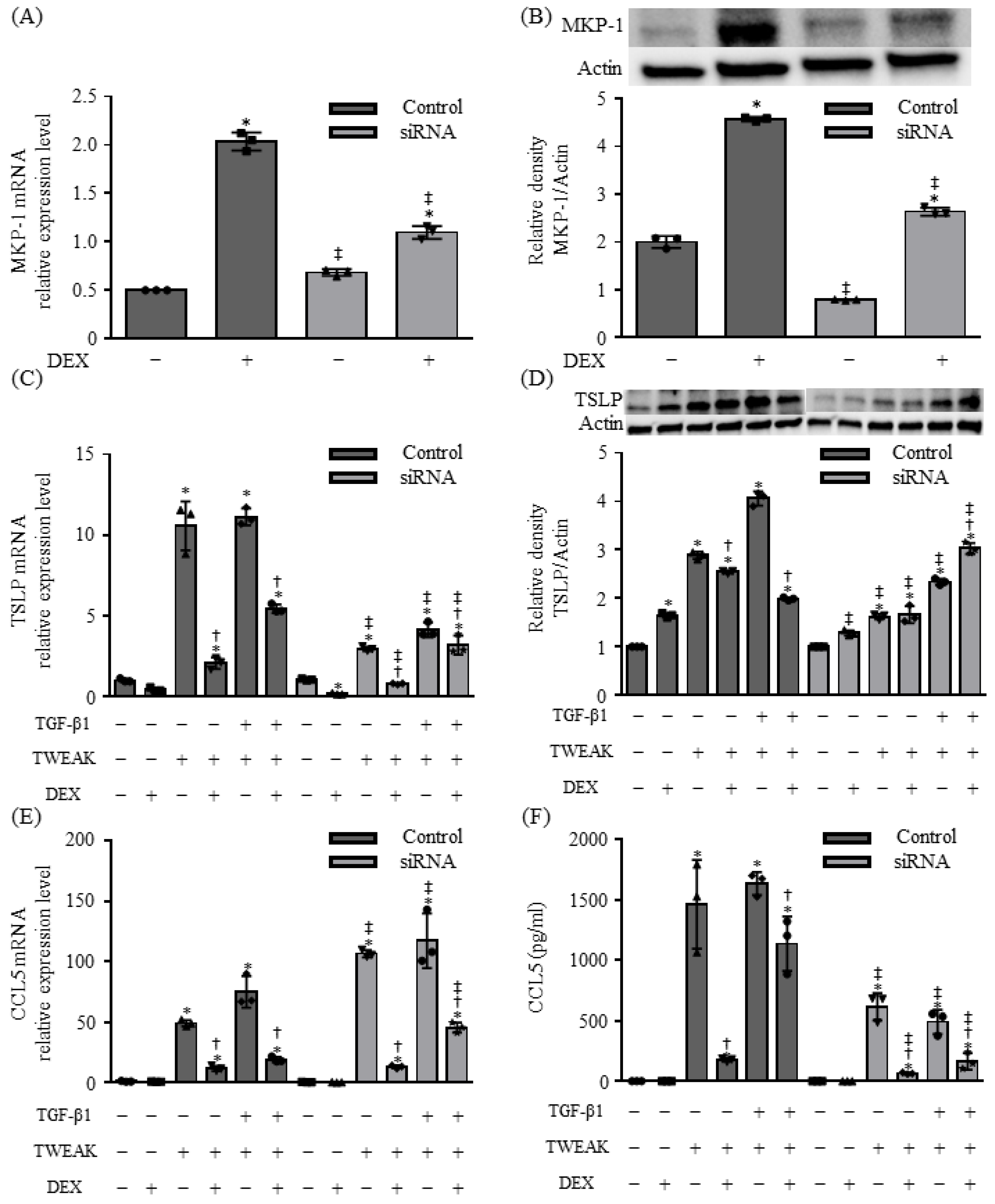
2.4. MKP-1 Is Involved in TSLP Production Induced by Co-Stimulation with TWEAK and TGF-β1 in BEAS-2B Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. RNA Isolation and Quantitative Real-Time Reverse Transcription–Polymerase Chain Reaction (qRT–PCR)
4.4. Preparation of Cell Lysates and Western Blotting
4.5. RNA Interference Assay
4.6. Measurement of Cytokine and Chemokine Levels
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ayroldi, E.; Riccardi, C. Glucocorticoid-induced leucine zipper (GILZ): A new important mediator of glucocorticoid action. FASEB J. 2009, 23, 3649–3658. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Glucocorticosteroids: Current and future directions. Br. J. Pharmacol. 2011, 163, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Rekers, N.V.; de Fijter, J.W.; Claas, F.H.; Eikmans, M. Mechanisms and risk assessment of steroid resistance in acute kidney transplant rejection. Transpl. Immunol. 2016, 38, 3–14. [Google Scholar] [CrossRef]
- Sevilla, L.M.; Jimenez-Panizo, A.; Alegre-Marti, A.; Estebanez-Perpina, E.; Caelles, C.; Perez, P. Glucocorticoid Resistance: Interference between the Glucocorticoid Receptor and the MAPK Signalling Pathways. Int. J. Mol. Sci. 2021, 22, 10049. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Kolls, J.K. Neutrophilic Inflammation in Asthma and Association with Disease Severity. Trends Immunol. 2017, 38, 942–954. [Google Scholar] [CrossRef]
- Manni, M.L.; Mandalapu, S.; McHugh, K.J.; Elloso, M.M.; Dudas, P.L.; Alcorn, J.F. Molecular Mechanisms of Airway Hyperresponsiveness in a Murine Model of Steroid-Resistant Airway Inflammation. J. Immunol. 2016, 196, 963–977. [Google Scholar] [CrossRef]
- McKinley, L.; Alcorn, J.F.; Peterson, A.; Dupont, R.B.; Kapadia, S.; Logar, A.; Henry, A.; Irvin, C.G.; Piganelli, J.D.; Ray, A.; et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J. Immunol. 2008, 181, 4089–4097. [Google Scholar] [CrossRef] [PubMed]
- Itoigawa, Y.; Harada, N.; Harada, S.; Katsura, Y.; Makino, F.; Ito, J.; Nurwidya, F.; Kato, M.; Takahashi, F.; Atsuta, R.; et al. TWEAK enhances TGF-beta-induced epithelial-mesenchymal transition in human bronchial epithelial cells. Respir. Res. 2015, 16, 48. [Google Scholar] [CrossRef]
- Matsuno, K.; Harada, N.; Harada, S.; Takeshige, T.; Ishimori, A.; Itoigawa, Y.; Henry, A.; Irvin, C.G.; Piganelli, J.D.; Ray, A.; et al. Combination of TWEAK and TGF-beta1 induces the production of TSLP, RANTES, and TARC in BEAS-2B human bronchial epithelial cells during epithelial-mesenchymal transition. Exp. Lung Res. 2018, 44, 332–343. [Google Scholar] [CrossRef]
- Takeshige, T.; Harada, N.; Harada, S.; Ishimori, A.; Katsura, Y.; Sasano, H.; Sandhu, Y.; Matsuno, K.; Makino, F.; Ito, J.; et al. Chitin induces steroid-resistant airway inflammation and airway hyperresponsiveness in mice. Allergol. Int. 2021, 70, 343–350. [Google Scholar] [CrossRef]
- Ijaz, T.; Pazdrak, K.; Kalita, M.; Konig, R.; Choudhary, S.; Tian, B.; Boldogh, I.; Brasier, A.R. Biology approaches to understanding Epithelial Mesenchymal Transition (EMT) in mucosal remodeling and signaling in asthma. World Allergy Organ. J. 2014, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, B.N.; Hammad, H. The airway epithelium in asthma. Nat. Med. 2012, 18, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R.; Roos, A.; Berg, T.; Nord, M.; Fuxe, J. Chronic respiratory aeroallergen exposure in mice induces epithelial-mesenchymal transition in the large airways. PLoS ONE 2011, 6, e16175. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, A.; Hosoki, K.; Toda, M.; Miyake, Y.; Matsushima, Y.; Matsumoto, T.; Boveda-Ruiz, D.; Gil-Bernabe, P.; Nagao, M.; Sugimoto, M.; et al. Eosinophils promote epithelial to mesenchymal transition of bronchial epithelial cells. PLoS ONE 2013, 8, e64281. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Puddicombe, S.M.; Polosa, R.; Richter, A.; Krishna, M.T.; Howarth, P.H.; Holgate, S.T.; Davies, D.E. Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J. 2000, 14, 1362–1374. [Google Scholar] [CrossRef]
- Burkly, L.C.; Michaelson, J.S.; Zheng, T.S. TWEAK/Fn14 pathway: An immunological switch for shaping tissue responses. Immunol. Rev. 2011, 244, 99–114. [Google Scholar] [CrossRef]
- Harada, N.; Nakayama, M.; Nakano, H.; Fukuchi, Y.; Yagita, H.; Okumura, K. Pro-inflammatory effect of TWEAK/Fn14 interaction on human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 2002, 299, 488–493. [Google Scholar] [CrossRef]
- Zhu, C.; Zhang, L.; Liu, Z.; Li, C.; Bai, Y. TWEAK/Fn14 interaction induces proliferation and migration in human airway smooth muscle cells via activating the NF-kappaB pathway. J. Cell. Biochem. 2018, 119, 3528–3536. [Google Scholar] [CrossRef] [PubMed]
- Soumelis, V.; Reche, P.A.; Kanzler, H.; Yuan, W.; Edward, G.; Homey, B.; Gilliet, M.; Ho, S.; Antonenko, S.; Lauerma, A.; et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat. Immunol. 2002, 3, 673–680. [Google Scholar] [CrossRef]
- Ziegler, S.F.; Artis, D. Sensing the outside world: TSLP regulates barrier immunity. Nat. Immunol. 2010, 11, 289–293. [Google Scholar] [CrossRef]
- Carr, T.F.; Berdnikovs, S.; Simon, H.U.; Bochner, B.S.; Rosenwasser, L.J. Eosinophilic bioactivities in severe asthma. World Allergy Organ. J. 2016, 9, 21. [Google Scholar] [CrossRef]
- Chvatchko, Y.; Proudfoot, A.E.; Buser, R.; Juillard, P.; Alouani, S.; Kosco-Vilbois, M.; Coyle, A.J.; Nibbs, R.J.; Graham, G.; Offorf, R.E.; et al. Inhibition of airway inflammation by amino-terminally modified RANTES/CC chemokine ligand 5 analogues is not mediated through CCR3. J. Immunol. 2003, 171, 5498–5506. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, J.D.; Sol, I.S.; Kim, M.J.; Kim, M.N.; Hong, J.Y.; Kim, H.R.; Kim, Y.H.; Lee, Y.J.; Kim, K.W.; et al. Sputum TWEAK expression correlates with severity and degree of control in non-eosinophilic childhood asthma. Pediatr. Allergy Immunol. 2018, 29, 42–49. [Google Scholar] [CrossRef]
- Cannarile, L.; Delfino, D.V.; Adorisio, S.; Riccardi, C.; Ayroldi, E. Implicating the Role of GILZ in Glucocorticoid Modulation of T-Cell Activation. Front. Immunol. 2019, 10, 1823. [Google Scholar] [CrossRef]
- Barnes, P.J. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2013, 131, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Li, L.B.; Leung, D.Y.; Martin, R.J.; Goleva, E. Inhibition of histone deacetylase 2 expression by elevated glucocorticoid receptor beta in steroid-resistant asthma. Am. J. Respir. Crit. Care Med. 2010, 182, 877–883. [Google Scholar] [CrossRef]
- Lehtola, T.; Nummenmaa, E.; Nieminen, R.; Hamalainen, M.; Vuolteenaho, K.; Moilanen, E. The glucocorticoid dexamethasone alleviates allergic inflammation through a mitogen-activated protein kinase phosphatase-1-dependent mechanism in mice. Basic Clin. Pharmacol. Toxicol. 2024, 134, 686–694. [Google Scholar] [CrossRef]
- Chen, T.; Guo, Z.P.; Li, M.M.; Li, J.Y.; Jiao, X.Y.; Zhang, Y.H.; Liu, H.J. Tumour necrosis factor-like weak inducer of apoptosis (TWEAK), an important mediator of endothelial inflammation, is associated with the pathogenesis of Henoch-Schonlein purpura. Clin. Exp. Immunol. 2011, 166, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.L.; McDonough, W.S.; Savitch, B.A.; Sawyer, T.F.; Winkles, J.A.; Berens, M.E. The tumor necrosis factor-like weak inducer of apoptosis (TWEAK)-fibroblast growth factor-inducible 14 (Fn14) signaling system regulates glioma cell survival via NFkappaB pathway activation and BCL-XL/BCL-W expression. J. Biol. Chem. 2005, 280, 3483–3492. [Google Scholar] [CrossRef] [PubMed]
- Vendrell, J.; Maymo-Masip, E.; Tinahones, F.; García-Espana, A.; Megia, A.; Caubet, E.; García-Fuentes, E.; Chacon, M.R. Tumor necrosis-like weak inducer of apoptosis as a proinflammatory cytokine in human adipocyte cells: Up-regulation in severe obesity is mediated by inflammation but not hypoxia. J. Clin. Endocrinol. Metab. 2010, 95, 2983–2992. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Okamoto, A.; Ichikawa, J.; Ando, T.; Tasaka, K.; Masuyama, K.; Ogawa, H.; Yagita, H.; Okumura, K.; Nakao, A. TWEAK/Fn14 interaction stimulates human bronchial epithelial cells to produce IL-8 and GM-CSF. Biochem. Biophys. Res. Commun. 2004, 318, 422–427. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Sense Primer (5′ 3′) | Antisense Primer (5′ 3′) |
|---|---|---|
| GILZ | CTTGGGAGAAGGCCGGAAG | CTGCTCTTGTCAGGGGTCTG |
| GRβ | GGCTATTCAAGCCCCAGCA | TTTGGGAGGTGGTCCTGTTG |
| HDAC2 | TCTGCTACTACTACGACGGTGATA | GTCATGCGGATTCTATGAGGCT |
| IL-8 | ACTGAGAGTGATTGAGAGTGGAC | AACCCTCTGCACCCAGTTTTC |
| CCL2 | CATGAAAGTCTCTGCCGCCC | GGGCATTGATTGCATCTGGCTG |
| MKP-1 | CCCTGAGTACTAGCGTCCCT | GGGCCACCCTGATCGTAGAG |
| CCL5 | AACCCAGCAGTCGTCCACAG | TTGTTCAGCCGGGAGTCATAC |
| CCL17 | CTTCAAGGGAGCCATTCCCC | TACAAAAACGATGGCATCCCTG |
| TSLP | CGCGTCGCTCGCCAAAGAAAT | TGAAGCGACGCCACAATCCTTG |
| GAPDH | GGTCTCCTCTGACTTCAACA | GTGAGGGTCTCTCTCTTCCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abe, S.; Harada, N.; Sandhu, Y.; Sasano, H.; Tanabe, Y.; Ueda, S.; Nishimaki, T.; Sato, Y.; Takeshige, T.; Harada, S.; et al. Co-Stimulation with TWEAK and TGF-β1 Induces Steroid-Insensitive TSLP and CCL5 Production in BEAS-2B Human Bronchial Epithelial Cells. Int. J. Mol. Sci. 2024, 25, 11625. https://doi.org/10.3390/ijms252111625
Abe S, Harada N, Sandhu Y, Sasano H, Tanabe Y, Ueda S, Nishimaki T, Sato Y, Takeshige T, Harada S, et al. Co-Stimulation with TWEAK and TGF-β1 Induces Steroid-Insensitive TSLP and CCL5 Production in BEAS-2B Human Bronchial Epithelial Cells. International Journal of Molecular Sciences. 2024; 25(21):11625. https://doi.org/10.3390/ijms252111625
Chicago/Turabian StyleAbe, Sumiko, Norihiro Harada, Yuuki Sandhu, Hitoshi Sasano, Yuki Tanabe, Shoko Ueda, Takayasu Nishimaki, Yoshihiko Sato, Tomohito Takeshige, Sonoko Harada, and et al. 2024. "Co-Stimulation with TWEAK and TGF-β1 Induces Steroid-Insensitive TSLP and CCL5 Production in BEAS-2B Human Bronchial Epithelial Cells" International Journal of Molecular Sciences 25, no. 21: 11625. https://doi.org/10.3390/ijms252111625
APA StyleAbe, S., Harada, N., Sandhu, Y., Sasano, H., Tanabe, Y., Ueda, S., Nishimaki, T., Sato, Y., Takeshige, T., Harada, S., Akiba, H., & Takahashi, K. (2024). Co-Stimulation with TWEAK and TGF-β1 Induces Steroid-Insensitive TSLP and CCL5 Production in BEAS-2B Human Bronchial Epithelial Cells. International Journal of Molecular Sciences, 25(21), 11625. https://doi.org/10.3390/ijms252111625