
Altered Steroidome in Women with Multiple Sclerosis


 , , ,
, , ,
Abstract
1. Introduction
1.1. Multiple Sclerosis and Steroids
1.1.1. Cortisol
1.1.2. Δ5 Steroids
1.1.3. Active Androgens
1.1.4. Estradiol
1.1.5. Progesterone and Its Metabolites
1.1.6. Previous Multi-Steroid Studies Focused on Circulating Steroids in Multiple Sclerosis
2. Results
2.1. Alterations in Steroid Levels and Their Correlations with the Severity of MS
2.1.1. Corticoids (C21 Δ4 Steroids) and 11β-Hydroxy-androstanes (C19 Δ4 and 5α/β Steroids) and Their Correlations with the Grade of MS
2.1.2. Δ5 and Δ4 Steroids and Their Correlations with the Grade of MS
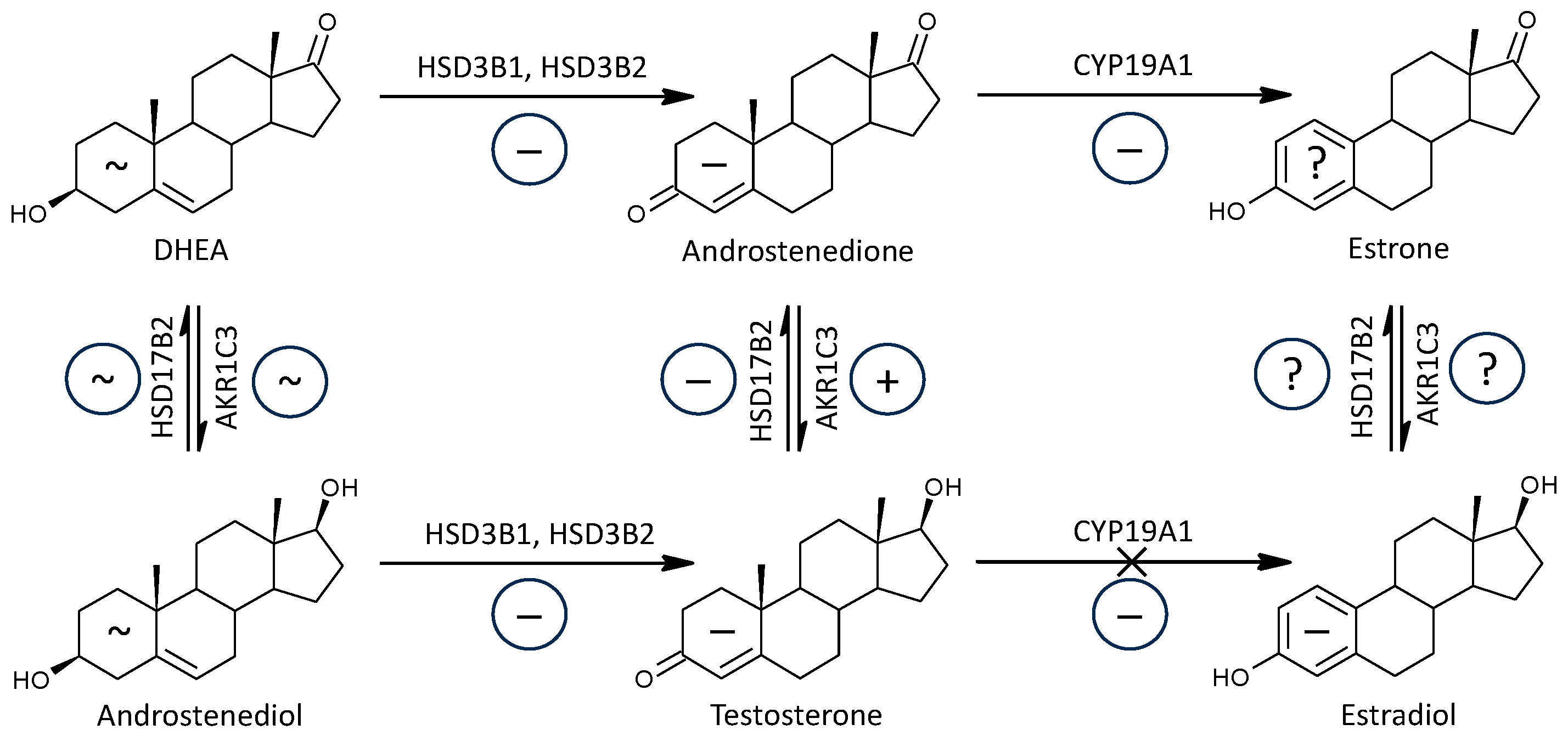
2.1.3. Active Androgens and Androstenedione
2.1.4. Estradiol and Its Precursors
2.1.5. Progesterone and Its Metabolites
2.2. Alterations in Steroid Molar Ratios
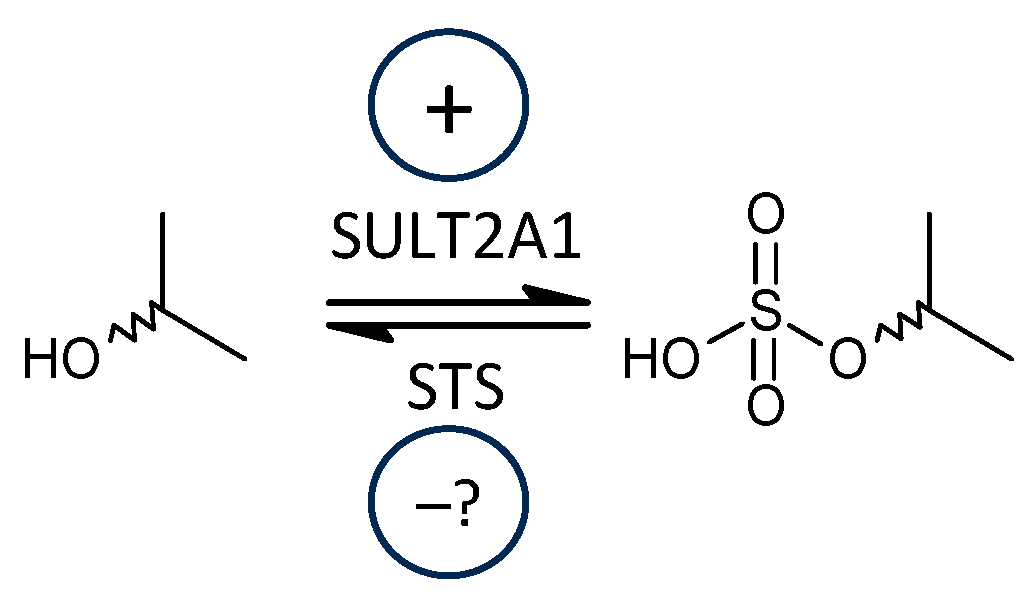
2.2.1. Steroid Sulfotransferase 2A1 (SULT2A1) vs. Steroid Sulfatase (STS)
2.2.2. C17-Hydroxylase, C17-20-Lyase (CYP17A1), Hydroxylase + Lyase Steps
2.2.3. C17-Hydroxylase, C17-20-Lyase (CYP17A1), Hydroxylase Step
2.2.4. C17-Hydroxylase, C17-20-Lyase (CYP17A1), Lyase Step
2.2.5. 3β-Hydroxysteroid Dehydrogenases (HSD3B1 and 2)
2.2.6. 11β-Hydroxylase (CYP11B1)
2.2.7. 11β-Hydroxysteroid Dehydrogenase, Type 1 (HSD11B1)
2.2.8. 7α-, 7β-, and 16α-Hydroxylating Enzymes (CYP7B1, CYP3A4, CYP3A7)
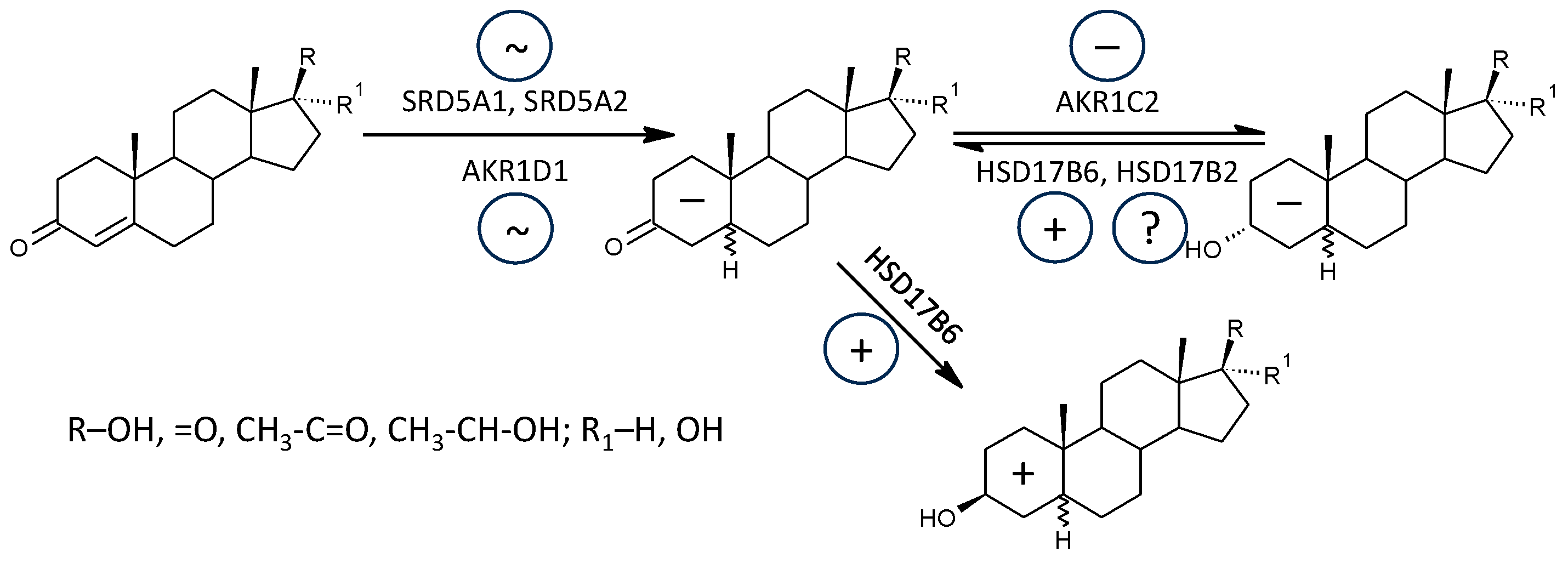
2.2.9. 5α-Reductase (SRD5A1, SRD5A2)
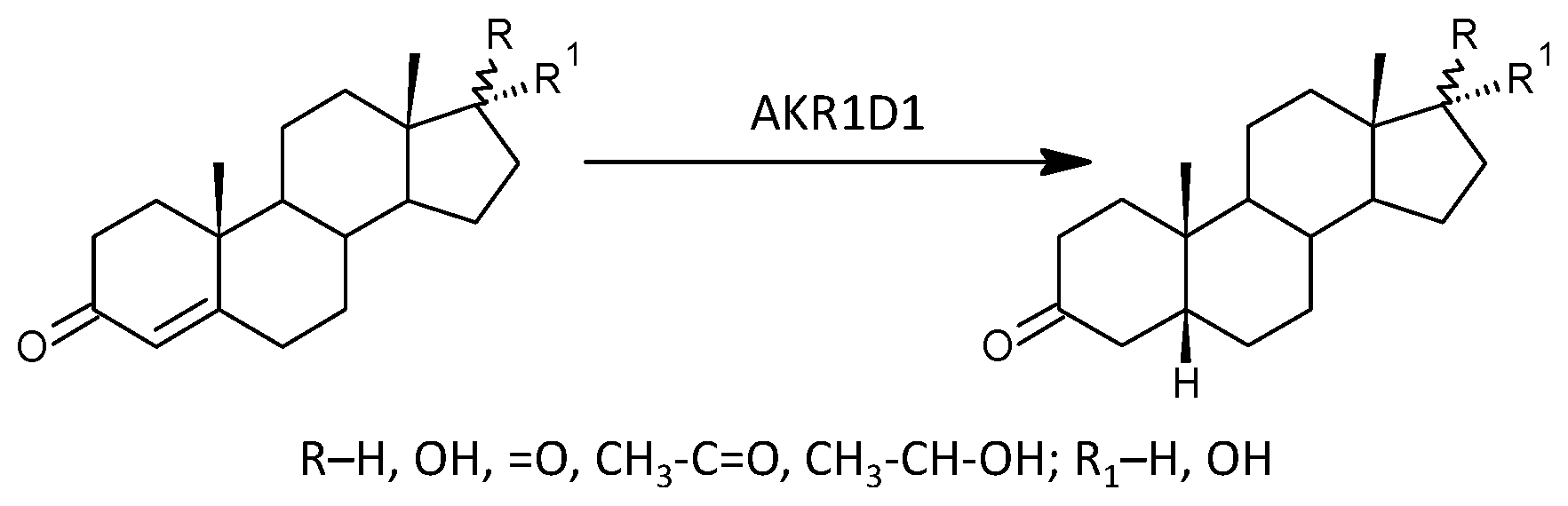
2.2.10. 5β-Reductase (AKR1D1)
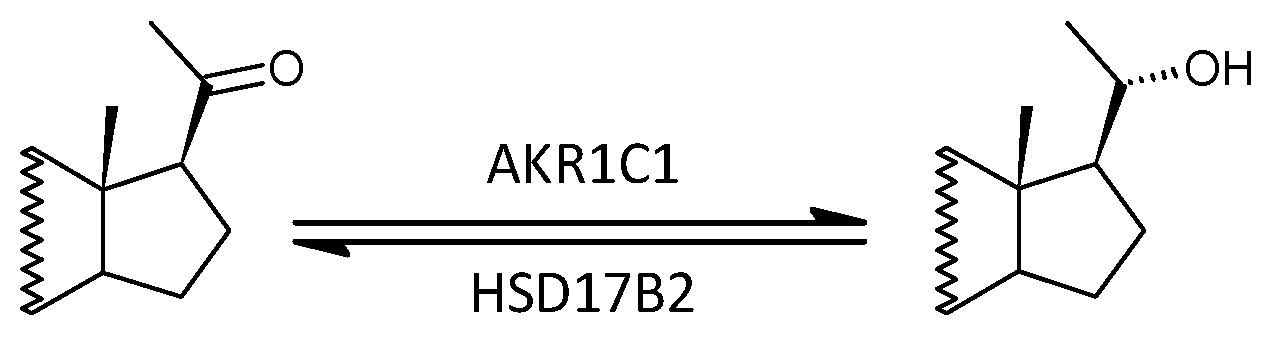
2.2.11. Aldoketoreductase 1C1 (AKR1C1) vs. 17β-Hydroxysteroid Dehydrogenase, Type 2 (HSD17B2)
2.2.12. Aldoketoreductase 1C2 (AKR1C2) vs. 17β-Hydroxysteroid dehydrogenase, Type 2 and 6 (HSD17B2, HSD17B6)
2.2.13. Aldoketoreductase 1C3 (AKR1C3) vs. 17β-Hydroxysteroid Dehydrogenase, Type 2 (HSD17B2)
3. Discussion
3.1. Altered Steroid Levels in Patients and Their Correlations with the Severity of MS
3.1.1. Corticoids (C21 Δ4 Steroids) and 11β-Hydroxy-androgens (C19 Δ4 and 5α/β Steroids)
3.1.2. Δ5 and Δ4 Steroids
3.1.3. Active Androgens and Androstenedione
3.1.4. Estrogens, Their Precursors, and Aromatase Functioning
3.1.5. Progesterone and Its Metabolites
3.2. Altered Steroid Molar Ratios in Patients
3.2.1. Steroid Sulfotransferase 2A1 (SULT2A1) vs. Steroid Sulfatase (STS)
3.2.2. C17-Hydroxylase, C17-20-Lyase (CYP17A1), Hydroxylase + Lyase Steps, and the Pathway to Cortisol Synthesis
3.2.3. C17-Hydroxylase, C17-20-Lyase (CYP17A1), Hydroxylase Step, and the Pathway to Cortisol Synthesis
3.2.4. C17-Hydroxylase, C17-20-Lyase (CYP17A1), Hydroxylase Step, and the Pathway to Cortisol Synthesis
3.2.5. 3β-Hydroxysteroid Dehydrogenases (HSD3B1 and 2)
3.2.6. 11β-Hydroxylase (CYP11B1)
3.2.7. 11β-Hydroxysteroid Dehydrogenase, Type 1 (HSD11B1)
3.2.8. 7α-, 7β-, and 16α-Hydroxylating Enzymes (CYP7B1, CYP3A4, CYP3A7)
3.2.9. 5α-Reductases (SRD5A1, SRD5A2)
3.2.10. 5β-Reductase (AKR1D1)
3.2.11. Aldoketoreductase 1C1 (AKR1C1) vs. 17β-Hydroxysteroid Dehydrogenase, Type 2 (HSD17B2)
3.2.12. Aldoketoreductase 1C2 (AKR1C2) vs. 17β-Hydroxysteroid Dehydrogenase, Type 2 and 6 (HSD17B2, HSD17B6)
3.2.13. Aldoketoreductase 1C3 (AKR1C3) vs. 17β-Hydroxysteroid Dehydrogenase, Type 2 (HSD17B2)
3.2.14. Potential Clinical Implications of the Findings
3.2.15. Future Directions
3.2.16. Limitations of the Study
4. Materials and Methods
4.1. Subjects
4.2. Steroid Analysis
4.3. Statistical Analysis
- Transformation of the original data to obtain the values with symmetric distribution and constant variance
- Checking the data homogeneity in predictors using Hotelling’s statistics and the eventual elimination of non-homogeneities
- Testing the relevance of predictors using variable importance statistics and the elimination of irrelevant predictors
- Calculating component loadings for individual variables to evaluate their correlations with the predictive component
- Calculating regression coefficients for the multiple regression model to evaluate the mutual independence of predictors after comparison with the corresponding component loadings from the OPLS model
- Calculating predicted values of the logarithm of the ratio of the probability of pathology presence to the probability of pathology absence (LLR)
- Calculating the probability of the pathology’s presence for individual subjects
- Calculating the sensitivity and specificity of the prediction
5. Conclusions
- (1)
- A comprehensive steroidomic analysis was performed in female MS patients compared to female age-matched controls. The MS patients included in this study were newly diagnosed (met the 2017 revised McDonald criteria) and had not yet been treated.
- (2)
- Associations between steroidomic data and indices of MS severity were evaluated.
- (3)
- Most steroids have been studied for the first time in terms of MS.
- (4)
- The results focused on differences between steroidomic data in MS patients and untreated controls, and the results focused on relationships between steroidomic data and MS severity, which were mostly discordant with a tendency to converge to the situation in controls with increasing severity of MS, which was interpreted (in the light of data from the literature) as an intensification of counter-regulatory mechanisms preventing the development of MS with increasing severity of the disease.
- (5)
- A significant trend towards higher ratios of conjugated steroids to their unconjugated counterparts was found in patients, indicating increased SULT2A1 sulfotransferase functioning, which is of particular interest in terms of the balance between excitatory and inhibitory steroid modulators of ionotropic receptors.
- (6)
- An altered metabolic pathway to cortisol was found in patients with decreased conversion of pregnenolone to 17-hydroxypregnenolone and 17-hydroxypregnenolone to 17-hydroxyprogesterone and increased conversion of 17-hydroxypregnenolone to DHEA, resulting in lower levels of 17-hydroxyprogesterone, as well as indications of impaired conversion of 11-deoxy-steroids to 11β-hydroxy-steroids and, finally, reduced conversion of the active glucocorticoid cortisol to its inactive metabolite cortisone.
- (7)
- Despite these metabolic barriers, both cortisol and cortisone levels were higher in patients, and therefore alterations in molar ratios in the cortisol pathway could be explained by depletion of enzymes involved in cortisol synthesis due to overactivation of HPAA, which has been described in MS patients.
- (8)
- Patients showed altered metabolic pathways to both the active androgen testosterone and the active estrogen estradiol, with decreased conversion of DHEA to androstenedione and androstenedione to testosterone, increased conversion of androstenedione to testosterone, and decreased conversion of androstenedione (via unmeasured estrone) to estradiol in the major pathway and testosterone to estradiol in the minor pathway of estradiol synthesis.
- (9)
- Reduced conversion of immunoprotective Δ5 androstanes to their more potent 7α/β-hydroxy metabolites was found in patients.
- (10)
- Patients showed lower levels of neuroprotective allopregnanolone compared to controls, as well as a higher ratio of antagonistic 3β-hydroxysteroids to their GABAergic neuroprotective 3α-hydroxy counterparts.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 11β-OH-3α,5α-THA | 11β-Hydroxyandrosterone |
| 11β-hydroxyandrostenedione | 11β-OH-A |
| 16α-OH-P | 16α-Hydroxyprogesterone |
| 16α-OH-Preg | 16α-Hydroxypregnenolone |
| 17-OH-P | 17-Hydroxyprogesterone |
| 17-OH-Preg | 17-Hydroxypregnenolone |
| 17-OH-20α-DHP | 17α-Hydroxy-20α-dihydroprogesterone |
| 5α-DHA | 5α-Androstane-3,17-dione |
| 5α-DHT | 5α-Dihydrotestosterone |
| 3α,5α,17-PD | 17-Hydroxyallopregnanolone |
| 3α,5α,17,20α-PT | 5α-Pregnane-3α,17,20α-triol |
| 3α,5α,20α-PD | 5α-Pregnane-3α, 20α-diol |
| 3α,5α-THA | Androsterone |
| 3α,5α-THP | 3α,5α-Tetrahydroprogesterone, allopregnanolone |
| 3β,5α,17,20α-PT | 5α-Pregnane-3β,17,20α-triol |
| 3β,5α-THA | Epiandrosterone |
| 3α,5β,17,20α-PT | 5β-Pregnane-3α,17,20α-triol |
| 3α,5β,17-PD | 17-Hydroxypregnanolone |
| 3α,5β-THA | Etiocholanolone |
| 3α,5β-THA | Epietiocholanolone |
| 8-iso-PGF2α | 8-iso-Prostaglandin F2α |
| σ1Rs | Sigma-1 receptors |
| A | Androstenedione |
| ACTH | Adrenocorticotropic hormone |
| AD | Androstanediol |
| ADIOL | Androstenediol |
| AKR1C1 | Aldo-keto reductase family 1 member C1 |
| AKR1C2 | Aldo-keto reductase family 1 member C2 |
| AKR1C3 | Aldo-keto reductase family 1 member C3 |
| AKR1D1 | 5β-Reductase |
| AMPARs | α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors |
| ANOVA | Analysis of variance |
| AT | Androstenetriol (5-androstene) |
| BBB | Blood-brain barrier |
| CI | Confidence interval |
| CNS | Central nervous system |
| CRH | Corticotropin-releasing hormone |
| CYP11A1 | Cholesterol desmolase |
| CYP11B1 | 11β-Hydroxylase |
| CYP17A1 | C17-hydroxylase-C17,20-lyase |
| CYP19A1 | Aromatase |
| CYP21A2 | 21-Hydroxylase |
| CYP3A4 | Cytochrome P450 3A4 |
| CYP3A7 | Cytochrome P450 3A7 |
| CYP7B1 | 7α-hydroxylase |
| DHEA | Dehydroepiandrosterone |
| DHEAS | Dehydroepiandrosterone sulfate |
| DHP | Dihydroprogesterone |
| EDSS | Expanded Disability Status Scale |
| ERα | Estrogen receptors α |
| ERβ | Estrogen receptors β |
| FSH | Follicle-stimulating hormone |
| GABA | γ-Aminobutyric acid |
| GABAARs | Type A γ-aminobutyric acid receptors |
| GR | Glucocorticoid receptors |
| GC-MS/MS | Gas chromatography tandem mass spectrometry |
| GPER | G protein-coupled estrogen receptors |
| HPAA | Hypothalamic-pituitary-adrenal axis |
| HSD3B1 | Type 1 3β-Hydroxysteroid dehydrogenase |
| HSD3B2 | Type 2 3β-Hydroxysteroid dehydrogenase |
| HSD3Bs | Type 1 and 2 3β-Hydroxysteroid dehydrogenases |
| HSD11B1 | Type 1 11β-Hydroxysteroid dehydrogenase |
| HSD11B2 | Type 2 11β-Hydroxysteroid dehydrogenase |
| HSD17B2 | Type 2 17β-Hydroxysteroid dehydrogenase |
| HSD17B3 | Type 3 17β-Hydroxysteroid dehydrogenase |
| HSD17B6 | Type 6 17β-Hydroxysteroid dehydrogenase (3α/β epimerase) |
| HPT9_R | 9-Hole Peg Test for right hand |
| HPT9_L | 9-Hole Peg Test for left hand |
| IFN-γ | Interferon γ |
| IL-1 | Interleukin 1 |
| IL-2 | Interleukin 2 |
| IL-4 | Interleukin 4 |
| IL-6 | Interleukin 6 |
| IL-10 | Interleukin 10 |
| IL-12 | Interleukin 12 |
| IL-17 | Interleukin 17 |
| LLR | Logarithm of likelihood ratio |
| mRNA | Messenger ribonucleic acid |
| NASs | Neuroactive steroids |
| NMDA | N-methyl-D-aspartate |
| NMDARs | N-methyl-D-aspartate receptors |
| OPLS | Orthogonal predictions to latent structure |
| P | Progesterone |
| PD | Pregnanediol |
| PGF2α | Prostaglandin F2α |
| PNS | Peripheral nervous system |
| Preg | Pregnenolone |
| PregS | Pregnenolone sulfate |
| PT | Pregnanetriol |
| ROS | Reactive oxygen species |
| SRD5A1 | Type 1 5α-reductase |
| SRD5A2 | Type 2 5α-reductase |
| SULT2A1 | Steroid sulfotransferase type 2A1 |
| STS | Steroid sulfatase |
| T | Testosterone |
| T25FWT | Timed 25-foot walk |
| THP | Tetrahydroprogesterone |
| TNF-α | Tumor necrosis factor α |
| TRPC5s | Short transient receptor potential channels 5 |
| TRPV1s | Vanilloid receptors |
| TRPM3s | Melastatin receptors |
| The letter C after the abbreviation of a steroid indicates its conjugated form | |
| The “, C” after the molar ratio of a steroid indicates that they are its conjugated forms. | |
References
- Ysrraelit, M.C.; Correale, J. Impact of sex hormones on immune function and multiple sclerosis development. Immunology 2019, 156, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Sparaco, M.; Bonavita, S. The role of sex hormones in women with multiple sclerosis: From puberty to assisted reproductive techniques. Front. Neuroendocrinol. 2021, 60, 100889. [Google Scholar] [CrossRef] [PubMed]
- Ngo, S.T.; Steyn, F.J.; McCombe, P.A. Gender differences in autoimmune disease. Front. Neuroendocrinol. 2014, 35, 347–369. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.; Studd, J.W. A pilot study of the effect upon multiple sclerosis of the menopause, hormone replacement therapy and the menstrual cycle. J. R. Soc. Med. 1992, 85, 612–613. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Makris, N. Multiple sclerosis and reproductive risks in women. Reprod. Sci. 2008, 15, 755–764. [Google Scholar] [CrossRef]
- Kamin, H.S.; Kertes, D.A. Cortisol and DHEA in development and psychopathology. Horm. Behav. 2017, 89, 69–85. [Google Scholar] [CrossRef]
- Groeneweg, F.L.; Karst, H.; de Kloet, E.R.; Joels, M. Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of nongenomic corticosteroid signalling. Mol. Cell. Endocrinol. 2012, 350, 299–309. [Google Scholar] [CrossRef]
- Mikulska, J.; Juszczyk, G.; Gawronska-Grzywacz, M.; Herbet, M. HPA Axis in the Pathomechanism of Depression and Schizophrenia: New Therapeutic Strategies Based on Its Participation. Brain Sci. 2021, 11, 1298. [Google Scholar] [CrossRef]
- Cherian, K.; Schatzberg, A.F.; Keller, J. HPA axis in psychotic major depression and schizophrenia spectrum disorders: Cortisol, clinical symptomatology, and cognition. Schizophr. Res. 2019, 213, 72–79. [Google Scholar] [CrossRef]
- Misiak, B.; Piotrowski, P.; Chec, M.; Samochowiec, J. Cortisol and dehydroepiandrosterone sulfate in patients with schizophrenia spectrum disorders with respect to cognitive performance. Compr. Psychoneuroendocrinol. 2021, 6, 100041. [Google Scholar] [CrossRef]
- Ritsner, M.; Maayan, R.; Gibel, A.; Strous, R.D.; Modai, I.; Weizman, A. Elevation of the cortisol/dehydroepiandrosterone ratio in schizophrenia patients. Eur. Neuropsychopharmacol. 2004, 14, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Ritsner, M.; Gibel, A.; Maayan, R.; Ratner, Y.; Ram, E.; Modai, I.; Weizman, A. State and trait related predictors of serum cortisol to DHEA(S) molar ratios and hormone concentrations in schizophrenia patients. Eur. Neuropsychopharmacol. 2007, 17, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Begemann, M.J.; Dekker, C.F.; van Lunenburg, M.; Sommer, I.E. Estrogen augmentation in schizophrenia: A quantitative review of current evidence. Schizophr. Res. 2012, 141, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Qaiser, M.Z.; Dolman, D.E.M.; Begley, D.J.; Abbott, N.J.; Cazacu-Davidescu, M.; Corol, D.I.; Fry, J.P. Uptake and metabolism of sulphated steroids by the blood-brain barrier in the adult male rat. J. Neurochem. 2017, 142, 672–685. [Google Scholar] [CrossRef]
- Cai, H.; Cao, T.; Zhou, X.; Yao, J.K. Neurosteroids in Schizophrenia: Pathogenic and Therapeutic Implications. Front. Psychiatry 2018, 9, 73. [Google Scholar] [CrossRef]
- Powrie, Y.S.L.; Smith, C. Central intracrine DHEA synthesis in ageing-related neuroinflammation and neurodegeneration: Therapeutic potential? J. Neuroinflammation 2018, 15, 289. [Google Scholar] [CrossRef]
- Honcu, P.; Hill, M.; Bicikova, M.; Jandova, D.; Velikova, M.; Kajzar, J.; Kolatorova, L.; Bestak, J.; Macova, L.; Kancheva, R.; et al. Activation of Adrenal Steroidogenesis and an Improvement of Mood Balance in Postmenopausal Females after Spa Treatment Based on Physical Activity. Int. J. Mol. Sci. 2019, 20, 3687. [Google Scholar] [CrossRef]
- MacKenzie, E.M.; Odontiadis, J.; Le Melledo, J.M.; Prior, T.I.; Baker, G.B. The relevance of neuroactive steroids in schizophrenia, depression, and anxiety disorders. Cell. Mol. Neurobiol. 2007, 27, 541–574. [Google Scholar] [CrossRef]
- Luu-The, V. Assessment of steroidogenesis and steroidogenic enzyme functions. J. Steroid Biochem. Mol. Biol. 2013, 137, 176–182. [Google Scholar] [CrossRef]
- Labrie, F. All sex steroids are made intracellularly in peripheral tissues by the mechanisms of intracrinology after menopause. J. Steroid Biochem. Mol. Biol. 2015, 145, 133–138. [Google Scholar] [CrossRef]
- Tomassini, V.; Pozzilli, C. Sex hormones, brain damage and clinical course of Multiple Sclerosis. J. Neurol. Sci. 2009, 286, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Foroughipour, A.; Norbakhsh, V.; Najafabadi, S.H.; Meamar, R. Evaluating sex hormone levels in reproductive age women with multiple sclerosis and their relationship with disease severity. J. Res. Med. Sci. 2012, 17, 882–885. [Google Scholar] [PubMed]
- Kanceva, R.; Starka, L.; Kancheva, L.; Hill, M.; Velikova, M.; Havrdova, E. Increased serum levels of C21 steroids in female patients with multiple sclerosis. Physiol. Res. 2015, 64 (Suppl. 2), S247–S254. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.; Parizek, A.; Kancheva, R.; Duskova, M.; Velikova, M.; Kriz, L.; Klimkova, M.; Paskova, A.; Zizka, Z.; Matucha, P.; et al. Steroid metabolome in plasma from the umbilical artery, umbilical vein, maternal cubital vein and in amniotic fluid in normal and preterm labor. J. Steroid Biochem. Mol. Biol. 2010, 121, 594–610. [Google Scholar] [CrossRef] [PubMed]
- Caruso, D.; Melis, M.; Fenu, G.; Giatti, S.; Romano, S.; Grimoldi, M.; Crippa, D.; Marrosu, M.G.; Cavaletti, G.; Melcangi, R.C. Neuroactive steroid levels in plasma and cerebrospinal fluid of male multiple sclerosis patients. J. Neurochem. 2014, 130, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Labrie, F.; Martel, C.; Belanger, A.; Pelletier, G. Androgens in women are essentially made from DHEA in each peripheral tissue according to intracrinology. J. Steroid Biochem. Mol. Biol. 2017, 168, 9–18. [Google Scholar] [CrossRef]
- Angeli, A.; Masera, R.G.; Sartori, M.L.; Fortunati, N.; Racca, S.; Dovio, A.; Staurenghi, A.; Frairia, R. Modulation by cytokines of glucocorticoid action. Ann. N. Y. Acad. Sci. 1999, 876, 210–220. [Google Scholar] [CrossRef]
- de Kloet, E.R.; Joels, M.; Holsboer, F. Stress and the brain: From adaptation to disease. Nat. Rev. Neurosci. 2005, 6, 463–475. [Google Scholar] [CrossRef]
- Hildebrandt, H.; Stachowiak, R.; Heber, I.; Schlake, H.P.; Eling, P. Relation between cognitive fatigue and circadian or stress related cortisol levels in MS patients. Mult. Scler. Relat. Disord. 2020, 45, 102440. [Google Scholar] [CrossRef]
- Slominski, R.M.; Tuckey, R.C.; Manna, P.R.; Jetten, A.M.; Postlethwaite, A.; Raman, C.; Slominski, A.T. Extra-adrenal glucocorticoid biosynthesis: Implications for autoimmune and inflammatory disorders. Genes Immun. 2020, 21, 150–168. [Google Scholar] [CrossRef]
- Tucha, L.; Fuermaier, A.B.; Koerts, J.; Buggenthin, R.; Aschenbrenner, S.; Weisbrod, M.; Thome, J.; Lange, K.W.; Tucha, O. Sustained attention in adult ADHD: Time-on-task effects of various measures of attention. J. Neural Transm. 2017, 124 (Suppl. S1), 39–53. [Google Scholar] [CrossRef] [PubMed]
- Vedhara, K.; Hyde, J.; Gilchrist, I.D.; Tytherleigh, M.; Plummer, S. Acute stress, memory, attention and cortisol. Psychoneuroendocrinology 2000, 25, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Padgett, D.A.; Glaser, R. How stress influences the immune response. Trends Immunol. 2003, 24, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.M.; Soares, N.M.; Souza, A.R.; Becker, J.; Finkelsztejn, A.; Almeida, R.M.M. Basal cortisol levels and the relationship with clinical symptoms in multiple sclerosis: A systematic review. Arq. Neuropsiquiatr. 2018, 76, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Heidbrink, C.; Hausler, S.F.; Buttmann, M.; Ossadnik, M.; Strik, H.M.; Keller, A.; Buck, D.; Verbraak, E.; van Meurs, M.; Krockenberger, M.; et al. Reduced cortisol levels in cerebrospinal fluid and differential distribution of 11beta-hydroxysteroid dehydrogenases in multiple sclerosis: Implications for lesion pathogenesis. Brain Behav. Immun. 2010, 24, 975–984. [Google Scholar] [CrossRef]
- Tomassini, V.; Onesti, E.; Mainero, C.; Giugni, E.; Paolillo, A.; Salvetti, M.; Nicoletti, F.; Pozzilli, C. Sex hormones modulate brain damage in multiple sclerosis: MRI evidence. J. Neurol. Neurosurg. Psychiatry 2005, 76, 272–275. [Google Scholar] [CrossRef]
- Grinsted, L.; Heltberg, A.; Hagen, C.; Djursing, H. Serum sex hormone and gonadotropin concentrations in premenopausal women with multiple sclerosis. J. Intern. Med. 1989, 226, 241–244. [Google Scholar] [CrossRef]
- Wei, T.; Lightman, S.L. The neuroendocrine axis in patients with multiple sclerosis. Brain 1997, 120 Pt 6, 1067–1076. [Google Scholar] [CrossRef]
- Bottasso, O.; Bay, M.L.; Besedovsky, H.; del Rey, A. The immuno-endocrine component in the pathogenesis of tuberculosis. Scand. J. Immunol. 2007, 66, 166–175. [Google Scholar] [CrossRef]
- Du, C.; Khalil, M.W.; Sriram, S. Administration of dehydroepiandrosterone suppresses experimental allergic encephalomyelitis in SJL/J mice. J. Immunol. 2001, 167, 7094–7101. [Google Scholar] [CrossRef]
- Rontzsch, A.; Thoss, K.; Petrow, P.K.; Henzgen, S.; Brauer, R. Amelioration of murine antigen-induced arthritis by dehydroepiandrosterone (DHEA). Inflamm. Res. 2004, 53, 189–198. [Google Scholar] [PubMed]
- Tan, X.D.; Dou, Y.C.; Shi, C.W.; Duan, R.S.; Sun, R.P. Administration of dehydroepiandrosterone ameliorates experimental autoimmune neuritis in Lewis rats. J. Neuroimmunol. 2009, 207, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.S.; Cui, Y.; Koh, Y.A.; Lee, H.C.; Cho, Y.B.; Won, Y.H. Effects of dehydroepiandrosterone on Th2 cytokine production in peripheral blood mononuclear cells from asthmatics. Korean J. Intern. Med. 2008, 23, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Sudo, N.; Yu, X.N.; Kubo, C. Dehydroepiandrosterone attenuates the spontaneous elevation of serum IgE level in NC/Nga mice. Immunol. Lett. 2001, 79, 177–179. [Google Scholar] [CrossRef]
- Noorbakhsh, F.; Ellestad, K.K.; Maingat, F.; Warren, K.G.; Han, M.H.; Steinman, L.; Baker, G.B.; Power, C. Impaired neurosteroid synthesis in multiple sclerosis. Brain 2011, 134 Pt 9, 2703–2721. [Google Scholar] [CrossRef]
- Gubbels Bupp, M.R.; Jorgensen, T.N. Androgen-Induced Immunosuppression. Front. Immunol. 2018, 9, 794. [Google Scholar] [CrossRef]
- Matuszewska, A.; Kowalski, K.; Jawien, P.; Tomkalski, T.; Gawel-Dabrowska, D.; Merwid-Lad, A.; Szelag, E.; Blaszczak, K.; Wiatrak, B.; Danielewski, M.; et al. The Hypothalamic-Pituitary-Gonadal Axis in Men with Schizophrenia. Int. J. Mol. Sci. 2023, 24, 6492. [Google Scholar] [CrossRef]
- Garcia-Estrada, J.; Del Rio, J.A.; Luquin, S.; Soriano, E.; Garcia-Segura, L.M. Gonadal hormones down-regulate reactive gliosis and astrocyte proliferation after a penetrating brain injury. Brain Res. 1993, 628, 271–278. [Google Scholar] [CrossRef]
- Bovolenta, P.; Wandosell, F.; Nieto-Sampedro, M. CNS glial scar tissue: A source of molecules which inhibit central neurite outgrowth. Prog. Brain Res. 1992, 94, 367–379. [Google Scholar]
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef]
- Dalal, M.; Kim, S.; Voskuhl, R.R. Testosterone therapy ameliorates experimental autoimmune encephalomyelitis and induces a T helper 2 bias in the autoantigen-specific T lymphocyte response. J. Immunol. 1997, 159, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Caruso, A.; Di Giorgi Gerevini, V.; Castiglione, M.; Marinelli, F.; Tomassini, V.; Pozzilli, C.; Caricasole, A.; Bruno, V.; Caciagli, F.; Moretti, A.; et al. Testosterone amplifies excitotoxic damage of cultured oligodendrocytes. J. Neurochem. 2004, 88, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Gogos, A.; Sbisa, A.M.; Sun, J.; Gibbons, A.; Udawela, M.; Dean, B. A Role for Estrogen in Schizophrenia: Clinical and Preclinical Findings. Int. J. Endocrinol. 2015, 2015, 615356. [Google Scholar] [CrossRef] [PubMed]
- McGregor, C.; Riordan, A.; Thornton, J. Estrogens and the cognitive symptoms of schizophrenia: Possible neuroprotective mechanisms. Front. Neuroendocr. 2017, 47, 19–33. [Google Scholar] [CrossRef]
- Rao, M.L.; Kolsch, H. Effects of estrogen on brain development and neuroprotection--implications for negative symptoms in schizophrenia. Psychoneuroendocrinology 2003, 28 (Suppl. 2), 83–96. [Google Scholar] [CrossRef]
- Melnikov, M.; Pashenkov, M.; Boyko, A. Dopaminergic Receptor Targeting in Multiple Sclerosis: Is There Therapeutic Potential? Int. J. Mol. Sci. 2021, 22, 5313. [Google Scholar] [CrossRef]
- Trenova, A.G.; Slavov, G.S.; Manova, M.G.; Kostadinova, I.I.; Vasileva, T.V. Female sex hormones and cytokine secretion in women with multiple sclerosis. Neurol. Res. 2013, 35, 95–99. [Google Scholar] [CrossRef]
- Ghoumari, A.M.; Ibanez, C.; El-Etr, M.; Leclerc, P.; Eychenne, B.; O’Malley, B.W.; Baulieu, E.E.; Schumacher, M. Progesterone and its metabolites increase myelin basic protein expression in organotypic slice cultures of rat cerebellum. J. Neurochem. 2003, 86, 848–859. [Google Scholar] [CrossRef]
- Bodhankar, S.; Wang, C.; Vandenbark, A.A.; Offner, H. Estrogen-induced protection against experimental autoimmune encephalomyelitis is abrogated in the absence of B cells. Eur. J. Immunol. 2011, 41, 1165–1175. [Google Scholar] [CrossRef]
- Noorbakhsh, F.; Baker, G.B.; Power, C. Allopregnanolone and neuroinflammation: A focus on multiple sclerosis. Front. Cell. Neurosci. 2014, 8, 134. [Google Scholar] [CrossRef]
- Balan, I.; Beattie, M.C.; O’Buckley, T.K.; Aurelian, L.; Morrow, A.L. Endogenous Neurosteroid (3 alpha, 5 alpha)3-Hydroxypregnan-20-one Inhibits Toll-like-4 Receptor Activation and Pro-inflammatory Signaling in Macrophages and Brain. Sci. Rep. 2019, 9, 1220. [Google Scholar] [CrossRef] [PubMed]
- Al-Shammri, S.; Rawoot, P.; Azizieh, F.; AbuQoora, A.; Hanna, M.; Saminathan, T.R.; Raghupathy, R. Th1/Th2 cytokine patterns and clinical profiles during and after pregnancy in women with multiple sclerosis. J. Neurol. Sci. 2004, 222, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Park-Chung, M.; Wu, F.S.; Purdy, R.H.; Malayev, A.A.; Gibbs, T.T.; Farb, D.H. Distinct sites for inverse modulation of N-methyl-D-aspartate receptors by sulfated steroids. Mol. Pharmacol. 1997, 52, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Park-Chung, M.; Malayev, A.; Purdy, R.H.; Gibbs, T.T.; Farb, D.H. Sulfated and unsulfated steroids modulate gamma-aminobutyric acidA receptor function through distinct sites. Brain Res. 1999, 830, 72–87. [Google Scholar] [CrossRef]
- Smejkalova, T.; Korinek, M.; Krusek, J.; Hrcka Krausova, B.; Candelas Serra, M.; Hajdukovic, D.; Kudova, E.; Chodounska, H.; Vyklicky, L. Endogenous neurosteroids pregnanolone and pregnanolone sulfate potentiate presynaptic glutamate release through distinct mechanisms. Br. J. Pharmacol. 2021, 178, 3888–3904. [Google Scholar] [CrossRef]
- Labrie, F. Adrenal androgens and intracrinology. Semin. Reprod. Med. 2004, 22, 299–309. [Google Scholar] [CrossRef]
- Majewska, M.D. Steroid regulation of the GABAA receptor: Ligand binding, chloride transport and behaviour. Ciba Found. Symp. 1990, 153, 83–97; Discussion 97–106. [Google Scholar]
- Petrovic, M.; Sedlacek, M.; Horak, M.; Chodounska, H.; Vyklicky, L., Jr. 20-oxo-5beta-pregnan-3alpha-yl sulfate is a use-dependent NMDA receptor inhibitor. J. Neurosci. 2005, 25, 8439–8450. [Google Scholar] [CrossRef]
- Johansson, T.; Le Greves, P. The effect of dehydroepiandrosterone sulfate and allopregnanolone sulfate on the binding of [(3)H]ifenprodil to the N-methyl-d-aspartate receptor in rat frontal cortex membrane. J. Steroid Biochem. Mol. Biol. 2005, 94, 263–266. [Google Scholar] [CrossRef]
- Gupta, M.K.; Guryev, O.L.; Auchus, R.J. 5alpha-reduced C21 steroids are substrates for human cytochrome P450c17. Arch. Biochem. Biophys. 2003, 418, 151–160. [Google Scholar] [CrossRef]
- Rege, J.; Nakamura, Y.; Wang, T.; Merchen, T.D.; Sasano, H.; Rainey, W.E. Transcriptome profiling reveals differentially expressed transcripts between the human adrenal zona fasciculata and zona reticularis. J. Clin. Endocrinol. Metab. 2014, 99, E518–E527. [Google Scholar] [CrossRef] [PubMed]
- Barnard, L.; Gent, R.; van Rooyen, D.; Swart, A.C. Adrenal C11-oxy C(21) steroids contribute to the C11-oxy C(19) steroid pool via the backdoor pathway in the biosynthesis and metabolism of 21-deoxycortisol and 21-deoxycortisone. J. Steroid Biochem. Mol. Biol. 2017, 174, 86–95. [Google Scholar] [CrossRef] [PubMed]
- do Nascimento, F.V.; Piccoli, V.; Beer, M.A.; von Frankenberg, A.D.; Crispim, D.; Gerchman, F. Association of HSD11B1 polymorphic variants and adipose tissue gene expression with metabolic syndrome, obesity and type 2 diabetes mellitus: A systematic review. Diabetol. Metab. Syndr. 2015, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.R.; Stewart, P.M.; Burt, D.; Brett, L.; McIntyre, M.A.; Sutanto, W.S.; de Kloet, E.R.; Monder, C. Localisation of 11 beta-hydroxysteroid dehydrogenase--tissue specific protector of the mineralocorticoid receptor. Lancet 1988, 2, 986–989. [Google Scholar] [CrossRef]
- Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E. 11beta-hydroxysteroid dehydrogenases: A growing multi-tasking family. Mol. Cell Endocrinol. 2021, 526, 111210. [Google Scholar] [CrossRef]
- Kern, S.; Krause, I.; Horntrich, A.; Thomas, K.; Aderhold, J.; Ziemssen, T. Cortisol awakening response is linked to disease course and progression in multiple sclerosis. PLoS ONE 2013, 8, e60647. [Google Scholar] [CrossRef]
- Kasperska-Zajac, A.; Brzoza, Z.; Rogala, B. Dehydroepiandrosterone and dehydroepiandrosterone sulphate in atopic allergy and chronic urticaria. Inflammation 2008, 31, 141–145. [Google Scholar] [CrossRef]
- Romagnani, S.; Kapsenberg, M.; Radbruch, A.; Adorini, L. Th1 and Th2 cells. Res. Immunol. 1998, 149, 871–873. [Google Scholar] [CrossRef]
- Pratschke, S.; von Dossow-Hanfstingl, V.; Dietz, J.; Schneider, C.P.; Tufman, A.; Albertsmeier, M.; Winter, H.; Angele, M.K. Dehydroepiandrosterone modulates T-cell response after major abdominal surgery. J. Surg. Res. 2014, 189, 117–125. [Google Scholar] [CrossRef]
- Sterzl, I.; Hampl, R.; Sterzl, J.; Votruba, J.; Starka, L. 7Beta-OH-DHEA counteracts dexamethasone induced suppression of primary immune response in murine spleenocytes. J. Steroid Biochem. Mol. Biol. 1999, 71, 133–137. [Google Scholar] [CrossRef]
- Hennebert, O.; Chalbot, S.; Alran, S.; Morfin, R. Dehydroepiandrosterone 7alpha-hydroxylation in human tissues: Possible interference with type 1 11beta-hydroxysteroid dehydrogenase-mediated processes. J. Steroid Biochem. Mol. Biol. 2007, 104, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Le Mee, S.; Hennebert, O.; Ferrec, C.; Wulfert, E.; Morfin, R. 7beta-Hydroxy-epiandrosterone-mediated regulation of the prostaglandin synthesis pathway in human peripheral blood monocytes. Steroids 2008, 73, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, H.; Lundqvist, J.; Norlin, M. Effects of CYP7B1-mediated catalysis on estrogen receptor activation. Biochim. Biophys. Acta 2010, 1801, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Eggertsen, G.; Chiang, J.Y.; Norlin, M. Estrogen-mediated regulation of CYP7B1: A possible role for controlling DHEA levels in human tissues. J. Steroid Biochem. Mol. Biol. 2006, 100, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Ahlem, C.N.; Page, T.M.; Auci, D.L.; Kennedy, M.R.; Mangano, K.; Nicoletti, F.; Ge, Y.; Huang, Y.; White, S.K.; Villegas, S.; et al. Novel components of the human metabolome: The identification, characterization and anti-inflammatory activity of two 5-androstene tetrols. Steroids 2011, 76, 145–155. [Google Scholar] [CrossRef]
- Reading, C.L.; Frincke, J.M.; White, S.K. Molecular targets for 17alpha-ethynyl-5-androstene-3beta,7beta,17beta-triol, an anti-inflammatory agent derived from the human metabolome. PLoS ONE 2012, 7, e32147. [Google Scholar] [CrossRef]
- Reddy, D.S. Neurosteroids: Endogenous role in the human brain and therapeutic potentials. Prog. Brain Res. 2010, 186, 113–137. [Google Scholar]
- Wang, M.D.; Borra, V.B.; Stromberg, J.; Lundgren, P.; Haage, D.; Backstrom, T. Neurosteroids 3beta, 20 (R/S)-pregnandiols decrease offset rate of the GABA-site activation at the recombinant GABA A receptor. Eur. J. Pharmacol. 2008, 586, 67–73. [Google Scholar] [CrossRef]
- Stromberg, J.; Lundgren, P.; Taube, M.; Backstrom, T.; Wang, M.; Haage, D. The effect of the neuroactive steroid 5 beta-pregnane-3 beta, 20(R)-diol on the time course of GABA evoked currents is different to that of pregnenolone sulphate. Eur. J. Pharmacol. 2009, 605, 78–86. [Google Scholar] [CrossRef]
- Lundgren, P.; Stromberg, J.; Backstrom, T.; Wang, M. Allopregnanolone-stimulated GABA-mediated chloride ion flux is inhibited by 3beta-hydroxy-5alpha-pregnan-20-one (isoallopregnanolone). Brain Res. 2003, 982, 45–53. [Google Scholar] [CrossRef]
- Tchernof, A.; Mansour, M.F.; Pelletier, M.; Boulet, M.M.; Nadeau, M.; Luu-The, V. Updated survey of the steroid-converting enzymes in human adipose tissues. J. Steroid Biochem. Mol. Biol. 2015, 147, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Hornsby, P.J.; Casson, P.; Morimoto, R.; Satoh, F.; Xing, Y.; Kennedy, M.R.; Sasano, H.; Rainey, W.E. Type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3) contributes to testosterone production in the adrenal reticularis. J. Clin. Endocrinol. Metab. 2009, 94, 2192–2198. [Google Scholar] [CrossRef] [PubMed]
- Ostinelli, G.; Vijay, J.; Vohl, M.C.; Grundberg, E.; Tchernof, A. AKR1C2 and AKR1C3 expression in adipose tissue: Association with body fat distribution and regulatory variants. Mol. Cell. Endocrinol. 2021, 527, 111220. [Google Scholar] [CrossRef] [PubMed]
- Rizner, T.L.; Penning, T.M. Role of aldo-keto reductase family 1 (AKR1) enzymes in human steroid metabolism. Steroids 2014, 79, 49–63. [Google Scholar] [CrossRef]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef]
- Murgia, F.; Giagnoni, F.; Lorefice, L.; Caria, P.; Dettori, T.; D’Alterio, M.N.; Angioni, S.; Hendren, A.J.; Caboni, P.; Pibiri, M.; et al. Sex Hormones as Key Modulators of the Immune Response in Multiple Sclerosis: A Review. Biomedicines 2022, 10, 3107. [Google Scholar] [CrossRef]
- Xu, C.; Liu, W.; You, X.; Leimert, K.; Popowycz, K.; Fang, X.; Wood, S.L.; Slater, D.M.; Sun, Q.; Gu, H.; et al. PGF2alpha modulates the output of chemokines and pro-inflammatory cytokines in myometrial cells from term pregnant women through divergent signaling pathways. Mol. Hum. Reprod. 2015, 21, 603–614. [Google Scholar] [CrossRef]
- Zheng, L.; Fei, J.; Feng, C.M.; Xu, Z.; Fu, L.; Zhao, H. Serum 8-iso-PGF2alpha Predicts the Severity and Prognosis in Patients With Community-Acquired Pneumonia: A Retrospective Cohort Study. Front. Med. 2021, 8, 633442. [Google Scholar] [CrossRef]
- Sharma, I.; Dhaliwal, L.K.; Saha, S.C.; Sangwan, S.; Dhawan, V. Role of 8-iso-prostaglandin F2alpha and 25-hydroxycholesterol in the pathophysiology of endometriosis. Fertil. Steril. 2010, 94, 63–70. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Hill, M.; Hana, V., Jr.; Velikova, M.; Parizek, A.; Kolatorova, L.; Vitku, J.; Skodova, T.; Simkova, M.; Simjak, P.; Kancheva, R.; et al. A method for determination of one hundred endogenous steroids in human serum by gas chromatography-tandem mass spectrometry. Physiol. Res. 2019, 68, 179–207. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable Type | Group | Subgroup | Parameter | Orthogonal Predictions to Latent Structure (OPLS) | Multiple Regression (MR) | ANOVA Factor MS (Multiple Sclerosis) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Component Loading | t-Statistics | Ra | Regression Coefficient | t-Statistics | Unit | MS− | MS+ | Effect size (ηp2) | p-Value | ||||||
| EXPLAINING VARIABLES | Steroids | Δ5 Steroids | Age | years | 39 (34, 43) | 39 (35, 43) | - | 0.898 | |||||||
| Preg | nM | 1.2 (0.93, 1.4) | 1.4 (1.2, 1.6) | 0.026 | 0.349 | ||||||||||
| PregS | nM | 110 (85, 140) | 130 (110, 160) | 0.022 | 0.363 | ||||||||||
| ↓ | 20α-DHPreg | nM | 4.8 (3.9, 5.7) | 3.4 (3, 3.9) | 0.119 | 0.046 | |||||||||
| 20α-DHPregS | μM | 0.83 (0.68, 1) | 0.62 (0.54, 0.72) | 0.084 | 0.097 | ||||||||||
| 17-OH-Preg | nM | 3.4 (2.6, 4.4) | 3.7 (3, 4.5) | 0.004 | 0.72 | ||||||||||
| 17-OH-PregS | nM | 7 (5.4, 8.9) | 8.6 (7.2, 10) | 0.031 | 0.315 | ||||||||||
| ↓ | 16α-OH-Preg | nM | 0.58 (0.46, 0.73) | 0.3 (0.26, 0.36) | 0.262 | 0.003 | |||||||||
| DHEA | nM | 5 (4, 6.2) | 5.6 (4.7, 6.6) | 0.011 | 0.539 | ||||||||||
| DHEAS | μM | 2.2 (1.6, 3) | 2.6 (2.1, 3.2) | 0.009 | 0.57 | ||||||||||
| 7α-OH-DHEA | nM | 1.2 (0.94, 1.4) | 0.95 (0.79, 1.1) | 0.032 | 0.309 | ||||||||||
| ↓ | 7β-OH-DHEA | nM | 0.69 (0.55, 0.87) | 0.36 (0.29, 0.44) | 0.233 | 0.005 | |||||||||
| Adiol | nM | 1.5 (1.2, 1.9) | 1.8 (1.5, 2.1) | 0.015 | 0.461 | ||||||||||
| AdiolS | μM | 1.1 (0.8, 1.5) | 0.68 (0.54, 0.86) | 0.088 | 0.084 | ||||||||||
| 3β,7α,17β-AT | nM | 0.19 (0.14, 0.25) | 0.2 (0.16, 0.24) | <0.001 | 0.912 | ||||||||||
| 3β,7β,17β-AT | nM | 0.19 (0.14, 0.25) | 0.18 (0.14, 0.22) | 0.001 | 0.837 | ||||||||||
| 3β,16α,17β-AT | pM | 160 (120, 220) | 170 (130, 220) | <0.001 | 0.899 | ||||||||||
| 3β,16α,17β-ATC | nM | 78 (59, 100) | 94 (75, 120) | 0.015 | 0.471 | ||||||||||
| Δ4 Steroids | P | pM | 180 (110, 310) | 130 (86, 190) | 0.014 | 0.476 | |||||||||
| 20α-DHP | nM | 0.23 (0.15, 0.33) | 0.25 (0.19, 0.34) | 0.003 | 0.722 | ||||||||||
| 20α-DHPC | nM | 1.6 (1.2, 2.2) | 1.6 (1.2, 2) | <0.001 | 0.995 | ||||||||||
| ↓ | 17-OH-P | −0.377 | −10.4 ** | −0.772 | −0.110 | −4.28 ** | nM | 1.2 (0.87, 1.6) | 0.67 (0.53, 0.86) | 0.106 | 0.046 | ||||
| 17-OH-20α-DHP | nM | 0.75 (0.56, 0.99) | 0.46 (0.37, 0.58) | 0.09 | 0.072 | ||||||||||
| 17-OH-20α-DHPC | nM | 6.5 (4.9, 8.6) | 8 (6.4, 9.9) | 0.02 | 0.412 | ||||||||||
| ↓ | 16α-OH-P | −0.351 | −3.88 ** | −0.720 | −0.092 | −4.26 ** | nM | 0.46 (0.35, 0.59) | 0.28 (0.22, 0.34) | 0.138 | 0.037 | ||||
| ↓ | A | nM | 3 (2.6, 3.4) | 2 (1.8, 2.3) | 0.215 | 0.005 | |||||||||
| ↓ | T | nM | 0.59 (0.48, 0.72) | 0.38 (0.31, 0.46) | 0.135 | 0.032 | |||||||||
| ↓ | E2 | pM | 260 (160, 430) | 100 (70, 150) | 0.123 | 0.033 | |||||||||
| EXPLAINING VARIABLES | Steroids | 20-oxo-5α/β-Reduced pregnanes | 5α-DHP | pM | 98 (62, 150) | 89 (61, 130) | 0.002 | 0.809 | |||||||
| ↓ | 3α,5α-THP | pM | 200 (160, 260) | 120 (100, 150) | 0.138 | 0.04 | |||||||||
| 3α,5α-THPC | nM | 7.2 (5.5, 9.6) | 5.8 (4.7, 7.1) | 0.024 | 0.346 | ||||||||||
| 3β,5α-THP | pM | 120 (97, 160) | 100 (86, 120) | 0.024 | 0.388 | ||||||||||
| 3β,5α-THPC | nM | 11 (9.6, 13) | 11 (10, 13) | <0.001 | 0.997 | ||||||||||
| 3α,5β-THP | pM | 73 (49, 110) | 67 (49, 92) | 0.002 | 0.813 | ||||||||||
| 3α,5β-THPC | nM | 19 (16, 23) | 15 (13, 17) | 0.061 | 0.153 | ||||||||||
| 3α,5β-THPC | nM | 2.5 (2.1, 3) | 3.2 (2.8, 3.7) | 0.075 | 0.117 | ||||||||||
| (3α,5α,17-PDC | nM | 2.1 (1.7, 2.6) | 2 (1.7, 2.4) | 0.001 | 0.846 | ||||||||||
| ↓ | 3α,5β,17-PD | −0.435 | −10.46 ** | −0.771 | −0.130 | −3.39 ** | pM | 56 (37, 85) | 24 (16, 36) | 0.137 | 0.04 | ||||
| ↓ | 3α,5β,17-PDC | nM | 14 (12, 15) | 8.4 (7.4, 9.5) | 0.328 | <0.001 | |||||||||
| 5α,20α-THP | pM | 150 (110, 200) | 140 (110, 180) | 0.001 | 0.839 | ||||||||||
| 5α,20α-THPC | nM | 0.39 (0.29, 0.52) | 0.29 (0.24, 0.37) | 0.032 | 0.28 | ||||||||||
| 20α-Hydroxy-5α/β-reduced pregnanes | 3α,5α,20α-PD | nM | 0.3 (0.22, 0.4) | 0.34 (0.27, 0.43) | 0.006 | 0.644 | |||||||||
| 3α,5α,20α-PDC | nM | 25 (18, 34) | 28 (22, 35) | 0.007 | 0.625 | ||||||||||
| 3β,5α,20α-PD | nM | 3.4 (2.5, 4.5) | 2.6 (2, 3.3) | 0.032 | 0.353 | ||||||||||
| 3β,5α,20α-PDC | nM | 530 (420, 680) | 390 (330, 460) | 0.072 | 0.126 | ||||||||||
| 5β,20α-THPC | nM | 0.64 (0.5, 0.86) | 0.5 (0.42, 0.61) | 0.033 | 0.281 | ||||||||||
| 3α,5β,20α-PD | pM | 110 (82, 150) | 90 (69, 120) | 0.019 | 0.427 | ||||||||||
| 3α,5β,20α-PDC | nM | 17 (13, 21) | 16 (14, 19) | <0.001 | 0.912 | ||||||||||
| 3β,5β,20α-PD | nM | 0.16 (0.12, 0.22) | 0.16 (0.13, 0.2) | <0.001 | 0.904 | ||||||||||
| 3β,5β,20α-PDC | nM | 10 (8.3, 13) | 15 (13, 19) | 0.105 | 0.066 | ||||||||||
| ↓ | 3α,5α,17,20α-PT | −0.389 | −10.24 ** | −0.796 | −0.083 | −7.01 ** | pM | 170 (110, 250) | 100 (71, 140) | 0.056 | 0.152 | ||||
| 3α,5α,17,20α-PTC | nM | 41 (29, 56) | 42 (32, 53) | <0.001 | 0.922 | ||||||||||
| 3β,5α,17,20α-PT | pM | 120 (76, 190) | 140 (99, 200) | 0.006 | 0.653 | ||||||||||
| ↑ | 3β,5α,17,20α-PTC | nM | 3.1 (2.2, 4.3) | 6 (4.4, 8.4) | 0.114 | 0.044 | |||||||||
| ↓ | 3α,5β,17,20α-PT | −0.411 | −7.43 ** | −0.841 | −0.091 | −3.95 ** | nM | 1.7 (1.3, 2.2) | 1.3 (0.98, 1.6) | 0.036 | 0.254 | ||||
| 3α,5β,17,20α-PTC | nM | 100 (80, 130) | 89 (72, 110) | 0.013 | 0.496 | ||||||||||
| EXPLAINING VARIABLES | Steroids | 5α/β-Reduced androstanes | ↓ | 5α-DHA | −0.384 | −7.2 ** | −0.786 | −0.101 | −3.78 ** | nM | 0.23 (0.2, 0.27) | 0.17 (0.15, 0.19) | 0.146 | 0.024 | |
| ↑ | 3α,5α-THA | nM | 0.32 (0.28, 0.38) | 0.49 (0.44, 0.55) | 0.256 | 0.003 | |||||||||
| 3α,5α-THAC | μM | 0.77 (0.58, 1) | 1 (0.79, 1.2) | 0.034 | 0.299 | ||||||||||
| 3β,5α-THA | nM | 0.22 (0.17, 0.27) | 0.21 (0.17, 0.25) | 0.002 | 0.808 | ||||||||||
| 3β,5α-THAC | nM | 300 (210, 410) | 320 (250, 410) | 0.002 | 0.806 | ||||||||||
| 3α,5β-THA | nM | 0.15 (0.12, 0.18) | 0.15 (0.13, 0.18) | <0.001 | 0.956 | ||||||||||
| 3α,5β-THAC | nM | 59 (44, 77) | 71 (57, 86) | 0.015 | 0.466 | ||||||||||
| 3β,5β-THAC | nM | 17 (12, 24) | 26 (20, 33) | 0.048 | 0.178 | ||||||||||
| 5α-DHT | nM | 0.16 (0.13, 0.19) | 0.16 (0.14, 0.19) | <0.001 | 0.889 | ||||||||||
| 5α-DHTC | nM | 1.4 (1.1, 1.6) | 1.5 (1.3, 1.7) | 0.006 | 0.65 | ||||||||||
| 3α,5α,17β-AD | pM | 66 (52, 84) | 56 (47, 68) | 0.016 | 0.452 | ||||||||||
| 3α,5α,17β-ADC | nM | 29 (22, 38) | 28 (23, 35) | <0.001 | 0.956 | ||||||||||
| 3β,5α,17β-AD | pM | 20 (16, 25) | 21 (18, 25) | 0.003 | 0.764 | ||||||||||
| 3β,5α,17β-ADC | nM | 64 (47, 84) | 67 (53, 82) | <0.001 | 0.864 | ||||||||||
| 3α,5β,17β-ADC | nM | 6.8 (5.3, 8.6) | 6.4 (5.2, 7.8) | 0.002 | 0.805 | ||||||||||
| ↑ | 3β,5β,17β-ADC | nM | 0.4 (0.3, 0.51) | 0.65 (0.53, 0.78) | 0.139 | 0.033 | |||||||||
| Corticoids + 11β-OH-androst. | ↑ | F | 0.162 | 0.99 | 0.330 | 0.087 | 1.6 | nM | 300 (250, 350) | 440 (380, 500) | 0.173 | 0.009 | |||
| ↑ | E | nM | 110 (100, 120) | 140 (120, 150) | 0.144 | 0.032 | |||||||||
| B | nM | 12 (8.8, 17) | 12 (9.7, 16) | <0.001 | 0.974 | ||||||||||
| 11β-OH-A | nM | 42 (36, 48) | 39 (35, 43) | 0.014 | 0.513 | ||||||||||
| ↓ | 11β-OH-3α,5α-THA | −0.304 | −5.24 ** | −0.625 | −0.073 | −3.45 ** | nM | 1.9 (1.5, 2.4) | 1.1 (0.91, 1.4) | 0.129 | 0.029 | ||||
| 11β-OH-3α,5α-THAC | nM | 33 (27, 40) | 27 (22, 31) | 0.045 | 0.223 | ||||||||||
| 11β-OH-3β,5α-THA | pM | 95 (67, 130) | 54 (41, 71) | 0.085 | 0.081 | ||||||||||
| 11β-OH-3β,5α-THAC | nM | 0.96 (0.76, 1.2) | 1.3 (1.1, 1.6) | 0.063 | 0.142 | ||||||||||
| 11β-OH-3α,5β-THA | nM | 1.7 (1.5, 2) | 1.3 (1.1, 1.5) | 0.073 | 0.105 | ||||||||||
| 11β-OH-3α,5β-THAC | nM | 13 (10, 17) | 8.5 (6.7, 11) | 0.088 | 0.075 | ||||||||||
| EXPLAINED VARIABLE | MS patient vs. control (LLRb) | 1.000 | 3.66 ** | 0.560 | |||||||||||
| Explained variability = 31.4% (28.2% after cross-validation), Sensitivity = 0.875(0.743–1), Specificity = 0.75(0.505–0.995) | |||||||||||||||
| EXPLAINING VARIABLES | CYP17A1 | hydroxylase + lyase | DHEA/Preg | 4.8 (4.1, 5.7) | 5.3 (4.7, 6) | 0.016 | 0.48 | ||||||||
| DHEA/PregC | 18 (16, 21) | 20 (18, 23) | 0.02 | 0.394 | |||||||||||
| ↑ | DHEA/20α-DHPreg | 0.272 | 3.49 ** | 0.583 | 0.063 | 2.8 * | 1.7 (1.5, 2) | 2 (1.8, 2.2) | 0.025 | 0.36 | |||||
| DHEA/20α-DHPregC | 2.8 (2.3, 3.4) | 3.8 (3.3, 4.4) | 0.073 | 0.097 | |||||||||||
| A/P | 13 (8.1, 21) | 18 (13, 25) | 0.017 | 0.432 | |||||||||||
| A/20α-DHP | 8.9 (6.1, 13) | 8.5 (6.3, 11) | <0.001 | 0.871 | |||||||||||
| 5α-DHA/5α-DHP | 2.3 (1.6, 3.2) | 2.9 (2.2, 4) | 0.022 | 0.444 | |||||||||||
| ↑ | 5α-DHA/5α,20α-THP | 1.3 (1.1, 1.6) | 1.9 (1.7, 2.2) | 0.137 | 0.026 | ||||||||||
| ↑ | 3α,5α-THA/3α,5α-THP | 2 (1.6, 2.5) | 3.1 (2.6, 3.8) | 0.116 | 0.046 | ||||||||||
| ↓ | 3α,5α-THA/3α,5α,20α-PD | −0.091 | −1.14 | −0.196 | −0.049 | −2.7 * | 1.6 (1.2, 2.1) | 1.2 (0.95, 1.5) | 0.035 | 0.256 | |||||
| ↑ | 3α,5α-THA/3α,5α-THP, C | 0.357 | 9.24 ** | 0.765 | 0.089 | 2.87 * | 89 (69, 110) | 180 (150, 220) | 0.241 | 0.002 | |||||
| ↑ | 3α,5α-THA/3α,5α,20α-PD, C | 0.394 | 14.89 ** | 0.846 | 0.081 | 3.12 ** | 20 (15, 26) | 36 (30, 43) | 0.173 | 0.013 | |||||
| 3β,5α-THA/3β,5α-THP | 2.2 (1.7, 2.8) | 2.1 (1.7, 2.5) | 0.002 | 0.81 | |||||||||||
| 3β,5β-THA/3α,5α,20α-PD | 103 | 94 (77, 120) | 69 (58, 82) | 0.107 | 0.104 | ||||||||||
| ↑ | 3β,5α-THA/3β,5α-THP, C | 0.393 | 11.86 ** | 0.843 | 0.098 | 5.56 ** | 27 (23, 32) | 34 (30, 38) | 0.076 | 0.12 | |||||
| ↑ | 3β,5β-THA/3α,5α,20α-PD, C | 0.400 | 6.59 ** | 0.859 | 0.086 | 4.48 ** | 0.73 (0.56, 0.93) | 0.82 (0.67, 0.99) | 0.008 | 0.602 | |||||
| 3α,5β-THA/3α,5β-THP | 2.7 (1.9, 3.7) | 2.8 (2.1, 3.6) | <0.001 | 0.89 | |||||||||||
| 3α,5β-THA/3α,5β,20α-PD | 1.3 (1.1, 1.6) | 1.9 (1.6, 2.2) | 0.096 | 0.074 | |||||||||||
| ↑ | 3α,5β-THA/3α,5β-THP, C | 0.393 | 11.86 ** | 0.843 | 0.098 | 5.56 ** | 2.6 (2, 3.3) | 4.3 (3.6, 5.1) | 0.136 | 0.021 | |||||
| ↑ | 3α,5β-THA/3α,5β,20α-PD, C | 0.400 | 6.59 ** | 0.859 | 0.086 | 4.48 ** | 2.6 (1.9, 3.4) | 3.9 (3.2, 4.7) | 0.079 | 0.087 | |||||
| ↑ | 3β,5β-THA/3β,5β-THP, C | 0.425 | 9.6 ** | 0.911 | 0.136 | 3.78 ** | 0.75 (0.62, 0.91) | 1.6 (1.4, 1.9) | 0.387 | <0.001 | |||||
| ↑ | 3β,5β-THA/3β,5β,20α-PD, C | 0.416 | 11.86 ** | 0.892 | 0.118 | 5.01 ** | 0.73 (0.52, 1) | 1.5 (1.2, 1.9) | 0.154 | 0.014 | |||||
| 3.8 (3, 4.8) | 4.1 (3.5, 4.9) | 0.005 | 0.669 | ||||||||||||
| EXPLAINED VARIABLE | MS patient vs. control (LLR) | 1.000 | 4.79 ** | 0.570 | |||||||||||
| Explained variability = 32.5% (29% after cross-validation), Sensitivity = 0.84(0.696–0.984), Specificity = 0.833(0.622–1) | |||||||||||||||
| EXPLAINING VARIABLES | CYP17A1 | hydroxylase | ↓ | 17-OH-Preg/Preg | 4.3 (3.4, 5.4) | 2.9 (2.5, 3.3) | 0.124 | 0.041 | |||||||
| 17-OH-Preg/Preg, C | 103 | 74 (63, 86) | 72 (63, 80) | 0.002 | 0.793 | ||||||||||
| 17-OH-P/P | 5.4 (3.6, 7.6) | 5 (3.7, 6.6) | 0.001 | 0.842 | |||||||||||
| ↓ | 17-OH-20α-DHP/20α-DHP | −0.599 | −10.72 ** | −0.800 | −0.174 | −2.67 * | 2.6 (1.9, 3.4) | 2 (1.6, 2.5) | 0.025 | 0.346 | |||||
| 17-OH-20α-DHP/20α-DHP, C | 4.6 (3.8, 5.6) | 5.5 (4.8, 6.3) | 0.032 | 0.309 | |||||||||||
| 3α,5α,17-PD/3α,5α-THP, C | 0.25 (0.2, 0.3) | 0.3 (0.26, 0.35) | 0.045 | 0.237 | |||||||||||
| ↓ | 3α,5β,17-PD/3α,5β-THP | −0.630 | −9.57 ** | −0.842 | −0.219 | −2.6 * | 0.93 (0.61, 1.5) | 0.4 (0.27, 0.6) | 0.145 | 0.046 | |||||
| 3α,5β,17-PD/3α,5β-THP, C | 0.68 (0.58, 0.81) | 0.52 (0.47, 0.58) | 0.109 | 0.061 | |||||||||||
| ↓ | 3α,5α,17,20α-PT/3α,5α,20α-PD | −0.499 | −5.21 ** | −0.677 | −0.160 | −6.61 ** | 0.6 (0.4, 0.88) | 0.31 (0.21, 0.43) | 0.094 | 0.073 | |||||
| 3α,5α,17,20α-PT/3α,5α,20α-PD, C | 0.95 (0.64, 1.4) | 1.4 (1, 1.8) | 0.032 | 0.286 | |||||||||||
| 3β,5α,17,20α-PT/3β,5α,20α-PD | 103 | 61 (40, 91) | 64 (45, 91) | <0.001 | 0.876 | ||||||||||
| ↑ | 3β,5α,17,20α-PT/3β,5α,20α-PD, C | 103 | 7.1 (4.8, 10) | 15 (11, 22) | 0.127 | 0.039 | |||||||||
| 3α,5β,17,20α-PT/3α,5β,20α-PD | 14 (11, 19) | 12 (9.5, 14) | 0.022 | 0.37 | |||||||||||
| 3α,5β,17,20α-PT/3α,5β,20α-PD, C | 5 (3.9, 6.3) | 5 (4.1, 5.9) | <0.001 | 0.98 | |||||||||||
| F/B | 28 (21, 35) | 36 (30, 43) | 0.039 | 0.234 | |||||||||||
| EXPLAINED VARIABLE | MS patient vs. control (LLR) | 1.000 | 2.34 * | 0.434 | |||||||||||
| Explained variability = 18.8% (14.5% after cross-validation), Sensitivity = 0.7(0.536–0.864), Specificity = 0.556(0.231–0.88) | |||||||||||||||
| EXPLAINING VARIABLES | CYP17A1 | lyase | ↑ | DHEA/17-OH-Preg | 0.375 | 7 ** | 0.662 | 0.122 | 2.78 * | 1.2 (1.1, 1.3) | 1.6 (1.4, 1.7) | 0.176 | 0.011 | ||
| DHEA/17-OH-Preg, C | 250 (210, 300) | 270 (230, 310) | 0.008 | 0.615 | |||||||||||
| ↑ | A/17-OH-P | 0.451 | 8.86 ** | 0.796 | 0.121 | 3.81 ** | 1.4 (1.1, 1.8) | 2.6 (2.1, 3.2) | 0.187 | 0.011 | |||||
| A/17-OH-20α-DHP | 3.6 (3, 4.5) | 4.2 (3.6, 5) | 0.016 | 0.442 | |||||||||||
| 3α,5α-THA/3α,5α,17-PD, C | 430 (320, 590) | 450 (350, 580) | <0.001 | 0.871 | |||||||||||
| ↓ | 3α,5β-THA/3α,5β,17-PD | −0.429 | −5.65 ** | −0.684 | −0.130 | −4.12 ** | 103 | 320 (220, 470) | 140 (98, 200) | 0.148 | 0.032 | ||||
| ↓ | 3α,5β-THA/3α,5β,17-PD, C | −0.424 | −9.39 ** | −0.748 | −0.113 | −3.17 ** | 0.24 (0.18, 0.31) | 0.13 (0.11, 0.16) | 0.159 | 0.015 | |||||
| ↓ | 3α,5α-THA/3α,5α,17,20α-PT | 0.46 (0.34, 0.6) | 0.24 (0.19, 0.31) | 0.148 | 0.022 | ||||||||||
| 3α,5α-THA/3α,5α,17,20α-PT, C | 103 | 51 (35, 74) | 41 (31, 56) | 0.01 | 0.545 | ||||||||||
| 3β,5α-THA/3β,5α,17,20α-PT | 0.54 (0.36, 0.79) | 0.79 (0.58, 1.1) | 0.035 | 0.271 | |||||||||||
| ↑ | 3β,5α-THA/3β,5α,17,20α-PT, C | 103 | 12 (8.8, 16) | 23 (18, 31) | 0.165 | 0.019 | |||||||||
| ↓ | 3α,5β-THA/3α,5β,17,20α-PT | −0.383 | −8.59 ** | −0.679 | −0.132 | −3.47 ** | 11 (9.3, 13) | 6 (5.2, 6.8) | 0.342 | <0.001 | |||||
| ↓ | 3α,5β-THA/3α,5β,17,20α-PT, C | −0.397 | −7.95 ** | −0.699 | −0.102 | −1.87 | 1.9 (1.5, 2.4) | 1.1 (0.96, 1.3) | 0.165 | 0.016 | |||||
| ↓ | 11β-OH-A/F | 103 | 130 (120, 150) | 110 (98, 120) | 0.131 | 0.039 | |||||||||
| EXPLAINED VARIABLE | MS patient vs. control (LLR) | 1.000 | 5.28 ** | 0.521 | |||||||||||
| Explained variability = 27.1% (21.3% after cross-validation), Sensitivity = 0.75(0.59–0.91), Specificity = 0.6(0.296–0.904) | |||||||||||||||
| EXPLAINING VARIABLES | HSD3B1, HSB3B2 | P/Preg | 103 | 140 (89, 230) | 93 (69, 130) | 0.028 | 0.312 | ||||||||
| ↓ | P/PregC | −0.351 | −7.09 ** | −0.614 | −0.110 | −2.88 * | 103 | 1.6 (0.94, 2.8) | 0.88 (0.59, 1.3) | 0.043 | 0.211 | ||||
| 20α-DHP/20α-DHPreg | 103 | 53 (42, 68) | 67 (56, 83) | 0.032 | 0.273 | ||||||||||
| 20α-DHP/20α-DHPreg, C | 103 | 0.3 (0.27, 0.32) | 0.3 (0.29, 0.32) | 0.006 | 0.649 | ||||||||||
| ↓ | 17-OH-P/17-OH-Preg | −0.459 | −7.62 ** | −0.803 | −0.158 | −3.14 ** | 0.24 (0.19, 0.31) | 0.18 (0.15, 0.21) | 0.061 | 0.153 | |||||
| ↓ | 17-OH-P/17-OH-Preg, C | −0.472 | −7.59 ** | −0.825 | −0.205 | −5.68 ** | 103 | 240 (170, 340) | 72 (57, 90) | 0.378 | <0.001 | ||||
| 16α-OH-P/16α-OH-Preg | 0.86 (0.69, 1.1) | 0.73 (0.62, 0.88) | 0.018 | 0.431 | |||||||||||
| ↓ | A/DHEA | −0.437 | −4.66 ** | −0.765 | −0.180 | −7.24 ** | 103 | 440 (390, 520) | 360 (330, 390) | 0.101 | 0.059 | ||||
| ↓ | A/DHEAC | −0.402 | −9.89 ** | −0.704 | −0.117 | −7.32 ** | 103 | 1.2 (0.9, 1.5) | 0.81 (0.67, 0.97) | 0.079 | 0.108 | ||||
| ↓ | T/Adiol | −0.338 | −5.7 ** | −0.591 | −0.150 | −3.41 ** | 103 | 320 (260, 390) | 270 (230, 310) | 0.032 | 0.294 | ||||
| T/AdiolC | 103 | 0.41 (0.31, 0.53) | 0.54 (0.44, 0.67) | 0.044 | 0.244 | ||||||||||
| EXPLAINED VARIABLE | MS patient vs. control (LLR) | 1.000 | 4.5 ** | 0.673 | |||||||||||
| Explained variability = 45.2% (41.2% after cross-validation), Sensitivity = 0.846(0.707–0.985), Specificity = 0.889(0.684–1) | |||||||||||||||
| EXPLAINING VARIABLES | CYP 19A1 | ↓ | E2/A | −0.579 | −73.7 ** | −0.987 | −0.152 | −4.35 ** | 103 | 99 (64, 160) | 40 (29, 55) | 0.149 | 0.02 | ||
| ↓ | E2/T | −0.569 | −41.6 ** | −0.969 | −0.153 | −6.07 ** | 103 | 500 (300, 860) | 200 (140, 290) | 0.107 | 0.048 | ||||
| ↓ | E2/(A+T) | −0.584 | −148 | −0.996 | −0.156 | −4.89 ** | 103 | 82.1 (52.6, 133) | 31.9 (23.1, 44.2) | 0.154 | 0.02 | ||||
| EXPLAINED VARIABLE | MS patient vs. control (LLR) | 1.000 | 3.57 ** | 0.454 | |||||||||||
| Explained variability = 20.6% (18.8% after cross-validation), Sensitivity = 0.708(0.526–0.89), Specificity = 0.583(0.304–0.862) | |||||||||||||||
| EXPLAINING VARIABLES | Conjugated/unconjugated steroids (C/U) | Δ5 + Δ4 Steroids | Preg | 99 (78, 130) | 100 (84, 120) | <0.001 | 0.955 | ||||||||
| 20α-DHPreg | 140 (120, 170) | 190 (170, 230) | 0.105 | 0.05 | |||||||||||
| 17-OH-Preg | 1.6 (1.2, 2.1) | 2.5 (2.1, 3) | 0.104 | 0.052 | |||||||||||
| DHEA | 0.196 | 1.21 | 0.319 | 0.117 | 1.84 | 430 (340, 540) | 370 (310, 450) | 0.015 | 0.481 | ||||||
| Adiol | 570 (420, 780) | 430 (350, 540) | 0.027 | 0.314 | |||||||||||
| 3β,16α,17β-AT | 590 (440, 790) | 440 (360, 550) | 0.038 | 0.255 | |||||||||||
| 20α-DHP | 6.8 (4.2, 11) | 7 (4.8, 10) | <0.001 | 0.954 | |||||||||||
| 17-OH-20α-DHP | 12 (7.8, 17) | 17 (13, 24) | 0.033 | 0.272 | |||||||||||
| 5α/β-Reduced pregnanes | 3α,5α-THP | 42 (31, 58) | 35 (28, 45) | 0.013 | 0.501 | ||||||||||
| 3β,5α-THP | 96 (72, 130) | 110 (86, 130) | 0.005 | 0.677 | |||||||||||
| 3α,5β-THP | 340 (240, 460) | 250 (180, 330) | 0.033 | 0.317 | |||||||||||
| ↑ | 3α,5β,17-PD | 0.452 | 5.83 ** | 0.648 | 0.151 | 3.17 ** | 210 (150, 300) | 360 (260, 520) | 0.082 | 0.112 | |||||
| 5α,20α-THP | 2.3 (1.7, 3) | 2.2 (1.8, 2.7) | <0.001 | 0.926 | |||||||||||
| ↓ | 3α,5α,20α-PD | −0.334 | −2.5 * | −0.544 | −0.111 | −1.72 | 130 (81, 210) | 79 (58, 110) | 0.039 | 0.239 | |||||
| 3β,5α,20α-PD | 160 (120, 220) | 230 (180, 300) | 0.05 | 0.237 | |||||||||||
| 3α,5β,20α-PD | 180 (130, 260) | 160 (130, 210) | 0.004 | 0.694 | |||||||||||
| ↑ | 3β,5β,20α-PD | 63 (45, 91) | 140 (100, 190) | 0.159 | 0.022 | ||||||||||
| ↑ | 3α,5α,17,20α-PT | 0.274 | 2.38 * | 0.448 | 0.112 | 1.91 * | 160 (110, 230) | 310 (240, 410) | 0.12 | 0.045 | |||||
| ↑ | 3β,5α,17,20α-PT | 22 (18, 26) | 41 (36, 47) | 0.331 | <0.001 | ||||||||||
| ↑ | 3α,5β,17,20α-PT | 0.403 | 3.49 ** | 0.656 | 0.138 | 4.49 ** | 60 (49, 73) | 81 (69, 95) | 0.075 | 0.096 | |||||
| 5α/β-Reduced androstanes | 3α,5α-THA | 10−3 | 1.6 (1.3, 2.1) | 1.9 (1.5, 2.3) | 0.014 | 0.496 | |||||||||
| 3β,5α-THA | 10−3 | 1.2 (1, 1.4) | 1.4 (1.2, 1.6) | 0.024 | 0.381 | ||||||||||
| 3α,5β-THA | 310 (260, 370) | 420 (370, 490) | 0.111 | 0.058 | |||||||||||
| 5α-DHT | 9.7 (7.3, 13) | 7.3 (5.9, 9.1) | 0.037 | 0.261 | |||||||||||
| 3α,5α,17β-AD | 10−3 | 0.48 (0.38, 0.61) | 0.45 (0.37, 0.55) | 0.004 | 0.711 | ||||||||||
| 3β,5α,17β-AD | 10−3 | 3.3 (2.5, 4.5) | 2.6 (2.1, 3.3) | 0.027 | 0.363 | ||||||||||
| ↑ | 11β-OH-3α,5α-THA | 0.457 | 6.1 ** | 0.746 | 0.173 | 4.61 ** | 9 (7, 11) | 18 (15, 22) | 0.237 | 0.003 | |||||
| ↑ | 11β-OH-3β,5α-THA | 0.469 | 7.08 ** | 0.767 | 0.195 | 2.99 * | 14 (12, 17) | 23 (21, 26) | 0.301 | <0.001 | |||||
| 11β-OH-3α,5β-THA | 6.4 (4.9, 8.3) | 7.1 (5.9, 8.6) | 0.007 | 0.632 | |||||||||||
| EXPLAINED VARIABLE | MS patient vs. control (LLR) | 1.000 | 4.33 ** | 0.630 | |||||||||||
| Explained variability = 39.7% (31.7% after cross-validation), Sensitivity = 0.875(0.743–1), Specificity = 0.714(0.478–0.951) | |||||||||||||||
| EXPLAINING VARIABLES | CYP11B1 | 11β-OH-A/A | 17 (14, 20) | 19 (16, 23) | 0.018 | 0.43 | |||||||||
| ↓ | 11β-OH-3α,5α-THA/3α,5α-THA | −0.693 | −11.66 ** | −0.883 | −0.245 | −4.67 ** | 5.2 (4.1, 6.4) | 3.3 (2.7, 4) | 0.119 | 0.034 | |||||
| 11β-OH-3α,5α-THA/3α,5α-THA, C | 103 | 45 (35, 59) | 35 (28, 43) | 0.038 | 0.265 | ||||||||||
| 11β-OH-3β,5α-THA/3β,5α-THA | 103 | 350 (270, 440) | 220 (180, 270) | 0.107 | 0.055 | ||||||||||
| 11β-OH-3β,5α-THA/3β,5α-THA, C | 103 | 3.5 (2.7, 4.6) | 3.7 (3, 4.7) | 0.002 | 0.805 | ||||||||||
| ↓ | 11β-OH-3α,5β-THA/3α,5β-THA | −0.722 | −11.82 ** | −0.905 | −0.274 | −3.99 ** | 10 (8.6, 12) | 7.5 (6.5, 8.7) | 0.102 | 0.057 | |||||
| ↓ | 11β-OH-3α,5β-THA/3α,5β-THA, C | 103 | 200 (150, 270) | 120 (94, 140) | 0.121 | 0.035 | |||||||||
| EXPLAINED VARIABLE | MS patient vs. control (LLR) | 1.000 | 2.05 * | 0.470 | |||||||||||
| Explained variability = 22% (19.6% after cross-validation), Sensitivity = 0.708(0.526–0.89), Specificity = 0.583(0.304–0.862) | |||||||||||||||
| EXPLAINING VARIABLES | CYP7B1, CYP3A4, CYP3A7 | ↓ | 7α-OH-DHEA/DHEA | −0.533 | −22.64 ** | −0.931 | −0.130 | −3.78 ** | 103 | 170 (140, 200) | 140 (120, 160) | 0.044 | 0.211 | ||
| ↓ | 3β,7α,17β-AT/Adiol | −0.497 | −12.96 ** | −0.869 | −0.126 | −5.34 ** | 103 | 220 (180, 260) | 170 (150, 190) | 0.076 | 0.116 | ||||
| ↓ | 7β-OH-DHEA/DHEA | −0.500 | −9.76 ** | −0.874 | −0.149 | −4.77 ** | 103 | 94 (76, 120) | 53 (44, 62) | 0.226 | 0.005 | ||||
| ↓ | 3β,7β,17β-AT/Adiol | −0.474 | −7.12 ** | −0.829 | −0.124 | −3.81 ** | 103 | 120 (110, 140) | 110 (94, 120) | 0.049 | 0.2 | ||||
| ↓ | 16α-OH-Preg/Preg | 103 | 400 (310, 510) | 210 (180, 240) | 0.248 | 0.003 | |||||||||
| 3β,16α,17β-AT/Adiol | 103 | 99 (80, 130) | 77 (66, 92) | 0.055 | 0.205 | ||||||||||
| ↑ | 3β,16α,17β-AT/Adiol, C | 103 | 60 (49, 75) | 98 (82, 120) | 0.164 | 0.017 | |||||||||
| 16α-OH-P/P | 1.8 (1.1, 2.7) | 1.7 (1.2, 2.3) | 0.001 | 0.845 | |||||||||||
| EXPLAINED VARIABLE | MS patient vs. control (LLR) | 1.000 | 3.8 ** | 0.463 | |||||||||||
| Explained variability = 21.4% (19.1% after cross-validation), Sensitivity = 0.68(0.497–0.863), Specificity = 0.455(0.16–0.749) | |||||||||||||||
| EXPLAINING VARIABLES | HSD11B1 | 7β-OH-DHEA/7α-OH-DHEA | 0.5 (0.43, 0.56) | 0.46 (0.41, 0.51) | 0.014 | 0.487 | |||||||||
| ↑ | 3β,7β,17β-AT/3β,7α,17β-AT/Adiol | 0.323 | 0.95 | 0.314 | 0.250 | 2.08 * | 0.59 (0.53, 0.65) | 0.73 (0.69, 0.78) | 0.191 | 0.011 | |||||
| ↑ | F/E | 0.948 | 7.82 ** | 0.921 | 0.609 | 6.96 ** | 2.6 (2.3, 2.9) | 3.6 (3.3, 3.9) | 0.239 | 0.003 | |||||
| EXPLAINED VARIABLE | MS patient vs. control (LLR) | 0.948 | 7.82 ** | 0.921 | |||||||||||
| Explained variability = 40.8% (38.2% after cross-validation), Sensitivity = 0.8(0.643–0.957), Specificity = 0.727(0.464–0.99) | |||||||||||||||
| EXPLAINING VARIABLES | SRD5A1, SRD5A2 | (5α-DHP+3α/β,5α-THP)/P | 2.9 (1.7, 4.8) | 2.3 (1.5, 3.4) | 0.011 | 0.583 | |||||||||
| 3α/β,5α-THP, C/P | 100 (60, 170) | 130 (89, 200) | 0.008 | 0.587 | |||||||||||
| (5α,20α-THP+3α/β,5α,20α-PD)/20α-DHP | 15 (11, 20) | 9.8 (7.6, 13) | 0.099 | 0.11 | |||||||||||
| (5α,20α-THP+3α/β,5α,20α-PD)/20α-DHP, C | 350 (250, 500) | 280 (220, 370) | 0.015 | 0.454 | |||||||||||
| 3α,5α,17-PDC/17-OH-P | 1.8 (1.4, 2.3) | 2.3 (1.9, 2.8) | 0.033 | 0.279 | |||||||||||
| 3α,5α,17,20α-PT/17-OH-20α-DHP | 0.47 (0.33, 0.66) | 0.5 (0.38, 0.65) | 0.001 | 0.824 | |||||||||||
| 3α,5α,17,20α-PT/17-OH-20α-DHP, C | 7.3 (4.8, 11) | 6.5 (4.6, 8.8) | 0.003 | 0.73 | |||||||||||
| (5α-DHA+3α/β,5α-THA)/A | 0.37 (0.32, 0.43) | 0.42 (0.38, 0.46) | 0.024 | 0.372 | |||||||||||
| ↑ | 3α/β,5α-THA, C/A | 400 (310, 520) | 700 (560, 870) | 0.151 | 0.025 | ||||||||||
| (5α-DHT+3α/β,5α-AD)/T | 0.54 (0.43, 0.67) | 0.58 (0.5, 0.68) | 0.005 | 0.674 | |||||||||||
| (5α-DHT+3α/β,5α-AD), C/T | 3 (2.2, 3.8) | 4 (3.3, 4.8) | 0.047 | 0.197 | |||||||||||
| ↓ | 11β-OH-3α/β,5α-THA/A | 103 | 45 (36, 57) | 27 (22, 33) | 0.151 | 0.023 | |||||||||
| 11β-OH-3α/β,5α-THA, C/A | 0.81 (0.62, 1) | 0.81 (0.66, 0.98) | <0.001 | 0.983 | |||||||||||
| EXPLAINED VARIABLE | MS patient vs. control (LLR) | ||||||||||||||
| Not relevant | |||||||||||||||
| EXPLAINING VARIABLES | AKR1D1 | 3α,5β-THP/P | 0.35 (0.21, 0.57) | 0.42 (0.28, 0.63) | 0.006 | 0.67 | |||||||||
| 3α/β,5β-THP, C/P | 140 (83, 220) | 140 (98, 210) | <0.001 | 0.905 | |||||||||||
| 3α/β,5β,20α-PD/20α-DHP | 1.3 (0.93, 1.8) | 1.4 (1.1, 1.8) | 0.002 | 0.784 | |||||||||||
| (5β,20α-THP+3α/β,5β,20α-PD)/20α-DHP, C | 30 (21, 42) | 22 (17, 29) | 0.026 | 0.325 | |||||||||||
| 3α,5β,17-PD/17-OH-P | 103 | 51 (35, 74) | 38 (27, 53) | 0.022 | 0.414 | ||||||||||
| 3α,5β,17-PDC/17-OH-P | 9.1 (7.8, 11) | 12 (11, 14) | 0.108 | 0.054 | |||||||||||
| 3α,5β,17,20α-PT/17-OH-20α-DHP | 2.2 (1.9, 2.6) | 2.5 (2.2, 2.8) | 0.024 | 0.387 | |||||||||||
| 3α,5β,17,20α-PT/17-OH-20α-DHP, C | 15 (11, 19) | 9.4 (7.9, 11) | 0.099 | 0.054 | |||||||||||
| 3α,5β-THA)/A | 103 | 61 (49, 74) | 78 (67, 90) | 0.058 | 0.168 | ||||||||||
| ↑ | 3α/β,5β-THA, C/A | 26 (19, 35) | 47 (39, 57) | 0.152 | 0.019 | ||||||||||
| ↑ | 3α/β,5β,17β-ADC/A | 10 (7.7, 14) | 17 (14, 21) | 0.134 | 0.036 | ||||||||||
| 11β-OH-3α,5β-THA/A | 103 | 43 (35, 50) | 34 (28, 39) | 0.05 | 0.183 | ||||||||||
| 11β-OH-3α,5β-THA, C/A | 103 | 250 (170, 330) | 220 (170, 280) | 0.004 | 0.707 | ||||||||||
| EXPLAINED VARIABLE | MS patient vs. control (LLR) | ||||||||||||||
| Not relevant | |||||||||||||||
| EXPLAINING VARIABLES | AKR1C1 vs. HSD17B2 | ↓ | 20α-DHPreg/Preg | −0.577 | −4.38 ** | −0.728 | −0.389 | −5.06 ** | 3.4 (2.9, 3.9) | 2.7 (2.3, 3) | 0.076 | 0.109 | |||
| ↓ | 20α-DHPreg/Preg, C | 7 (6.1, 8) | 5.1 (4.7, 5.5) | 0.22 | 0.005 | ||||||||||
| ↑ | 20α-DHP/P | 0.332 | 2.46 * | 0.407 | 0.054 | 0.71 | 1.3 (0.88, 1.7) | 2.2 (1.8, 2.8) | 0.109 | 0.046 | |||||
| ↑ | 20α-DHPC/P | 0.312 | 3.3 ** | 0.385 | −0.083 | −0.87 | 7.7 (4.1, 14) | 13 (7.8, 20) | 0.021 | 0.38 | |||||
| 17-OH-20α-DHP/17-OH-P | 0.55 (0.43, 0.68) | 0.76 (0.65, 0.89) | 0.079 | 0.092 | |||||||||||
| ↑ | 17-OH-20α-DHPC/17-OH-P | 0.537 | 7.77 ** | 0.668 | 0.139 | 1.35 | 6.3 (4.3, 9.2) | 11 (8.2, 15) | 0.07 | 0.103 | |||||
| 5α,20α-THP/5α-DHP | 1.5 (1.3, 1.8) | 1.7 (1.5, 2) | 0.015 | 0.522 | |||||||||||
| 5α,20α-THPC/5α-DHP | 3.5 (2.4, 5) | 3.9 (2.9, 5.3) | 0.004 | 0.724 | |||||||||||
| 3α,5α,20α-PD/3α,5α-THP | 1.7 (1.2, 2.4) | 1.6 (1.2, 2.2) | <0.001 | 0.905 | |||||||||||
| 3α,5α,20α-PD/3α,5α-THP, C | 4.7 (4.1, 5.4) | 4.7 (4.3, 5.3) | <0.001 | 0.98 | |||||||||||
| 3β,5α,20α-PD/3β,5α-THP | 24 (18, 31) | 22 (17, 27) | 0.006 | 0.679 | |||||||||||
| 3β,5α,20α-PD/3β,5α-THP, C | 44 (37, 53) | 45 (39, 51) | <0.001 | 0.963 | |||||||||||
| 3α,5β,20α-PD/3α,5β-THP | 1.8 (1.3, 2.4) | 1.2 (0.95, 1.6) | 0.059 | 0.188 | |||||||||||
| 3α,5β,20α-PD/3α,5β-THP, C | 1 (0.88, 1.3) | 1.1 (0.93, 1.2) | <0.001 | 0.934 | |||||||||||
| 3β,5β,20α-PD/3β,5β-THP, C | 5.1 (4.2, 6.2) | 5.3 (4.6, 6.3) | 0.002 | 0.776 | |||||||||||
| 3α,5α,17,20α-PT/3α,5α,17-PD, C | 22 (16, 30) | 21 (17, 27) | <0.001 | 0.864 | |||||||||||
| ↑ | 3α,5β,17,20α-PT/3α,5β,17-PD | 0.433 | 2.21 * | 0.586 | 0.312 | 1.76 | 32 (23, 44) | 61 (45, 84) | 0.135 | 0.042 | |||||
| 3α,5β,17,20α-PT/3α,5β,17-PD, C | 8.7 (7.1, 11) | 10 (9, 12) | 0.03 | 0.306 | |||||||||||
| EXPLAINED VARIABLE | MS patient vs. control (LLR) | 1.000 | 3.33 ** | 0.528 | |||||||||||
| Explained variability = 31.2% (18% after cross-validation), Sensitivity = 0.786(0.634–0.938), Specificity = 0.727(0.464–0.99) | |||||||||||||||
| EXPLAINING VARIABLES | AKR1C2 vs. HSD17B2,6 | 3α/β | 3α,5α-THP/3β,5α-THP | 1.7 (1.4, 2.1) | 1.5 (1.3, 1.8) | 0.018 | 0.461 | ||||||||
| ↓ | 3α,5α-THP/3β,5α-THP, C | −0.220 | −1.87 | −0.343 | −0.114 | −2.51 * | 0.82 (0.69, 1) | 0.53 (0.47, 0.59) | 0.22 | 0.003 | |||||
| ↓ | 3α,5β-THP/3β,5β-THP, C | −0.435 | −5.75 ** | −0.677 | −0.169 | −2.8 * | 7.2 (6.4, 8) | 4.9 (4.5, 5.3) | 0.311 | <0.001 | |||||
| 3α,5α,20α-PD/3β,5α,20α-PD | 103 | 95 (65, 140) | 110 (76, 150) | 0.003 | 0.756 | ||||||||||
| 3α,5α,20α-PD/3β,5α,20α-PD, C | 103 | 58 (43, 80) | 58 (47, 72) | <0.001 | 0.992 | ||||||||||
| ↓ | 3α,5β,20α-PD/3β,5β,20α-PD, C | −0.506 | −6.05 ** | −0.788 | −0.209 | −5.29 ** | 1.7 (1.4, 1.9) | 1 (0.89, 1.1) | 0.3 | <0.001 | |||||
| ↓ | 3α,5α,17,20α-PT/3β,5α,17,20α-PT | −0.482 | −4.84 ** | −0.750 | −0.174 | −3.13 ** | 1.2 (1, 1.5) | 0.75 (0.63, 0.88) | 0.176 | 0.011 | |||||
| ↓ | 3α,5α,17,20α-PT/3β,5α,17,20α-PT, C | 13 (9.2, 19) | 6.2 (4.7, 8.1) | 0.147 | 0.021 | ||||||||||
| 3α,5α-THA/3β,5α-THA | 1.8 (1.5, 2) | 2 (1.8, 2.3) | 0.023 | 0.36 | |||||||||||
| 3α,5α-THA/3β,5α-THA, C | 2.6 (2.4, 2.9) | 2.7 (2.5, 2.9) | 0.001 | 0.848 | |||||||||||
| 3α,5β-THA/3β,5β-THA, C | 3.4 (2.9, 4.1) | 2.7 (2.4, 3.1) | 0.067 | 0.117 | |||||||||||
| 3α,5α,17β-AD/3β,5α,17β-AD | 2.7 (2.2, 3.4) | 3 (2.6, 3.6) | 0.009 | 0.586 | |||||||||||
| 3α,5α,17β-AD/3β,5α,17β-AD, C | 0.52 (0.45, 0.59) | 0.51 (0.46, 0.57) | <0.001 | 0.926 | |||||||||||
| ↓ | 3α,5β,17β-AD/3β,5β,17β-AD, C | −0.273 | −2.21 * | −0.425 | −0.136 | −2.26 * | 15 (13, 18) | 12 (11, 13) | 0.084 | 0.073 | |||||
| 11β-OH-3α,5α-THA/11β-OH-3β,5α-THA | 25 (20, 31) | 22 (19, 26) | 0.013 | 0.509 | |||||||||||
| ↓ | 11β-OH-3α,5α-THA/11β-OH-3β,5α-THA, C | −0.335 | −3.08 ** | −0.521 | −0.129 | −2.16 * | 34 (29, 40) | 24 (21, 28) | 0.134 | 0.033 | |||||
| 3α/oxo | 3α,5α-THP/5α-DHP | 1.7 (1.3, 2.1) | 2.2 (1.8, 2.6) | 0.048 | 0.237 | ||||||||||
| 3α,5α-THPC/5α-DHP | 77 (51, 120) | 71 (51, 100) | 0.002 | 0.833 | |||||||||||
| ↑ | 3α,5α,20α-PD/5α,20α-THP | 0.315 | 2.86 * | 0.490 | 0.116 | 2.24 * | 1.8 (1.3, 2.4) | 2.3 (1.8, 2.9) | 0.024 | 0.364 | |||||
| 3α,5α,20α-PD/5α,20α-THP, C | 86 (67, 110) | 96 (81, 110) | 0.01 | 0.565 | |||||||||||
| 3α,5β,20α-PD/5β,20α-THP, C | 50 (40, 61) | 35 (30, 41) | 0.103 | 0.056 | |||||||||||
| ↑ | 3α,5α-THA/5α-DHA | 1.7 (1.5, 1.9) | 2.5 (2.3, 2.8) | 0.297 | 0.001 | ||||||||||
| ↑ | 3α,5α-THAC/5α-DHA | 10−3 | 3.6 (2.8, 4.6) | 5.9 (4.9, 7) | 0.138 | 0.028 | |||||||||
| EXPLAINED VARIABLE | MS patient vs. control (LLR) | 1.000 | 4.88 ** | 0.631 | |||||||||||
| Explained variability = 39.8% (31.8% after cross-validation), Sensitivity = 0.767(0.615–0.918), Specificity = 0.7(0.416–0.984) | |||||||||||||||
| EXPLAINING VARIABLES | AKR1C3 vs. HSD17B2 | Adiol/DHEA | 0.29 (0.26, 0.33) | 0.29 (0.26, 0.32) | <0.001 | 0.934 | |||||||||
| Adiol/DHEA, C | 0.44 (0.36, 0.55) | 0.36 (0.3, 0.42) | 0.034 | 0.27 | |||||||||||
| 3β,7α,17β-AT/7α-OH-DHEA | 0.32 (0.28, 0.38) | 0.32 (0.29, 0.36) | <0.001 | 0.98 | |||||||||||
| ↑ | 3β,7β,17β-AT/7β-OH-DHEA | 0.33 (0.28, 0.4) | 0.48 (0.41, 0.55) | 0.129 | 0.029 | ||||||||||
| ↑ | T/A | 0.18 (0.16, 0.22) | 0.25 (0.22, 0.29) | 0.114 | 0.047 | ||||||||||
| ↑ | 5α-DHT/5α-DHA | 0.791 | 4.06 ** | 0.826 | 0.392 | 3.73 ** | 0.72 (0.61, 0.85) | 1.1 (0.93, 1.2) | 0.181 | 0.012 | |||||
| ↑ | 5α-DHTC/5α-DHA | 0.613 | 2.25 * | 0.634 | 0.282 | 1.83 | 6.2 (5.2, 7.5) | 8.8 (7.5, 10) | 0.112 | 0.046 | |||||
| 3α,5α,17β-AD/3α,5α-THA | 103 | 140 (120, 170) | 130 (120, 150) | 0.01 | 0.573 | ||||||||||
| 3α,5α,17β-AD/3α,5α-THA, C | 103 | 39 (33, 45) | 32 (28, 36) | 0.054 | 0.178 | ||||||||||
| 3β,5α,17β-AD/3β,5α-THA | 103 | 82 (60, 110) | 96 (78, 120) | 0.012 | 0.525 | ||||||||||
| 3β,5α,17β-AD/3β,5α-THA, C | 103 | 210 (180, 240) | 200 (180, 220) | 0.003 | 0.735 | ||||||||||
| 3α,5β,17β-AD/3α,5β-THA, C | 103 | 110 (100, 130) | 110 (97, 120) | 0.008 | 0.603 | ||||||||||
| 3β,5β,17β-AD/3β,5β-THA, C | 103 | 22 (19, 26) | 25 (22, 28) | 0.02 | 0.413 | ||||||||||
| EXPLAINED VARIABLE | MS patient vs. control (LLR) | 1.000 | 2 * | 0.500 | |||||||||||
| Explained variability = 25% (17% after cross-validation), Sensitivity = 0.679(0.506–0.852), Specificity = 0.455(0.16–0.749) | |||||||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kancheva, R.; Hill, M.; Velíková, M.; Kancheva, L.; Včelák, J.; Ampapa, R.; Židó, M.; Štětkářová, I.; Libertínová, J.; Vosátková, M.; et al. Altered Steroidome in Women with Multiple Sclerosis. Int. J. Mol. Sci. 2024, 25, 12033. https://doi.org/10.3390/ijms252212033
Kancheva R, Hill M, Velíková M, Kancheva L, Včelák J, Ampapa R, Židó M, Štětkářová I, Libertínová J, Vosátková M, et al. Altered Steroidome in Women with Multiple Sclerosis. International Journal of Molecular Sciences. 2024; 25(22):12033. https://doi.org/10.3390/ijms252212033
Chicago/Turabian StyleKancheva, Radmila, Martin Hill, Marta Velíková, Ludmila Kancheva, Josef Včelák, Radek Ampapa, Michal Židó, Ivana Štětkářová, Jana Libertínová, Michala Vosátková, and et al. 2024. "Altered Steroidome in Women with Multiple Sclerosis" International Journal of Molecular Sciences 25, no. 22: 12033. https://doi.org/10.3390/ijms252212033
APA StyleKancheva, R., Hill, M., Velíková, M., Kancheva, L., Včelák, J., Ampapa, R., Židó, M., Štětkářová, I., Libertínová, J., Vosátková, M., & Kubala Havrdová, E. (2024). Altered Steroidome in Women with Multiple Sclerosis. International Journal of Molecular Sciences, 25(22), 12033. https://doi.org/10.3390/ijms252212033