
SARS-CoV-2 Infection and Alpha-Synucleinopathies: Potential Links and Underlying Mechanisms



Abstract
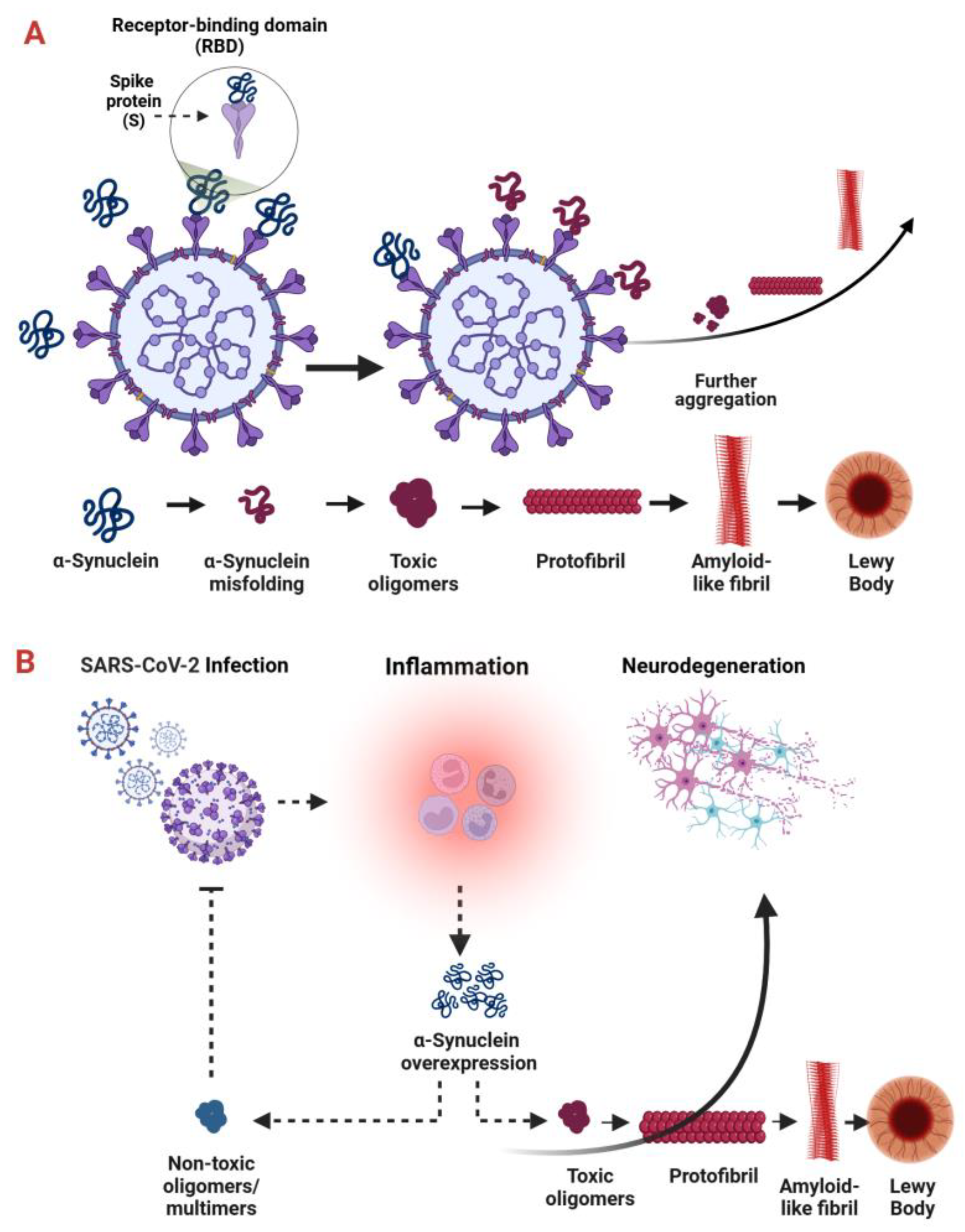
:1. Introduction
2. The Relationship Between COVID-19 and PD
2.1. Parkinsonism as a Consequense of COVID-19

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aim | Method/Subject of Analysis | Results | Reference |
|---|---|---|---|
| Meta-analysis of serum/plasma proteomic data from COVID-19 patients assaying the links between SARS-CoV-2 infection and neurological disorders, specifically AD and PD. | Mass spectrometry-based proteomics data with a total of 538 COVID-19 patients and 523 healthy controls. | Analysis confirmed a direct correlation in the expression patterns of 24 proteins implicated in AD and 23 proteins involved in PD with COVID-19. A protein–protein interaction network and cluster analysis revealed direct correlation in differential expression between COVID-19 and PD and identified SNCA as a hub protein. | [50] |
| Meta-analysis answering the question: Are the new-onset neurodegenerative diseases long-term sequelae of the SARS-CoV-2 infection? | Articles published up to 10 January 2023. Twelve studies involving 33,146,809 individuals (2,688,417 post-COVID-19 cases and 30,458,392 controls). | The significant correlation between SARS-CoV-2 infection and increased risk for new-onset AD (Hazard Ratio [HR] = 1.50), dementia (HR = 1.66), and PD (HR = 1.44). | [57] |
| The systematic review concentrated on the impact of post-SARS-CoV-2 immune-mediated responses/the host’s altered immune counter-offensive on the occurrence of neurodegenerative diseases like PD in a complex interrelation between genetic and epigenetic risk factors. | A synthetic and systematic literature review based on the “Preferred Reporting Items for Systematic Principles Reviews and Meta-Analyses” (PRISMA) methodology; 104 papers were finally selected. | It is too early to establish if the neuroinflammatory events accompanied by COVID-19 could activate long-term neurodegenerative consequences and lead to new cases of PD occurrence and the worsening of the existing disease outcome. Further clinical and prospective longitudinal cohort studies are required. | [58] |
| The analysis of the possible mechanisms involved in COVID-19-induced neuropathology like PD. The analysis of pathways involved in the downregulation of ACE2 following SARS-CoV-2 infection and its effect on PD progression. | The analysis of the pathways involved in the downregulation of ACE2 following SARS-CoV-2 infection and its effect on PD progression. The molecules and chemicals associated with COVID-19 and PD were subjected to Ingenuity Pathway Analysis (IPA) “Grow”; 81 overlapping molecules between COVID-19 and PD were further subjected to IPA’s “Core Analysis” tool to identify the upstream regulators and signaling pathways. | Core Analysis revealed the neuroinflammation signaling pathway (NISP) to be one of the principal signaling pathways involved and SNCA as the top upstream controller associated with both COVID-19 and PD. A network connectivity pathway map of the downstream effects of COVID-19 revealed that ACE2 blocking upregulates SNCA expression, potentially accelerating PD progression. | [59] |
| A systematic review and meta-analysis of studies reporting parkinsonism cases among patients recovering from COVID-19. | Research from seven major databases covering a timeline of 1 January 2020 to 1 January 2022. Ten studies met the inclusion criteria and covered thirteen patients with a median age of 60.0. There were eight males (61.5% of patients), and 53.8% of individuals were documented to have at least one comorbidity. Fisher’s exact test was used to examine the factors connected with COVID-19 and parkinsonism as its results. | Indication of parkinsonism as post-COVID-19 neurological sequelae. Cogwheel rigidity was the most common manifestation of parkinsonism in eleven patients. The most standard medicine modality used was Levodopa (76.9% of cases). Ten patients (76.9%) with bradykinesia achieved a complete recovery. | [60] |
2.2. The Prevalence, Outcomes, and Prognosis of COVID-19 in Patients Diagnosed with PD
2.3. The Theory of SARS-CoV-2 Neuroinvasion in PD
2.4. The Role of SARS-CoV-2 in α-Syn Alterations
2.5. Promising Therapeutic Targets for Both PD and COVID-19
3. The Connection Between COVID-19 and α-Synucleinopathies Other Than PD
3.1. DLB
3.2. MSA
3.3. PAF
3.4. RBD
4. Age and Gender Aspects of Relationships Between SARS-CoV-2 Infection and α-Synucleinopathies
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Calabresi, P.; Mechelli, A.; Natale, G.; Volpicelli-Daley, L.; Di Lazzaro, G.; Ghiglieri, V. Alpha-Synuclein in Parkinson’s Disease and Other Synucleinopathies: From Overt Neurodegeneration Back to Early Synaptic Dysfunction. Cell Death Dis. 2023, 14, 176. [Google Scholar] [CrossRef] [PubMed]
- Nardone, R.; Höller, Y.; Brigo, F.; Versace, V.; Sebastianelli, L.; Florea, C.; Schwenker, K.; Golaszewski, S.; Saltuari, L.; Trinka, E. Spinal Cord Involvement in Lewy Body-Related α-Synucleinopathies. J. Spinal Cord Med. 2020, 43, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Ryman, S.; Vakhtin, A.A.; Richardson, S.P.; Lin, H.C. Microbiome-Gut-Brain Dysfunction in Prodromal and Symptomatic Lewy Body Diseases. J. Neurol. 2023, 270, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Motyl, J.A.; Strosznajder, J.B.; Wencel, A.; Strosznajder, R.P. Recent Insights into the Interplay of Alpha-Synuclein and Sphingolipid Signaling in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 6277. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-Synuclein in Lewy Bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. Alpha-Synuclein in Filamentous Inclusions of Lewy Bodies from Parkinson’s Disease and Dementia with Lewy Bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. The Alpha-Synucleinopathies: Parkinson’s Disease, Dementia with Lewy Bodies, and Multiple System Atrophy. Ann. N. Y. Acad. Sci. 2000, 920, 16–27. [Google Scholar] [CrossRef]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.Y.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of Alpha-Synuclein in Lewy Bodies of Sporadic Parkinson’s Disease and Dementia with Lewy Bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar]
- Deleidi, M.; Gasser, T. The Role of Inflammation in Sporadic and Familial Parkinson’s Disease. Cell. Mol. Life Sci. 2013, 70, 4259–4273. [Google Scholar] [CrossRef]
- Marogianni, C.; Sokratous, M.; Dardiotis, E.; Hadjigeorgiou, G.M.; Bogdanos, D.; Xiromerisiou, G. Neurodegeneration and Inflammation-An Interesting Interplay in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8421. [Google Scholar] [CrossRef]
- Sheng, Z.-M.; Chertow, D.S.; Ambroggio, X.; McCall, S.; Przygodzki, R.M.; Cunningham, R.E.; Maximova, O.A.; Kash, J.C.; Morens, D.M.; Taubenberger, J.K. Autopsy Series of 68 Cases Dying before and during the 1918 Influenza Pandemic Peak. Proc. Natl. Acad. Sci. USA 2011, 108, 16416–16421. [Google Scholar] [CrossRef] [PubMed]
- Marreiros, R.; Müller-Schiffmann, A.; Trossbach, S.V.; Prikulis, I.; Hänsch, S.; Weidtkamp-Peters, S.; Moreira, A.R.; Sahu, S.; Soloviev, I.; Selvarajah, S.; et al. Disruption of Cellular Proteostasis by H1N1 Influenza A Virus Causes α-Synuclein Aggregation. Proc. Natl. Acad. Sci. USA 2020, 117, 6741–6751. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Li, X.; Cui, J. Evolutionary Perspectives on Novel Coronaviruses Identified in Pneumonia Cases in China. Natl. Sci. Rev. 2020, 7, 239–242. [Google Scholar] [CrossRef]
- Dhama, K.; Khan, S.; Tiwari, R.; Sircar, S.; Bhat, S.; Malik, Y.S.; Singh, K.P.; Chaicumpa, W.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. Coronavirus Disease 2019-COVID-19. Clin. Microbiol. Rev. 2020, 33, e00028-20. [Google Scholar] [CrossRef]
- Available online: https://www.who.int/Emergencies/Diseases/Novel-Coronavirus-2019 (accessed on 30 October 2024).
- El-Maradny, Y.A.; Badawy, M.A.; Mohamed, K.I.; Ragab, R.F.; Moharm, H.M.; Abdallah, N.A.; Elgammal, E.M.; Rubio-Casillas, A.; Uversky, V.N.; Redwan, E.M. Unraveling the Role of the Nucleocapsid Protein in SARS-CoV-2 Pathogenesis: From Viral Life Cycle to Vaccine Development. Int. J. Biol. Macromol. 2024, 279, 135201. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Yang, L.; Kim, T.W.; Han, Y.; Nair, M.S.; Harschnitz, O.; Zhu, J.; Wang, P.; Koo, S.Y.; Lacko, L.A.; Chandar, V.; et al. SARS-CoV-2 Infection Causes Dopaminergic Neuron Senescence. Cell Stem Cell 2024, 31, 196–211.e6. [Google Scholar] [CrossRef]
- Jia, F.; Han, J. COVID-19 Related Neurological Manifestations in Parkinson’s Disease: Has Ferroptosis Been a Suspect? Cell Death Discov. 2024, 10, 146. [Google Scholar] [CrossRef]
- Hernández, V.S.; Zetter, M.A.; Guerra, E.C.; Hernández-Araiza, I.; Karuzin, N.; Hernández-Pérez, O.R.; Eiden, L.E.; Zhang, L. ACE2 Expression in Rat Brain: Implications for COVID-19 Associated Neurological Manifestations. Exp. Neurol. 2021, 345, 113837. [Google Scholar] [CrossRef] [PubMed]
- Raisinghani, N.; Alshahrani, M.; Gupta, G.; Verkhivker, G. AlphaFold2 Modeling and Molecular Dynamics Simulations of the Conformational Ensembles for the SARS-CoV-2 Spike Omicron JN.1, KP.2 and KP.3 Variants: Mutational Profiling of Binding Energetics Reveals Epistatic Drivers of the ACE2 Affinity and Escape Hotspots of Antibody Resistance. Viruses 2024, 16, 1458. [Google Scholar] [CrossRef] [PubMed]
- Raisinghani, N.; Alshahrani, M.; Gupta, G.; Verkhivker, G. Atomistic Prediction of Structures, Conformational Ensembles and Binding Energetics for the SARS-CoV-2 Spike JN.1, KP.2 and KP.3 Variants Using AlphaFold2 and Molecular Dynamics Simulations: Mutational Profiling and Binding Free Energy Analysis Reveal Epistatic Hotspots of the ACE2 Affinity and Immune Escape. Chapman University School of Pharmacy, Irvine, CA, United States of America. bioRxiv Prepr. Serv. Biol. 2024; manuscript to be submitted. [Google Scholar] [CrossRef]
- Flaherty, G.T.; Hession, P.; Liew, C.H.; Lim, B.C.W.; Leong, T.K.; Lim, V.; Sulaiman, L.H. COVID-19 in Adult Patients with Pre-Existing Chronic Cardiac, Respiratory and Metabolic Disease: A Critical Literature Review with Clinical Recommendations. Trop. Dis. Travel Med. Vaccines 2020, 6, 16. [Google Scholar] [CrossRef]
- Brola, W.; Wilski, M. Neurological Consequences of COVID-19. Pharmacol. Rep. 2022, 74, 1208–1222. [Google Scholar] [CrossRef]
- Zaib, S.; Javed, H.; Khan, I.; Jaber, F.; Sohail, A.; Zaib, Z.; Mehboob, T.; Tabassam, N.; Ogaly, H.A. Neurodegenerative Diseases: Their Onset, Epidemiology, Causes and Treatment. ChemistrySelect 2023, 8, e202300225. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of the Parkinson Pandemic. J. Park. Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef]
- Antony, P.M.A.; Diederich, N.J.; Krüger, R.; Balling, R. The Hallmarks of Parkinson’s Disease. FEBS J. 2013, 280, 5981–5993. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Braak, H.; de Vos, R.A.I.; Bohl, J.; Del Tredici, K. Gastric Alpha-Synuclein Immunoreactive Inclusions in Meissner’s and Auerbach’s Plexuses in Cases Staged for Parkinson’s Disease-Related Brain Pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef]
- Dogra, N.; Mani, R.J.; Katare, D.P. The Gut-Brain Axis: Two Ways Signaling in Parkinson’s Disease. Cell. Mol. Neurobiol. 2022, 42, 315–332. [Google Scholar] [CrossRef]
- Roos, D.S.; Klein, M.; Deeg, D.J.H.; Doty, R.L.; Berendse, H.W. Prevalence of Prodromal Symptoms of Parkinson’s Disease in the Late Middle-Aged Population. J. Parkinsons. Dis. 2022, 12, 967–974. [Google Scholar] [CrossRef]
- Leite Silva, A.B.R.; Gonçalves de Oliveira, R.W.; Diógenes, G.P.; de Castro Aguiar, M.F.; Sallem, C.C.; Lima, M.P.P.; de Albuquerque Filho, L.B.; Peixoto de Medeiros, S.D.; Penido de Mendonça, L.L.; de Santiago Filho, P.C.; et al. Premotor, Nonmotor and Motor Symptoms of Parkinson’s Disease: A New Clinical State of the Art. Ageing Res. Rev. 2023, 84, 101834. [Google Scholar] [CrossRef]
- Leta, V.; Urso, D.; Batzu, L.; Lau, Y.H.; Mathew, D.; Boura, I.; Raeder, V.; Falup-Pecurariu, C.; van Wamelen, D.; Ray Chaudhuri, K. Viruses, Parkinsonism and Parkinson’s Disease: The Past, Present and Future. J. Neural Transm. 2022, 129, 1119–1132. [Google Scholar] [CrossRef]
- Méndez-Guerrero, A.; Laespada-García, M.I.; Gómez-Grande, A.; Ruiz-Ortiz, M.; Blanco-Palmero, V.A.; Azcarate-Diaz, F.J.; Rábano-Suárez, P.; Álvarez-Torres, E.; de Fuenmayor-Fernández de la Hoz, C.P.; Vega Pérez, D.; et al. Acute Hypokinetic-Rigid Syndrome Following SARS-CoV-2 Infection. Neurology 2020, 95, e2109–e2118. [Google Scholar] [CrossRef]
- Cohen, M.E.; Eichel, R.; Steiner-Birmanns, B.; Janah, A.; Ioshpa, M.; Bar-Shalom, R.; Paul, J.J.; Gaber, H.; Skrahina, V.; Bornstein, N.M.; et al. A Case of Probable Parkinson’s Disease after SARS-CoV-2 Infection. Lancet. Neurol. 2020, 19, 804–805. [Google Scholar] [CrossRef]
- Faber, I.; Brandão, P.R.P.; Menegatti, F.; de Carvalho Bispo, D.D.; Maluf, F.B.; Cardoso, F. Coronavirus Disease 2019 and Parkinsonism: A Non-Post-Encephalitic Case. Mov. Disord. 2020, 35, 1721–1722. [Google Scholar] [CrossRef]
- Idrees, D.; Kumar, V. SARS-CoV-2 Spike Protein Interactions with Amyloidogenic Proteins: Potential Clues to Neurodegeneration. Biochem. Biophys. Res. Commun. 2021, 554, 94–98. [Google Scholar] [CrossRef]
- Sinha, S.; Mittal, S.; Roy, R. Parkinson’s Disease and the COVID-19 Pandemic: A Review Article on the Association between SARS-CoV-2 and α-Synucleinopathy. J. Mov. Disord. 2021, 14, 184–192. [Google Scholar] [CrossRef]
- Conte, C. Possible Link between SARS-CoV-2 Infection and Parkinson’s Disease: The Role of Toll-Like Receptor 4. Int. J. Mol. Sci. 2021, 22, 7135. [Google Scholar] [CrossRef]
- Semerdzhiev, S.A.; Fakhree, M.A.A.; Segers-Nolten, I.; Blum, C.; Claessens, M.M.A.E. Interactions between SARS-CoV-2 N-Protein and α-Synuclein Accelerate Amyloid Formation. ACS Chem. Neurosci. 2022, 13, 143–150. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, X.; Huang, Z.; Ma, K. SARS-CoV-2 Proteins Interact with Alpha Synuclein and Induce Lewy Body-like Pathology In Vitro. Int. J. Mol. Sci. 2022, 23, 3394. [Google Scholar] [CrossRef]
- Philippens, I.H.C.H.M.; Böszörményi, K.P.; Wubben, J.A.M.; Fagrouch, Z.C.; van Driel, N.; Mayenburg, A.Q.; Lozovagia, D.; Roos, E.; Schurink, B.; Bugiani, M.; et al. Brain Inflammation and Intracellular α-Synuclein Aggregates in Macaques after SARS-CoV-2 Infection. Viruses 2022, 14, 776. [Google Scholar] [CrossRef]
- Mesias, V.S.D.; Zhu, H.; Tang, X.; Dai, X.; Liu, W.; Guo, Y.; Huang, J. Moderate Binding between Two SARS-CoV-2 Protein Segments and α-Synuclein Alters Its Toxic Oligomerization Propensity Differently. J. Phys. Chem. Lett. 2022, 13, 10642–10648. [Google Scholar] [CrossRef]
- Datta, A.K.; Mukherjee, A.; Biswas, A. Gastrointestinal, Respiratory, and Olfactory Neurotropism of Sars-Cov2 as a Possible Trigger of Parkinson’s Disease: Is a Multi-Hit Multi-Step Process on the Cards. Ann. Indian Acad. Neurol. 2023, 26, 127–136. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Kaushik, A.; Kujawska, M.; Ahmed, E.A.; Batiha, G.E.-S. SARS-COV-2 Infection and Parkinson’s Disease: Possible Links and Perspectives. J. Neurosci. Res. 2023, 101, 952–975. [Google Scholar] [CrossRef]
- Dulski, J.; Sławek, J. Incidence and Characteristics of Post-COVID-19 Parkinsonism and Dyskinesia Related to COVID-19 Vaccines. Neurol. Neurochir. Pol. 2023, 57, 53–62. [Google Scholar] [CrossRef]
- Sandeep; Subba, R.; Mondal, A.C. Does COVID-19 Trigger the Risk for the Development of Parkinson’s Disease? Therapeutic Potential of Vitamin C. Mol. Neurobiol. 2023. online ahead of print. [Google Scholar] [CrossRef]
- Mahin, A.; Soman, S.P.; Modi, P.K.; Raju, R.; Keshava Prasad, T.S.; Abhinand, C.S. Meta-Analysis of the Serum/Plasma Proteome Identifies Significant Associations between COVID-19 with Alzheimer’s/Parkinson’s Diseases. J. Neurovirol. 2024, 30, 57–70. [Google Scholar] [CrossRef]
- Iravanpour, F.; Farrokhi, M.R.; Jafarinia, M.; Oliaee, R.T. The Effect of SARS-CoV-2 on the Development of Parkinson’s Disease: The Role of α-Synuclein. Hum. Cell 2024, 37, 1–8. [Google Scholar] [CrossRef]
- Boura, I.; Qamar, M.A.; Daddoveri, F.; Leta, V.; Poplawska-domaszewicz, K.; Falup-pecurariu, C.; Chaudhuri, K.R. SARS-CoV-2 and Parkinson’s Disease: A Review of Where We Are Now. Biomedicines 2023, 11, 2524. [Google Scholar] [CrossRef]
- Rao, A.R.; Hidayathullah, S.M.; Hegde, K.; Adhikari, P. Parkinsonism: An Emerging Post COVID Sequelae. IDCases 2022, 27, e01388. [Google Scholar] [CrossRef]
- Calculli, A.; Bocci, T.; Porcino, M.; Avenali, M.; Casellato, C.; Arceri, S.; Regalbuto, S.; Priori, A.; Pisani, A. Parkinson Disease Following COVID-19: Report of Six Cases. Eur. J. Neurol. 2023, 30, 1272–1280. [Google Scholar] [CrossRef]
- Cavallieri, F.; Fioravanti, V.; Bove, F.; Del Prete, E.; Meoni, S.; Grisanti, S.; Zedde, M.; Pascarella, R.; Moro, E.; Valzania, F. COVID-19 and Parkinsonism: A Critical Appraisal. Biomolecules 2022, 12, 970. [Google Scholar] [CrossRef]
- A Albornoz, E.A.; Amarilla, A.A.; Modhiran, N.; Parker, S.; Li, X.X.; Wijesundara, D.K.; Aguado, J.; Zamora, A.P.; McMillan, C.L.D.; Liang, B.; et al. SARS-CoV-2 Drives NLRP3 Inflammasome Activation in Human Microglia through Spike Protein. Mol. Psychiatry 2023, 28, 2878–2893. [Google Scholar] [CrossRef]
- Rahmati, M.; Yon, D.K.; Lee, S.W.; Soysal, P.; Koyanagi, A.; Jacob, L.; Li, Y.; Park, J.M.; Kim, Y.W.; Shin, J.I.; et al. New-Onset Neurodegenerative Diseases as Long-Term Sequelae of SARS-CoV-2 Infection: A Systematic Review and Meta-Analysis. J. Med. Virol. 2023, 95, e28909. [Google Scholar] [CrossRef]
- Anghelescu, A.; Onose, G.; Popescu, C.; Băilă, M.; Stoica, S.I.; Postoiu, R.; Brumă, E.; Petcu, I.R.; Ciobanu, V.; Munteanu, C. Parkinson’s Disease and SARS-CoV-2 Infection: Particularities of Molecular and Cellular Mechanisms Regarding Pathogenesis and Treatment. Biomedicines 2022, 10, 1000. [Google Scholar] [CrossRef]
- Zhang, J.; Bishir, M.; Barbhuiya, S.; Chang, S.L. Meta-Analysis of the Mechanisms Underlying COVID-19 Modulation of Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 13554. [Google Scholar] [CrossRef]
- Ali, S.S.; Mumtaz, A.; Qamar, M.A.; Tebha, S.S.; Parhin, A.; Butt, M.; Essar, M.Y. New-Onset Parkinsonism as a COVID-19 Infection Sequela: A Systematic Review and Meta-Analysis. Ann. Med. Surg. 2022, 80, 104281. [Google Scholar] [CrossRef]
- Crunfli, F.; Carregari, V.C.; Veras, F.P.; Silva, L.S.; Nogueira, M.H.; Antunes, A.S.L.M.; Vendramini, P.H.; Valença, A.G.F.; Brandão-Teles, C.; Zuccoli, G.d.S.; et al. Morphological, Cellular, and Molecular Basis of Brain Infection in COVID-19 Patients. Proc. Natl. Acad. Sci. USA 2022, 119, e2200960119. [Google Scholar] [CrossRef]
- Rota, S.; Boura, I.; Wan, Y.-M.; Lazcano-Ocampo, C.; Rodriguez-Violante, M.; Antonini, A.; Chaudhuri, K.R. Spotlight on Non-Motor Symptoms and COVID-19. Int. Rev. Neurobiol. 2022, 165, 103–133. [Google Scholar] [CrossRef]
- Bougea, A.; Georgakopoulou, V.E.; Palkopoulou, M.; Efthymiopoulou, E.; Angelopoulou, E.; Spandidos, D.A.; Zikos, P. New-onset Non-motor Symptoms in Patients with Parkinson’s Disease and Post-COVID-19 Syndrome: A Prospective Cross-sectional Study. Med. Int. 2023, 3, 23. [Google Scholar] [CrossRef]
- Xian, W.; Lin, L.; Wu, W.; Su, F.; Pei, Z. Fatigue and Long Duration of Infection Are Associated with Worsen Motor and Non-motor Symptoms in Parkinson’s Disease Following Omicron COVID-19 Pandemic. Brain Behav. 2024, 14, e3396. [Google Scholar] [CrossRef]
- Mameli, F.; Zirone, E.; Capetti, B.; Mellace, D.; Ferrucci, R.; Franco, G.; Di Fonzo, A.; Barbieri, S.; Ruggiero, F. Changes in Non-Motor Symptoms in Patients with Parkinson’s Disease Following COVID-19 Pandemic Restrictions: A Systematic Review. Front. Psychol. 2022, 13, 939520. [Google Scholar] [CrossRef]
- van der Heide, A.; Meinders, M.J.; Bloem, B.R.; Helmich, R.C. The Impact of the COVID-19 Pandemic on Psychological Distress, Physical Activity, and Symptom Severity in Parkinson’s Disease. J. Parkinsons. Dis. 2020, 10, 1355–1364. [Google Scholar] [CrossRef]
- D’Iorio, A.; Baiano, C.; Maraucci, G.; Vitale, C.; Amboni, M.; Santangelo, G. A Longitudinal Study on the Effects of COVID-19 Pandemic on Non-Motor Symptoms in Parkinson’s Disease. Neurol. Sci. 2022, 43, 4605–4609. [Google Scholar] [CrossRef]
- El-Qushayri, A.E.; Ghozy, S.; Reda, A.; Kamel, A.M.A.; Abbas, A.S.; Dmytriw, A.A. The Impact of Parkinson’s Disease on Manifestations and Outcomes of Covid-19 Patients: A Systematic Review and Meta-Analysis. Rev. Med. Virol. 2022, 32, e2278. [Google Scholar] [CrossRef]
- Afraie, M.; Moradi, G.; Mohammadzedeh, P.; Azami, M.; Riyahifar, S.; Moradi, Y. COVID-19 and Parkinson’s Disease: A Systematic Review and Meta-Analysis. Acta Neurol. Belg. 2023, 123, 1209–1223. [Google Scholar] [CrossRef]
- Park, J.M.; Woo, W.; Lee, S.C.; Park, S.; Yon, D.K.; Lee, S.W.; Smith, L.; Koyanagi, A.; Shin, J.I.; Kim, Y.W. Prevalence and Mortality Risk of Neurological Disorders during the COVID-19 Pandemic: An Umbrella Review of the Current Evidence. Neuroepidemiology 2023, 57, 129–147. [Google Scholar] [CrossRef]
- Smadi, M.; Kaburis, M.; Schnapper, Y.; Reina, G.; Molero, P.; Molendijk, M.L. SARS-CoV-2 Susceptibility and COVID-19 Illness Course and Outcome in People with Pre-Existing Neurodegenerative Disorders: Systematic Review with Frequentist and Bayesian Meta-Analyses. Br. J. Psychiatry 2023, 223, 348–361. [Google Scholar] [CrossRef]
- Boruah, A.P.; Thakur, K.T.; Gadani, S.P.; Kothari, K.U.; Chomba, M.; Guekht, A.; Heydari, K.; Hoo, F.K.; Hwang, S.; Michael, B.D.; et al. Pre-Existing Neurological Conditions and COVID-19 Co-Infection: Data from Systematic Reviews, Meta-Analyses, and Scoping Reviews. J. Neurol. Sci. 2023, 455, 120858. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, M.P.B.; de Castro, A.E.F.; Miri, A.L.; Lima, C.R.; Truax, B.D.; Probst, V.S.; Smaili, S.M. The Impact of the COVID-19 Pandemic on Neuropsychiatric and Sleep Disorders, and Quality of Life in Individuals with Neurodegenerative and Demyelinating Diseases: A Systematic Review and Meta-Analysis of Observational Studies. BMC Neurol. 2023, 23, 150. [Google Scholar] [CrossRef] [PubMed]
- Mai, A.S.; Yong, J.H.; Tan, B.J.-W.; Xiao, B.; Tan, E.-K. Impact of COVID-19 Pandemic on Patients with Parkinson’s Disease: A Meta-Analysis of 13,878 Patients. Ann. Clin. Transl. Neurol. 2022, 9, 1504–1513. [Google Scholar] [CrossRef] [PubMed]
- Khoshnood, R.J.; Zali, A.; Tafreshinejad, A.; Ghajarzadeh, M.; Ebrahimi, N.; Safari, S.; Mirmosayyeb, O. Parkinson’s Disease and COVID-19: A Systematic Review and Meta-Analysis. Neurol. Sci. 2022, 43, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Chambergo-Michilot, D.; Barros-Sevillano, S.; Rivera-Torrejón, O.; De la Cruz-Ku, G.A.; Custodio, N. Factors Associated with COVID-19 in People with Parkinson’s Disease: A Systematic Review and Meta-Analysis. Eur. J. Neurol. 2021, 28, 3467–3477. [Google Scholar] [CrossRef]
- Putri, C.; Hariyanto, T.I.; Hananto, J.E.; Christian, K.; Situmeang, R.F.V.; Kurniawan, A. Parkinson’s Disease May Worsen Outcomes from Coronavirus Disease 2019 (COVID-19) Pneumonia in Hospitalized Patients: A Systematic Review, Meta-Analysis, and Meta-Regression. Park. Relat. Disord. 2021, 87, 155–161. [Google Scholar] [CrossRef]
- Xiang, P.; Xu, X.; Lu, X.; Gao, L.; Wang, H.; Li, Z.; Xiong, H.; Li, R.; Xiong, Y.; Pu, L.; et al. Case Report: Identification of SARS-CoV-2 in Cerebrospinal Fluid by Ultrahigh-Depth Sequencing in a Patient With Coronavirus Disease 2019 and Neurological Dysfunction. Front. Med. 2021, 8, 629828. [Google Scholar] [CrossRef]
- Achar, A.; Ghosh, C. COVID-19-Associated Neurological Disorders: The Potential Route of CNS Invasion and Blood-Brain Relevance. Cells 2020, 9, 2360. [Google Scholar] [CrossRef]
- Emmi, A.; Sandre, M.; Porzionato, A.; Antonini, A. Smell Deficits in COVID-19 and Possible Links with Parkinson’s Disease. Int. Rev. Neurobiol. 2022, 165, 91–102. [Google Scholar] [CrossRef]
- Butowt, R.; Bilinska, K.; von Bartheld, C.S. Olfactory Dysfunction in COVID-19: New Insights into the Underlying Mechanisms. Trends Neurosci. 2023, 46, 75–90. [Google Scholar] [CrossRef]
- Klingenstein, M.; Klingenstein, S.; Neckel, P.H.; Mack, A.F.; Wagner, A.P.; Kleger, A.; Liebau, S.; Milazzo, A. Evidence of SARS-CoV2 Entry Protein ACE2 in the Human Nose and Olfactory Bulb. Cells. Tissues. Organs 2020, 209, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Bryche, B.; St Albin, A.; Murri, S.; Lacôte, S.; Pulido, C.; Ar Gouilh, M.; Lesellier, S.; Servat, A.; Wasniewski, M.; Picard-Meyer, E.; et al. Massive Transient Damage of the Olfactory Epithelium Associated with Infection of Sustentacular Cells by SARS-CoV-2 in Golden Syrian Hamsters. Brain. Behav. Immun. 2020, 89, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Shen, W.; Rowan, N.R.; Kulaga, H.; Hillel, A.; Ramanathan, M.; Lane, A.P. Elevated ACE-2 Expression in the Olfactory Neuroepithelium: Implications for Anosmia and Upper Respiratory SARS-CoV-2 Entry and Replication. Eur. Respir. J. 2020, 56, 2001948. [Google Scholar] [CrossRef] [PubMed]
- Fodoulian, L.; Tuberosa, J.; Rossier, D.; Boillat, M.; Kan, C.; Pauli, V.; Egervari, K.; Lobrinus, J.A.; Landis, B.N.; Carleton, A.; et al. SARS-CoV-2 Receptors and Entry Genes Are Expressed in the Human Olfactory Neuroepithelium and Brain. iScience 2020, 23, 101839. [Google Scholar] [CrossRef]
- Brann, D.H.; Tsukahara, T.; Weinreb, C.; Lipovsek, M.; Van den Berge, K.; Gong, B.; Chance, R.; Macaulay, I.C.; Chou, H.-J.; Fletcher, R.B.; et al. Non-Neuronal Expression of SARS-CoV-2 Entry Genes in the Olfactory System Suggests Mechanisms Underlying COVID-19-Associated Anosmia. Sci. Adv. 2020, 6, eabc5801. [Google Scholar] [CrossRef]
- Bilinska, K.; Jakubowska, P.; Von Bartheld, C.S.; Butowt, R. Expression of the SARS-CoV-2 Entry Proteins, ACE2 and TMPRSS2, in Cells of the Olfactory Epithelium: Identification of Cell Types and Trends with Age. ACS Chem. Neurosci. 2020, 11, 1555–1562. [Google Scholar] [CrossRef]
- Divani, A.A.; Andalib, S.; Biller, J.; Di Napoli, M.; Moghimi, N.; Rubinos, C.A.; Nobleza, C.O.; Sylaja, P.N.; Toledano, M.; Lattanzi, S.; et al. Central Nervous System Manifestations Associated with COVID-19. Curr. Neurol. Neurosci. Rep. 2020, 20, 60. [Google Scholar] [CrossRef]
- Torres-Pasillas, G.; Chi-Castañeda, D.; Carrillo-Castilla, P.; Marín, G.; Hernández-Aguilar, M.E.; Aranda-Abreu, G.E.; Manzo, J.; García, L.I. Olfactory Dysfunction in Parkinson’s Disease, Its Functional and Neuroanatomical Correlates. NeuroSci 2023, 4, 134–151. [Google Scholar] [CrossRef]
- Li, H.; Qian, J.; Wang, Y.; Wang, J.; Mi, X.; Qu, L.; Song, N.; Xie, J. Potential Convergence of Olfactory Dysfunction in Parkinson’s Disease and COVID-19: The Role of Neuroinflammation. Ageing Res. Rev. 2024, 97, 102288. [Google Scholar] [CrossRef]
- Käufer, C.; Schreiber, C.S.; Hartke, A.-S.; Denden, I.; Stanelle-Bertram, S.; Beck, S.; Kouassi, N.M.; Beythien, G.; Becker, K.; Schreiner, T.; et al. Microgliosis and Neuronal Proteinopathy in Brain Persist beyond Viral Clearance in SARS-CoV-2 Hamster Model. EBioMedicine 2022, 79, 103999. [Google Scholar] [CrossRef]
- Schreiber, C.S.; Wiesweg, I.; Stanelle-Bertram, S.; Beck, S.; Kouassi, N.M.; Schaumburg, B.; Gabriel, G.; Richter, F.; Käufer, C. Sex-Specific Biphasic Alpha-Synuclein Response and Alterations of Interneurons in a COVID-19 Hamster Model. eBioMedicine 2024, 105, 105191. [Google Scholar] [CrossRef] [PubMed]
- Emmi, A.; Rizzo, S.; Barzon, L.; Sandre, M.; Carturan, E.; Sinigaglia, A.; Riccetti, S.; Della Barbera, M.; Boscolo-Berto, R.; Cocco, P.; et al. Detection of SARS-CoV-2 Viral Proteins and Genomic Sequences in Human Brainstem Nuclei. NPJ Park. Dis. 2023, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Laurent, S.; Onur, O.A.; Kleineberg, N.N.; Fink, G.R.; Schweitzer, F.; Warnke, C. A Systematic Review of Neurological Symptoms and Complications of COVID-19. J. Neurol. 2021, 268, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Khalefah, M.M.; Khalifah, A.M. Determining the Relationship between SARS-CoV-2 Infection, Dopamine, and COVID-19 Complications. J. Taibah Univ. Med. Sci. 2020, 15, 550–553. [Google Scholar] [CrossRef]
- Riederer, P.; Monoranu, C.; Strobel, S.; Iordache, T.; Sian-Hülsmann, J. Iron as the Concert Master in the Pathogenic Orchestra Playing in Sporadic Parkinson’s Disease. J. Neural Transm. 2021, 128, 1577–1598. [Google Scholar] [CrossRef]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s Disease: Why Is Advancing Age the Biggest Risk Factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef]
- Wang, Q.; Oyarzabal, E.; Wilson, B.; Qian, L.; Hong, J.-S. Substance P Enhances Microglial Density in the Substantia Nigra through Neurokinin-1 Receptor/NADPH Oxidase-Mediated Chemotaxis in Mice. Clin. Sci. (Lond.) 2015, 129, 757–767. [Google Scholar] [CrossRef]
- Tulisiak, C.T.; Mercado, G.; Peelaerts, W.; Brundin, L.; Brundin, P. Can Infections Trigger Alpha-Synucleinopathies? Prog. Mol. Biol. Transl. Sci. 2019, 168, 299–322. [Google Scholar] [CrossRef]
- Philippens, I.H.C.H.M.; Böszörményi, K.P.; Wubben, J.A.; Fagrouch, Z.C.; van Driel, N.; Mayenburg, A.Q.; Lozovagia, D. SARS-CoV-2 Causes Brain Inflammation and Induces Lewy Body Formation in Macaques. bioRxiv, 2021; manuscript to be submitted. [Google Scholar] [CrossRef]
- Chang, M.H.; Park, J.H.; Lee, H.K.; Choi, J.Y.; Koh, Y.H. SARS-CoV-2 Spike Protein 1 Causes Aggregation of α-Synuclein via Microglia-Induced Inflammation and Production of Mitochondrial ROS: Potential Therapeutic Applications of Metformin. Biomedicines 2024, 12, 1223. [Google Scholar] [CrossRef]
- Semerdzhiev, S.A.; Segers-Nolten, I.; van der Schoot, P.; Blum, C.; Claessens, M.M.A.E. SARS-CoV-2 N-Protein Induces the Formation of Composite α-Synuclein/N-Protein Fibrils That Transform into a Strain of α-Synuclein Fibrils. Nanoscale 2023, 15, 18337–18346. [Google Scholar] [CrossRef] [PubMed]
- Arawaka, S.; Sato, H.; Sasaki, A.; Koyama, S.; Kato, T. Mechanisms Underlying Extensive Ser129-Phosphorylation in α-Synuclein Aggregates. Acta Neuropathol. Commun. 2017, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Santerre, M.; Arjona, S.P.; Allen, C.N.; Callen, S.; Buch, S.; Sawaya, B.E. HIV-1 Vpr Protein Impairs Lysosome Clearance Causing SNCA/Alpha-Synuclein Accumulation in Neurons. Autophagy 2021, 17, 1768–1782. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome Inhibition Prevents α-Synuclein Pathology and Dopaminergic Neurodegeneration in Mice. Sci. Transl. Med. 2018, 10, 139–148. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Pavel, A.; Murray, D.K.; Stoessl, A.J. COVID-19 and Selective Vulnerability to Parkinson’s Disease. Lancet. Neurol. 2020, 19, 719. [Google Scholar] [CrossRef]
- Rosen, B.; Kurtishi, A.; Vazquez-Jimenez, G.R.; Møller, S.G. The Intersection of Parkinson’s Disease, Viral Infections, and COVID-19. Mol. Neurobiol. 2021, 58, 4477–4486. [Google Scholar] [CrossRef]
- Beatman, E.L.; Massey, A.; Shives, K.D.; Burrack, K.S.; Chamanian, M.; Morrison, T.E.; Beckham, J.D. Alpha-Synuclein Expression Restricts RNA Viral Infections in the Brain. J. Virol. 2015, 90, 2767–2782. [Google Scholar] [CrossRef]
- Jang, H.; Boltz, D.; Sturm-Ramirez, K.; Shepherd, K.R.; Jiao, Y.; Webster, R.; Smeyne, R.J. Highly Pathogenic H5N1 Influenza Virus Can Enter the Central Nervous System and Induce Neuroinflammation and Neurodegeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 14063–14068. [Google Scholar] [CrossRef]
- Massey, A.R.; Beckham, J.D. Alpha-Synuclein, a Novel Viral Restriction Factor Hiding in Plain Sight. DNA Cell Biol. 2016, 35, 643–645. [Google Scholar] [CrossRef]
- Limanaqi, F.; Zecchini, S.; Saulle, I.; Strizzi, S.; Vanetti, C.; Garziano, M.; Cappelletti, G.; Parolin, D.; Caccia, S.; Trabattoni, D.; et al. Alpha-Synuclein Dynamics Bridge Type-I Interferon Response and SARS-CoV-2 Replication in Peripheral Cells. Biol. Res. 2024, 57, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Liu, H.; Gao, S.; Jiang, N.; Chen, S.; Xie, W. Effect of Salidroside on Neuroprotection and Psychiatric Sequelae during the COVID-19 Pandemic: A Review. Biomed. Pharmacother. 2024, 170, 115999. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Feng, Y.; Yang, R.; Wu, L.; Li, R.; Huang, L.; Yang, Q.; Chen, J. Salidroside Promotes the Pathological α-Synuclein Clearance Through Ubiquitin-Proteasome System in SH-SY5Y Cells. Front. Pharmacol. 2018, 9, 377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Y.; Li, R.; Zhu, L.; Fu, B.; Yan, T. Salidroside Ameliorates Parkinson’s Disease by Inhibiting NLRP3-Dependent Pyroptosis. Aging (Albany NY) 2020, 12, 9405–9426. [Google Scholar] [CrossRef]
- Halma, M.T.J.; Marik, P.E.; Saleeby, Y.M. Exploring Autophagy in Treating SARS-CoV-2 Spike Protein-Related Pathology. Endocr. Metab. Sci. 2024, 14, 100163. [Google Scholar] [CrossRef]
- Liu, T.; Wang, P.; Yin, H.; Wang, X.; Lv, J.; Yuan, J.; Zhu, J.; Wang, Y. Rapamycin Reverses Ferroptosis by Increasing Autophagy in MPTP/MPP+-Induced Models of Parkinson’s Disease. Neural Regen. Res. 2023, 18, 2514–2519. [Google Scholar] [CrossRef]
- Gopar-Cuevas, Y.; Saucedo-Cardenas, O.; Loera-Arias, M.J.; Montes-de-Oca-Luna, R.; Rodriguez-Rocha, H.; Garcia-Garcia, A. Metformin and Trehalose-Modulated Autophagy Exerts a Neurotherapeutic Effect on Parkinson’s Disease. Mol. Neurobiol. 2023, 60, 7253–7273. [Google Scholar] [CrossRef]
- Hasan, D.; Shono, A.; van Kalken, C.K.; van der Spek, P.J.; Krenning, E.P.; Kotani, T. A Novel Definition and Treatment of Hyperinflammation in COVID-19 Based on Purinergic Signalling. Purinergic Signal. 2022, 18, 13–59. [Google Scholar] [CrossRef]
- Simões, J.L.B.; de Carvalho Braga, G.; Eichler, S.W.; da Silva, G.B.; Bagatini, M.D. Implications of COVID-19 in Parkinson’s Disease: The Purinergic System in a Therapeutic-Target Perspective to Diminish Neurodegeneration. Purinergic Signal. 2024, 20, 487–507. [Google Scholar] [CrossRef]
- Gan, M.; Moussaud, S.; Jiang, P.; McLean, P.J. Extracellular ATP Induces Intracellular Alpha-Synuclein Accumulation via P2X1 Receptor-Mediated Lysosomal Dysfunction. Neurobiol. Aging 2015, 36, 1209–1220. [Google Scholar] [CrossRef]
- Mizoguchi, K.; Yokoo, H.; Yoshida, M.; Tanaka, T.; Tanaka, M. Amantadine Increases the Extracellular Dopamine Levels in the Striatum by Re-Uptake Inhibition and by N-Methyl-D-Aspartate Antagonism. Brain Res. 1994, 662, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Rascol, O.; Fabbri, M.; Poewe, W. Amantadine in the Treatment of Parkinson’s Disease and Other Movement Disorders. Lancet. Neurol. 2021, 20, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Marmol, S.; Feldman, M.; Singer, C.; Margolesky, J. Amantadine Revisited: A Contender for Initial Treatment in Parkinson’s Disease? CNS Drugs 2021, 35, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.D.; Lyons, K.E.; Pahwa, R. Amantadine Extended-Release Capsules for Levodopa-Induced Dyskinesia in Patients with Parkinson’s Disease. Ther. Clin. Risk Manag. 2018, 14, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.P.; Sodhi, K.K.; Bali, A.K.; Shree, P. Influenza A Virus and Its Antiviral Drug Treatment Options. Med. Microecol. 2023, 16, 100083. [Google Scholar] [CrossRef]
- Butterworth, R.F. Potential for the Repurposing of Adamantane Antivirals for COVID-19. Drugs R D 2021, 21, 267–272. [Google Scholar] [CrossRef]
- Cortés-Borra, A.; Aranda-Abreu, G.E. Amantadine in the Prevention of Clinical Symptoms Caused by SARS-CoV-2. Pharmacol. Rep. 2021, 73, 962–965. [Google Scholar] [CrossRef]
- Rejdak, K.; Grieb, P. Adamantanes Might Be Protective from COVID-19 in Patients with Neurological Diseases: Multiple Sclerosis, Parkinsonism and Cognitive Impairment. Mult. Scler. Relat. Disord. 2020, 42, 102163. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Castaño-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Severe Acute Respiratory Syndrome Coronavirus E Protein Transports Calcium Ions and Activates the NLRP3 Inflammasome. Virology 2015, 485, 330–339. [Google Scholar] [CrossRef]
- Toft-Bertelsen, T.L.; Jeppesen, M.G.; Tzortzini, E.; Xue, K.; Giller, K.; Becker, S.; Mujezinovic, A.; Bentzen, B.H.; Andreas, L.B.; Kolocouris, A.; et al. Amantadine Has Potential for the Treatment of COVID-19 Because It Inhibits Known and Novel Ion Channels Encoded by SARS-CoV-2. Commun. Biol. 2021, 4, 1347. [Google Scholar] [CrossRef]
- Fink, K.; Nitsche, A.; Neumann, M.; Grossegesse, M.; Eisele, K.-H.; Danysz, W. Amantadine Inhibits SARS-CoV-2 In Vitro. Viruses 2021, 13, 539. [Google Scholar] [CrossRef] [PubMed]
- Przytuła, F.; Kasprzak, J.; Dulski, J.; Koziorowski, D.; Kwaśniak-Butowska, M.; Sołtan, W.; Roszmann, A.; Śmiłowska, K.; Schinwelski, M.; Sławek, J. Morbidity and Severity of COVID-19 in Patients with Parkinson’s Disease Treated with Amantadine—A Multicenter, Retrospective, Observational Study. Park. Relat. Disord. 2023, 106, 105238. [Google Scholar] [CrossRef] [PubMed]
- Weis, N.; Bollerup, S.; Sund, J.D.; Glamann, J.B.; Vinten, C.; Jensen, L.R.; Sejling, C.; Kledal, T.N.; Rosenkilde, M.M. Amantadine for COVID-19 Treatment (ACT) Study: A Randomized, Double-Blinded, Placebo-Controlled Clinical Trial. Clin. Microbiol. Infect. 2023, 29, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Kamel, W.A.; Kamel, M.I.; Alhasawi, A.; Elmasry, S.; AlHamdan, F.; Al-Hashel, J.Y. Effect of Pre-Exposure Use of Amantadine on COVID-19 Infection: A Hospital-Based Cohort Study in Patients With Parkinson’s Disease or Multiple Sclerosis. Front. Neurol. 2021, 12, 704186. [Google Scholar] [CrossRef] [PubMed]
- Harandi, A.A.; Pakdaman, H.; Medghalchi, A.; Kimia, N.; Kazemian, A.; Siavoshi, F.; Barough, S.S.; Esfandani, A.; Hosseini, M.H.; Sobhanian, S.A. A Randomized Open-Label Clinical Trial on the Effect of Amantadine on Post Covid 19 Fatigue. Sci. Rep. 2024, 14, 1343. [Google Scholar] [CrossRef]
- Müller, T.; Riederer, P.; Kuhn, W. Aminoadamantanes: From Treatment of Parkinson’s and Alzheimer’s Disease to Symptom Amelioration of Long COVID-19 Syndrome? Expert Rev. Clin. Pharmacol. 2023, 16, 101–107. [Google Scholar] [CrossRef]
- Khayachi, A.; Ase, A.; Liao, C.; Kamesh, A.; Kuhlmann, N.; Schorova, L.; Chaumette, B.; Dion, P.; Alda, M.; Séguéla, P.; et al. Chronic Lithium Treatment Alters the Excitatory/Inhibitory Balance of Synaptic Networks and Reduces MGluR5-PKC Signalling in Mouse Cortical Neurons. J. Psychiatry Neurosci. 2021, 46, E402–E414. [Google Scholar] [CrossRef]
- Singulani, M.P.; Ferreira, A.F.F.; Figueroa, P.S.; Cuyul-Vásquez, I.; Talib, L.L.; Britto, L.R.; Forlenza, O.V. Lithium and Disease Modification: A Systematic Review and Meta-Analysis in Alzheimer’s and Parkinson’s Disease. Ageing Res. Rev. 2024, 95, 102231. [Google Scholar] [CrossRef]
- De Picker, L.J.; Leboyer, M.; Geddes, J.R.; Morrens, M.; Harrison, P.J.; Taquet, M. Association between Serum Lithium Level and Incidence of COVID-19 Infection. Br. J. Psychiatry 2022, 221, 425–427. [Google Scholar] [CrossRef]
- Garg, A.; Thapliyal, K.; Singh, V.P. Naltrexone beyond Psychiatric Domain. Indian J. Med. Spec. 2022, 13, 211–215. [Google Scholar] [CrossRef]
- Kučić, N.; Rački, V.; Šverko, R.; Vidović, T.; Grahovac, I.; Mršić-Pelčić, J. Immunometabolic Modulatory Role of Naltrexone in BV-2 Microglia Cells. Int. J. Mol. Sci. 2021, 22, 8429. [Google Scholar] [CrossRef] [PubMed]
- Choubey, A.; Dehury, B.; Kumar, S.; Medhi, B.; Mondal, P. Naltrexone a Potential Therapeutic Candidate for COVID-19. J. Biomol. Struct. Dyn. 2022, 40, 963–970. [Google Scholar] [CrossRef]
- Lin, K.-J.; Chen, S.-D.; Lin, K.-L.; Liou, C.-W.; Lan, M.-Y.; Chuang, Y.-C.; Wang, P.-W.; Lee, J.-J.; Wang, F.-S.; Lin, H.-Y.; et al. Iron Brain Menace: The Involvement of Ferroptosis in Parkinson Disease. Cells 2022, 11, 3829. [Google Scholar] [CrossRef] [PubMed]
- Chaubey, G.K.; Dilawari, R.; Modanwal, R.; Talukdar, S.; Dhiman, A.; Raje, C.I.; Raje, M. Excess Iron Aggravates the Severity of COVID-19 Infection. Free Radic. Biol. Med. 2023, 208, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Zeinivand, M.; Sharifi, M.; Hassanshahi, G.; Nedaei, S.E. Deferoxamine Has the Potential to Improve the COVID-19-Related Inflammatory Response in Diabetic Patients. Int. J. Pept. Res. Ther. 2023, 29, 63. [Google Scholar] [CrossRef] [PubMed]
- Naidu, S.A.G.; Clemens, R.A.; Naidu, A.S. SARS-CoV-2 Infection Dysregulates Host Iron (Fe)-Redox Homeostasis (Fe-R-H): Role of Fe-Redox Regulators, Ferroptosis Inhibitors, Anticoagulants, and Iron-Chelators in COVID-19 Control. J. Diet. Suppl. 2023, 20, 312–371. [Google Scholar] [CrossRef]
- Almutary, A.M.; Althunayyan, S.; Bagalb, A.S.; Mady, A.F.; Alenazi, L.; Mumtaz, S.A.; Al-Hammad, Z.; Abdulrahman, B.; Al-Odat, M.A.; Mhawish, H.; et al. Deferoxamine in the Management of COVID-19 Adult Patients Admitted to ICU: A Prospective Observational Cohort Study. Ann. Med. Surg. 2023, 85, 1468–1474. [Google Scholar] [CrossRef]
- Qiu, B.; Zandkarimi, F.; Saqi, A.; Castagna, C.; Tan, H.; Sekulic, M.; Miorin, L.; Hibshoosh, H.; Toyokuni, S.; Uchida, K.; et al. Fatal COVID-19 Pulmonary Disease Involves Ferroptosis. Nat. Commun. 2024, 15, 3816. [Google Scholar] [CrossRef]
- Soni, S.; Lukhey, M.S.; Thawkar, B.S.; Chintamaneni, M.; Kaur, G.; Joshi, H.; Ramniwas, S.; Tuli, H.S. A Current Review on P2X7 Receptor Antagonist Patents in the Treatment of Neuroinflammatory Disorders: A Patent Review on Antagonists. Naunyn. Schmiedebergs. Arch. Pharmacol. 2024, 397, 4643–4656. [Google Scholar] [CrossRef]
- Yu, H.-G.; Sizemore, G.; Martinez, I.; Perrotta, P. Inhibition of SARS-CoV-2 Viral Channel Activity Using FDA-Approved Channel Modulators Independent of Variants. Biomolecules 2022, 12, 1673. [Google Scholar] [CrossRef]
- Alzaabi, M.M.; Hamdy, R.; Ashmawy, N.S.; Hamoda, A.M.; Alkhayat, F.; Khademi, N.N.; Al Joud, S.M.A.; El-Keblawy, A.A.; Soliman, S.S.M. Flavonoids Are Promising Safe Therapy against COVID-19. Phytochem. Rev. 2022, 21, 291–312. [Google Scholar] [CrossRef] [PubMed]
- Melrose, J.; Smith, M.M. Natural and Semi-Synthetic Flavonoid Anti-SARS-CoV-2 Agents for the Treatment of Long COVID-19 Disease and Neurodegenerative Disorders of Cognitive Decline. Front. Biosci. 2022, 14, 27. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.V.; Chan, M.; Banadyga, L.; He, S.; Zhu, W.; Chrétien, M.; Mbikay, M. Quercetin Inhibits SARS-CoV-2 Infection and Prevents Syncytium Formation by Cells Co-Expressing the Viral Spike Protein and Human ACE2. Virol. J. 2024, 21, 29. [Google Scholar] [CrossRef] [PubMed]
- Tanibuchi, M.; Ueda, S.; Kouno, M.; Otani, K. Dementia with Lewy Bodies after COVID-19 Infection with Catatonia: A Case Report. Psychiatry Clin. Neurosci. Rep. 2023, 2, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.; Heckelmann, E.; Zhou, Y.; Lin, L.; Cuccurullo, S. New-Onset Multiple System Atrophy With Hot Cross Buns Sign Presenting in a Patient With COVID-19. Am. J. Phys. Med. Rehabil. 2023, 102, e54–e55. [Google Scholar] [CrossRef]
- Heidbreder, A.; Sonnweber, T.; Stefani, A.; Ibrahim, A.; Cesari, M.; Bergmann, M.; Brandauer, E.; Tancevski, I.; Löffler-Ragg, J.; Högl, B. Video-Polysomnographic Findings after Acute COVID-19: REM Sleep without Atonia as Sign of CNS Pathology? Sleep Med. 2021, 80, 92–95. [Google Scholar] [CrossRef]
- Steele, T.; Bauer, D.; Cesarone, O.; Lovold, K.; Paule, G.; Bibi, N.; Strainis, E.; Williams, J.; Jagielski, J.; Feemster, J.; et al. Isolated REM Sleep Without Atonia Following COVID-19 Infection: A Case-Control Study. Sleep 2022, 45, A244–A245. [Google Scholar] [CrossRef]
- Armstrong, M.J. Advances in Dementia with Lewy Bodies. Ther. Adv. Neurol. Disord. 2021, 14, 17562864211057666. [Google Scholar] [CrossRef]
- Jellinger, K.A.; Korczyn, A.D. Are Dementia with Lewy Bodies and Parkinson’s Disease Dementia the Same Disease? BMC Med. 2018, 16, 34. [Google Scholar] [CrossRef]
- Jellinger, K.A. Are There Morphological Differences between Parkinson’s Disease-Dementia and Dementia with Lewy Bodies? Parkinsonism Relat. Disord. 2022, 100, 24–32. [Google Scholar] [CrossRef]
- Beach, S.R.; Luccarelli, J.; Praschan, N.; Fusunyan, M.; Fricchione, G.L. Molecular and Immunological Origins of Catatonia. Schizophr. Res. 2024, 263, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Dawood, A.S.; Dawood, A.; Dawood, S. Catatonia after COVID-19 Infection: Scoping Review. BJPsych Bull. 2023, 47, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Torboli, D.; Mioni, G.; Bussé, C.; Cagnin, A.; Vallesi, A. Subjective Experience of Time in Dementia with Lewy Bodies during COVID-19 Lockdown. Curr. Psychol. 2023, 42, 4653–4662. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-C.; Liu, S.; Gan, J.; Ma, L.; Du, X.; Zhu, H.; Han, J.; Xu, J.; Wu, H.; Fei, M.; et al. The Impact of the COVID-19 Pandemic and Lockdown on Mild Cognitive Impairment, Alzheimer’s Disease and Dementia With Lewy Bodies in China: A 1-Year Follow-Up Study. Front. Psychiatry 2021, 12, 711658. [Google Scholar] [CrossRef] [PubMed]
- Łuc, M.; Szcześniak, D.; Trypka, E.; Mazurek, J.; Rymaszewska, J. SARS-CoV-2 Pandemic and the Population with Dementia. Recommendations under the Auspices of the Polish Psychiatric Association. Psychiatr. Pol. 2020, 54, 421–436. [Google Scholar] [CrossRef]
- Killen, A.; Olsen, K.; McKeith, I.G.; Thomas, A.J.; O’Brien, J.T.; Donaghy, P.; Taylor, J.-P. The Challenges of COVID-19 for People with Dementia with Lewy Bodies and Family Caregivers. Int. J. Geriatr. Psychiatry 2020, 35, 1431–1436. [Google Scholar] [CrossRef]
- Jellinger, K.A. Multiple System Atrophy—A Clinicopathological Update. Free Neuropathol. 2020, 1, 1–17. [Google Scholar] [CrossRef]
- Bagchi, A.D. Multiple System Atrophy. J. Nurse Pract. 2022, 18, 951–956. [Google Scholar] [CrossRef]
- Stefanova, N.; Wenning, G.K. Multiple System Atrophy: At the Crossroads of Cellular, Molecular and Genetic Mechanisms. Nat. Rev. Neurosci. 2023, 24, 334–346. [Google Scholar] [CrossRef]
- Matschke, J.; Lütgehetmann, M.; Hagel, C.; Sperhake, J.P.; Schröder, A.S.; Edler, C.; Mushumba, H.; Fitzek, A.; Allweiss, L.; Dandri, M.; et al. Neuropathology of Patients with COVID-19 in Germany: A Post-Mortem Case Series. Lancet. Neurol. 2020, 19, 919–929. [Google Scholar] [CrossRef]
- Stankovic, I.; Fanciulli, A.; Sidoroff, V.; Wenning, G.K. A Review on the Clinical Diagnosis of Multiple System Atrophy. Cerebellum 2023, 22, 825–839. [Google Scholar] [CrossRef] [PubMed]
- Polverino, P.; De Santis, T.; Perdixi, E.; Chiò, A.; Albanese, A. Case Report: Atypical Parkinsonism Following SARS-CoV-2 Infection. Front. Neurol. 2023, 14, 1208213. [Google Scholar] [CrossRef] [PubMed]
- Hague, K.; Lento, P.; Morgello, S.; Caro, S.; Kaufmann, H. The Distribution of Lewy Bodies in Pure Autonomic Failure: Autopsy Findings and Review of the Literature. Acta Neuropathol. 1997, 94, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Pavy-Le Traon, A.; Foubert-Samier, A.; Fabbri, M. An Overview on Pure Autonomic Failure. Rev. Neurol. (Paris) 2024, 180, 94–100. [Google Scholar] [CrossRef]
- Allendes, F.J.; Díaz, H.S.; Ortiz, F.C.; Marcus, N.J.; Quintanilla, R.; Inestrosa, N.C.; Del Rio, R. Cardiovascular and Autonomic Dysfunction in Long-COVID Syndrome and the Potential Role of Non-Invasive Therapeutic Strategies on Cardiovascular Outcomes. Front. Med. 2022, 9, 1095249. [Google Scholar] [CrossRef]
- Umapathi, T.; Poh, M.Q.W.; Fan, B.E.; Li, K.F.C.; George, J.; Tan, J.Y.L. Acute Hyperhidrosis and Postural Tachycardia in a COVID-19 Patient. Clin. Auton. Res. 2020, 30, 571–573. [Google Scholar] [CrossRef]
- Barbic, F.; Minonzio, M.; Cairo, B.; Shiffer, D.; Zamuner, A.R.; Cavalieri, S.; Dipaola, F.; Magnavita, N.; Porta, A.; Furlan, R. Work Ability Assessment and Its Relationship with Cardiovascular Autonomic Profile in Postural Orthostatic Tachycardia Syndrome. Int. J. Environ. Res. Public Health 2020, 17, 7836. [Google Scholar] [CrossRef]
- Zamunér, A.R.; Minonzio, M.; Shiffer, D.; Fornerone, R.; Cairo, B.; Porta, A.; Rigo, S.; Furlan, R.; Barbic, F. Relationships Between Cardiovascular Autonomic Profile and Work Ability in Patients With Pure Autonomic Failure. Front. Hum. Neurosci. 2021, 15, 761501. [Google Scholar] [CrossRef]
- Rinaldi, L.; Rigo, S.; Pani, M.; Bisoglio, A.; Khalaf, K.; Minonzio, M.; Shiffer, D.; Romeo, M.A.; Verzeletti, P.; Ciccarelli, M.; et al. Long-COVID Autonomic Syndrome in Working Age and Work Ability Impairment. Sci. Rep. 2024, 14, 11835. [Google Scholar] [CrossRef]
- Sixel-Döring, F.; Zimmermann, J.; Wegener, A.; Mollenhauer, B.; Trenkwalder, C. The Evolution of REM Sleep Behavior Disorder in Early Parkinson Disease. Sleep 2016, 39, 1737–1742. [Google Scholar] [CrossRef]
- Feuerstein, J.S.; Amara, A. REM Behavior Disorder: Implications for PD Therapeutics. Curr. Neurol. Neurosci. Rep. 2023, 23, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Diaconu, Ș.; Falup-Pecurariu, O.; Țînț, D.; Falup-Pecurariu, C. REM Sleep Behaviour Disorder in Parkinson’s Disease (Review). Exp. Ther. Med. 2021, 22, 812. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Partinen, E.; Chan, N.Y.; Dauvilliers, Y.; Inoue, Y.; De Gennaro, L.; Plazzi, G.; Bolstad, C.J.; Nadorff, M.R.; Merikanto, I.; et al. Dream-Enactment Behaviours during the COVID-19 Pandemic: An International COVID-19 Sleep Study. J. Sleep Res. 2023, 32, e13613. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef]
- Onder, G.; Rezza, G.; Brusaferro, S. Case-Fatality Rate and Characteristics of Patients Dying in Relation to COVID-19 in Italy. JAMA 2020, 323, 1775–1776. [Google Scholar] [CrossRef]
- Chen, Y.; Klein, S.L.; Garibaldi, B.T.; Li, H.; Wu, C.; Osevala, N.M.; Li, T.; Margolick, J.B.; Pawelec, G.; Leng, S.X. Aging in COVID-19: Vulnerability, Immunity and Intervention. Ageing Res. Rev. 2021, 65, 101205. [Google Scholar] [CrossRef]
- Li, H.; Manwani, B.; Leng, S.X. Frailty, Inflammation, and Immunity. Aging Dis. 2011, 2, 466–473. [Google Scholar]
- Pawelec, G. Age and Immunity: What Is “Immunosenescence”? Exp. Gerontol. 2018, 105, 4–9. [Google Scholar] [CrossRef]
- Sallard, E.; Lescure, F.-X.; Yazdanpanah, Y.; Mentre, F.; Peiffer-Smadja, N. Type 1 Interferons as a Potential Treatment against COVID-19. Antiviral Res. 2020, 178, 104791. [Google Scholar] [CrossRef]
- Kim, J.-A.; Seong, R.-K.; Shin, O.S. Enhanced Viral Replication by Cellular Replicative Senescence. Immune Netw. 2016, 16, 286–295. [Google Scholar] [CrossRef]
- Thakar, J.; Mohanty, S.; West, A.P.; Joshi, S.R.; Ueda, I.; Wilson, J.; Meng, H.; Blevins, T.P.; Tsang, S.; Trentalange, M.; et al. Aging-Dependent Alterations in Gene Expression and a Mitochondrial Signature of Responsiveness to Human Influenza Vaccination. Aging (Albany NY) 2015, 7, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Kadambari, S.; Klenerman, P.; Pollard, A.J. Why the Elderly Appear to Be More Severely Affected by COVID-19: The Potential Role of Immunosenescence and CMV. Rev. Med. Virol. 2020, 30, e2144. [Google Scholar] [CrossRef] [PubMed]
- Moss, P. “The Ancient and the New”: Is There an Interaction between Cytomegalovirus and SARS-CoV-2 Infection? Immun. Ageing 2020, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Nicoli, F.; Solis-Soto, M.T.; Paudel, D.; Marconi, P.; Gavioli, R.; Appay, V.; Caputo, A. Age-Related Decline of de Novo T Cell Responsiveness as a Cause of COVID-19 Severity. GeroScience 2020, 42, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Lu, L.; Cao, W.; Li, T. Hypothesis for Potential Pathogenesis of SARS-CoV-2 Infection-a Review of Immune Changes in Patients with Viral Pneumonia. Emerg. Microbes Infect. 2020, 9, 727–732. [Google Scholar] [CrossRef]
- Mackman, N.; Antoniak, S.; Wolberg, A.S.; Kasthuri, R.; Key, N.S. Coagulation Abnormalities and Thrombosis in Patients Infected with SARS-CoV-2 and Other Pandemic Viruses. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2033–2044. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement Associated Microvascular Injury and Thrombosis in the Pathogenesis of Severe COVID-19 Infection: A Report of Five Cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Noris, M.; Benigni, A.; Remuzzi, G. The Case of Complement Activation in COVID-19 Multiorgan Impact. Kidney Int. 2020, 98, 314–322. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, S.; Leng, S.X. Chronic Low-Grade Inflammatory Phenotype (CLIP) and Senescent Immune Dysregulation. Clin. Ther. 2019, 41, 400–409. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-Aging. An Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. A. Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Radzikowska, U.; Ding, M.; Tan, G.; Zhakparov, D.; Peng, Y.; Wawrzyniak, P.; Wang, M.; Li, S.; Morita, H.; Altunbulakli, C.; et al. Distribution of ACE2, CD147, CD26, and Other SARS-CoV-2 Associated Molecules in Tissues and Immune Cells in Health and in Asthma, COPD, Obesity, Hypertension, and COVID-19 Risk Factors. Allergy 2020, 75, 2829–2845. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.-M.; Kotzbauer, P.T.; Uryu, K.; Leight, S.; Trojanowski, J.Q.; Lee, V.M.-Y. Neuroinflammation and Oxidation/Nitration of Alpha-Synuclein Linked to Dopaminergic Neurodegeneration. J. Neurosci. 2008, 28, 7687–7698. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Savolainen, M.; Chu, Y.; Halliday, G.M.; Kordower, J.H. Temporal Evolution of Microglia and α-Synuclein Accumulation Following Foetal Grafting in Parkinson’s Disease. Brain 2019, 142, 1690–1700. [Google Scholar] [CrossRef]
- Bae, E.-J.; Choi, M.; Kim, J.T.; Kim, D.-K.; Jung, M.K.; Kim, C.; Kim, T.-K.; Lee, J.S.; Jung, B.C.; Shin, S.J.; et al. TNF-α Promotes α-Synuclein Propagation through Stimulation of Senescence-Associated Lysosomal Exocytosis. Exp. Mol. Med. 2022, 54, 788–800. [Google Scholar] [CrossRef]
- Kim, C.; Ho, D.-H.; Suk, J.-E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.-J.; et al. Neuron-Released Oligomeric α-Synuclein Is an Endogenous Agonist of TLR2 for Paracrine Activation of Microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef]
- Kim, T.-K.; Bae, E.-J.; Jung, B.C.; Choi, M.; Shin, S.J.; Park, S.J.; Kim, J.T.; Jung, M.K.; Ulusoy, A.; Song, M.-Y.; et al. Inflammation Promotes Synucleinopathy Propagation. Exp. Mol. Med. 2022, 54, 2148–2161. [Google Scholar] [CrossRef]
- Thorne, N.J.; Tumbarello, D.A. The Relationship of Alpha-Synuclein to Mitochondrial Dynamics and Quality Control. Front. Mol. Neurosci. 2022, 15, 947191. [Google Scholar] [CrossRef]
- Esteves, A.R.; Arduíno, D.M.; Silva, D.F.F.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial Dysfunction: The Road to Alpha-Synuclein Oligomerization in PD. Park. Dis. 2011, 2011, 693761. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy Proteins Regulate Innate Immune Responses by Inhibiting the Release of Mitochondrial DNA Mediated by the NALP3 Inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Moreno Fernández-Ayala, D.J.; Navas, P.; López-Lluch, G. Age-Related Mitochondrial Dysfunction as a Key Factor in COVID-19 Disease. Exp. Gerontol. 2020, 142, 111147. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shoemaker, R.; Thatcher, S.E.; Batifoulier-Yiannikouris, F.; English, V.L.; Cassis, L.A. Administration of 17β-Estradiol to Ovariectomized Obese Female Mice Reverses Obesity-Hypertension through an ACE2-Dependent Mechanism. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E1066–E1075. [Google Scholar] [CrossRef] [PubMed]
- Stelzig, K.E.; Canepa-Escaro, F.; Schiliro, M.; Berdnikovs, S.; Prakash, Y.S.; Chiarella, S.E. Estrogen Regulates the Expression of SARS-CoV-2 Receptor ACE2 in Differentiated Airway Epithelial Cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L1280–L1281. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Chen, J.; Wang, X.; Zhang, F.; Liu, Y. Age- and Gender-Related Difference of ACE2 Expression in Rat Lung. Life Sci. 2006, 78, 2166–2171. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.W.; Mao, H.J.; Wu, Y.L.; Tanaka, Y.; Zhang, W. TMPRSS2: A Potential Target for Treatment of Influenza Virus and Coronavirus Infections. Biochimie 2017, 142, 1–10. [Google Scholar] [CrossRef]
- Lau, E.H.Y.; Hsiung, C.A.; Cowling, B.J.; Chen, C.-H.; Ho, L.-M.; Tsang, T.; Chang, C.-W.; Donnelly, C.A.; Leung, G.M. A Comparative Epidemiologic Analysis of SARS in Hong Kong, Beijing and Taiwan. BMC Infect. Dis. 2010, 10, 50. [Google Scholar] [CrossRef]
- Wambier, C.G.; Goren, A. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection Is Likely to Be Androgen Mediated. J. Am. Acad. Dermatol. 2020, 83, 308–309. [Google Scholar] [CrossRef]
- McMurray, R.W.; Suwannaroj, S.; Ndebele, K.; Jenkins, J.K. Differential Effects of Sex Steroids on T and B Cells: Modulation of Cell Cycle Phase Distribution, Apoptosis and Bcl-2 Protein Levels. Pathobiology 2001, 69, 44–58. [Google Scholar] [CrossRef]
- Folstad, I.; Karter, A.J. Parasites, Bright Males, and the Immunocompetence Handicap. Am. Nat. 1992, 139, 603–622. [Google Scholar] [CrossRef]
- Zeng, F.; Dai, C.; Cai, P.; Wang, J.; Xu, L.; Li, J.; Hu, G.; Wang, Z.; Zheng, F.; Wang, L. A Comparison Study of SARS-CoV-2 IgG Antibody between Male and Female COVID-19 Patients: A Possible Reason Underlying Different Outcome between Sex. J. Med. Virol. 2020, 92, 2050–2054. [Google Scholar] [CrossRef]
- Hepworth, M.R.; Hardman, M.J.; Grencis, R.K. The Role of Sex Hormones in the Development of Th2 Immunity in a Gender-Biased Model of Trichuris Muris Infection. Eur. J. Immunol. 2010, 40, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.T.; Gray, A.; Higgins, S.A.; Hubby, B.; Kast, W.M. Androgen Ablation Augments Prostate Cancer Vaccine Immunogenicity Only When Applied after Immunization. Prostate 2009, 69, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Nicola, W.G.; Khayria, M.I.; Osfor, M.M. Plasma Testosterone Level and the Male Genital System after Chloroquine Therapy. Boll. Chim. Farm. 1997, 136, 39–43. [Google Scholar] [PubMed]
- Kovats, S. Estrogen Receptors Regulate Innate Immune Cells and Signaling Pathways. Cell. Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Lasagna, A.; Zuccaro, V.; Ferraris, E.; Corbella, M.; Bruno, R.; Pedrazzoli, P. COVID-19 and Breast Cancer: May the Microbiome Be the Issue? Future Oncol. 2021, 17, 123–126. [Google Scholar] [CrossRef]
- Goren, A.; Vaño-Galván, S.; Wambier, C.G.; McCoy, J.; Gomez-Zubiaur, A.; Moreno-Arrones, O.M.; Shapiro, J.; Sinclair, R.D.; Gold, M.H.; Kovacevic, M.; et al. A Preliminary Observation: Male Pattern Hair Loss among Hospitalized COVID-19 Patients in Spain - A Potential Clue to the Role of Androgens in COVID-19 Severity. J. Cosmet. Dermatol. 2020, 19, 1545–1547. [Google Scholar] [CrossRef]
- Schlenker, E.H.; Hansen, S.N. Sex-Specific Densities of Estrogen Receptors Alpha and Beta in the Subnuclei of the Nucleus Tractus Solitarius, Hypoglossal Nucleus and Dorsal Vagal Motor Nucleus Weanling Rats. Brain Res. 2006, 1123, 89–100. [Google Scholar] [CrossRef]
- Faas, M.; Bouman, A.; Moesa, H.; Heineman, M.J.; de Leij, L.; Schuiling, G. The Immune Response during the Luteal Phase of the Ovarian Cycle: A Th2-Type Response? Fertil. Steril. 2000, 74, 1008–1013. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fett, C.; Mack, M.; Ten Eyck, P.P.; Meyerholz, D.K.; Perlman, S. Sex-Based Differences in Susceptibility to Severe Acute Respiratory Syndrome Coronavirus Infection. J. Immunol. 2017, 198, 4046–4053. [Google Scholar] [CrossRef]
- Li, J.; Ming, Z.; Yang, L.; Wang, T.; Liu, G.; Ma, Q. Long Noncoding RNA XIST: Mechanisms for X Chromosome Inactivation, Roles in Sex-Biased Diseases, and Therapeutic Opportunities. Genes Dis. 2022, 9, 1478–1492. [Google Scholar] [CrossRef]
- Libert, C.; Dejager, L.; Pinheiro, I. The X Chromosome in Immune Functions: When a Chromosome Makes the Difference. Nat. Rev. Immunol. 2010, 10, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Wang, Y.-H.; Liu, Y.-J. Plasmacytoid Dendritic Cell Precursors/Type I Interferon-Producing Cells Sense Viral Infection by Toll-like Receptor (TLR) 7 and TLR9. Springer Semin. Immunopathol. 2005, 26, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Souyris, M.; Cenac, C.; Azar, P.; Daviaud, D.; Canivet, A.; Grunenwald, S.; Pienkowski, C.; Chaumeil, J.; Mejía, J.E.; Guéry, J.-C. TLR7 Escapes X Chromosome Inactivation in Immune Cells. Sci. Immunol. 2018, 3, eaap8855. [Google Scholar] [CrossRef] [PubMed]
- Karpuzoglu, E.; Phillips, R.A.; Gogal, R.M.; Ansar Ahmed, S. IFN-Gamma-Inducing Transcription Factor, T-Bet Is Upregulated by Estrogen in Murine Splenocytes: Role of IL-27 but Not IL-12. Mol. Immunol. 2007, 44, 1808–1814. [Google Scholar] [CrossRef]
- Allahverdiyeva, S.; Geyer, C.E.; Veth, J.; de Vries, L.M.; de Taeye, S.W.; van Gils, M.J.; den Dunnen, J.; Chen, H. Testosterone and Estradiol Reduce Inflammation of Human Macrophages Induced by Anti-SARS-CoV-2 IgG. Eur. J. Immunol. 2024, 9, e2451226. [Google Scholar] [CrossRef]
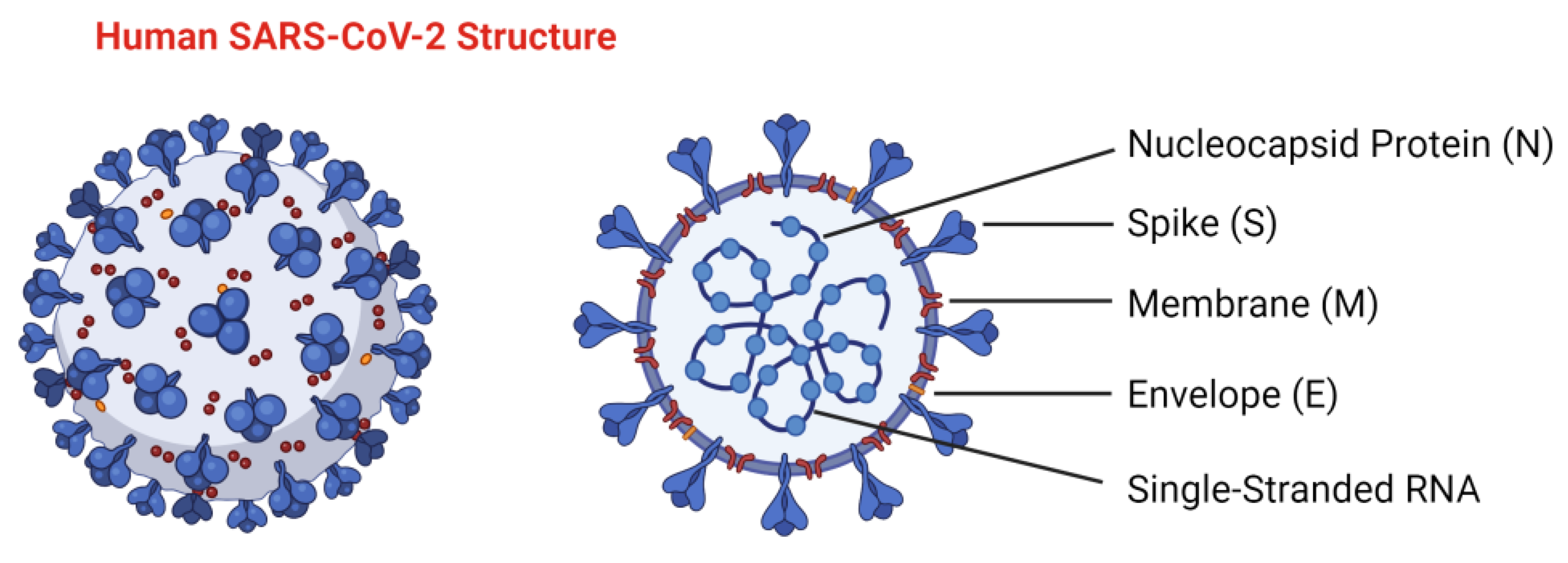
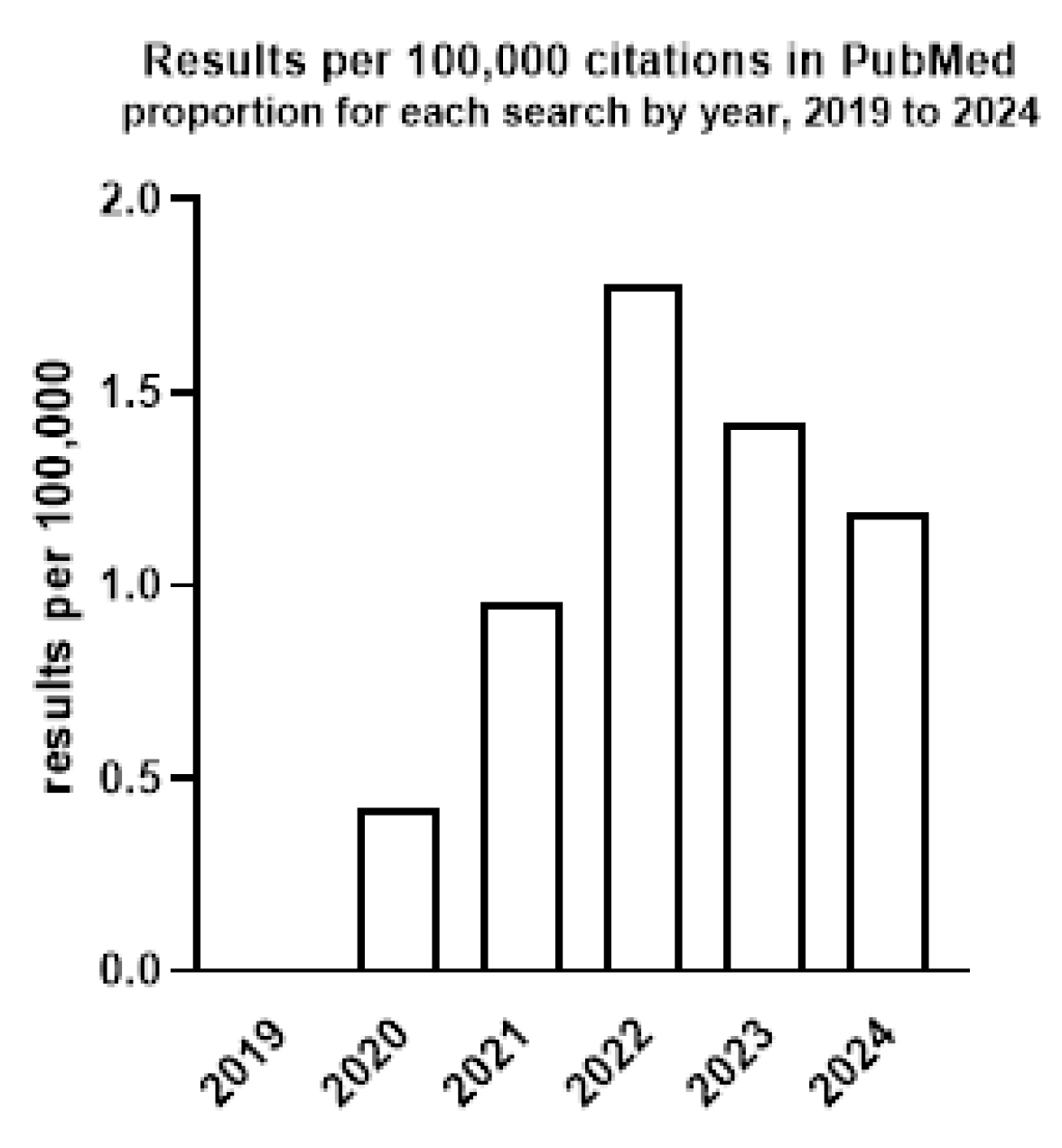
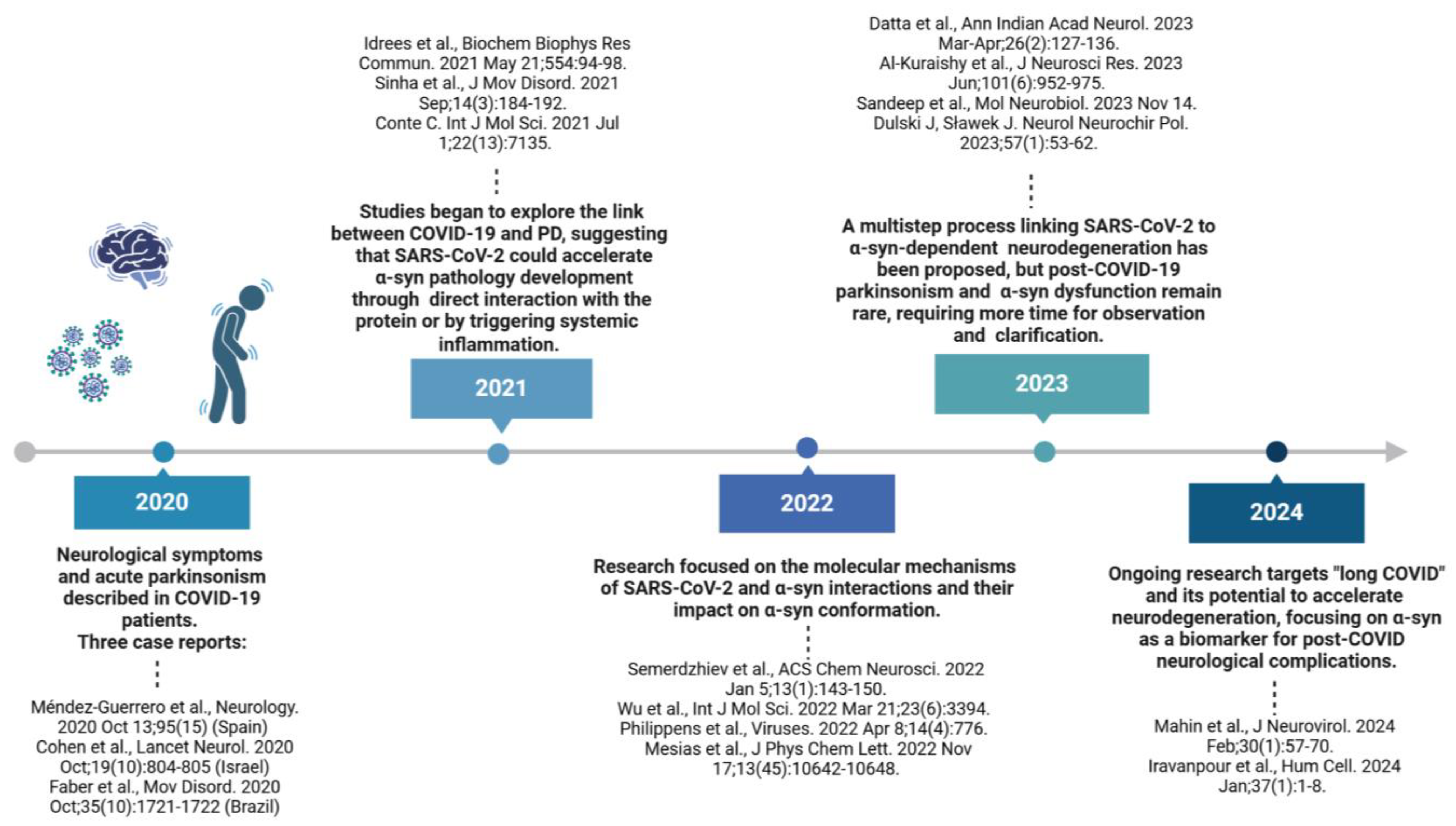
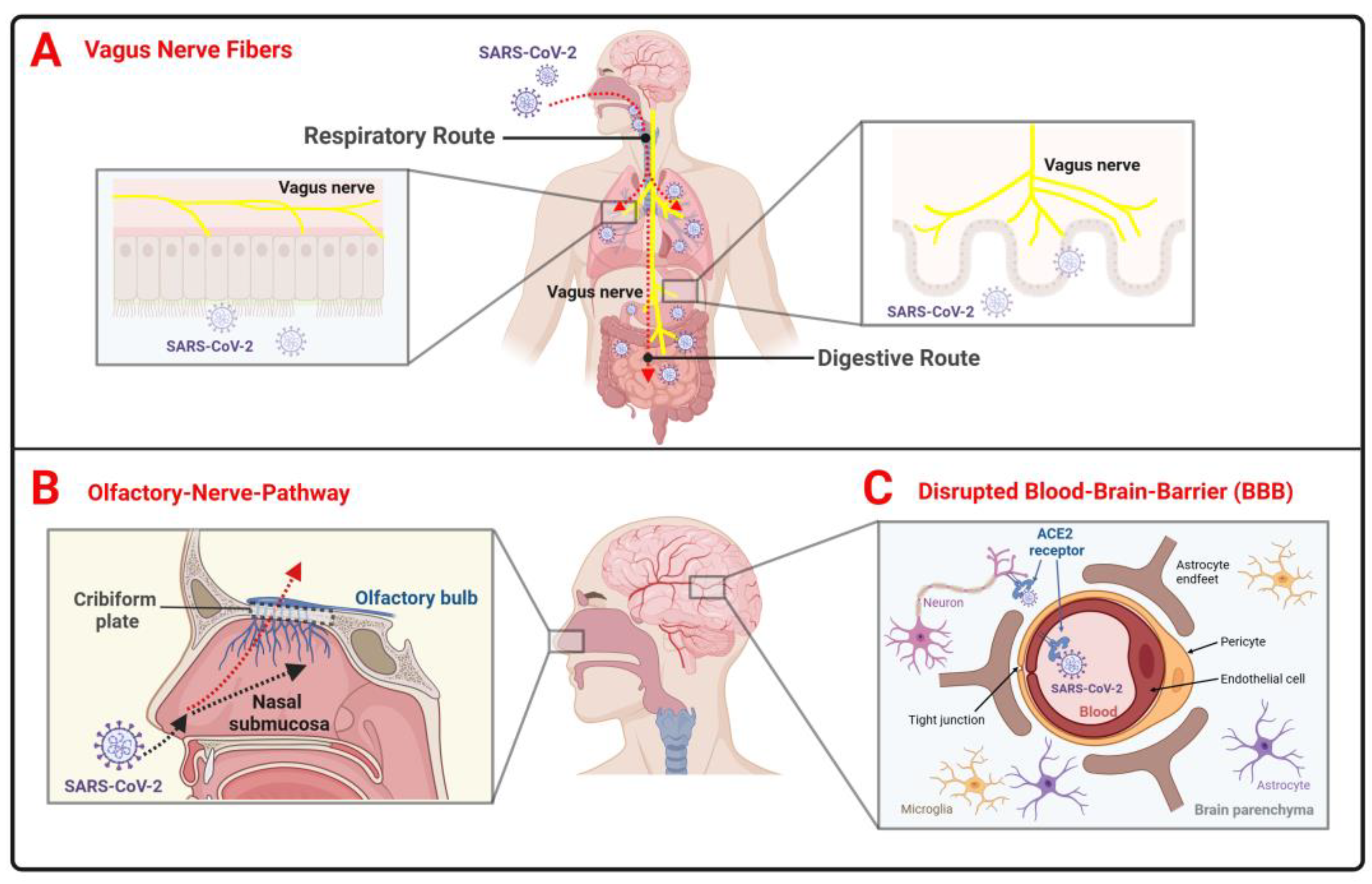
| Aim | Method/Subject of Analysis | Results | Reference |
|---|---|---|---|
| Meta-analysis aimed to determine the prevalence of COVID-19, its symptoms in elderly patients with PD and the association between PD and COVID-19. | Twenty articles were selected from January 2019 to 20 October 2021. | The prevalence of COVID-19 and the hospitalization of patients with PD was 1.06% and 0.98%, respectively; the prevalence of depression and anxiety during the pandemic in these groups was 46% and 43%, respectively; the risk of COVID-19 infection was equal in the PD patients and healthy controls. | [69] |
| Analysis of the prevalence of neurological disorders (including PD) in COVID-19 without overlapping meta-analysis errors. | Four meta-analyses involving 182,386 COVID-19 patients, published from November 2019 to September 2021. | The combined prevalence of PD during the COVID-19 pandemic was 0.67%; PD was not a statistically significant risk of mortality in COVID-19 patients (Odds Ratio [OR] = 3.94). | [70] |
| Review of all qualified studies to quantify the strength of affinities between pre-existing neurodegenerative diseases, SARS-CoV-2 vulnerability, and COVID-19 outcome. | Pre-registered systematic review with frequentist and Bayesian meta-analyses; 9 January 2023 was the final search date; 136 primary studies (total sample size n = 97,643,494), reporting on 268 effect-size estimates, met the inclusion criteria. | The odds for a positive SARS-CoV-2 test result were raised for individuals with pre-existing AD (OR = 2.86), dementia (OR = 1.83), and PD (OR = 1.65). People with pre-existing AD were at a higher risk for COVID-19-related hospital admission (OR = 3.72), but people with MCI, PD, or mixed dementia were not. People with AD and PD were at a higher risk for COVID-19-related intensive care unit admissions (pooled OR range: 1.55–1.65). All neurodegenerative disorders were at a higher risk for COVID-19-related mortality (pooled OR range: 1.56–2.27). In general, people with neurodegenerative disease and MCI are at a disproportionally high risk of acquiring COVID-19 and have a poor outcome once infected. | [71] |
| The assessment of the association between pre-existing neurological conditions and COVID-19 outcomes. | Literature review of systematic reviews, meta-analyses, and scoping reviews published between 1 January 2020 and 1 January 2023. Thirty-nine articles fulfilled the inclusion criteria, with data estimating >3 million people from 51 countries. | In total, 92.3% of the articles suggested a significant link between pre-existing neurological disorders including cerebrovascular disease, PD, AD and other dementias, and epilepsy and an increased risk of severe COVID-19 and mortality in the acute infectious period. | [72] |
| The systematic review and meta-analysis aimed to investigate the influence of the COVID-19 pandemic on neuropsychiatric disorders (depression, anxiety, stress) and sleep disturbances (sleep quality, insomnia), as well as the quality of life among patients with PD, MS, and AD compared with healthy people. | Observational studies (i.e., cross-sectional, case–control, cohort) raised from the research of 7 databases between March 2020 and December 2022. An analysis of eighteen studies (PD = 7, MS = 11) with a total of 627 individuals with PD (healthy controls = 857) and 3923 individuals with MS (healthy controls = 2432); twelve studies (PD = 4, MS = 8) were included in the meta-analysis. | The COVID-19 pandemic negatively affected people with PD, evidenced by significantly higher levels of depression and stress, measured by standardized mean differences (SMD) = 0.40 and 0.60, respectively. MS patients also presented higher levels of depression/stress, and additionally lower quality of life compared with the healthy control groups. | [73] |
| Meta-analysis of factors that affect the well-being of PD individuals from diverse populations during the pandemic. | Research of articles published between 2020 and 2022; the analysis includes twenty-seven studies involving 13,878 patients from America, Europe, Asia, and Africa. | High prevalence of diminished physical activity and exercise, and aggravating motor and neuropsychiatric symptoms (17–56%) during the COVID-19 pandemic, with patients in lower-income countries being exceptionally vulnerable, i.e., anxiety (adjusted Odds Ratios, [aOR] = 8.94), sleep (aOR = 5.16), and PD symptoms (aOR = 3.57). Younger age correlated with decreased physical activity, exercise, sleep, and worsening PD symptoms. Female PD patients reported a more pronounced decrease in physical activity and sleep disturbances. | [74] |
| Systematic review and meta-analysis aimed to determine the pooled prevalence of COVID-19 in PD patients. | Thirty articles for meta-analysis with the number of included patients differed between 10 and 64,434; published before Sep 2021. | The pooled prevalence of COVID-19 infection in PD cases was 5% besides mortality and hospitalization rates were 12% and 49%, respectively. The pooled prevalence of fever and cough in cases with PD was 4% and 3%, respectively. | [75] |
| Systematic review aimed to determine the impact of PD on the COVID-19 prevalence and patient prognosis. | Thirteen papers including 8649 PD patients and 88,710 control subjects/till 12 March 2021. | The pooled prevalence rate of COVID-19 among PD patients was 2.12%. The hospitalization rate for PD patients with COVID-19 was 39.89%, while the total mortality rate was 25.1%. There were no significant differences in hospitalization and mortality rates among COVID-19 patients with and those without PD. Fever, cough, fatigue, and anorexia existed as the most common manifestations with rates of 72.72%, 66.99%, 61.58%, and 52.55%, respectively. | [68] |
| Systematic review aimed to determine the influence of factors connected with COVID-19 in PD patients. | Literature research up to November 2020 (updated until 1 April 2021); finally, six studies (four case–control studies and two cross-sectional studies) in the qualitative and quantitative syntheses. | The following factors were connected with COVID-19 in PD patients: obesity (OR: 1.79) and pulmonary disease (OR: 1.92), COVID-19 contact (OR: 41.77), vitamin D supplementation (OR: 0.50), hospitalization (OR: 11.78), and death (OR: 11.23). The authors did not find any significant correlation between COVID-19 and hypertension, diabetes, cardiopathy, cancer, any cognitive problem, dementia, chronic obstructive pulmonary disease, renal or hepatic disease, smoking, and tremor. | [76] |
| Analysis of the relationship between PD and in-hospital outcomes of COVID-19. | A total of 12 studies with 103,874 COVID-19 patients. | PD was connected with poor in-hospital outcomes, OR = 2.64. Subgroup analysis showed that PD was connected with severe COVID-19 OR = 2.61, and mortality from COVID-19 Relative Risk (RR) = 2.63. Meta-regression showed that the association between PD and in-hospital outcomes of COVID-19 was influenced by age, but not by gender, dementia, hypertension, and diabetes. | [77] |
| Molecular Target | Example of Drug | Reference |
|---|---|---|
| Inhibition of NLRP3-dependent programed cell death, called pyroptosis and autophagy regulation, promotion of α-syn clearance, and restoration of proteasome 20 S activity. Reduction in α-syn (Ser129) phosphorylation. | Salidroside | [113,114,115] |
| Blocking of the P2X7R/NLRP3 axis triggering a cytokine storm. Reduction in ATP concentration and the activation of IL-1β and IL-6. Prevention of the influx of Ca2+ and the occasion of α-syn mutations. | Lidocaine | [119,150] |
| Counteraction glutamine-mediated excitotoxicity by inhibition of calcium influx into the cells by NMDA receptor channel blocking. Inhibition of SARS-CoV-2 viral channel activity, like the Protein E (envelope) cation channel, representing SARS-CoV-2 viroporin involved in its virulence. | Amantadine, Memantine | [131,151] |
| Regulation of calcium homeostasis by mGluR5. | Lithium | [138,139,140] |
| Switch microglia to anti-inflammatory and neuroprotective M2-phenotype. TLR-4 antagonism, ameliorating cytokine storm. Disruption of SARS-CoV-2 S protein binding to ACE2. Reduction in the phosphorylation/activity of ERK1/2. | Low-Dose Naltrexone | [141,142,143] |
| Prevention of the Fenton reaction and the ferroptosis inhibition. | Deferoxamine, Phyto-chelators like Caffeic acid, Curcumin, α-Lipoic acid (ALA), and Phytic acid | [144,146,147,148] |
| Scavenging of lipid peroxides and prevention of oxidative damage by lipophilic antioxidants. | Ferrostatin-1, Liproxstatin-1 | [144,149] |
| Anti-oxidative effects on the DA neurons in PD by increasing Nrf2 expression, and inhibition of the interaction of ACE2 with the S protein of SARS-CoV-2. | Flavones (Chrysin, Quercetin) | [152,153,154] |
| Disease | Case of Patient Description | Ref. |
|---|---|---|
| DLB | A 68-year-old man after COVID-19 with catatonia symptoms, like sub-stupor, immobility, catalepsy, and rejection. Catatonia appeared for the first time after SARS-CoV-2 infection and did not respond to lorazepam, though the ECT provided relief. Diagnostic imaging, including a DAT scan and 123I-meta-iodobenzylguanidine imaging, showed reduced uptake, leading to the final diagnosis of DLB instead of delirium. | [155] |
| MSA | A 65-year-old woman hospitalized for COVID-19 developed ataxia, progressive dizziness, and blurry vision. The evaluation noted rightward nystagmus, resting tremor of the right hand, slowed finger tapping bilaterally, right dysmetria, and a shuffling gait. Lumbar puncture was negative. Brain MRI indicated moderate cerebellar and pontine volume loss with crossed hyperintensity of the pons, known as the “hot cross buns sign”. Videonystagmography proved the cerebellar etiology of her symptoms. MSA with parkinsonian components (MSA-P) was finally diagnosed. The patient reacted positively to therapy with amantadine and carbidopa/levodopa, as well as vestibular rehabilitation and meclizine. Given the close temporal link between SARS-CoV-2 infection and the emergence of MSA features, the authors suggest that this MSA-P case may be related to the COVID-19 infection. | [156] |
| RBD | Patients with suspected sleep disorders after acute COVID-19 underwent video-polysomnography (v-PSG). At 60 days post-diagnosis, 4/11 patients (36%) were diagnosed with obstructive sleep apnea (OSA). Also, 4/11 patients showed REM sleep without atonia (RWA), a recognized prodromal stage of RBD, and two additional patients showed an RWA index within the highest range of normality. | [157] |
| The results of the case–control studies of 25 patients with previous COVID-19 infection compared with 25 age–sex matched controls who tested negative for COVID-19 before polysomnography. Isolated RWA occurred more frequently in the COVID-19 (9/25 patients, 36%) patients than in the controls (3/25 patients, 12%). | [158] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motyl, J.A.; Gromadzka, G.; Czapski, G.A.; Adamczyk, A. SARS-CoV-2 Infection and Alpha-Synucleinopathies: Potential Links and Underlying Mechanisms. Int. J. Mol. Sci. 2024, 25, 12079. https://doi.org/10.3390/ijms252212079
Motyl JA, Gromadzka G, Czapski GA, Adamczyk A. SARS-CoV-2 Infection and Alpha-Synucleinopathies: Potential Links and Underlying Mechanisms. International Journal of Molecular Sciences. 2024; 25(22):12079. https://doi.org/10.3390/ijms252212079
Chicago/Turabian StyleMotyl, Joanna Agata, Grażyna Gromadzka, Grzegorz Arkadiusz Czapski, and Agata Adamczyk. 2024. "SARS-CoV-2 Infection and Alpha-Synucleinopathies: Potential Links and Underlying Mechanisms" International Journal of Molecular Sciences 25, no. 22: 12079. https://doi.org/10.3390/ijms252212079
APA StyleMotyl, J. A., Gromadzka, G., Czapski, G. A., & Adamczyk, A. (2024). SARS-CoV-2 Infection and Alpha-Synucleinopathies: Potential Links and Underlying Mechanisms. International Journal of Molecular Sciences, 25(22), 12079. https://doi.org/10.3390/ijms252212079