Abstract
Obesity is a common risk factor in multiple tumor types, including prostate cancer. Obesity has been associated with driving metastasis, therapeutic resistance, and increased mortality. The effect of adipose tissue on the tumor microenvironment is still poorly understood. This review aims to highlight the work conducted in the field of obesity and prostate cancer and bring attention to areas where more research is needed. In this review, we have described key differences between healthy adipose tissues and obese adipose tissues, as they relate to the tumor microenvironment, focusing on mechanisms related to metabolic changes, abnormal adipokine secretion, altered immune cell presence, and heightened oxidative stress as drivers of prostate cancer formation and progression. Interestingly, common treatment options for prostate cancer ignore the adipose tissue located near the site of the tumor. Because of this, we have outlined how excess adipose tissue potentially affects therapeutics’ efficacy, such as androgen deprivation, chemotherapy, and radiation treatment, and identified possible drug targets to increase prostate cancer responsiveness to clinical treatments. Understanding how obesity affects the tumor microenvironment will pave the way for understanding why some prostate cancers become metastatic or treatment-resistant, and why patients experience recurrence.
1. Introduction
Prostate cancer is the third most diagnosed cancer in the United States behind only skin cancer and breast cancer [1,2] and is the second most diagnosed cancer in males around the world [3]. A total of 1 in 8 men will be diagnosed with prostate cancer during their lifetime. Of those men, 1 in 41 will die due to complications related to prostate cancer [2,4]. Therapeutic options include surgery, radiation, hormonal-based therapies, and chemotherapy [5]. While the five-year survival rate for prostate cancer is quite high, metastasis, castration resistance, and recurrence result in prostate cancer mortality [2,4]. In a prospective study on men with prostate cancer, adiposity was evaluated and deemed to be associated with characteristics of a higher Gleason Score in patients diagnosed with prostate cancer [6].
Obesity is one of the most common diseases in the world, with 1 in 8 people, or 39.2% of males, living with obesity [7]. Obesity, induced by poor diet, is characterized by large amounts of white adipose tissue (WAT) accumulation [8]. WAT has a large number of lipid droplets, which can aid in the formation of tumors by providing the components needed for metabolism, membrane biogenesis, and signaling molecules. Obesity is also associated with chronic adipose tissue inflammation and oxidative stress that leads to changes in metabolism, immune cell recruitment, and cytokine secretion, which creates a pro-tumorigenic landscape for cancer cells to thrive [9]. Not only is obesity linked to a worse prognosis in prostate cancer, but many other cancers as well, including, endometrial and ovarian cancer, breast cancer, and colon cancer [10,11].
In recent years, there have been a few published papers suggesting that abdominal radiation therapy causes dysregulation of adipokine secretion and changes in the immune system toward favoring tumor growth [12,13,14]. Similarly, some studies have identified that excess adipose tissue in the tumor microenvironment (TME), such as seen in obesity, impacts the efficacy of chemotherapeutic agents and androgen deprivation treatments [15,16]. However, the full impact of aberrant adipose tissue on the TME remains unclear.
This review describes changes in adipose tissue that occur during obesity and aims to provide insight as to how common therapeutic options for prostate cancer are affected by obesity and dysregulated adipocyte signaling, and in turn how the therapeutics affect adipose tissue. Adipocytes affect the TME through cytokines, adipokines, and lipid release. Obesity creates a more oxidatively stressed and pro-inflammatory environment, which promotes tumor initiation and progression. As obesity rates continue to rise, a larger portion of prostate cancer patients will be obese and it is unclear how visceral and subcutaneous adipose tissues and obesity affect standard treatments for prostate cancer such as ADT, radiation, and chemotherapy. The goal of this review is to magnify these areas that need more work carried out to improve prostate cancer patient outcomes.
2. Adipose Tissue Composition
In humans, the abdominal regions have two main types of WAT, subcutaneous and visceral fat [17]. Visceral adipose tissue differs from subcutaneous fat in the type of adipocyte, endocrine function, lipolytic activity, and associated inflammatory cells [18].
Structurally, WAT is composed of white adipocytes containing one large lipid droplet, which flattens and pushes the nucleus to the edge of the cell, and very few mitochondria. Adipocytes have two main functions, lipid handling and endocrine functions. Besides adipocytes, WAT is comprised of numerous other cell types, such as adipose stem cells (ASCs), immune, stromal, endothelial, and nerve cells; WAT is considered to be well vascularized and innervated (Figure 1) [19]. The complexity of the adipose tissue results in complex systemic signaling through the secretion of adipokines. This complex endocrine signaling allows adipose tissue to maintain energy homeostasis and communicate its nutrient status to the rest of the body.
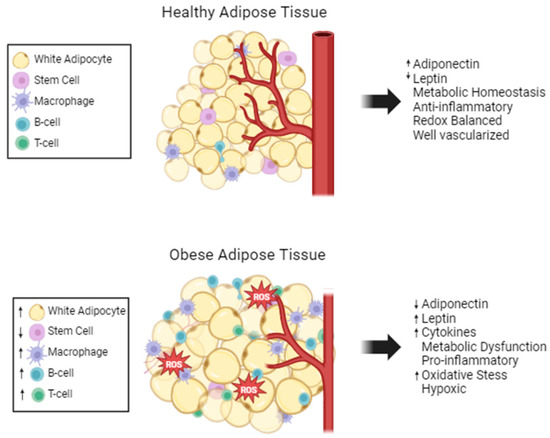
Figure 1.
Adipose tissue composition changes with obesity. Healthy adipose tissue is well maintained and regulated. Stem cells and a few anti-inflammatory immune cells are present. Obese adipose tissue has fewer mature adipocytes, but the adipocytes are larger in size. There is excess ROS and limited vascularization in obese adipose tissue. Many pro-inflammatory cells are present, and the metabolism and cytokine secretions are dysregulated, increased (↑) and decreased (↓).
Within the adipocyte, the large lipid droplet holds fatty acids in the form of triglycerides within the lumen, making the lipid droplet an energy-storing organelle that maintains energy homeostasis. When there is excess energy, fatty acids are converted to acyl-coA, which can then be metabolized into triglycerides and stored within the lipid droplet. Conversely, when energy is needed by cells, fatty acids are mobilized through lipolysis. Lipolysis involves lipase enzymes cleaving triglycerides to free the glycerol head from the three fatty acid tails [20]. Lipolysis is a heavily regulated process [21,22], and once hydrolyzed, free fatty acids are dispersed from the adipocyte to enter the vasculature [23]. Lipogenesis and lipolysis work together to maintain metabolic homeostasis.
As part of the endocrine function of adipose tissue, adipocytes release fatty acid binding protein 4 (FABP4) at the same time lipolysis occurs [24]. FABP4 is a lipid chaperone secreted from adipocytes that regulates glucose homeostasis [25]. Additionally, adipocytes secrete insulin-like growth factor-1 (IGF-1) that stimulates angiogenic growth factors in endothelial cells and ASCs to proliferate and survive [26,27].
The subcutaneous and visceral adipose depots play a crucial role in maintaining energy homeostasis by storing and releasing fatty acids as needed [28]. Within WAT, there is heterogeneity in the capacity to store lipids and the rate of fatty acids release, which has strongly been linked to the location of the adipose tissue [29]. For example, preadipocytes express gene signatures indicative of their depot of origin [30]. Mature adipocytes from the visceral WAT have significant enhancement of pathways related to glucose and lipid metabolism and stress response as compared to abdominal subcutaneous WAT [31]. Functionally, visceral WAT is more metabolically active than subcutaneous WAT [32].
Visceral adipose tissue contains more androgen and adrenergic receptors than subcutaneous fat, which maintains homeostatic lipolysis levels and, in turn, makes visceral adipocytes more metabolically active [33,34]. Additionally, subcutaneous and visceral adipose tissues differ in the synthesis and secretion of adipokines. For example, visceral fat is more heavily associated with pro-inflammatory cytokine secretion.
3. Effect of Obesity on Adipose Tissue
Alterations to WAT structure and function are major contributors to many diseases. Adipose tissue changes drastically with obesity. Adipose tissue structure and function are changed by increased adipocyte size and the induction of chronic low-grade inflammation [35]. Hypertrophy of adipocytes is linked to adipose tissue dysfunction and increased risk of metabolic syndromes [36]. In obesity, fat accumulates around organs and in the pelvic region.
The differentiation status of pre-adipocytes found in WAT is determined by metabolic status [37]. In an obese state, visceral WAT has reduced adipogenic capacity and current literature suggests this is due to enhanced inflammation and oxidation, which induces senescence of ASCs [38,39]. An in vitro study by Silva and collaborators noted that obese ASCs secrete more pro-inflammatory cytokines than mature adipocytes or ASCs from lean patients [40]. Additionally, obese ASCs proliferate less and undergo early senescence as compared to non-obese ASCs [41]. Obese adipose tissue is hypoxic due to enlarged adipocytes blocking vascularization. This causes fibroblast activation and excessive accumulation of the extracellular matrix [42]. Activated fibroblasts promote cancer development and progression by secreting factors that induce angiogenesis and immune evasion.
Adipose tissue maintains its immune cell population through the secretion of adipokines. Obesity causes the hypertrophy-induced cell death of adipocytes, which increases monocyte chemoattractant protein 1 (MCP1) expression [43]. Therefore, a hallmark of obesity is dead or dying adipocytes surrounded by macrophages producing crown-like structures [43]. B-cell and T-cell levels are also increased in obese adipose tissue. Another inflammatory marker, C-reactive protein (CRP) is positively correlated with abdominal obesity in a clinical study [44]. CRP modulates the expression of matrix metalloproteinases (MMPs) and MMP inhibitors, which include extracellular matrix remodeling in adipose tissue [45]. Overall, obesity creates a vicious cycle where adipose tissue structure is altered leading to increased inflammation and oxidative stress, and the inflammation and oxidative stress causes further dysregulation of adipocyte structure and function (Figure 1).
Oxidative stress is another distinct feature of obesity [46]. Reactive oxygen species (ROS) are highly reactive molecules that when elevated damage proteins, lipids, and DNA. High levels of ROS are linked to dysfunctional cellular homeostasis and tissue damage [47,48]. Calcium exchange, dysregulated by obesity-associated hyperlipidemia, leads to mitochondrial Ca2+ overload and subsequently increases mitochondrial ROS production [49]. Additionally, obese patients have less glutathione peroxidase 3 (GPx3), a hydrogen peroxide scavenger, in their adipose tissues [50]. GPx3 expression is attenuated by high levels of tumor necrosis factor-α (TNFα) and hypoxia and depressed levels lead to a rise in systemic oxidative stress and increased occurrence of metabolic complications [50]. An activated immune system further feeds the obesity–oxidative stress cycle, creating increased ROS production while dampening antioxidant capabilities.
Even in a normal, healthy state, adipocytes are vulnerable to oxidative stress. Due to the abundance of lipids found in the membrane and lipid droplets of adipocytes, adipose tissue is highly susceptible to lipid peroxidation [51]. Lipid peroxidation is generally described as the process in which oxidants attack lipids and convert them into peroxide and hydroperoxide derivatives [52]. These lipid peroxides can undergo further reactions to modify DNA and proteins and induce more oxidative damage. Carbon–carbon double bond lipids, such as polyunsaturated fatty acids (PUFAs), are prone to lipid peroxidation [51]. Mature adipocytes have reduced DNA repair mechanisms making these cells more vulnerable to DNA oxidation, as well [53].
4. Obesity and Prostate Cancer
A characteristic of cancer progression is the epithelial-to-mesenchymal transition (EMT), which enables cancer cells to leave the primary TME. During EMT, E-cadherin is downregulated, and N-cadherin and vimentin are upregulated loosening cell–cell contacts and resulting in a migratory phenotype [54]. Obesity enhances EMT in cancer cells. Specifically, prostate cancer cells cultured in serum from obese and normal-weight mice showed that obese serum induced EMT [55], supporting the idea that obesity enhances cancer cell invasiveness. A possible mechanism is that adipose tissue releases the chemokine (C-X-C motif) ligand 12, CXCL12, otherwise known as stromal cell-derived factor 12. CXCL12 enhances migration through the activation of STAT3, NF-κB, and MAPK signaling pathways [56,57]. CXCL12 and its receptor, chemokine (C-X-C motif) receptor 4 (CXCR4), are found in high concentrations in the stromal compartment of prostate cancer tumors [58,59]. The use of a CXCR4 antagonistic blocker, AMD3100, or CRISPR-Cas9 knockout of CXCR4 decreased the metastatic potential of prostate cancer cells [59]. In addition, the chemokine (C-C motif) ligand 7 (CCL7) is another factor that directs the migration of prostate cancer cells harboring its respective receptor, C-C chemokine receptor type 3 (CCR3), which is linked to poor prognosis [60]. Obese adipocytes release more CCL7 increasing this migratory signaling pathway when compared to normal adipocytes [60].
Along with CXCL12/CXCR4 and CCL7/CCR3 signaling, FABPs and peroxisome proliferator-activated receptors (PPARs) are established biomarkers for malignant prostate cancers; elevated expression levels of both FABPs and PPARs are strongly correlated with higher Gleason scores [61,62]. FABP4+ prostate cancer cells were significantly more invasive as compared to cells treated with a FABP inhibitor, and treatment with a fatty acid, oleic acid, was able to further enhance the invasiveness of FABP4+ prostate cancer cells [63]. FABP and fatty acids secreted from adipocytes in the TME aid in the ability of cancer cells to invade tissue by providing energy to cancer cells. PPAR-γ is highly expressed in adipose tissue where it plays a role in differentiating preadipocytes into mature adipocytes [64]. PPAR-γ mRNA and protein expression are elevated in prostate cancer biopsies [65]. PUFAs activate PPAR-γ, which in turn upregulates vascular endothelial growth factor (VEGF), aiding in the creation of a blood supply, rich with nutrients, within the TME [65]. VEGF also acts upon the Akt3 serine/threonine kinase to retain the PPAR-γ coactivator 1a (PGC1a) in the nucleus, leading to mitochondrial biogenesis and switching to a primarily oxidative form of cellular metabolism [66]. Increasing electron transport chain activity provides cancer cells with more energy to proliferate and migrate to other areas of the body [66]. Obesity leads to the increased presence of free fatty acids, chemokines, and cytokines that can activate cellular pathways, such as PPAR-γ, which participate in high-energy processes allowing for cancer cells to thrive.
5. Adipose Tissue Effects on Cancer
5.1. Adipose-Regulated Prostate Cancer Metabolism
5.1.1. Adipose as an Energy Source for Cancer Cells
In patients with prostate cancer, those with increased peri-prostatic fat are more likely to experience relapsed cancer [67]. The metabolic flux of prostate cancer is dynamic and provides the framework for migration and proliferation and is affected by lipids in the TME. In the early stages, prostate cancer cells rely on glycolysis, but when cells become castration-resistant, they rely more on oxidative phosphorylation. As the cells metastasize, they utilize oxidative phosphorylation, glycolysis, and β-oxidation, increasing the reliance on free fatty acids [68,69]. Also, prostate cancer cells dysregulate their lipid metabolism to increase the production of proteins needed for de novo lipogenesis [70,71,72]. Apart from the de novo synthesis, excess adipose tissue supplies TME of breast cancer and leukemia cells with free fatty acids, adipokines, and immune cells to stimulate the cancer cells to proliferate and metastasize to distant organs and aid in the signaling process of many biological pathways, such as anti-apoptosis [73,74]. The vascularization of WAT keeps a rich supply of nutrients entering the adipose tissue, which can be secreted to cancer cells to help drive proliferation and metastasis.
The TME and oxygen levels have known effects on prostate cancer metabolism [75]. Prostate cancer’s hypoxic environment promotes lipid accumulation [76]. A portion of the TME consists of adipocytes [77,78]. Prostate cancer cells overexpress fatty acid synthase protein (FASN) and monoacylglycerol lipase (MAGL), both metabolic enzymes that promote the formation and breakdown of lipids, which are now thought to be metabolic oncogenes [79,80,81]. Both in vitro and in vivo models confirmed that FASN and MAGL, in the presence of FABP5, enhance the metastatic potential of prostate cancer [82,83]. Specifically, FABP5 interacts with lipases to control the efflux of fatty acids from adipocytes by driving lipolysis [84], and the increased availability of fatty acids provides cancer cells with the energy needed to sustain continuous movement through the cell cycle.
Since adipocytes are a major contributor to the TME, there is an assumed large number of fatty acids available for energy consumption in prostate cancer cells. Fatty acids are broken down by the β-oxidation metabolic pathway, which connects to other pathways synthesizing metabolic products such as ATP, acetyl-coA, and steroid hormones. Gene analysis of prostate cancer cells revealed increased expression of branched-chain fatty acid β-oxidation enzymes that break down long-chain fatty acids in the peroxisome [85].
Another regulator of prostate cancer metabolism is the mTOR pathway [86,87], which serves as a hub for the integration of metabolism, cell growth, and disease [88,89]. Activation of mTOR mediates the activity of sterol regulatory element-binding protein (SREBP1), a master regulator of lipid metabolism [90]. SREBP1 binds transcriptional start sites of de novo fatty acid synthesis genes and recruits transcriptional machinery to enhance expression [91]. Along with elevated expression of FASN, stearoyl-CoA dehydrogenase (SCD) also exhibited increased expression when SREBP1 was overexpressed in prostate cancer [92,93]. Many cancer cells overexpress SCD to avoid lipotoxicity by maintaining homeostasis between lipid synthesis and lipid oxidation [94,95]. Silencing SCD would disrupt the homeostatic nature of the TME making it a potential therapeutic target [93,96].
5.1.2. Adipose as a Source of Building Blocks for Proteins in Cancer Cells
Many cancer cells utilize amino acids, nucleic acids, and lipids from their environment to maintain the necessary molecules required for proliferation [97,98,99]. To metabolize these molecules into the oxidized biomass precursors, nicotinamide adenine dinucleotide (NAD+) is needed to maintain metabolic pathways such as glycolysis, TCA cycle, and oxidative phosphorylation [100]. Limited availability of NAD+ decreases the accessibility of biomass substrates and cellular proliferation both in vitro and in vivo [101,102,103]. Alternatively, the synthesis of highly unsaturated fatty acids (HUFA) from PUFA aids in the regeneration of NAD+; thus, helping the cell to maintain a proliferative state [104]. During hypoxia, the de novo synthesis of fatty acids is repressed while the exogenous uptake of lipids is increased [105]. The increased availability of lipids leads to the activation of fatty acid β-oxidation pathways that help to maintain the NAD+/NADH pool needed for the synthesis of biomolecules, such as proteins.
Several groups have identified that the lipid-rich environment surrounding the tumor results in the upregulation of fatty acid β-oxidation in prostate cancer cells [106,107,108]. This metabolic phenotype of prostate cancer makes the inhibition of fatty acid β-oxidation an interesting target for limiting tumor growth and sensitizing cells to hormone-based therapies such as enzalutamide [108,109]. Nasser and co-workers identified 2,4 dienoyl-CoA reductase (DECR1), a rate-limiting enzyme in the β-oxidation of PUFAs, as being upregulated in patient prostate cancer samples and correlated with shorter relapse-free survival rates [110]. Knockdown of DECR1 in androgen receptor sensitive and resistant cell lines resulted in decreased cell viability, colony formation, invasion, and migration [110], suggesting a plausible therapeutic target of β-oxidation. The presence of lipids in TME provides the tools for cancer cells to maintain energy and biomolecule levels needed for rapid proliferation.
5.1.3. Adipose as a Source for Maintaining Membranes in Cancer Cells
Metabolically, lipids are needed for more than just energy; they are needed to help maintain cellular composition. There are three main lipid classes found in the plasma membrane: glycerophospholipids, sphingolipids, and sterols [111,112]. In mammalian cells, cholesterol is the main plasma membrane sterol and is synthesized using the mevalonate pathway of isoprenoid metabolism [113]. Lipids are first converted to acetyl-CoA by β-oxidation. In the mevalonate pathway, the addition of two acetyl-CoAs leads to the formation of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA), which is formed into sterols including cholesterol to be used to form new membranes. Two mevalonate pathway enzymes, HMG-CoA synthase 1 and HMG-CoA reductase, are overexpressed in prostate cancer, especially at the early stage [114]. Studies have shown that inhibition of the mevalonate pathway, using statin drugs, enhances tumor suppression [115,116,117]. In prostate cancer, blocking the formation of cholesterol with statins induces apoptosis [114,118].
In cancer, membrane lipids are often abnormal in that they contain a lower amount of oxidizable fatty acids, increasing resistance to oxidative damage and chemotherapy treatments [112]. Cellular membranes are comprised of phospholipids, which are synthesized in the endoplasmic reticulum through the process of fusing glycerol-3-phosphate and fatty acyl-CoA [119]. Hobby et al. discovered that exogenous fatty acids can supply the fatty acyl chains needed for membrane biosynthesis [120], establishing the idea that adipocytes secrete fatty acids for cancer cells to take up and utilize for membrane biosynthesis.
5.1.4. Obesity Effects on Metabolism, Building Blocks, and Membranes
Obesity is a risk factor for many metabolic diseases due to the development of insulin resistance. Elevated free fatty acid levels are the primary cause of insulin resistance, as are dysregulated adipokines and oxidative stress [121]. In the late 1990s, fatty acids were found to inhibit the action of insulin by dampening the signaling propagation from insulin receptors [122,123]. Decreased insulin signaling in adipose tissue results in increased lipolysis and elevated free fatty acid release [124]. The mitochondria of adipocytes are dysregulated in an obese state, and genes involved in fatty acid synthesis, uptake, and β-oxidation are upregulated [125,126]. Augmentation of mitochondrial energy homeostasis, through decreased fatty acid oxidation, leads to cellular dysfunction and lipotoxicity by synthesizing excessive levels of fatty acids [127,128].
In obesity, the accumulation of ROS byproducts leads to impaired translation and an enhanced ribosomal stress response [129]. Impairment of NAD+ synthesis affects the homeostatic nature of protein, carbohydrate, and lipid metabolism contributing to both the development of obesity and the resulting complications [130]. In adipose tissue where the availability of lipids is high, oxidative conditions lead to the production of reactive aldehydes such as trans-4-hydroxy-2-nonenal (4-HNE) through stress-induced lipid peroxidation [53]. 4-HNE in adipose tissue increases protein carbonylation, which is the ROS-related oxidation of protein sidechains [131]. The same study identified adipose-specific FABP as being carbonylated, which results in reduced activity. Thus, increased protein carbonylation is correlated with increased occurrence of metabolic disorders during obesity [132].
While the availability of lipids is needed for the biosynthesis of cellular membranes, the accumulation of lipids, like in obesity, affects this biosynthesis. Deficiency in enzymes related to phospholipid synthesis protects mice from diet-induced obesity [133], and abnormally high or low concentrations of phospholipids alter glucose and lipid metabolism [134]. Increased exogenous saturated fatty acid uptake decreases membrane fluidity [135]. Decreased fluidity is caused by ROS linked to metabolic dysfunction in obesity [136]. Therefore, in obesity, the excess accumulation of lipids allows for membrane biosynthesis to continue and new adipocytes to be synthesized.
Obese adipose tissue is highly vascularized. The vascularization of WAT keeps a rich supply of nutrients entering the adipose tissue, which can be secreted to cancer cells to help drive proliferation and metastasis. The notable innervation of WAT provides a pathway for cancer cells to enter the adipose tissue through a process called perineural invasion (PNI) [137]. This is another way in which cancer cells are able to utilize adipose tissue.
5.2. Adipokine Signaling and Cancer Progression
Obesity dysregulates adipocyte metabolism systemically leading to the aberrant release of adipokines. Adipokines released from adipose tissue, such as leptin and adiponectin, play an active role in the proliferation of prostate cancer cells. Since a large population of the world is considered obese, there has been a vast amount of research conducted on the effect of obesity on adipokine secretion, specifically leptin, adiponectin, visfatin, apelin, and resistin. Leptin is an adipocyte-specific hormone that when released systemically, activates the hypothalamus, the brain region responsible for satiety, to control appetite and energy expenditure [138,139]. Adiponectin, also secreted from adipocytes, has been established to regulate energy homeostasis through lipid and carbohydrate metabolism and exert anti-inflammatory effects [140]. Leptin is greatly upregulated, and adiponectin is downregulated during obesity [141]. Not only is leptin upregulated in patients with obesity, but high leptin expression is also correlated to poor prognosis in several cancers, including thyroid, ovarian, prostate, and breast [141,142,143,144]. One study elucidated the antagonistic effects of adiponectin and leptin on carcinogenesis and discovered adiponectin was able to inhibit cell migration, invasion, and proliferation, while increased leptin levels induced protumorigenic properties, such as increased proliferation and migration [141]. Overall, increased amounts of leptin have been attributed to tumorigenic characteristics while decreased amounts of adiponectin are tied to a lack of activated anti-tumor pathways playing an influential role in the crosstalk between dysregulated adipose tissue and cancer.
5.2.1. Adipokine Secretion Is Associated with Cancer Cell Progression
Leptin is an upstream activator of JNK and causes proliferation in an androgen-independent prostate cancer cell [145]. During obesity, leptin expression is increased, which shortens the G1 phase by promoting the transcription of cyclin D1, the regulator of the transition from the G1 to S phase [146]. The expression of cyclin D1 activates cyclin-dependent kinase 4/6 (CDK4/6), which results in quick entry to the S phase, resulting in faster DNA replication [147]. Leptin and its receptor, Ob-R, are expressed in the hypothalamus, human vasculature, and endothelial cells [138,148]. In vivo, corneal vascularization was observed in WT but not in Ob-R deficient rats [148], substantiating the notion that an increased leptin expression could enable cancer cells to grow blood vessels. In gastric and ovarian cancers, leptin can induce migration and decrease cell-to-cell adhesion by activating the JAK-STAT and MEK pathways [149,150]. In vitro studies using intestinal epithelial cells identified leptin as a contributing factor in lipid droplet formation through the mTOR pathway, a prominent cellular growth pathway [151]. In many cancers, the mTOR pathway is upregulated, possibly due to the dysregulated presence of leptin [152]. The observed results of leptin have not been characterized in prostate cancer; however, given the high correlation between obesity and prostate cancer, it is assumed that leptin will have similar effects on prostate TME.
Visfatin, also known as nicotinamide phosphoribosyl transferase (NAMPT), is an adipokine secreted from visceral adipose tissue. Visfatin is the rate-limiting enzyme in the biosynthesis of NAD, which plays a role in cellular homeostasis and maintaining cell viability [153]. Visfatin is upregulated in the serum of patients with different types of cancer including prostate, endometrial, and liver [154,155,156,157]. In the context of cancer, visfatin has been found to promote malignancy through increasing angiogenesis via VEGF [155], mediating chemotherapy resistance, and inducing cellular proliferation.
Other adipokines that have been shown to play a role in prostate cancer progression include apelin and resistin. Apelin is a ligand to the apelin receptor (APJ) and is expressed by both the prostate and adipose tissue [158]. Through the APJ axis, apelin downregulates tissue inhibitors of matrix metalloproteinases (TIMP2) expression to increase prostate cancer progression and metastasis [159,160]. In support of this study, elevated apelin expression in the serum of cancer patients has been reported [158]. Additionally, the adipokine resistin induces prostate cancer migration, invasion, and proliferation [161,162].
5.2.2. Adiponectin Is Associated with Anti-Tumor Properties
Adiponectin inhibits cell migration, invasion, and proliferation and has many anti-inflammatory effects. Mice with diet-induced obesity were given exogenous adiponectin leading to decreases in colon cancer lesion formation as compared to mice with obesity that were not given adiponectin [163]. This same study determined that adiponectin inhibits mTOR through adenosine monophosphate-dependent kinase (AMPK) activation, both of which control cell growth and metabolism [163]. Disulfide bond A oxidoreductase-like protein (DsbA-L) regulates adiponectin multimerization in 3T3 cells and, thus, adiponectin activity [164,165]. High molecular weight adiponectin is the most biologically active form, mediating insulin sensitivity and glucose homeostasis [166]. Deficiency of DsbA-L not only leads to the misfolding and subsequent removal of adiponectin through the ER-associated degradation (ERAD) pathway [164], but it also suppresses the production of interferon-γ, a cytokine with important anti-tumor effects [167].
AMPK is a known repressor of mTOR [168,169]. Reduced levels of adiponectin result in unchecked mTOR regulation and enhanced lipid metabolism, which together promote cancer proliferation and progression. Thus, the lack of adiponectin caused by obesity plays an active role in cancer. This effect is potentially working with insulin resistance and subsequent hyperinsulinemia often found in obese patients. Insulin is a signaling hormone in the body that is released from pancreatic beta cells when blood glucose levels are high triggering the activation of enzymes associated with glycolysis and glycogenesis and blocking fatty acid oxidation [170]. Insulin resistance, therefore, promotes the metabolism of fatty acids providing cancer cells with the building blocks needed for proliferation. Additionally, adiponectin is a positive regulator of apoptosis [171,172]. Thus, decreased adiponectin levels promote the proliferation of cancer cells by limiting apoptotic cell death.
5.3. Adipose Tissue Inflammation and Cancer Progression
Over the past decade, many reviews have suggested that adipocytes are a major regulator of the immune system. However, very few studies have linked the complexity of the adipose tissue environment to the TME, specifically in terms of the immune cell composition and how it regulates the tumor’s activity. Co-culturing of adipocytes and cancer cells leads to the upregulation of many pro-inflammatory cytokines such as family members of interleukin-6 (IL-6), a multifunctional cytokine in host defense, and CXCL1, which is a chemoattractant for many immune cells, including neutrophils [173,174]. These same cytokines also polarize macrophages toward an anti-inflammatory, M2 phenotype, which is responsible for tissue healing, or, in the context of cancer, the creation of a favorable TME [175]. With obesity-related inflamed adipose tissue, other aberrantly regulated adipokines aid in the recruitment of immune cells and the survival of cancer cells by activating proliferation and cellular growth pathways [173,174,176]. Most studies investigating the link between adipose tissue and cancer have been conducted in the context of breast cancer. However, since obesity is one of the leading risk factors for prostate cancer, chronic inflammation in the adipose tissue is possibly one factor aiding in the creation of prostate TME [6,9]. Local inflammation is a shared quality in both obese and cancer patients. This crosstalk between the tumor and the adipose tissue and vice versa provides many avenues to explore how secreted proteins found within, surrounding, and distant from the primary tumor affect progression and metastasis (Figure 2).
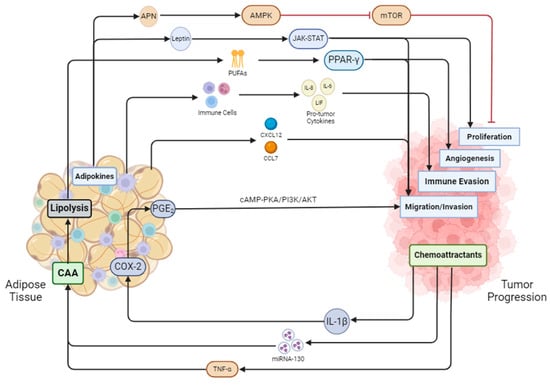
Figure 2.
Adipose tissue and cancer are in constant crosstalk with one another. Adiponectin secreted by adipose tissue inhibits mTOR and in turn blocks proliferation, while leptin activates the JAK-STAT pathway to promote proliferation. Adipose tissue undergoes lipolysis secreting fatty acids that activate PPAR-γ, initiating angiogenesis in tumor cells. Immune cells from adipose tissue secrete pro-tumor factors aiding the tumor in evading the immune system. Adipose tissue also secretes cytokines such as CXCL12 and CCL7 to induce prostate cancer cell migration. Prostate cancer also releases cytokines such as IL-1B, which activates COX-2 and PGE2. PGE2 then cycles back to the tumor to promote migration and invasion through the cAMP-PKA/PI3K/AKT pathway. MiRNA-130 and TNF-α from cancer cells promote the dedifferentiation of adipocytes into cancer-associated adipocytes (CAAs). CAAs undergo enhanced lipolysis, feeding the cancer cells fatty acids.
5.3.1. Adipocyte Secreted Cytokines and Tumor Progression
Adipose tissue surrounding the tumor and surrounding the organs that contain tumors play a unique role in tumor formation and progression. Obese adipose tissue is rich in T cells, being the second most numerous leukocyte population in adipose tissue behind macrophages [177]. T cells in adipose tissue release leukemia inhibitory factor (LIF), a cytokine from the IL-6 family, which controls stem cell self-renewal, inflammatory response, and cancer progression [178,179]. LIF induces breast cancer migration through the activation of the ERK1/STAT pathway [173]. CXCL1, CXCL2, and CXCL3 are chemo-attractants released by tumor cells that stimulate white adipose tissue to release adipose stromal cells, as well as activate the ERK/STAT/LIF pathway [173,174]. Inhibition of these pathways leads to reduced metastatic and angiogenic properties of cancer cells [180]. In obese prostate cancer patients, not only is there an abundance of WAT, but CXCL1 is upregulated in epithelial cells giving more opportunity for the movement of adipocytes to the TME [174]. This suggests that blocking the CXCL family could potentially reduce the ability of prostate cancer to metastasize.
Another chemoattractant that plays a significant role in tumor progression is interleukin-8 (IL-8) secretion. Lipid droplet accumulation in adipocytes leads to the release of IL-8, which consequently is significantly upregulated in obesity [181]. In metastatic breast cancer cells, IL-8 is upregulated compared to the primary breast cancer cells [182], causing pro-angiogenic effects [183]. In another study, in vitro and in vivo models confirmed that IL-6 is overexpressed in adipose tissue that surrounds breast cancer tumors. Additionally, higher adipocyte IL-6 expression correlates with worse prognosis [184], and increased total body adipose tissue corresponds to increased levels of serum IL-6 [185,186]. In patients, elevated levels of IL-6 are more likely to be produced from omental adipose tissue as compared to subcutaneous tissues, where most fat deposits reside in obesity [186]. IL-6 signaling also plays a role in the polarization of macrophages and the upstream activation of interleukin-4, which is responsible for adipose tissue macrophage proliferation [187], as well as regulating metabolic homeostasis [186]. Thus, adipocyte-derived IL-6 has a multifaceted role in the TME.
5.3.2. Macrophages
Macrophages are a group of diverse myeloid cells that are a part of the innate immune response and clear dead cells and pathogens [188]. In normal tissue remodeling, macrophages can recognize the presence of phosphatidylserine on the extracellular side of the plasma membrane, which is a signal for the removal of apoptotic cells through phagocytosis [189,190]. However, macrophages are extremely plastic cells and can change phenotypically when exposed to certain stimuli [191,192]. There are two well-established classifications for macrophages: classically activated (M1) and alternatively activated [188,193,194]. M1 macrophages are characterized as being pro-inflammatory and M2 macrophages are considered anti-inflammatory [192,195]. However, this is an oversimplification and there are other classifications of macrophages beyond M1 and M2.
In obesity, the number of macrophages in the adipose tissue is often very high causing chronic, local inflammation [196]. Obesity causes decreased adiponectin levels leading to adipocyte death, which stimulates the macrophages in the adipose tissue to polarize toward a pro-inflammatory phenotype [195] to enhance the influx of bone marrow-derived monocyte progenitor cells differentiating into macrophages [197]. In creating an inflammatory environment, macrophages secrete cytokines, such as TNFα, IL-6, and IL-8, which spread throughout the body creating chronic, systemic inflammation [197]. This is how the chronic inflammation of obese adipose tissue is established and sustained.
Tumor-associated macrophages (TAMs) found within the TME have a phenotype that is not M1 or M2 but rather an ability to transition between the two [176]. TAMs are speculated to aid in tumor progression by using the M1 phenotype to infiltrate the TME [198]. Once the macrophages are in the TME, colony-stimulating factor (CSF-1) is released from the tumor cells to trigger the phenotypic change of the M1 macrophages to M2 [199,200]. The M2 phenotype is special in its ability to promote tumor cell proliferation and invasion by secreting factors, such as epithelial growth factor and TGF-β, as well as increasing the activation of pathways such as MAPK [193,200,201]. In an in vivo model using diphtheria toxin to deplete macrophages, it was established that macrophages are needed for angiogenesis to occur, further solidifying the need for macrophages in the creation and maintenance of the TME [176]. In patients with ovarian cancer, the presence of M2-like macrophages was strongly correlated with a worse prognosis [202], solidifying the idea that an aim for cancer therapies could be to block the phenotypic switching through CSF-1 inhibitors.
Macrophages isolated from adipose tissue contain excessive amounts of lipid droplets driving inflammation and altering the metabolism of the neighboring cells [203]. TAMs from breast cancer tumors contain increased expression of FABPs [204]. Macrophages collected from a mouse lung metastasis contained an abundance of lipid droplets [193]. Likewise, macrophages co-cultured with tumor cells had a higher accumulation of lipids than macrophages cultured with normal cells. Interestingly, macrophages co-cultured with tumors had similar levels of lipids observed in adipose tissue-associated macrophages [191,203]. In an analysis of primary and metastatic samples taken from patients with prostate cancer, a cluster of FABP genes was significantly upregulated in the metastatic samples [205], supporting the idea that fatty acid utilization is not only needed at the primary tumor but also during metastasis. Such high expression levels of lipids in the TAMs suggest that obesity could accelerate the formation of tumors by recruiting macrophages containing energy-filled lipid droplets to the TME. In conclusion, macrophages from obese adipose tissue drive tumor formation and cancer progression.
5.4. Oxidative Stress from Obesity Drives Cancer Progression
Another hallmark of obesity is increased oxidative stress. The overabundant supply of energy substrates in obesity is thought to drive mitochondrial dysfunction and ROS signaling [206]. While ROS normally function as signaling molecules, if uncontrolled, they can damage DNA, lipids, and proteins. Since adipocytes are full of triglyceride lipids, adipose tissue is especially vulnerable to the effects of oxidative stress, which results in adipose tissue remodeling, aberrant signal transduction, and heightened inflammation [207].
Prostate cancer exposure to an adipocyte-rich environment, in vitro and in vivo, induced the upregulation of heme-oxygenase (HO-1), an oxidative stress enzyme [208]. Increased HO-1 levels can increase the metastatic potential, as well as promote growth and invasiveness in prostate cancer cells [208,209]. Paracrine signaling by periprostatic adipose tissue (PPAT) is upregulated in obesity. Laurent et al. elucidated that free fatty acids released by the PPAT stimulate the expression of NADPH oxidase 5 (NOX5) in prostate cancer cells residing nearest PPAT [210]. NOX5 expression plays a role in inducing proliferation and survival in prostate cancer [211]. Additionally, NOX5 is implicated in activating HIF-1α in normoxic conditions [212], and obesity induces a signaling cascade in adipocytes that leads to the activation of HIF-1α [213]. In prostate cancer, HIF-1α functions to drive angiogenesis, cell proliferation, and metastasis [214].
Decades ago, prostate cancer tumorigenesis was linked to increased levels of ROS and an impaired ability to maintain redox homeostasis. Abundant ROS induces spontaneous mutagenesis by damaging cellular DNA [215,216,217], suggesting a possible mechanism for ROS-mediated tumorigenesis. In addition, the presence of lipid peroxidation products such as malondialdehyde and acrolein in plasma and tumor tissues can form DNA adducts, which have been positively correlated with prostate cancer progression [218]. Obesity accompanied by oxidative stress drives DNA mutation and dysregulated signaling pathways, supporting cancer progression.
6. Cancer Effects on Adipose Tissue
Both tumor cells and adipocytes secrete proteins and small molecules that function as signaling molecules. Tumor cells can secrete cytokines that recruit adipose stromal cells to the microenvironment, and on the other hand, adipocytes can secrete adipokines that aid in the development of the TME [174,182]. Tumor cells can attract nearby adipocytes by releasing TNF-α and exosomes containing miRNA-130, both of which inhibit the transcription of PPAR-γ, leading to the dedifferentiation of mature adipocytes into cancer-associated adipocytes (CAAs) [208,219]. CAAs provide the tumor with fatty acids for energy requirements, lipids for membrane biosynthesis, and protect the growing tumor from immune infiltration. Cancer cells and adipocytes are in constant crosstalk with one another to modulate the expression of proteins, cytokines, and other cells within the TME [220] (Figure 2).
Prostate cancer is known to metastasize to regions rich in adipocytes, such as bone marrow. Bone marrow adipocyte numbers increase with age and obesity, both of which are risk factors for metastatic cancer [221]. As previously stated in this review, lipids derived from adipose tissue can help supply cancer cells with the energy needed to maintain proliferation. Alternatively, cancer cells can trigger the activation of lipolysis pathways to increase the release of lipids from adipocytes [222].
Herron et al. determined that IL-1β secreted from prostate cancer cells interacts with bone marrow adipocytes to increase the expression of cyclooxygenase-2 (COX-2), an enzyme needed to form prostaglandin E2 (PGE2), promoting both survival and proliferation of metastatic cells [223,224]. In prostate cancer, PGE2 phosphorylates STAT3, which induces a mesenchymal phenotype [225] and promotes invasion via the cAMP-PKA/PI3K-Akt signaling pathway [226]. At the molecular level, few studies have elucidated the effects that cancer formation and progression have on surrounding adipose tissue and adipocytes.
From a clinical standpoint, the indirect impact of cancer on adipose tissue is well documented and described. The treatment of prostate cancer can require the use of chemotherapeutics. Such treatments are known to affect adipose tissue composition and distribution within the human body [227]. Another study found that the peri-prostatic adipose tissue area is increased in patients who develop castration-resistant prostate cancer (CRPC) [228], suggesting that the development of CRPC alters the adipose distribution in the abdominal cavity. Vertulli et al. found a strong correlation between recurrence after radical prostatectomy and levels of abdominal adipose tissue [229]. Overall, these findings suggest that incomplete removal of prostate tumors in a patient with heightened adiposity can increase the risk of recurrence in the same manner that adipose tissue can induce primary tumor formation.
7. Cancer Treatments Cause Changes in Adipocytes
7.1. Hormone-Based Therapy: ADT
Many prostate cancer tumors express androgen receptors (ARs) and prostate-specific androgens. Hormonal-based therapies, such as androgen deprivation therapies (ADT), are frequently used in prostate cancer to reduce the number of androgens circulating throughout the prostate gland to reduce prostate cancer growth [230,231]. Although most prostate cancers initially respond to ADT, long-term treatment results in a cell population that can survive in low levels of androgen [232]. More and more studies demonstrate that lipids play a role in castration resistance.
Clinical studies show that the use of ADT is associated with changes to adipose tissue composition, specifically increased fat mass [233,234]. Such an increase often leads to a second co-morbidity, obesity, which as shown in this review and others, increases the risk of prostate cancer progression. While the connection between prostate cancer, ADT, and obesity has been well established in the clinical setting, less can be said about the impact of the three at the molecular level. For example, ADT increases body fat mass, the risk of insulin resistance, and serum adiponectin levels [235,236]. In one study, periprostatic fat collected from patients who received six months of ADT treatments showed a significant trend of pro-inflammatory, obesity-like adipose tissue [236]. Differential tissue composition and gene-enrichment analysis pointed toward the infiltration of immune cells in the adipose tissue following ADT treatment [237]. Taken with the notion that obesity is a risk factor for prostate cancer, ADT could potentially be causing adverse effects on the adipose tissue linking it to tumor progression after treatment.
Testosterone is the main androgen that is repressed in prostate cancer patients who receive ADT. Testosterone is a type of steroid hormone that regulates male and female sexual characteristics, maintains the strength of bones and muscles, and stimulates erythropoiesis [238]. The synthesis of testosterone starts with the oxidative cleavage of lipid cholesterol and ends with the androstenedione being reduced by 17β-hydroxysteroid dehydrogenase to yield testosterone [239]. Decreased testosterone is associated with an increase in total cholesterol levels and low-density lipoprotein (LDL) [240], both are positively correlated to an increased risk for incidence and progression of cancer [241,242,243]. Elevated cholesterol is important for protein transport and for the synthesis of new membranes [244,245], and elevated LDL levels can cause an increase in sensitivity to oxidative stress [246].
Thioesterase superfamily member 6 (THEM6), which is highly expressed in adipose tissue where it regulates intracellular fatty acid homeostasis, is upregulated in ADT-resistant prostate cancers, and activates the unfolded protein response (UPR) [247]. Blomme et al. found that depletion of THEM6 leads to lipid remodeling in cancer cells, particularly the loss of ether triglycerides, and re-sensitizes prostate cancer cells to ADT [247]. Additionally, yes-associated protein 1 (YAP1) is found to be overexpressed in enzalutamide-, an ADT drug, resistant prostate cancers [248,249]. YAP1 maintains ADT resistance by contributing to cancer stemness and lipid metabolism, in which YAP1+/+, ADT-resistant cells had high levels of free fatty acids and increased expression of genes related to lipid metabolism compared to YAP1−/−, ADT-resistant cells [250]. Both THEM6 and YAP1 are proteins that regulate lipids and are intertwined with ADT resistance at a molecular level (Figure 3A).
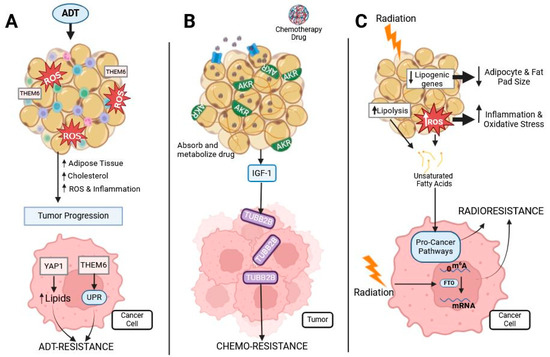
Figure 3.
Prostate cancer treatments’ effect on adipose and cancer. Prostate cancer is commonly treated with ADT, chemotherapies, and radiotherapy, all of which interact with adipose tissue: (A) ADT treatment induces oxidative stress and inflammation in adipose tissue. This causes an increase in adipose tissue, cholesterol, oxidative stress, and inflammation, leading to tumor progression. Obesity-related proteins THEM6 and YAP1 are associated with ADT resistance in prostate cancer by increasing cellular lipid content and activating the UPR. (B) Chemotherapy is often lipophilic and, therefore, easily sequestered and metabolized by adipocytes containing the AKR protein, reducing the availability of the drug for prostate tumors. Chemotherapy induces dysregulated cytokine secretion from adipose tissue, such as an increased release of IGF-1, causing the upregulation of TUBB2B, which is associated with chemo-resistance. (C) Radiation to adipose tissue decreases lipogenic gene expression and increases ROS and lipolysis. Both ROS and lipolysis lead to the release of unsaturated fatty acids from the adipocytes, which cancer cells use to activate pro-cancer pathways. Additionally, radiation activates FTO, which demethylates mRNA to promote radioresistance.
7.2. Chemotherapy
As stated above, obesity has been linked to poor outcomes in patients with multiple different cancers [6,10,11], but the exact mechanism is unknown. Clinically, increased adiposity is positively associated with worse survival in men undergoing ADT and docetaxel treatment [251,252]. Adipocytes are known to sequester lipophilic molecules [253]. Since most chemotherapeutic agents are hydrophobic, they are readily taken up by adipocytes, which contributes to the development of chemoresistance (Figure 3B). Additionally, in vitro and in vivo studies demonstrate that adipocytes secrete factors that induce chemo-resistance in prostate cancer [254,255].
Some of the mechanisms through which adipocytes are believed to induce chemotherapeutic resistance are by metabolizing drugs and altering the redox status of the cancer cells. Cancer cells that express high levels of CD36, a fatty acid transporter, have a more drug-resistant profile than cancer cells that do not express CD36 [256], suggesting a potential interplay between fatty acid metabolism and drug resistance. A pivotal study from 2017, provides strong evidence that adipocytes can absorb and metabolize daunorubicin, a chemotherapeutic drug commonly used to treat leukemias, altering its pharmacokinetics [257]. Adipocytes express an abundant amount of aldo-keto reductase (AKR) enzymes [257,258]. Additionally, AKR is expressed at higher levels in obese individuals and is thought to play a role in obesity-associated chemoresistance [257]. AKRs metabolize many chemotherapeutics including anthracyclines, mitomycin, cisplatins, antitubulin agents, vinca alkaloids, and cyclophosphamides [259]. From this study, we can infer that obese subjects will have less chemotherapeutic efficacy, which is one possible mechanism for obesity-related chemotherapeutic resistance. Abundant adipose tissue and lipids not only promote pro-tumor properties but also disrupt the treatment of cancers leading to chemoresistance and increased likelihood of recurrence. Thus, there is a need to better understand the role that adipocytes have on the TME (Figure 3B).
In prostate cancer, docetaxel is the first line of defense for patients with CRPC [260]. Insulin-like growth factor-1 (IGF-1) secreted from peri-prostatic fat significantly increases prostate cancer cell expression of TUBB2B, β-tubulin isoform 2B [261]. TUBB2B is known to be upregulated in highly metastatic prostate cancer and promotes docetaxel resistance by preventing drug-induced microtubule stabilization [262]. Cabazitaxel is a chemotherapeutic commonly prescribed to metastatic CRPC patients. Like docetaxel, cabazitaxel binds to tubulins impairing microtubule formation, blocking mitosis, and initiating apoptosis [263]. In the presence of ASCs, prostate cancer cells were chemo-resistant to docetaxel and cabazitaxel [255]. Mechanistically, ASCs decrease ROS levels found within the TME, reducing the effect of chemotherapeutic drugs [255]. Therefore, depletion of ASCs from tumors may re-sensitize cancer cells to chemotherapy (Figure 3B).
7.3. Radiotherapy
Another common treatment option for those with prostate cancer is external beam radiation therapy (EBRT). EBRT uses high-energy X-rays or electron beams to kill cancer cells by damaging DNA and hindering the ability to replicate [264]. At the cellular level, radiation plays both a direct and an indirect role in subduing DNA replication processes. Radiation can directly cause DNA damage through single- and double-stranded breaks and with the formation of adducts to certain bases [265,266]. Indirectly, the release of radiation energy generates free radicals and ROS through water ionization hours to days after exposure. Increased ROS increases the susceptibility of DNA, protein, and lipid damage [266,267,268]. It has been documented that obese men with prostate cancer are more likely to experience treatment failure following radiotherapy. However, the effects of radiation on adipose tissue are not well understood and have not been extensively described.
When mice receive pelvic radiation, their fat pads atrophy, which correlates to an increase in fibrosis, and skin damage [269,270]. Poglio et al. are one of very few papers outlining the effect of radiation on adipose tissue [271]. Using both sublethal and lethal doses of radiation, they observed pronounced morphological changes to subcutaneous adipose tissue. Within 7 days after radiation exposure, adipocytes begin apoptosis, and a decrease in the mean size of adipocytes is observed. Not only were the mature adipocytes affected by the radiation, but the stromal vascular fraction (SVF) containing adipocyte precursors was damaged [271], and the stroma of adipose tissue underwent rapid extracellular matrix remodeling [272]. Following radiation treatment, the SVF exhibited decreased proliferative capacity and inhibited differentiation potential. Oxidative stress in adipocytes was observed in a dose-dependent manner, through the increase in NOX expression and decrease in manganese superoxide dismutase activity [271], suggesting a link between oxidative stress and adipocyte cell death.
Metabolically, radiation disrupts the usual metabolic profile of cancer cells. In 2011, Jo et al. aimed to elucidate the effects of whole-body radiation on lipogenic-related gene expression in gonadal white adipose tissue in mice [13]. The expression levels of lipogenic genes such as FASN, acetyl-CoA carboxylase, and glucose-6-pyruvate dehydrogenase decreased following radiation suggesting a possible reason for a decreased fat pad size following radiation [13]. After radiation, cancer cells can be found to have increased lipogenic substrates and intracellular unsaturated fatty acids [273,274]. Unsaturated fatty acids are used within the cells to maintain membrane fluidity, decrease the secretion of pro-inflammatory cytokines, improve the function of vascular endothelial cells, and inhibit pro-inflammatory processes [275]. In patient samples, lipogenic pathways have been found to be enriched in those with glioblastoma recurrence following radiation treatment [273]. The increased biosynthesis of free fatty acids protects cells against ER stress and subsequent apoptosis [273], protecting the cells from the damage inflicted by radiation therapy. These metabolic alterations driven by lipids are one way cancer cells can evade radiotherapy leading to higher recurrence rates.
On the protein level, ROS levels can determine protein activation and inhibition through post-translational adducts. Therefore, radiation-induced ROS levels can have a dominant role in cellular signaling. Fat mass and obesity-associated (FTO) protein, whose expression is elevated in obese subjects, is known to play a prominent role in human obesity, energy homeostasis, and pre-adipogenic regulation [276,277], as well as being upregulated in certain cancers [278,279,280]. FTO is an RNA N6-methyladenosine (m6A) demethylase that regulates the amount of m6A RNA present in nuclear DNA [276,281]. In the presence of radiation, FTO removes m6A RNA methylation leading to the promotion of radiotherapy-resistant cancers by blocking ferroptosis potentially through the demethylation of ubiquitin thioesterase, suggesting a possible synergistic mechanism for radiation resistance in obese patients [279,282,283,284] (Figure 3C).
The demonstration of the effects of radiation on the TME has implicated radiation as a culprit in the recurrence of several cancers including gliomas, breast, and thyroid cancers. Radiation damage to adipose tissue also persists years after the initial radiation treatment as documented in a study that followed adult childhood cancer survivors [14]. The same study was able to establish striking similarities in adipose tissue dysfunction between irradiated and obese adipose tissues including activation of inflammatory pathways and altered adipokines [14]. Irradiating the brain, which contains virtually no triglycerides but vast amounts of lipid-derived cholesterol, with 20 Gy of radiation prior to implantation with human xenograft glioblastomas, significantly increases tumor burden and leads to faster mortality due to alteration in metabolic profiles that favored the proliferation of cancer cells such as rises in ATP/GTP and a decrease in antioxidants [274], suggesting that oxidative stress induced through radiation alters lipid-rich microenvironments to promote tumor progression. Another explanation for radiation-induced cancer recurrence is through the enhancement of cancer stem cells following sublethal doses of radiation and subsequent re-initiation of tumors [285,286,287]. Furthermore, persistent remodeling of the extracellular matrix following radiation of adipose tissue could enable the creation of a pro-tumorigenic microenvironment [272]. However, this is an understudied area, and more work needs to be performed to determine the effect radiation has on adipocytes and how this contributes to the TME.
Adipose tissue and lipids play a multifaceted role in promoting tumor progression. After ADT and radiation, adipose tissue becomes inflamed and oxidatively stressed, like obese adipose tissue. Thus, some therapies may induce a pro-tumor microenvironment. Additionally, adipose tissue sequesters and metabolizes lipophilic chemotherapies, reducing efficacy in tumors with an adipose-rich landscape. On the other hand, obese adipose tissue signals to tumor cells to increase proteins and other factors to protect against ADT, chemotherapy, and radiation (Figure 3). Overall, more research into the effect of therapeutics on adipose tissue is needed to fully elucidate the effect that dysregulated adipose tissue can have on the TME and tumor progression.
7.4. Strategies to Target Adipose Tissue
Obesity drives prostate cancer progression through the presence of chronic inflammation, aberrant adipokine section, increased ROS, and elevated whole-body fatty acid release. The presence of excess adipose tissue also alters the common treatment options for prostate cancer. To combat the progression of cancer, these pathways related to obesity and adipocytes need to be targeted.
Disrupting lipid metabolism within the TME decreases the availability of free fatty acids that the tumor uses to maintain energy, create membranes, and as signaling molecules. SCD converts saturated fatty acids into monosaturated fatty acids and promotes cancer cell proliferation, migration, and metastasis [94,95]. The SCD inhibitor, CAY10566, has been proven to be effective at reducing oleate levels and blocking the growth of tumors [288]. Blocking the circulation of cholesterol using statins, cholesterol-lowering drugs, may help decrease the formation of new membranes, and thus, reduce the ability of cancer cells to proliferate [114,118]. To decrease the uptake of fatty acids by the cancer cells, CD36 could be targeted and blocked. Sulfo-N-hydroxysuccinimidyl (NHS) ester of oleate (SSO) irreversibly binds CD36 and may work as a drug treatment for cancers [289]. To reduce the rate of β-oxidation, targeting DECR1 has been shown to decrease cell viability, migration, and colony formation [110]. Overall, inhibiting any aspect of lipid metabolism may work to reduce the progression of prostate cancer.
Other potential targets for reducing adipose-related cancer progression include altering adipokine secretions. Obese adipose tissue secretes increased leptin and decreased adiponectin. The pro-tumor qualities induced by leptin would be minimized by inhibiting leptin secretion. In breast cancer, LCF1, a leptin activity inhibitor, decreases cancer cell growth and motility [290]. On the other hand, increasing adiponectin, using metformin, enhances anti-tumorigenic properties [291,292]. Therefore, using LCF1 or metformin in combination with other chemotherapeutic agents may be beneficial in lowering overall tumor burden.
Obesity reduces the efficiency of common therapeutic practices to work on prostate cancer. Both ADT and radiation treatments increase the oxidative stress within adipocytes. Adipocytes need to maintain low levels of oxidative stress to function properly. Therefore, the reduction in ROS within adipose tissue specifically would minimize changes in metabolism, inflammation, and cytokine secretion [9]. Vitamin E scavenges lipid peroxyl radicals, so targeting vitamin E directly to the adipose tissue may help reduce damages associated with excess ROS. Additionally, MnTE-2-PyP (T2E), a manganese porphyrin mimetic, protects fat from radiation damage by scavenging ROS [293]. Within the tumor, excess lipid peroxides induce ferroptosis; however, in the presence of radiation, FTO blocks ferroptosis, promoting radiation resistance [279]. Currently, Bisantrene (CS1) and Brequinar (CS2) are two small-molecule FTO inhibitors shown to reduce RNA methyltransferase activity in acute myeloid leukemia patients but have not been tested in prostate cancer [281].
In prostate cancer, THEM6 and YAP1 play integral roles in contributing to ADT resistance. Depleting THEM6 activity re-sensitizes prostate cancer cells to ADT and induces lipid remodeling [247]. YAP1 inhibition in tumors reduces cancer stemness and minimizes lipid metabolism. Blocking both YAP1 and THEM6 proteins re-sensitizes prostate cancer cells to ADT treatment, and therefore, may be a viable option for CRPCs. Docetaxel is the most prescribed chemotherapy for patients with CRPC [260]. TUBB2B prevents drug-induced microtubule stabilization [262]. Thus, depletion or inhibition of TUBB2B from cancer cells, potentially by minimizing IGF-1 secretion, reduces docetaxel resistance.
Finally, since obesity is strongly correlated with an increased risk of developing cancer, weight loss is thought to be a powerful tool for minimizing the chances of developing cancer. Glucagon-like peptide 1 (GLP-1) receptor agonists, like Ozempic, are prescription medications for patients with type 2 diabetes, heart disease, and more recently, obesity. GLP-1 receptor agonists work by stimulating insulin production and reducing blood sugar levels, promoting weight loss. The current literature is divided on how GLP-1 receptor agonist usage is associated with the risk of cancer development. Some studies have noted that GLP-1 agonist usage increased the risk of thyroid cancers [294,295], while others have demonstrated a decreased risk of prostate cancer progression [296,297]. This is an area in which more research is needed.
Table 1 systematically lays out targets to decrease prostate cancer progression, the potential ways to target those biomolecules, and the impact they have on cancer cells (Table 1).

Table 1.
Potential ways to target adipose-driven pathways in prostate cancer.
8. Conclusions and Future Perspectives
It is well documented that obese individuals are at a greater risk of developing cancers, including prostate cancer. Current research has shown that adipose tissue and adipocytes play an intimate role in the progression of prostate cancer. In obese adipose tissue, numerous pro-inflammatory cytokines, as well as abundant amounts of ROS are present and secreted into the microenvironment nearby, altering the function of the adipose tissue [44]. Through many reported mechanisms, obese adipocytes induce the progression of prostate cancer by supplying the cancer cells with building blocks necessary for proliferation, migration, and overall survival [183]. Adipocytes secrete adipokines such as leptin, adiponectin, and CCL2 that recruit pro-inflammatory cells to the microenvironment, helping cancer cells evade the immune system [141]. Conversely, the formation of cancer can also drive inflammation in adipose tissue triggering cancer progression [222].
Obese patients do not respond to prostate cancer therapy very well and have higher mortality rates as compared to non-obese prostate cancer patients. Following radiotherapy, obese cancer patients more frequently report urinary and erectile dysfunction compared to their non-obese counterparts [298]. Additionally, the use of combination treatments was found to exacerbate poor quality of life symptoms [299]. It is currently not known why this occurs. In this review, we have demonstrated that adipose tissue can contribute to resistance to standard treatments used in prostate cancer, such as ADT, chemotherapy, and radiation. Elucidating the cellular and molecular mechanisms of adipose-related ADT- and chemo-resistance, and the effect of radiation on large depots of adipocytes, could improve the standard treatment options for those with prostate cancer.
Author Contributions
The conceptualization of this article was done by K.T.L.-W. and R.E.O.-D.; K.T.L.-W. drafted the article; Review and editing were completed by E.A.K. and R.E.O.-D. All authors have read and agreed to the published version of the manuscript.
Funding
This project was supported, in part, by the Nebraska Center for the Prevention of Obesity and Diseases NPOD Pilot Grant P20 GM104320 and by the National Institute of Health grants R01CA255618 and P30CA036727.
Acknowledgments
All figures were generated with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Key Statistics for Prostate Cancer. All About Cancer 01/12/2023. Available online: https://www.cancer.org/cancer/types/prostate-cancer/about/key-statistics.html (accessed on 30 May 2024).
- World Cancer Research Fund International. Prostate Cancer Statistics 2024. Available online: https://www.wcrf.org/cancer-trends/prostate-cancer-statistics/ (accessed on 30 May 2024).
- Cancer of the Prostate—Cancer Stat Facts. Surveillance, Epidemiology, and End Results Program. Available online: https://seer.cancer.gov/statfacts/html/prost.html (accessed on 11 September 2024).
- Turini, M.; Redaelli, A.; Gramegna, P.; Radice, D. Quality of life and economic considerations in the management of prostate cancer. Pharmacoeconomics 2003, 21, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Di Sebastiano, K.M.; Pinthus, J.H.; Duivenvoorden, W.C.; Patterson, L.; Dubin, J.A.; Mourtzakis, M. Elevated C-Peptides, Abdominal Obesity, and Abnormal Adipokine Profile are Associated With Higher Gleason Scores in Prostate Cancer. Prostate 2017, 77, 211–221. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Obesity and Overweight 2024. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 12 September 2024).
- Zhang, Y.; Daquinag, A.C.; Amaya-Manzanares, F.; Sirin, O.; Tseng, C.; Kolonin, M.G. Stromal progenitor cells from endogenous adipose tissue contribute to pericytes and adipocytes that populate the tumor microenvironment. Cancer Res. 2012, 72, 5198–5208. [Google Scholar] [CrossRef] [PubMed]
- Cozzo, A.J.; Fuller, A.M.; Makowski, L. Contribution of Adipose Tissue to Development of Cancer. Compr. Physiol. 2017, 8, 237–282. [Google Scholar]
- Cheng, Y.; Wang, Z.; Jia, X.; Zhou, R.; Wang, J. Association Between Abdominal Adipose Tissue Distribution and Risk of Endometrial Cancer: A Case-Control Study. Clin. Med. Insights Oncol. 2022, 16, 11795549221140776. [Google Scholar] [CrossRef]
- Jansen, M.P.; Knijnenburg, T.; Reijm, E.A.; Simon, I.; Kerkhoven, R.; Droog, M.; Velds, A.; van Laere, S.; Dirix, L.; Alexi, X.; et al. Hallmarks of aromatase inhibitor drug resistance revealed by epigenetic profiling in breast cancer. Cancer Res. 2013, 73, 6632–6641. [Google Scholar] [CrossRef]
- Bacarella, N.; Ruggiero, A.; Davis, A.T.; Uberseder, B.; Davis, M.A.; Bracy, D.P.; Wasserman, D.H.; Cline, J.M.; Sherrill, C.; Kavanagh, K. Whole Body Irradiation Induces Diabetes and Adipose Insulin Resistance in Nonhuman Primates. Int. J. Radiat. Oncol. Biol. Phys. 2020, 106, 878–886. [Google Scholar] [CrossRef]
- Jo, S.K.; Seol, M.A.; Park, H.R.; Jung, U.; Roh, C. Ionising radiation triggers fat accumulation in white adipose tissue. Int. J. Radiat. Biol. 2011, 87, 302–310. [Google Scholar] [CrossRef]
- Huang, X.; Maguire, O.A.; Walker, J.M.; Jiang, C.S.; Carroll, T.S.; Luo, J.D.; Tonorezos, E.; Friedman, D.N.; Cohen, P. Therapeutic radiation exposure of the abdomen during childhood induces chronic adipose tissue dysfunction. JCI Insight 2021, 6, e153586. [Google Scholar] [CrossRef]
- Mentoor, I.; Engelbrecht, A.M.; van Jaarsveld, P.J.; Nell, T. Chemoresistance: Intricate Interplay Between Breast Tumor Cells and Adipocytes in the Tumor Microenvironment. Front. Endocrinol. 2018, 9, 758. [Google Scholar] [CrossRef]
- Wade, C.A.; Kyprianou, N. Adipose tissue: Enabler of prostate cancer aggressive behavior. Transl. Androl. Urol. 2019, 8, S242–S245. [Google Scholar] [CrossRef]
- Hamilton, G.; Schlein, A.N.; Middleton, M.S.; Hooker, C.A.; Wolfson, T.; Gamst, A.C.; Loomba, R.; Sirlin, C.B. In vivo triglyceride composition of abdominal adipose tissue measured by (1) H MRS at 3T. J. Magn. Reson. Imaging 2017, 45, 1455–1463. [Google Scholar] [CrossRef]
- Bruun, J.M.; Lihn, A.S.; Pedersen, S.B.; Richelsen, B. Monocyte chemoattractant protein-1 release is higher in visceral than subcutaneous human adipose tissue (AT): Implication of macrophages resident in the AT. J. Clin. Endocrinol. Metab. 2005, 90, 2282–2289. [Google Scholar] [CrossRef]
- Yang Loureiro, Z.; Solivan-Rivera, J.; Corvera, S. Adipocyte Heterogeneity Underlying Adipose Tissue Functions. Endocrinology 2022, 163, bqab138. [Google Scholar] [CrossRef]
- Lake, A.C.; Sun, Y.; Li, J.L.; Kim, J.E.; Johnson, J.W.; Li, D.; Revett, T.; Shih, H.H.; Liu, W.; Paulsen, J.E.; et al. Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. J. Lipid Res. 2005, 46, 2477–2487. [Google Scholar] [CrossRef]
- Holm, C. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Biochem. Soc. Trans. 2003, 31, 1120–1124. [Google Scholar] [CrossRef]
- Jenkins, C.M.; Mancuso, D.J.; Yan, W.; Sims, H.F.; Gibson, B.; Gross, R.W. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J. Biol. Chem. 2004, 279, 48968–48975. [Google Scholar] [CrossRef]
- Harada, K.; Shen, W.J.; Patel, S.; Natu, V.; Wang, J.; Osuga, J.; Ishibashi, S.; Kraemer, F.B. Resistance to high-fat diet-induced obesity and altered expression of adipose-specific genes in HSL-deficient mice. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E1182–E1195. [Google Scholar] [CrossRef]
- Prentice, K.J.; Saksi, J.; Robertson, L.T.; Lee, G.Y.; Inouye, K.E.; Eguchi, K.; Lee, A.; Cakici, O.; Otterbeck, E.; Cedillo, P.; et al. A hormone complex of FABP4 and nucleoside kinases regulates islet function. Nature 2021, 600, 720–726. [Google Scholar] [CrossRef]
- Cao, H.; Sekiya, M.; Ertunc, M.E.; Burak, M.F.; Mayers, J.R.; White, A.; Inouye, K.; Rickey, L.M.; Ercal, B.C.; Furuhashi, M.; et al. Adipocyte lipid chaperone AP2 is a secreted adipokine regulating hepatic glucose production. Cell Metab. 2013, 17, 768–778. [Google Scholar] [CrossRef]
- Huat, T.J.; Khan, A.A.; Pati, S.; Mustafa, Z.; Abdullah, J.M.; Jaafar, H. IGF-1 enhances cell proliferation and survival during early differentiation of mesenchymal stem cells to neural progenitor-like cells. BMC Neurosci. 2014, 15, 91. [Google Scholar] [CrossRef]
- Lin, S.; Zhang, Q.; Shao, X.; Zhang, T.; Xue, C.; Shi, S.; Zhao, D.; Lin, Y. IGF-1 promotes angiogenesis in endothelial cells/adipose-derived stem cells co-culture system with activation of PI3K/Akt signal pathway. Cell Prolif. 2017, 50, e12390. [Google Scholar] [CrossRef]
- Rosen, E.D.; Spiegelman, B.M. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006, 444, 847–853. [Google Scholar] [CrossRef]
- Kelley, D.E.; Thaete, F.L.; Troost, F.; Huwe, T.; Goodpaster, B.H. Subdivisions of subcutaneous abdominal adipose tissue and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E941–E948. [Google Scholar] [CrossRef]
- Macotela, Y.; Emanuelli, B.; Mori, M.A.; Gesta, S.; Schulz, T.J.; Tseng, Y.H.; Kahn, C.R. Intrinsic differences in adipocyte precursor cells from different white fat depots. Diabetes 2012, 61, 1691–1699. [Google Scholar] [CrossRef]
- Raajendiran, A.; Krisp, C.; Souza, D.P.; Ooi, G.; Burton, P.R.; Taylor, R.A.; Molloy, M.P.; Watt, M.J. Proteome analysis of human adipocytes identifies depot-specific heterogeneity at metabolic control points. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E1068–E1084. [Google Scholar] [CrossRef]
- Ibrahim, M.M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes. Rev. 2010, 11, 11–18. [Google Scholar] [CrossRef]
- Hellmer, J.; Marcus, C.; Sonnenfeld, T.; Arner, P. Mechanisms for differences in lipolysis between human subcutaneous and omental fat cells. J. Clin. Endocrinol. Metab. 1992, 75, 15–20. [Google Scholar]
- Arner, P.; Hellstrom, L.; Wahrenberg, H.; Bronnegard, M. Beta-adrenoceptor expression in human fat cells from different regions. J. Clin. Investig. 1990, 86, 1595–1600. [Google Scholar] [CrossRef]
- Stenkula, K.G.; Erlanson-Albertsson, C. Adipose cell size: Importance in health and disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R284–R295. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [PubMed]
- Rossmeislova, L.; Malisova, L.; Kracmerova, J.; Tencerova, M.; Kovacova, Z.; Koc, M.; Siklova-Vitkova, M.; Viquerie, N.; Langin, D.; Stich, V. Weight loss improves the adipogenic capacity of human preadipocytes and modulates their secretory profile. Diabetes 2013, 62, 1990–1995. [Google Scholar] [CrossRef]
- Andersen, E.; Ingerslev, L.R.; Fabre, O.; Donkin, I.; Altintas, A.; Versteyhe, S.; Bisgaard, T.; Kristiansen, V.B.; Simar, D.; Barres, R. Preadipocytes from obese humans with type 2 diabetes are epigenetically reprogrammed at genes controlling adipose tissue function. Int. J. Obes. 2019, 43, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Tchoukalova, Y.; Koutsari, C.; Jensen, M. Committed subcutaneous preadipocytes are reduced in human obesity. Diabetologia 2007, 50, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Silva, K.R.; Cortes, I.; Liechocki, S.; Carneiro, J.R.; Souza, A.A.; Borojevic, R.; Maya-Monteiro, C.M.; Baptista, L.S. Characterization of stromal vascular fraction and adipose stem cells from subcutaneous, preperitoneal and visceral morbidly obese human adipose tissue depots. PLoS ONE 2017, 12, e0174115. [Google Scholar] [CrossRef] [PubMed]
- Roldan, M.; Macias-Gonzalez, M.; Garcia, R.; Tinahones, F.J.; Martin, M. Obesity short-circuits stemness gene network in human adipose multipotent stem cells. FASEB J. 2011, 25, 4111–4126. [Google Scholar] [CrossRef]
- Divoux, A.; Tordjman, J.; Lacasa, D.; Veyrie, N.; Hugol, D.; Aissat, A.; Basdevant, A.; Guerre-Millo, M.; Poitou, C.; Zucker, J.D.; et al. Fibrosis in human adipose tissue: Composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes 2010, 59, 2817–2825. [Google Scholar] [CrossRef]
- Tourniaire, F.; Romier-Crouzet, B.; Lee, J.H.; Marcotorchino, J.; Gouranton, E.; Salles, J.; Malezet, C.; Astier, J.; Darmon, P.; Blouin, E.; et al. Chemokine Expression in Inflamed Adipose Tissue Is Mainly Mediated by NF-kappaB. PLoS ONE 2013, 8, e66515. [Google Scholar] [CrossRef]
- Lapice, E.; Maione, S.; Patti, L.; Cipriano, P.; Rivellese, A.A.; Riccardi, G.; Vaccaro, O. Abdominal adiposity is associated with elevated C-reactive protein independent of BMI in healthy nonobese people. Diabetes Care 2009, 32, 1734–1736. [Google Scholar] [CrossRef]
- Nakai, K.; Tanaka, H.; Yamanaka, K.; Takahashi, Y.; Murakami, F.; Matsuike, R.; Sekino, J.; Tanabe, N.; Morita, T.; Yamazaki, Y.; et al. Effects of C-reactive protein on the expression of matrix metalloproteinases and their inhibitors via Fcgamma receptors on 3T3-L1 adipocytes. Int. J. Med. Sci. 2017, 14, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Jakubiak, G.K.; Osadnik, K.; Lejawa, M.; Kasperczyk, S.; Osadnik, T.; Pawlas, N. Oxidative Stress in Association with Metabolic Health and Obesity in Young Adults. Oxid. Med. Cell Longev. 2021, 2021, 9987352. [Google Scholar] [CrossRef] [PubMed]
- Fernando, R.; Wardelmann, K.; Deubel, S.; Kehm, R.; Jung, T.; Mariotti, M.; Vasilaki, A.; Gladyshev, V.N.; Kleinridders, A.; Grune, T.; et al. Low steady-state oxidative stress inhibits adipogenesis by altering mitochondrial dynamics and decreasing cellular respiration. Redox Biol. 2020, 32, 101507. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Bravo-San Pedro, J.M. Targeting Autophagy to Counteract Obesity-Associated Oxidative Stress. Antioxidants 2021, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- Arruda, A.P.; Pers, B.M.; Parlakgul, G.; Guney, E.; Inouye, K.; Hotamisligil, G.S. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 2014, 20, 1427–1435. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, A.Y.; Choi, J.W.; Kim, M.; Yasue, S.; Son, H.J.; Masuzaki, H.; Park, K.S.; Kim, J.B. Dysregulation of adipose glutathione peroxidase 3 in obesity contributes to local and systemic oxidative stress. Mol. Endocrinol. 2008, 22, 2176–2189. [Google Scholar] [CrossRef]
- Girotti, A.W. Mechanisms of lipid peroxidation. J. Free Radic. Biol. Med. 1985, 1, 87–95. [Google Scholar] [CrossRef]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Murdolo, G.; Bartolini, D.; Tortoioli, C.; Vermigli, C.; Piroddi, M.; Galli, F. Accumulation of 4-Hydroxynonenal Characterizes Diabetic Fat and Modulates Adipogenic Differentiation of Adipose Precursor Cells. Int. J. Mol. Sci. 2023, 24, 16645. [Google Scholar] [CrossRef]
- Khan, M.I.; Hamid, A.; Adhami, V.M.; Lall, R.K.; Mukhtar, H. Role of epithelial mesenchymal transition in prostate tumorigenesis. Curr. Pharm. Des. 2015, 21, 1240–1248. [Google Scholar] [CrossRef]
- Price, R.S.; Cavazos, D.A.; De Angel, R.E.; Hursting, S.D.; deGraffenried, L.A. Obesity-related systemic factors promote an invasive phenotype in prostate cancer cells. Prostate Cancer Prostatic Dis. 2012, 15, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, J.; Yoon, J.H.; Ghim, J.; Yea, K.; Song, P.; Park, S.; Lee, A.; Hong, C.P.; Jang, M.S.; et al. CXCL12 secreted from adipose tissue recruits macrophages and induces insulin resistance in mice. Diabetologia 2014, 57, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Ahn, S.; Blando, J.; Su, F.; Kolonin, M.G.; DiGiovanni, J. Proinflammatory CXCL12-CXCR4/CXCR7 Signaling Axis Drives Myc-Induced Prostate Cancer in Obese Mice. Cancer Res. 2017, 77, 5158–5168. [Google Scholar] [CrossRef] [PubMed]
- Lavaee, F.; Zare, S.; Mojtahedi, Z.; Malekzadeh, M.; Khademi, B.; Ghaderi, A. Serum CXCL12, but not CXCR4, Is Associated with Head and Neck Squamous Cell Carcinomas. Asian Pac. J. Cancer Prev. 2018, 19, 901–904. [Google Scholar]
- Su, F.; Daquinag, A.C.; Ahn, S.; Saha, A.; Dai, Y.; Zhao, Z.; DiGiovanni, J.; Kolonin, M.G. Progression of prostate carcinoma is promoted by adipose stromal cell-secreted CXCL12 signaling in prostate epithelium. NPJ Precis. Oncol. 2021, 5, 26. [Google Scholar] [CrossRef]
- Laurent, V.; Guerard, A.; Mazerolles, C.; Le Gonidec, S.; Toulet, A.; Nieto, L.; Zaidi, F.; Majed, B.; Garandeau, D.; Socrier, Y.; et al. Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat. Commun. 2016, 7, 10230. [Google Scholar] [CrossRef]
- Forootan, F.S.; Forootan, S.S.; Malki, M.I.; Chen, D.; Li, G.; Lin, K.; Rudland, P.S.; Foster, C.S.; Ke, Y. The expression of C-FABP and PPARgamma and their prognostic significance in prostate cancer. Int. J. Oncol. 2014, 44, 265–275. [Google Scholar] [CrossRef]
- Segawa, Y.; Yoshimura, R.; Hase, T.; Nakatani, T.; Wada, S.; Kawahito, Y.; Kishimoto, T.; Sano, H. Expression of peroxisome proliferator-activated receptor (PPAR) in human prostate cancer. Prostate 2002, 51, 108–116. [Google Scholar] [CrossRef]
- Uehara, H.; Takahashi, T.; Oha, M.; Ogawa, H.; Izumi, K. Exogenous fatty acid binding protein 4 promotes human prostate cancer cell progression. Int. J. Cancer 2014, 135, 2558–2568. [Google Scholar] [CrossRef]
- Rosen, E.D.; Hsu, C.H.; Wang, X.; Sakai, S.; Freeman, M.W.; Gonzalez, F.J.; Spiegelman, B.M. C/EBPalpha induces adipogenesis through PPARgamma: A unified pathway. Genes. Dev. 2002, 16, 22–26. [Google Scholar] [CrossRef]
- Forootan, F.S.; Forootan, S.S.; Gou, X.; Yang, J.; Liu, B.; Chen, D.; Al Fayi, M.S.; Al-Jameel, W.; Rudland, P.S.; Hussain, S.A.; et al. Fatty acid activated PPARgamma promotes tumorigenicity of prostate cancer cells by up regulating VEGF via PPAR responsive elements of the promoter. Oncotarget 2016, 7, 9322–9339. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, L.C.A.; Mui, E.; Nixon, C.; Hedley, A.; Strachan, D.; MacKay, G.; Sumpton, D.; Sansom, O.J.; Leung, H.Y.; Ahmad, I. PPAR-gamma induced AKT3 expression increases levels of mitochondrial biogenesis driving prostate cancer. Oncogene 2021, 40, 2355–2366. [Google Scholar] [CrossRef] [PubMed]
- Salji, M.; Hendry, J.; Patel, A.; Ahmad, I.; Nixon, C.; Leung, H.Y. Peri-prostatic Fat Volume Measurement as a Predictive Tool for Castration Resistance in Advanced Prostate Cancer. Eur. Urol. Focus 2018, 4, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Centenera, M.M.; Swinnen, J.V. Androgen control of lipid metabolism in prostate cancer: Novel insights and future applications. Endocr. Relat. Cancer 2016, 23, R219–R227. [Google Scholar] [CrossRef]
- Van de Sande, T.; Roskams, T.; Lerut, E.; Joniau, S.; Van Poppel, H.; Verhoeven, G.; Swinnen, J.V. High-level expression of fatty acid synthase in human prostate cancer tissues is linked to activation and nuclear localization of Akt/PKB. J. Pathol. 2005, 206, 214–219. [Google Scholar] [CrossRef]
- Diedrich, J.D.; Rajagurubandara, E.; Herroon, M.K.; Mahapatra, G.; Huttemann, M.; Podgorski, I. Bone marrow adipocytes promote the Warburg phenotype in metastatic prostate tumors via HIF-1alpha activation. Oncotarget 2016, 7, 64854–64877. [Google Scholar] [CrossRef]
- Chen, J.; Guccini, I.; Di Mitri, D.; Brina, D.; Revandkar, A.; Sarti, M.; Pasquini, E.; Alajati, A.; Pinton, S.; Losa, M.; et al. Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nat. Genet. 2018, 50, 219–228. [Google Scholar] [CrossRef]
- Taylor, R.A.; Lo, J.; Ascui, N.; Watt, M.J. Linking obesogenic dysregulation to prostate cancer progression. Endocr. Connect. 2015, 4, R68–R80. [Google Scholar] [CrossRef]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef]
- dos Santos, C.R.; Domingues, G.; Matias, I.; Matos, J.; Fonseca, I.; de Almeida, J.M.; Dias, S. LDL-cholesterol signaling induces breast cancer proliferation and invasion. Lipids Health Dis. 2014, 13, 16. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Nambiar, D.K.; Ramteke, A.; Kumar, R.; Dhar, D.; Agarwal, C.; Bergman, B.; Graner, M.; Maroni, P.; Singh, R.P.; et al. Hypoxia induces triglycerides accumulation in prostate cancer cells and extracellular vesicles supporting growth and invasiveness following reoxygenation. Oncotarget 2015, 6, 22836–22856. [Google Scholar] [CrossRef] [PubMed]
- Arner, P.; Bernard, S.; Salehpour, M.; Possnert, G.; Liebl, J.; Steier, P.; Buchholz, B.A.; Eriksson, M.; Arner, E.; Hauner, H.; et al. Dynamics of human adipose lipid turnover in health and metabolic disease. Nature 2011, 478, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Ryden, M.; Andersson, D.P.; Bernard, S.; Spalding, K.; Arner, P. Adipocyte triglyceride turnover and lipolysis in lean and overweight subjects. J. Lipid Res. 2013, 54, 2909–2913. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhu, Y.; Lin, X.; Tan, X.; Lu, B.; Li, Y. Stabilization of FASN by ACAT1-mediated GNPAT acetylation promotes lipid metabolism and hepatocarcinogenesis. Oncogene 2020, 39, 2437–2449. [Google Scholar] [CrossRef]
- Li, X.; Gao, S.; Li, W.; Liu, Z.; Shi, Z.; Qiu, C.; Jiang, J. Effect of monoacylglycerol lipase on the tumor growth in endometrial cancer. J. Obstet. Gynaecol. Res. 2019, 45, 2043–2054. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, B.; Seviour, E.G.; Tao, K.X.; Liu, X.H.; Ling, Y.; Chen, J.Y.; Wang, G.B. Monoacylglycerol lipase (MAGL) knockdown inhibits tumor cells growth in colorectal cancer. Cancer Lett. 2011, 307, 6–17. [Google Scholar] [CrossRef]
- Carbonetti, G.; Wilpshaar, T.; Kroonen, J.; Studholme, K.; Converso, C.; d’Oelsnitz, S.; Kaczocha, M. FABP5 coordinates lipid signaling that promotes prostate cancer metastasis. Sci. Rep. 2019, 9, 18944. [Google Scholar] [CrossRef]
- Nomura, D.K.; Lombardi, D.P.; Chang, J.W.; Niessen, S.; Ward, A.M.; Long, J.Z.; Hoover, H.H.; Cravatt, B.F. Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem. Biol. 2011, 18, 846–856. [Google Scholar] [CrossRef]
- Hertzel, A.V.; Bennaars-Eiden, A.; Bernlohr, D.A. Increased lipolysis in transgenic animals overexpressing the epithelial fatty acid binding protein in adipose cells. J. Lipid Res. 2002, 43, 2105–2111. [Google Scholar] [CrossRef]
- Zha, S.; Ferdinandusse, S.; Hicks, J.L.; Denis, S.; Dunn, T.A.; Wanders, R.J.; Luo, J.; De Marzo, A.M.; Isaacs, W.B. Peroxisomal branched chain fatty acid beta-oxidation pathway is upregulated in prostate cancer. Prostate 2005, 63, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Edlind, M.P.; Hsieh, A.C. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J. Androl. 2014, 16, 378–386. [Google Scholar] [PubMed]
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crosby, K.; Sheen, J.H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 2009, 15, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Nada, S.; Kimura, H.; Tajima, S.; Takahashi, Y.; Kitamura, A.; Oneyama, C.; Okada, M. The mTOR pathway controls cell proliferation by regulating the FoxO3a transcription factor via SGK1 kinase. PLoS ONE 2014, 9, e88891. [Google Scholar] [CrossRef]
- Han, J.; Li, E.; Chen, L.; Zhang, Y.; Wei, F.; Liu, J.; Deng, H.; Wang, Y. The CREB coactivator CRTC2 controls hepatic lipid metabolism by regulating SREBP1. Nature 2015, 524, 243–246. [Google Scholar] [CrossRef]
- Wu, S.; Naar, A.M. SREBP1-dependent de novo fatty acid synthesis gene expression is elevated in malignant melanoma and represents a cellular survival trait. Sci. Rep. 2019, 9, 10369. [Google Scholar] [CrossRef]
- Lounis, M.A.; Bergeron, K.F.; Burhans, M.S.; Ntambi, J.M.; Mounier, C. Oleate activates SREBP-1 signaling activity in SCD1-deficient hepatocytes. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E710–E720. [Google Scholar] [CrossRef]
- Guan, M.; Fousek, K.; Chow, W.A. Nelfinavir inhibits regulated intramembrane proteolysis of sterol regulatory element binding protein-1 and activating transcription factor 6 in castration-resistant prostate cancer. FEBS J. 2012, 279, 2399–2411. [Google Scholar] [CrossRef]
- Kim, S.J.; Choi, H.; Park, S.S.; Chang, C.; Kim, E. Stearoyl CoA desaturase (SCD) facilitates proliferation of prostate cancer cells through enhancement of androgen receptor transactivation. Mol. Cells 2011, 31, 371–377. [Google Scholar] [CrossRef]
- Roongta, U.V.; Pabalan, J.G.; Wang, X.; Ryseck, R.P.; Fargnoli, J.; Henley, B.J.; Yang, W.P.; Zhu, J.; Madireddi, M.T.; Lawrence, R.M.; et al. Cancer cell dependence on unsaturated fatty acids implicates stearoyl-CoA desaturase as a target for cancer therapy. Mol. Cancer Res. 2011, 9, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Lopez, E.F.; Cruz-Hernandez, C.D.; Cortes-Ramirez, S.A.; Ramirez-Higuera, A.; Pena-Montes, C.; Rodriguez-Dorantes, M.; Oliart-Ros, R.M. Inhibition of Stearoyl-CoA Desaturase by Sterculic Oil Reduces Proliferation and Induces Apoptosis in Prostate Cancer Cell Lines. Nutr. Cancer 2022, 74, 1308–1321. [Google Scholar] [CrossRef] [PubMed]
- Hosios, A.M.; Hecht, V.C.; Danai, L.V.; Johnson, M.O.; Rathmell, J.C.; Steinhauser, M.L.; Manalis, S.R.; Vander Heiden, M.G. Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev. Cell 2016, 36, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.H.; Fowle-Grider, R.; Mahieu, N.G.; Liu, G.Y.; Chen, Y.J.; Wang, R.; Singh, M.; Potter, G.S.; Gross, R.W.; Schaefer, J.; et al. Exogenous Fatty Acids Are the Preferred Source of Membrane Lipids in Proliferating Fibroblasts. Cell Chem. Biol. 2016, 23, 483–493. [Google Scholar] [CrossRef]
- Li, Z.; Ji, B.W.; Dixit, P.D.; Tchourine, K.; Lien, E.C.; Hosios, A.M.; Abbott, K.L.; Rutter, J.C.; Westermark, A.M.; Gorodetsky, E.F.; et al. Cancer cells depend on environmental lipids for proliferation when electron acceptors are limited. Nat. Metab. 2022, 4, 711–723. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef]
- Diehl, F.F.; Lewis, C.A.; Fiske, B.P.; Vander Heiden, M.G. Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nat. Metab. 2019, 1, 861–867. [Google Scholar] [CrossRef]
- Titov, D.V.; Cracan, V.; Goodman, R.P.; Peng, J.; Grabarek, Z.; Mootha, V.K. Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science 2016, 352, 231–235. [Google Scholar] [CrossRef]
- Kim, W.; Deik, A.; Gonzalez, C.; Gonzalez, M.E.; Fu, F.; Ferrari, M.; Churchhouse, C.L.; Florez, J.C.; Jacobs, S.B.R.; Clish, C.B.; et al. Polyunsaturated Fatty Acid Desaturation Is a Mechanism for Glycolytic NAD(+) Recycling. Cell Metab. 2019, 29, 856–870.e7. [Google Scholar] [CrossRef]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Balaban, S.; Nassar, Z.D.; Zhang, A.Y.; Hosseini-Beheshti, E.; Centenera, M.M.; Schreuder, M.; Lin, H.M.; Aishah, A.; Varney, B.; Liu-Fu, F.; et al. Extracellular Fatty Acids Are the Major Contributor to Lipid Synthesis in Prostate Cancer. Mol. Cancer Res. 2019, 17, 949–962. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijon, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glode, L.M.; Eckel, R.H.; et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Itkonen, H.M.; Brown, M.; Urbanucci, A.; Tredwell, G.; Ho Lau, C.; Barfeld, S.; Hart, C.; Guldvik, I.J.; Takhar, M.; Heemers, H.V.; et al. Lipid degradation promotes prostate cancer cell survival. Oncotarget 2017, 8, 38264–38275. [Google Scholar] [CrossRef]
- Flaig, T.W.; Salzmann-Sullivan, M.; Su, L.J.; Zhang, Z.; Joshi, M.; Gijon, M.A.; Kim, J.; Arcaroli, J.J.; Van Bokhoven, A.; Lucia, M.S.; et al. Lipid catabolism inhibition sensitizes prostate cancer cells to antiandrogen blockade. Oncotarget 2017, 8, 56051–56065. [Google Scholar] [CrossRef]
- Nassar, Z.D.; Mah, C.Y.; Dehairs, J.; Burvenich, I.J.; Irani, S.; Centenera, M.M.; Helm, M.; Shrestha, R.K.; Moldovan, M.; Don, A.S.; et al. Human DECR1 is an androgen-repressed survival factor that regulates PUFA oxidation to protect prostate tumor cells from ferroptosis. eLife 2020, 9, e54166. [Google Scholar] [CrossRef]
- Szlasa, W.; Zendran, I.; Zalesinska, A.; Tarek, M.; Kulbacka, J. Lipid composition of the cancer cell membrane. J. Bioenerg. Biomembr. 2020, 52, 321–342. [Google Scholar] [CrossRef]
- Kopecka, J.; Trouillas, P.; Gasparovic, A.C.; Gazzano, E.; Assaraf, Y.G.; Riganti, C. Phospholipids and cholesterol: Inducers of cancer multidrug resistance and therapeutic targets. Drug Resist. Updat. 2020, 49, 100670. [Google Scholar] [CrossRef]
- Dufourc, E.J. Sterols and membrane dynamics. J. Chem. Biol. 2008, 1, 63–77. [Google Scholar] [CrossRef]
- Ashida, S.; Kawada, C.; Inoue, K. Stromal regulation of prostate cancer cell growth by mevalonate pathway enzymes HMGCS1 and HMGCR. Oncol. Lett. 2017, 14, 6533–6542. [Google Scholar] [CrossRef]
- Gong, L.; Xiao, Y.; Xia, F.; Wu, P.; Zhao, T.; Xie, S.; Wang, R.; Wen, Q.; Zhou, W.; Xu, H.; et al. The mevalonate coordinates energy input and cell proliferation. Cell Death Dis. 2019, 10, 327. [Google Scholar] [CrossRef] [PubMed]
- Greenaway, J.B.; Virtanen, C.; Osz, K.; Revay, T.; Hardy, D.; Shepherd, T.; DiMattia, G.; Petrik, J. Ovarian tumour growth is characterized by mevalonate pathway gene signature in an orthotopic, syngeneic model of epithelial ovarian cancer. Oncotarget 2016, 7, 47343–47365. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.H.; Huang, C.H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P.t.; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2019, 176, 564–580.e19. [Google Scholar] [CrossRef] [PubMed]
- Longo, J.; Mullen, P.J.; Yu, R.; van Leeuwen, J.E.; Masoomian, M.; Woon, D.T.S.; Wang, Y.; Chen, E.X.; Hamilton, R.J.; Sweet, J.M.; et al. An actionable sterol-regulated feedback loop modulates statin sensitivity in prostate cancer. Mol. Metab. 2019, 25, 119–130. [Google Scholar] [CrossRef]
- Fagone, P.; Jackowski, S. Membrane phospholipid synthesis and endoplasmic reticulum function. J. Lipid Res. 2009, 50, S311–S316. [Google Scholar] [CrossRef]
- Hobby, C.R.; Herndon, J.L.; Morrow, C.A.; Peters, R.E.; Symes, S.J.K.; Giles, D.K. Exogenous fatty acids alter phospholipid composition, membrane permeability, capacity for biofilm formation, and antimicrobial peptide susceptibility in Klebsiella pneumoniae. Microbiologyopen 2019, 8, e00635. [Google Scholar] [CrossRef]
- Boden, G. Obesity, insulin resistance and free fatty acids. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 139–143. [Google Scholar] [CrossRef]
- Boden, G.; Chen, X. Effects of fat on glucose uptake and utilization in patients with non-insulin-dependent diabetes. J. Clin. Investig. 1995, 96, 1261–1268. [Google Scholar] [CrossRef]
- Dresner, A.; Laurent, D.; Marcucci, M.; Griffin, M.E.; Dufour, S.; Cline, G.W.; Slezak, L.A.; Andersen, D.K.; Hundal, R.S.; Rothman, D.L.; et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J. Clin. Investig. 1999, 103, 253–259. [Google Scholar] [CrossRef]
- Reynisdottir, S.; Langin, D.; Carlstrom, K.; Holm, C.; Rossner, S.; Arner, P. Effects of weight reduction on the regulation of lipolysis in adipocytes of women with upper-body obesity. Clin. Sci. 1995, 89, 421–429. [Google Scholar] [CrossRef]
- Li, Q.; Guo, S.; Yang, C.; Liu, X.; Chen, X.; He, J.; Tong, C.; Ding, Y.; Peng, C.; Geng, Y.; et al. High-fat diet-induced obesity primes fatty acid beta-oxidation impairment and consequent ovarian dysfunction during early pregnancy. Ann. Transl. Med. 2021, 9, 887. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Wang, Y.; Zhang, X.; Li, P.; Shang, L.; Chen, X.; Zeng, J. Targeting peroxisomal fatty acid oxidation improves hepatic steatosis and insulin resistance in obese mice. J. Biol. Chem. 2023, 299, 102845. [Google Scholar] [CrossRef] [PubMed]
- Dobbins, R.L.; Szczepaniak, L.S.; Bentley, B.; Esser, V.; Myhill, J.; McGarry, J.D. Prolonged inhibition of muscle carnitine palmitoyltransferase-1 promotes intramyocellular lipid accumulation and insulin resistance in rats. Diabetes 2001, 50, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Leamy, A.K.; Egnatchik, R.A.; Young, J.D. Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease. Prog. Lipid Res. 2013, 52, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Snieckute, G.; Ryder, L.; Vind, A.C.; Wu, Z.; Arendrup, F.S.; Stoneley, M.; Chamois, S.; Martinez-Val, A.; Leleu, M.; Dreos, R.; et al. ROS-induced ribosome impairment underlies ZAKalpha-mediated metabolic decline in obesity and aging. Science 2023, 382, eadf3208. [Google Scholar] [CrossRef]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef]
- Grimsrud, P.A.; Picklo, M.J., Sr.; Griffin, T.J.; Bernlohr, D.A. Carbonylation of adipose proteins in obesity and insulin resistance: Identification of adipocyte fatty acid-binding protein as a cellular target of 4-hydroxynonenal. Mol. Cell Proteom. 2007, 6, 624–637. [Google Scholar] [CrossRef]
- Curtis, J.M.; Hahn, W.S.; Long, E.K.; Burrill, J.S.; Arriaga, E.A.; Bernlohr, D.A. Protein carbonylation and metabolic control systems. Trends Endocrinol. Metab. 2012, 23, 399–406. [Google Scholar] [CrossRef]
- Jacobs, R.L.; Zhao, Y.; Koonen, D.P.; Sletten, T.; Su, B.; Lingrell, S.; Cao, G.; Peake, D.A.; Kuo, M.S.; Proctor, S.D.; et al. Impaired de novo choline synthesis explains why phosphatidylethanolamine N-methyltransferase-deficient mice are protected from diet-induced obesity. J. Biol. Chem. 2010, 285, 22403–22413. [Google Scholar] [CrossRef]
- van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef]
- Bakan, E.; Yildirim, A.; Kurtul, N.; Polat, M.F.; Dursun, H.; Cayir, K. Effects of type 2 diabetes mellitus on plasma fatty acid composition and cholesterol content of erythrocyte and leukocyte membranes. Acta Diabetol. 2006, 43, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Pilon, M. Revisiting the membrane-centric view of diabetes. Lipids Health Dis. 2016, 15, 167. [Google Scholar] [CrossRef] [PubMed]
- Ayala, G.E.; Wheeler, T.M.; Shine, H.D.; Schmelz, M.; Frolov, A.; Chakraborty, S.; Rowley, D. In vitro dorsal root ganglia and human prostate cell line interaction: Redefining perineural invasion in prostate cancer. Prostate 2001, 49, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Senaris, R.; Garcia-Caballero, T.; Casabiell, X.; Gallego, R.; Castro, R.; Considine, R.V.; Dieguez, C.; Casanueva, F.F. Synthesis of leptin in human placenta. Endocrinology 1997, 138, 4501–4504. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.; Kim, K.W.; Kim, M.S. Leptin signalling pathways in hypothalamic neurons. Cell Mol. Life Sci. 2016, 73, 1457–1477. [Google Scholar] [CrossRef]
- Nguyen, T.M.D. Adiponectin: Role in Physiology and Pathophysiology. Int. J. Prev. Med. 2020, 11, 136. [Google Scholar] [CrossRef]
- Nigro, E.; Orlandella, F.M.; Polito, R.; Mariniello, R.M.; Monaco, M.L.; Mallardo, M.; De Stefano, A.E.; Iervolino, P.L.C.; Salvatore, G.; Daniele, A. Adiponectin and leptin exert antagonizing effects on proliferation and motility of papillary thyroid cancer cell lines. J. Physiol. Biochem. 2021, 77, 237–248. [Google Scholar] [CrossRef]
- Assidi, M.; Yahya, F.M.; Al-Zahrani, M.H.; Elkhatib, R.; Zari, A.; Elaimi, A.; Al-Maghrabi, J.; Dallol, A.; Buhmeida, A.; Abu-Elmagd, M. Leptin Protein Expression and Promoter Methylation in Ovarian Cancer: A Strong Prognostic Value with Theranostic Promises. Int. J. Mol. Sci. 2021, 22, 12872. [Google Scholar] [CrossRef]
- Chen, X.; Zha, X.; Chen, W.; Zhu, T.; Qiu, J.; Roe, O.D.; Li, J.; Wang, Z.; Yin, Y. Leptin attenuates the anti-estrogen effect of tamoxifen in breast cancer. Biomed. Pharmacother. 2013, 67, 22–30. [Google Scholar] [CrossRef]
- Gorrab, A.; Pagano, A.; Ayed, K.; Chebil, M.; Derouiche, A.; Kovacic, H.; Gati, A. Leptin Promotes Prostate Cancer Proliferation and Migration by Stimulating STAT3 Pathway. Nutr. Cancer 2021, 73, 1217–1227. [Google Scholar] [CrossRef]
- Onuma, M.; Bub, J.D.; Rummel, T.L.; Iwamoto, Y. Prostate cancer cell-adipocyte interaction: Leptin mediates androgen-independent prostate cancer cell proliferation through c-Jun NH2-terminal kinase. J. Biol. Chem. 2003, 278, 42660–42667. [Google Scholar] [CrossRef] [PubMed]
- Saxena, N.K.; Vertino, P.M.; Anania, F.A.; Sharma, D. leptin-induced growth stimulation of breast cancer cells involves recruitment of histone acetyltransferases and mediator complex to CYCLIN D1 promoter via activation of Stat3. J. Biol. Chem. 2007, 282, 13316–13325. [Google Scholar] [CrossRef] [PubMed]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [PubMed]
- Sierra-Honigmann, M.R.; Nath, A.K.; Murakami, C.; Garcia-Cardena, G.; Papapetropoulos, A.; Sessa, W.C.; Madge, L.A.; Schechner, J.S.; Schwabb, M.B.; Polverini, P.J.; et al. Biological action of leptin as an angiogenic factor. Science 1998, 281, 1683–1686. [Google Scholar] [CrossRef] [PubMed]
- Park, K.B.; Kim, E.Y.; Chin, H.; Yoon, D.J.; Jun, K.H. Leptin stimulates migration and invasion and maintains cancer stem-like properties in gastric cancer cells. Oncol. Rep. 2022, 48, 21100. [Google Scholar] [CrossRef]
- Chen, C.; Chang, Y.C.; Lan, M.S.; Breslin, M. Leptin stimulates ovarian cancer cell growth and inhibits apoptosis by increasing cyclin D1 and Mcl-1 expression via the activation of the MEK/ERK1/2 and PI3K/Akt signaling pathways. Int. J. Oncol. 2013, 42, 1113–1119. [Google Scholar] [CrossRef]
- Fazolini, N.P.; Cruz, A.L.; Werneck, M.B.; Viola, J.P.; Maya-Monteiro, C.M.; Bozza, P.T. Leptin activation of mTOR pathway in intestinal epithelial cell triggers lipid droplet formation, cytokine production and increased cell proliferation. Cell Cycle 2015, 14, 2667–2676. [Google Scholar] [CrossRef]
- Easton, J.B.; Houghton, P.J. mTOR and cancer therapy. Oncogene 2006, 25, 6436–6446. [Google Scholar] [CrossRef]
- Revollo, J.R.; Grimm, A.A.; Imai, S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 2004, 279, 50754–50763. [Google Scholar] [CrossRef]
- Cymbaluk-Ploska, A.; Chudecka-Glaz, A.; Pius-Sadowska, E.; Sompolska-Rzechula, A.; Machalinski, B.; Menkiszak, J. Circulating Serum Level of Visfatin in Patients with Endometrial Cancer. BioMed Res. Int. 2018, 2018, 8576179. [Google Scholar] [CrossRef]
- Huang, C.L.; Achudhan, D.; Liu, P.I.; Lin, Y.Y.; Liu, S.C.; Guo, J.H.; Liu, C.L.; Wu, C.Y.; Wang, S.W.; Tang, C.H. Visfatin upregulates VEGF-C expression and lymphangiogenesis in esophageal cancer by activating MEK1/2-ERK and NF-kappaB signaling. Aging 2023, 15, 4774–4793. [Google Scholar] [PubMed]
- Sun, Y.; Zhu, S.; Wu, Z.; Huang, Y.; Liu, C.; Tang, S.; Wei, L. Elevated serum visfatin levels are associated with poor prognosis of hepatocellular carcinoma. Oncotarget 2017, 8, 23427–23435. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hasan, M.K.; Alvarado, E.; Yuan, H.; Wu, H.; Chen, W.Y. NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene 2011, 30, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Zeng, Z.C.; Xi, M.; Wan, S.; Hua, W.; Liu, Y.L.; Zhou, Y.L.; Luo, H.W.; Jiang, F.N.; Zhong, W.D. Dysregulated microRNA-224/apelin axis associated with aggressive progression and poor prognosis in patients with prostate cancer. Hum. Pathol. 2015, 46, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.H.; Chang, S.L.; Khanh, P.M.; Trang, N.T.N.; Liu, S.C.; Tsai, H.C.; Chang, A.C.; Lin, J.Y.; Chen, P.C.; Liu, J.F.; et al. Apelin Promotes Prostate Cancer Metastasis by Downregulating TIMP2 via Increases in miR-106a-5p Expression. Cells 2022, 11, 3285. [Google Scholar] [CrossRef]
- Soylu, H.; Unal, B.; Aksu, K.; Avci, S.; Caylan, A.E.; Ustunel, I.I. Evaluation of angiogenic apelin/apelin receptor axis in normal prostate, high grade prostatic intraepithelial neoplasia and prostatic adenocarcinoma. Malays. J. Pathol. 2022, 44, 461–467. [Google Scholar]
- Liu, C.W.; Peng, H.Y.; Siao, A.C.; Tsuei, Y.W.; Lin, Y.Y.; Shiah, S.G.; Shih, L.J.; Yeh, C.C.; Lee, S.W.; Kao, Y.H. Resistin stimulates PC-3 prostate cancer cell growth through stimulation of SOCS3 and SOCS5 genes. Exp. Biol. Med. 2023, 248, 1695–1707. [Google Scholar] [CrossRef]
- Oregel-Cortez, M.I.; Frayde-Gomez, H.; Quintana-Gonzalez, G.; Garcia-Gonzalez, V.; Vazquez-Jimenez, J.G.; Galindo-Hernandez, O. Resistin Induces Migration and Invasion in PC3 Prostate Cancer Cells: Role of Extracellular Vesicles. Life 2023, 13, 2321. [Google Scholar] [CrossRef]
- Fujisawa, T.; Endo, H.; Tomimoto, A.; Sugiyama, M.; Takahashi, H.; Saito, S.; Inamori, M.; Nakajima, N.; Watanabe, M.; Kubota, N.; et al. Adiponectin suppresses colorectal carcinogenesis under the high-fat diet condition. Gut 2008, 57, 1531–1538. [Google Scholar] [CrossRef]
- Liu, M.; Zhou, L.; Xu, A.; Lam, K.S.; Wetzel, M.D.; Xiang, R.; Zhang, J.; Xin, X.; Dong, L.Q.; Liu, F. A disulfide-bond A oxidoreductase-like protein (DsbA-L) regulates adiponectin multimerization. Proc. Natl. Acad. Sci. USA 2008, 105, 18302–18307. [Google Scholar] [CrossRef]
- Liu, M.; Xiang, R.; Wilk, S.A.; Zhang, N.; Sloane, L.B.; Azarnoush, K.; Zhou, L.; Chen, H.; Xiang, G.; Walter, C.A.; et al. Fat-specific DsbA-L overexpression promotes adiponectin multimerization and protects mice from diet-induced obesity and insulin resistance. Diabetes 2012, 61, 2776–2786. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Pankow, J.S.; Ballantyne, C.M.; Couper, D.; Hoogeveen, R.C.; Pereira, M.; Duncan, B.B.; Schmidt, M.I. High-molecular-weight adiponectin and the risk of type 2 diabetes in the ARIC study. J. Clin. Endocrinol. Metab. 2010, 95, 5097–5104. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yang, J.; Cai, Y.; Fu, S.; Zhang, N.; Fu, X.; Li, L. IFN-gamma-mediated inhibition of lung cancer correlates with PD-L1 expression and is regulated by PI3K-AKT signaling. Int. J. Cancer 2018, 143, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Bucher, F.; Ryu, S.; Jeong, J.; Lee, B.Y.; Jeong, Y.; Kim, M.S.; Oh, Y.S.; Baek, M.C.; Yoon, J.H.; et al. An adiponectin receptor agonist antibody stimulates glucose uptake and fatty-acid oxidation by activating AMP-activated protein kinase. Cytokine 2020, 126, 154863. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Wagg, C.S.; Altamimi, T.R.; Uddin, G.M.; Ho, K.L.; Darwesh, A.M.; Seubert, J.M.; Lopaschuk, G.D. Insulin directly stimulates mitochondrial glucose oxidation in the heart. Cardiovasc. Diabetol. 2020, 19, 207. [Google Scholar] [CrossRef]
- Brakenhielm, E.; Veitonmaki, N.; Cao, R.; Kihara, S.; Matsuzawa, Y.; Zhivotovsky, B.; Funahashi, T.; Cao, Y. Adiponectin-induced antiangiogenesis and antitumor activity involve caspase-mediated endothelial cell apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 2476–2481. [Google Scholar] [CrossRef]
- Xing, S.Q.; Zhang, C.G.; Yuan, J.F.; Yang, H.M.; Zhao, S.D.; Zhang, H. Adiponectin induces apoptosis in hepatocellular carcinoma through differential modulation of thioredoxin proteins. Biochem. Pharmacol. 2015, 93, 221–231. [Google Scholar] [CrossRef]
- Zhou, C.; He, X.; Tong, C.; Li, H.; Xie, C.; Wu, Y.; Wang, L.; Yan, X.; Luo, D.; Tang, Y.; et al. Cancer-associated adipocytes promote the invasion and metastasis in breast cancer through LIF/CXCLs positive feedback loop. Int. J. Biol. Sci. 2022, 18, 1363–1380. [Google Scholar] [CrossRef]
- Zhang, T.; Tseng, C.; Zhang, Y.; Sirin, O.; Corn, P.G.; Li-Ning-Tapia, E.M.; Troncoso, P.; Davis, J.; Pettaway, C.; Ward, J.; et al. CXCL1 mediates obesity-associated adipose stromal cell trafficking and function in the tumour microenvironment. Nat. Commun. 2016, 7, 11674. [Google Scholar] [CrossRef]
- Zhu, Z.; Ding, J.; Ma, Z.; Iwashina, T.; Tredget, E.E. Systemic depletion of macrophages in the subacute phase of wound healing reduces hypertrophic scar formation. Wound Repair. Regen. 2016, 24, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Arendt, L.M.; McCready, J.; Keller, P.J.; Baker, D.D.; Naber, S.P.; Seewaldt, V.; Kuperwasser, C. Obesity promotes breast cancer by CCL2-mediated macrophage recruitment and angiogenesis. Cancer Res. 2013, 73, 6080–6093. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ghosh, S.; Perrard, X.D.; Feng, L.; Garcia, G.E.; Perrard, J.L.; Sweeney, J.F.; Peterson, L.E.; Chan, L.; Smith, C.W.; et al. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation 2007, 115, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Takahashi, M.; Carpino, N.; Jou, S.T.; Chao, J.R.; Tanaka, S.; Shigeyoshi, Y.; Parganas, E.; Ihle, J.N. Leukemia inhibitory factor regulates trophoblast giant cell differentiation via Janus kinase 1-signal transducer and activator of transcription 3-suppressor of cytokine signaling 3 pathway. Mol. Endocrinol. 2008, 22, 1673–1681. [Google Scholar] [CrossRef]
- Gearing, D.P.; Gough, N.M.; King, J.A.; Hilton, D.J.; Nicola, N.A.; Simpson, R.J.; Nice, E.C.; Kelso, A.; Metcalf, D. Molecular cloning and expression of cDNA encoding a murine myeloid leukaemia inhibitory factor (LIF). EMBO J. 1987, 6, 3995–4002. [Google Scholar] [CrossRef]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef]
- Auguet, T.; Bertran, L.; Binetti, J.; Aguilar, C.; Martinez, S.; Sabench, F.; Lopez-Dupla, J.M.; Porras, J.A.; Riesco, D.; Del Castillo, D.; et al. Relationship between IL-8 Circulating Levels and TLR2 Hepatic Expression in Women with Morbid Obesity and Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2020, 21, 4189. [Google Scholar] [CrossRef]
- De Larco, J.E.; Wuertz, B.R.; Rosner, K.A.; Erickson, S.A.; Gamache, D.E.; Manivel, J.C.; Furcht, L.T. A potential role for interleukin-8 in the metastatic phenotype of breast carcinoma cells. Am. J. Pathol. 2001, 158, 639–646. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef]
- Cuzzone, D.A.; Weitman, E.S.; Albano, N.J.; Ghanta, S.; Savetsky, I.L.; Gardenier, J.C.; Joseph, W.J.; Torrisi, J.S.; Bromberg, J.F.; Olszewski, W.L.; et al. IL-6 regulates adipose deposition and homeostasis in lymphedema. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1426–H1434. [Google Scholar] [CrossRef] [PubMed]
- Fried, S.K.; Bunkin, D.A.; Greenberg, A.S. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: Depot difference and regulation by glucocorticoid. J. Clin. Endocrinol. Metab. 1998, 83, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Braune, J.; Weyer, U.; Hobusch, C.; Mauer, J.; Bruning, J.C.; Bechmann, I.; Gericke, M. IL-6 Regulates M2 Polarization and Local Proliferation of Adipose Tissue Macrophages in Obesity. J. Immunol. 2017, 198, 2927–2934. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef]
- Anderson, H.A.; Maylock, C.A.; Williams, J.A.; Paweletz, C.P.; Shu, H.; Shacter, E. Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells. Nat. Immunol. 2003, 4, 87–91. [Google Scholar] [CrossRef]
- Fadok, V.A.; Voelker, D.R.; Campbell, P.A.; Cohen, J.J.; Bratton, D.L.; Henson, P.M. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 1992, 148, 2207–2216. [Google Scholar] [CrossRef]
- Su, P.; Wang, Q.; Bi, E.; Ma, X.; Liu, L.; Yang, M.; Qian, J.; Yi, Q. Enhanced Lipid Accumulation and Metabolism Are Required for the Differentiation and Activation of Tumor-Associated Macrophages. Cancer Res. 2020, 80, 1438–1450. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Huggins, D.N.; LaRue, R.S.; Wang, Y.; Knutson, T.P.; Xu, Y.; Williams, J.W.; Schwertfeger, K.L. Characterizing Macrophage Diversity in Metastasis-Bearing Lungs Reveals a Lipid-Associated Macrophage Subset. Cancer Res. 2021, 81, 5284–5295. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Ohashi, K.; Parker, J.L.; Ouchi, N.; Higuchi, A.; Vita, J.A.; Gokce, N.; Pedersen, A.A.; Kalthoff, C.; Tullin, S.; Sams, A.; et al. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J. Biol. Chem. 2010, 285, 6153–6160. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Sun, Q. Macrophage recruitment in obese adipose tissue. Obes. Rev. 2015, 16, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Li, X.; Tan, S.; Zhou, H.J.; Ji, W.; Bellone, S.; Xu, X.; Zhang, H.; Santin, A.D.; Lou, G.; et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J. Clin. Investig. 2016, 126, 4157–4173. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Komohara, Y.; Takaishi, K.; Katabuchi, H.; Takeya, M. Detection of M2 macrophages and colony-stimulating factor 1 expression in serous and mucinous ovarian epithelial tumors. Pathol. Int. 2009, 59, 300–305. [Google Scholar] [CrossRef]
- Zeng, X.Y.; Xie, H.; Yuan, J.; Jiang, X.Y.; Yong, J.H.; Zeng, D.; Dou, Y.Y.; Xiao, S.S. M2-like tumor-associated macrophages-secreted EGF promotes epithelial ovarian cancer metastasis via activating EGFR-ERK signaling and suppressing lncRNA LIMT expression. Cancer Biol. Ther. 2019, 20, 956–966. [Google Scholar] [CrossRef]
- Zhu, C.; Mustafa, D.; Zheng, P.P.; van der Weiden, M.; Sacchetti, A.; Brandt, M.; Chrifi, I.; Tempel, D.; Leenen, P.J.M.; Duncker, D.J.; et al. Activation of CECR1 in M2-like TAMs promotes paracrine stimulation-mediated glial tumor progression. Neuro Oncol. 2017, 19, 648–659. [Google Scholar] [CrossRef]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian Res. 2014, 7, 19. [Google Scholar] [CrossRef]
- van Dierendonck, X.; de la Rosa Rodriguez, M.A.; Georgiadi, A.; Mattijssen, F.; Dijk, W.; van Weeghel, M.; Singh, R.; Borst, J.W.; Stienstra, R.; Kersten, S. HILPDA Uncouples Lipid Droplet Accumulation in Adipose Tissue Macrophages from Inflammation and Metabolic Dysregulation. Cell Rep. 2020, 30, 1811–1822.e6. [Google Scholar] [CrossRef]
- Hao, J.; Yan, F.; Zhang, Y.; Triplett, A.; Zhang, Y.; Schultz, D.A.; Sun, Y.; Zeng, J.; Silverstein, K.A.T.; Zheng, Q.; et al. Expression of Adipocyte/Macrophage Fatty Acid-Binding Protein in Tumor-Associated Macrophages Promotes Breast Cancer Progression. Cancer Res. 2018, 78, 2343–2355. [Google Scholar] [CrossRef]
- Liu, R.Z.; Godbout, R. An Amplified Fatty Acid-Binding Protein Gene Cluster in Prostate Cancer: Emerging Roles in Lipid Metabolism and Metastasis. Cancers 2020, 12, 3823. [Google Scholar] [CrossRef] [PubMed]
- Bournat, J.C.; Brown, C.W. Mitochondrial dysfunction in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [PubMed]
- Herroon, M.K.; Rajagurubandara, E.; Diedrich, J.D.; Heath, E.I.; Podgorski, I. Adipocyte-activated oxidative and ER stress pathways promote tumor survival in bone via upregulation of Heme Oxygenase 1 and Survivin. Sci. Rep. 2018, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D.; Abrahamsson, P.A. Expression of heme oxygenase-1 (HSP32) in human prostate: Normal, hyperplastic, and tumor tissue distribution. Urology 1996, 47, 727–733. [Google Scholar] [CrossRef]
- Laurent, V.; Toulet, A.; Attane, C.; Milhas, D.; Dauvillier, S.; Zaidi, F.; Clement, E.; Cinato, M.; Le Gonidec, S.; Guerard, A.; et al. Periprostatic Adipose Tissue Favors Prostate Cancer Cell Invasion in an Obesity-Dependent Manner: Role of Oxidative Stress. Mol. Cancer Res. 2019, 17, 821–835. [Google Scholar] [CrossRef]
- Holl, M.; Koziel, R.; Schafer, G.; Pircher, H.; Pauck, A.; Hermann, M.; Klocker, H.; Jansen-Durr, P.; Sampson, N. ROS signaling by NADPH oxidase 5 modulates the proliferation and survival of prostate carcinoma cells. Mol. Carcinog. 2016, 55, 27–39. [Google Scholar] [CrossRef]
- Antony, S.; Jiang, G.; Wu, Y.; Meitzler, J.L.; Makhlouf, H.R.; Haines, D.C.; Butcher, D.; Hoon, D.S.; Ji, J.; Zhang, Y.; et al. NADPH oxidase 5 (NOX5)-induced reactive oxygen signaling modulates normoxic HIF-1alpha and p27(Kip1) expression in malignant melanoma and other human tumors. Mol. Carcinog. 2017, 56, 2643–2662. [Google Scholar] [CrossRef]
- Fontana, F.; Anselmi, M.; Carollo, E.; Sartori, P.; Procacci, P.; Carter, D.; Limonta, P. Adipocyte-Derived Extracellular Vesicles Promote Prostate Cancer Cell Aggressiveness by Enabling Multiple Phenotypic and Metabolic Changes. Cells 2022, 11, 2388. [Google Scholar] [CrossRef]
- Mohamed, O.A.A.; Tesen, H.S.; Hany, M.; Sherif, A.; Abdelwahab, M.M.; Elnaggar, M.H. The role of hypoxia on prostate cancer progression and metastasis. Mol. Biol. Rep. 2023, 50, 3873–3884. [Google Scholar] [CrossRef]
- Sakai, A.; Nakanishi, M.; Yoshiyama, K.; Maki, H. Impact of reactive oxygen species on spontaneous mutagenesis in Escherichia coli. Genes Cells 2006, 11, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Du, M.Q.; Carmichael, P.L.; Phillips, D.H. Induction of activating mutations in the human c-Ha-ras-1 proto-oncogene by oxygen free radicals. Mol. Carcinog. 1994, 11, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Higinbotham, K.G.; Rice, J.M.; Diwan, B.A.; Kasprzak, K.S.; Reed, C.D.; Perantoni, A.O. GGT to GTT transversions in codon 12 of the K-ras oncogene in rat renal sarcomas induced with nickel subsulfide or nickel subsulfide/iron are consistent with oxidative damage to DNA. Cancer Res. 1992, 52, 4747–4751. [Google Scholar] [PubMed]
- Custovic, Z.; Zarkovic, K.; Cindric, M.; Cipak, A.; Jurkovic, I.; Sonicki, Z.; Uchida, K.; Zarkovic, N. Lipid peroxidation product acrolein as a predictive biomarker of prostate carcinoma relapse after radical surgery. Free Radic. Res. 2010, 44, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, J.; Tobar, N.; Caceres, M.; Espinoza, L.; Escobar, P.; Dotor, J.; Smith, P.C.; Martinez, J. Soluble factors derived from tumor mammary cell lines induce a stromal mammary adipose reversion in human and mice adipose cells. Possible role of TGF-beta1 and TNF-alpha. Breast Cancer Res. Treat. 2010, 119, 497–508. [Google Scholar] [CrossRef]
- Tokuda, Y.; Satoh, Y.; Fujiyama, C.; Toda, S.; Sugihara, H.; Masaki, Z. Prostate cancer cell growth is modulated by adipocyte-cancer cell interaction. BJU Int. 2003, 91, 716–720. [Google Scholar] [CrossRef]
- Hardaway, A.L.; Herroon, M.K.; Rajagurubandara, E.; Podgorski, I. Marrow adipocyte-derived CXCL1 and CXCL2 contribute to osteolysis in metastatic prostate cancer. Clin. Exp. Metastasis 2015, 32, 353–368. [Google Scholar] [CrossRef]
- Duncan, R.E.; Ahmadian, M.; Jaworski, K.; Sarkadi-Nagy, E.; Sul, H.S. Regulation of lipolysis in adipocytes. Annu. Rev. Nutr. 2007, 27, 79–101. [Google Scholar] [CrossRef]
- Liu, Q.; Russell, M.R.; Shahriari, K.; Jernigan, D.L.; Lioni, M.I.; Garcia, F.U.; Fatatis, A. Interleukin-1beta promotes skeletal colonization and progression of metastatic prostate cancer cells with neuroendocrine features. Cancer Res. 2013, 73, 3297–3305. [Google Scholar] [CrossRef]
- Gartung, A.; Zhao, J.; Chen, S.; Mottillo, E.; VanHecke, G.C.; Ahn, Y.H.; Maddipati, K.R.; Sorokin, A.; Granneman, J.; Lee, M.J. Characterization of Eicosanoids Produced by Adipocyte Lipolysis: Implication of Cyclooxygenase-2 in Adipose Inflammation. J. Biol. Chem. 2016, 291, 16001–16010. [Google Scholar] [CrossRef]
- Tong, D.; Liu, Q.; Liu, G.; Xu, J.; Lan, W.; Jiang, Y.; Xiao, H.; Zhang, D.; Jiang, J. Metformin inhibits castration-induced EMT in prostate cancer by repressing COX2/PGE2/STAT3 axis. Cancer Lett. 2017, 389, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhou, W.; Ge, J.; Zhang, Z. Prostaglandin E2 receptor EP4 is involved in the cell growth and invasion of prostate cancer via the cAMP-PKA/PI3K-Akt signaling pathway. Mol. Med. Rep. 2018, 17, 4702–4712. [Google Scholar] [CrossRef] [PubMed]
- Price, R.G.; Lloyd, S.; Wang, X.; Haaland, B.; Nelson, G.; Salter, B. Adipose Tissue Distribution and Body Mass Index (BMI) Correlation With Daily Image-Guided Radiotherapy (IGRT) Shifts of Abdominal Radiation Therapy Patients. Cureus 2023, 15, e40979. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, C.M.; Howard, L.E.; Oyekunle, T.; De Hoedt, A.M.; Salama, J.K.; Song, H.; Freedland, S.J.; Allott, E.H. Abdominal and pelvic adipose tissue distribution and risk of prostate cancer recurrence after radiation therapy. Prostate 2020, 80, 1244–1252. [Google Scholar] [CrossRef]
- Vertulli, D.; Santucci, D.; Esperto, F.; Beomonte Zobel, B.; Grasso, R.F.; Faiella, E. Impact of adipose tissue distribution on prostate cancer recurrence after radical prostatectomy. Actas Urol. Esp. (Engl. Ed.) 2023, 47, 104–110. [Google Scholar] [CrossRef]
- Simon, N.I.; Parker, C.; Hope, T.A.; Paller, C.J. Best Approaches and Updates for Prostate Cancer Biochemical Recurrence. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 1–8. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef]
- Sharifi, N.; Gulley, J.L.; Dahut, W.L. Androgen deprivation therapy for prostate cancer. JAMA 2005, 294, 238–244. [Google Scholar] [CrossRef]
- Stone, P.; Hardy, J.; Huddart, R.; A’Hern, R.; Richards, M. Fatigue in patients with prostate cancer receiving hormone therapy. Eur. J. Cancer 2000, 36, 1134–1141. [Google Scholar] [CrossRef]
- Haseen, F.; Murray, L.J.; Cardwell, C.R.; O’Sullivan, J.M.; Cantwell, M.M. The effect of androgen deprivation therapy on body composition in men with prostate cancer: Systematic review and meta-analysis. J. Cancer Surviv. 2010, 4, 128–139. [Google Scholar] [CrossRef]
- Choi, S.M.; Kam, S.C. Metabolic effects of androgen deprivation therapy. Korean J. Urol. 2015, 56, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.; Birzniece, V.; McLean, M.; Gurney, H.; Hayden, A.; Cheema, B.S. The Adverse Effects of Androgen Deprivation Therapy in Prostate Cancer and the Benefits and Potential Anti-oncogenic Mechanisms of Progressive Resistance Training. Sports Med. Open 2020, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Mangiola, S.; Stuchbery, R.; McCoy, P.; Chow, K.; Kurganovs, N.; Kerger, M.; Papenfuss, A.; Hovens, C.M.; Corcoran, N.M. Androgen deprivation therapy promotes an obesity-like microenvironment in periprostatic fat. Endocr. Connect. 2019, 8, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Mudali, S.; Dobs, A.S. Effects of testosterone on body composition of the aging male. Mech. Ageing Dev. 2004, 125, 297–304. [Google Scholar] [CrossRef]
- Brooks, R.V. Androgens. Clin. Endocrinol. Metab. 1975, 4, 503–520. [Google Scholar] [CrossRef]
- Wolny-Rokicka, E.; Tukiendorf, A.; Wydmanski, J.; Ostrowska, M.; Zembron-Lacny, A. Lipid Status During Combined Treatment in Prostate Cancer Patients. Am. J. Mens. Health 2019, 13, 1557988319876488. [Google Scholar] [CrossRef]
- Jun, S.Y.; Brown, A.J.; Chua, N.K.; Yoon, J.Y.; Lee, J.J.; Yang, J.O.; Jang, I.; Jeon, S.J.; Choi, T.I.; Kim, C.H.; et al. Reduction of Squalene Epoxidase by Cholesterol Accumulation Accelerates Colorectal Cancer Progression and Metastasis. Gastroenterology 2021, 160, 1194–1207.e28. [Google Scholar] [CrossRef]
- Wang, X.; Sun, B.; Wei, L.; Jian, X.; Shan, K.; He, Q.; Huang, F.; Ge, X.; Gao, X.; Feng, N.; et al. Cholesterol and saturated fatty acids synergistically promote the malignant progression of prostate cancer. Neoplasia 2022, 24, 86–97. [Google Scholar] [CrossRef]
- Yang, L.; Sun, J.; Li, M.; Long, Y.; Zhang, D.; Guo, H.; Huang, R.; Yan, J. Oxidized Low-Density Lipoprotein Links Hypercholesterolemia and Bladder Cancer Aggressiveness by Promoting Cancer Stemness. Cancer Res. 2021, 81, 5720–5732. [Google Scholar] [CrossRef]
- Banker, D.E.; Mayer, S.J.; Li, H.Y.; Willman, C.L.; Appelbaum, F.R.; Zager, R.A. Cholesterol synthesis and import contribute to protective cholesterol increments in acute myeloid leukemia cells. Blood 2004, 104, 1816–1824. [Google Scholar] [CrossRef]
- Hughes-Fulford, M.; Chen, Y.; Tjandrawinata, R.R. Fatty acid regulates gene expression and growth of human prostate cancer PC-3 cells. Carcinogenesis 2001, 22, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Muldoon, M.F.; Kritchevsky, S.B.; Evans, R.W.; Kagan, V.E. Serum total antioxidant activity in relative hypo- and hypercholesterolemia. Free Radic. Res. 1996, 25, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Blomme, A.; Peter, C.; Mui, E.; Rodriguez Blanco, G.; An, N.; Mason, L.M.; Jamieson, L.E.; McGregor, G.H.; Lilla, S.; Ntala, C.; et al. THEM6-mediated reprogramming of lipid metabolism supports treatment resistance in prostate cancer. EMBO Mol. Med. 2022, 14, e14764. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Hjorth-Jensen, K.; Hekmat, O.; Iglesias-Gato, D.; Kruse, T.; Wang, C.; Wei, W.; Ke, B.; Yan, B.; Niu, Y.; et al. In vivo quantitative phosphoproteomic profiling identifies novel regulators of castration-resistant prostate cancer growth. Oncogene 2015, 34, 2764–2776. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, S.; Chen, X.; Stauffer, S.; Yu, F.; Lele, S.M.; Fu, K.; Datta, K.; Palermo, N.; Chen, Y.; et al. The hippo pathway effector YAP regulates motility, invasion, and castration-resistant growth of prostate cancer cells. Mol. Cell Biol. 2015, 35, 1350–1362. [Google Scholar] [CrossRef]
- Lee, H.C.; Ou, C.H.; Huang, Y.C.; Hou, P.C.; Creighton, C.J.; Lin, Y.S.; Hu, C.Y.; Lin, S.C. YAP1 overexpression contributes to the development of enzalutamide resistance by induction of cancer stemness and lipid metabolism in prostate cancer. Oncogene 2021, 40, 2407–2421. [Google Scholar] [CrossRef]
- Stangl-Kremser, J.; Suarez-Ibarrola, R.; Andrea, D.; Korn, S.M.; Pones, M.; Kramer, G.; Marhold, M.; Krainer, M.; Enikeev, D.V.; Glybochko, P.V.; et al. Assessment of body composition in the advanced stage of castration-resistant prostate cancer: Special focus on sarcopenia. Prostate Cancer Prostatic Dis. 2020, 23, 309–315. [Google Scholar] [CrossRef]
- Cushen, S.J.; Power, D.G.; Murphy, K.P.; McDermott, R.; Griffin, B.T.; Lim, M.; Daly, L.; MacEneaney, P.; O’Sullivan, K.; Prado, C.M.; et al. Impact of body composition parameters on clinical outcomes in patients with metastatic castrate-resistant prostate cancer treated with docetaxel. Clin. Nutr. ESPEN 2016, 13, e39–e45. [Google Scholar] [CrossRef]
- Kwak, J.G.; Lee, J. Bone Marrow Adipocytes Contribute to Tumor Microenvironment-Driven Chemoresistance via Sequestration of Doxorubicin. Cancers 2023, 15, 2737. [Google Scholar] [CrossRef]
- Herroon, M.K.; Diedrich, J.D.; Rajagurubandara, E.; Martin, C.; Maddipati, K.R.; Kim, S.; Heath, E.I.; Granneman, J.; Podgorski, I. Prostate Tumor Cell-Derived IL1beta Induces an Inflammatory Phenotype in Bone Marrow Adipocytes and Reduces Sensitivity to Docetaxel via Lipolysis-Dependent Mechanisms. Mol. Cancer Res. 2019, 17, 2508–2521. [Google Scholar] [CrossRef]
- Su, F.; Ahn, S.; Saha, A.; DiGiovanni, J.; Kolonin, M.G. Adipose stromal cell targeting suppresses prostate cancer epithelial-mesenchymal transition and chemoresistance. Oncogene 2019, 38, 1979–1988. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Parmentier, J.H.; Tucci, J.; Pei, H.; Cortez-Toledo, O.; Dieli-Conwright, C.M.; Oberley, M.J.; Neely, M.; Orgel, E.; Louie, S.G.; et al. Adipocytes Sequester and Metabolize the Chemotherapeutic Daunorubicin. Mol. Cancer Res. 2017, 15, 1704–1713. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhao, L.; Zhang, H.; Liu, X.; Zhao, L.; Zhao, X.; Li, Y.; Li, J. AKR1C3 overexpression may serve as a promising biomarker for prostate cancer progression. Diagn. Pathol. 2014, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M.; Jonnalagadda, S.; Trippier, P.C.; Rizner, T.L. Aldo-Keto Reductases and Cancer Drug Resistance. Pharmacol. Rev. 2021, 73, 1150–1171. [Google Scholar] [CrossRef]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Theodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef]
- Liotti, A.; La Civita, E.; Cennamo, M.; Crocetto, F.; Ferro, M.; Guadagno, E.; Insabato, L.; Imbimbo, C.; Palmieri, A.; Mirone, V.; et al. Periprostatic adipose tissue promotes prostate cancer resistance to docetaxel by paracrine IGF-1 upregulation of TUBB2B beta-tubulin isoform. Prostate 2021, 81, 407–417. [Google Scholar] [CrossRef]
- Nami, B.; Wang, Z. Genetics and Expression Profile of the Tubulin Gene Superfamily in Breast Cancer Subtypes and Its Relation to Taxane Resistance. Cancers 2018, 10, 274. [Google Scholar] [CrossRef]
- Paller, C.J.; Antonarakis, E.S. Cabazitaxel: A novel second-line treatment for metastatic castration-resistant prostate cancer. Drug Des. Dev. Ther. 2011, 5, 117–124. [Google Scholar]
- Koka, K.; Verma, A.; Dwarakanath, B.S.; Papineni, R.V.L. Technological Advancements in External Beam Radiation Therapy (EBRT): An Indispensable Tool for Cancer Treatment. Cancer Manag. Res. 2022, 14, 1421–1429. [Google Scholar] [CrossRef]
- Cadet, J.; Davies, K.J.A. Oxidative DNA damage & repair: An introduction. Free Radic. Biol. Med. 2017, 107, 2–12. [Google Scholar] [PubMed]
- Nakano, T.; Xu, X.; Salem, A.M.H.; Shoulkamy, M.I.; Ide, H. Radiation-induced DNA-protein cross-links: Mechanisms and biological significance. Free Radic. Biol. Med. 2017, 107, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Barker, S.; Weinfeld, M.; Murray, D. DNA-protein crosslinks: Their induction, repair, and biological consequences. Mutat. Res. 2005, 589, 111–135. [Google Scholar] [CrossRef] [PubMed]
- Riley, P.A. Free radicals in biology: Oxidative stress and the effects of ionizing radiation. Int. J. Radiat. Biol. 1994, 65, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Tirado, F.R.; Bhanja, P.; Castro-Nallar, E.; Olea, X.D.; Salamanca, C.; Saha, S. Radiation-induced toxicity in rectal epithelial stem cell contributes to acute radiation injury in rectum. Stem Cell Res. Ther. 2021, 12, 63. [Google Scholar] [CrossRef]
- Takemura, N.; Kurashima, Y.; Mori, Y.; Okada, K.; Ogino, T.; Osawa, H.; Matsuno, H.; Aayam, L.; Kaneto, S.; Park, E.J.; et al. Eosinophil depletion suppresses radiation-induced small intestinal fibrosis. Sci. Transl. Med. 2018, 10, eaan0333. [Google Scholar] [CrossRef]
- Poglio, S.; Galvani, S.; Bour, S.; Andre, M.; Prunet-Marcassus, B.; Penicaud, L.; Casteilla, L.; Cousin, B. Adipose tissue sensitivity to radiation exposure. Am. J. Pathol. 2009, 174, 44–53. [Google Scholar] [CrossRef]
- Barcellos-Hoff, M.H.; Ravani, S.A. Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer Res. 2000, 60, 1254–1260. [Google Scholar]
- De Martino, M.; Daviaud, C.; Minns, H.E.; Lazarian, A.; Wacker, A.; Costa, A.P.; Attarwala, N.; Chen, Q.; Choi, S.W.; Rabadan, R.; et al. Radiation therapy promotes unsaturated fatty acids to maintain survival of glioblastoma. Cancer Lett. 2023, 570, 216329. [Google Scholar] [CrossRef]
- Gupta, K.; Vuckovic, I.; Zhang, S.; Xiong, Y.; Carlson, B.L.; Jacobs, J.; Olson, I.; Petterson, X.M.; Macura, S.I.; Sarkaria, J.; et al. Radiation Induced Metabolic Alterations Associate With Tumor Aggressiveness and Poor Outcome in Glioblastoma. Front. Oncol. 2020, 10, 535. [Google Scholar] [CrossRef]
- Wiktorowska-Owczarek, A.; Berezinska, M.; Nowak, J.Z. PUFAs: Structures, Metabolism and Functions. Adv. Clin. Exp. Med. 2015, 24, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yang, Y.; Sun, B.F.; Shi, Y.; Yang, X.; Xiao, W.; Hao, Y.J.; Ping, X.L.; Chen, Y.S.; Wang, W.J.; et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014, 24, 1403–1419. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Closas, M.; Couch, F.J.; Lindstrom, S.; Michailidou, K.; Schmidt, M.K.; Brook, M.N.; Orr, N.; Rhie, S.K.; Riboli, E.; Feigelson, H.S.; et al. Genome-wide association studies identify four ER negative-specific breast cancer risk loci. Nat. Genet. 2013, 45, 392–398, 398e1–398e2. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.M.; Li, Z.X.; Wu, Y.H.; Shi, Z.L.; Mi, J.L.; Hu, K.; Wang, R.S. m6A demethylase FTO renders radioresistance of nasopharyngeal carcinoma via promoting OTUB1-mediated anti-ferroptosis. Transl. Oncol. 2023, 27, 101576. [Google Scholar] [CrossRef]
- Iles, M.M.; Law, M.H.; Stacey, S.N.; Han, J.; Fang, S.; Pfeiffer, R.; Harland, M.; Macgregor, S.; Taylor, J.C.; Aben, K.K.; et al. A variant in FTO shows association with melanoma risk not due to BMI. Nat. Genet. 2013, 45, 428–432, 432e421. [Google Scholar]
- Su, R.; Dong, L.; Li, Y.; Gao, M.; Han, L.; Wunderlich, M.; Deng, X.; Li, H.; Huang, Y.; Gao, L.; et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell 2020, 38, 79–96.e11. [Google Scholar] [CrossRef]
- Fitzsimmons, C.M.; Batista, P.J. It’s complicated… m(6)A-dependent regulation of gene expression in cancer. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 382–393. [Google Scholar] [CrossRef]
- Relier, S.; Ripoll, J.; Guillorit, H.; Amalric, A.; Achour, C.; Boissiere, F.; Vialaret, J.; Attina, A.; Debart, F.; Choquet, A.; et al. FTO-mediated cytoplasmic m(6)A(m) demethylation adjusts stem-like properties in colorectal cancer cell. Nat. Commun. 2021, 12, 1716. [Google Scholar] [CrossRef]
- Xiang, Y.; Laurent, B.; Hsu, C.H.; Nachtergaele, S.; Lu, Z.; Sheng, W.; Xu, C.; Chen, H.; Ouyang, J.; Wang, S.; et al. RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature 2017, 543, 573–576. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Casal, R.; Bhattacharya, C.; Ganesh, N.; Bailey, L.; Basse, P.; Gibson, M.; Epperly, M.; Levina, V. Non-small cell lung cancer cells survived ionizing radiation treatment display cancer stem cell and epithelial-mesenchymal transition phenotypes. Mol. Cancer 2013, 12, 94. [Google Scholar] [CrossRef] [PubMed]
- Oatman, N.; Dasgupta, N.; Arora, P.; Choi, K.; Gawali, M.V.; Gupta, N.; Parameswaran, S.; Salomone, J.; Reisz, J.A.; Lawler, S.; et al. Mechanisms of stearoyl CoA desaturase inhibitor sensitivity and acquired resistance in cancer. Sci. Adv. 2021, 7, eabd7459. [Google Scholar] [CrossRef] [PubMed]
- Kuda, O.; Pietka, T.A.; Demianova, Z.; Kudova, E.; Cvacka, J.; Kopecky, J.; Abumrad, N.A. Sulfo-N-succinimidyl oleate (SSO) inhibits fatty acid uptake and signaling for intracellular calcium via binding CD36 lysine 164: SSO also inhibits oxidized low density lipoprotein uptake by macrophages. J. Biol. Chem. 2013, 288, 15547–15555. [Google Scholar] [CrossRef]
- Gelsomino, L.; Giordano, C.; Camera, G.; Sisci, D.; Marsico, S.; Campana, A.; Tarallo, R.; Rinaldi, A.; Fuqua, S.; Leggio, A.; et al. Leptin Signaling Contributes to Aromatase Inhibitor Resistant Breast Cancer Cell Growth and Activation of Macrophages. Biomolecules 2020, 10, 543. [Google Scholar] [CrossRef]
- Su, J.R.; Lu, Z.H.; Su, Y.; Zhao, N.; Dong, C.L.; Sun, L.; Zhao, S.F.; Li, Y. Relationship of Serum Adiponectin Levels and Metformin Therapy in Patients with Type 2 Diabetes. Horm. Metab. Res. 2016, 48, 92–98. [Google Scholar] [CrossRef]
- Kim, K.; Yang, W.H.; Jung, Y.S.; Cha, J.H. A new aspect of an old friend: The beneficial effect of metformin on anti-tumor immunity. BMB Rep. 2020, 53, 512–520. [Google Scholar] [CrossRef]
- Shrishrimal, S.; Kosmacek, E.A.; Chatterjee, A.; Tyson, M.J.; Oberley-Deegan, R.E. The SOD Mimic, MnTE-2-PyP, Protects from Chronic Fibrosis and Inflammation in Irradiated Normal Pelvic Tissues. Antioxidants 2017, 6, 87. [Google Scholar] [CrossRef]
- Bezin, J.; Gouverneur, A.; Penichon, M.; Mathieu, C.; Garrel, R.; Hillaire-Buys, D.; Pariente, A.; Faillie, J.L. GLP-1 Receptor Agonists and the Risk of Thyroid Cancer. Diabetes Care 2023, 46, 384–390. [Google Scholar] [CrossRef]
- Elashoff, M.; Matveyenko, A.V.; Gier, B.; Elashoff, R.; Butler, P.C. Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology 2011, 141, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Nomiyama, T.; Kawanami, T.; Irie, S.; Hamaguchi, Y.; Terawaki, Y.; Murase, K.; Tsutsumi, Y.; Nagaishi, R.; Tanabe, M.; Morinaga, H.; et al. Exendin-4, a GLP-1 receptor agonist, attenuates prostate cancer growth. Diabetes 2014, 63, 3891–3905. [Google Scholar] [CrossRef] [PubMed]
- Shigeoka, T.; Nomiyama, T.; Kawanami, T.; Hamaguchi, Y.; Horikawa, T.; Tanaka, T.; Irie, S.; Motonaga, R.; Hamanoue, N.; Tanabe, M.; et al. Activation of overexpressed glucagon-like peptide-1 receptor attenuates prostate cancer growth by inhibiting cell cycle progression. J. Diabetes Investig. 2020, 11, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Depotte, L.; Caroux, M.; Gligorov, J.; Canoui-Poitrine, F.; Belkacemi, Y.; De La Taille, A.; Tournigand, C.; Kempf, E. Association between overweight, obesity, and quality of life of patients receiving an anticancer treatment for prostate cancer: A systematic literature review. Health Qual. Life Outcomes 2023, 21, 11. [Google Scholar] [CrossRef]
- Sanda, M.G.; Dunn, R.L.; Michalski, J.; Sandler, H.M.; Northouse, L.; Hembroff, L.; Lin, X.; Greenfield, T.K.; Litwin, M.S.; Saigal, C.S.; et al. Quality of life and satisfaction with outcome among prostate-cancer survivors. N. Engl. J. Med. 2008, 358, 1250–1261. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).