
T-Type Voltage-Gated Calcium Channels: Potential Regulators of Smooth Muscle Contractility


Abstract
:1. Introduction
2. The Mechanism of Smooth Muscle Contraction Initiated by Calcium Influx
3. Subtypes of T-Type VGCCs
4. Structural Properties of T-Type VGCCs
5. Electrophysiology and Biophysics of T-Type VGCCs
6. Regulation of T-Type Voltage-Gated Calcium Channel Expression
6.1. T-Type VGCC-Expressing Cells
6.2. Regulation by Transcription Factors
6.3. Epigenetic Regulation
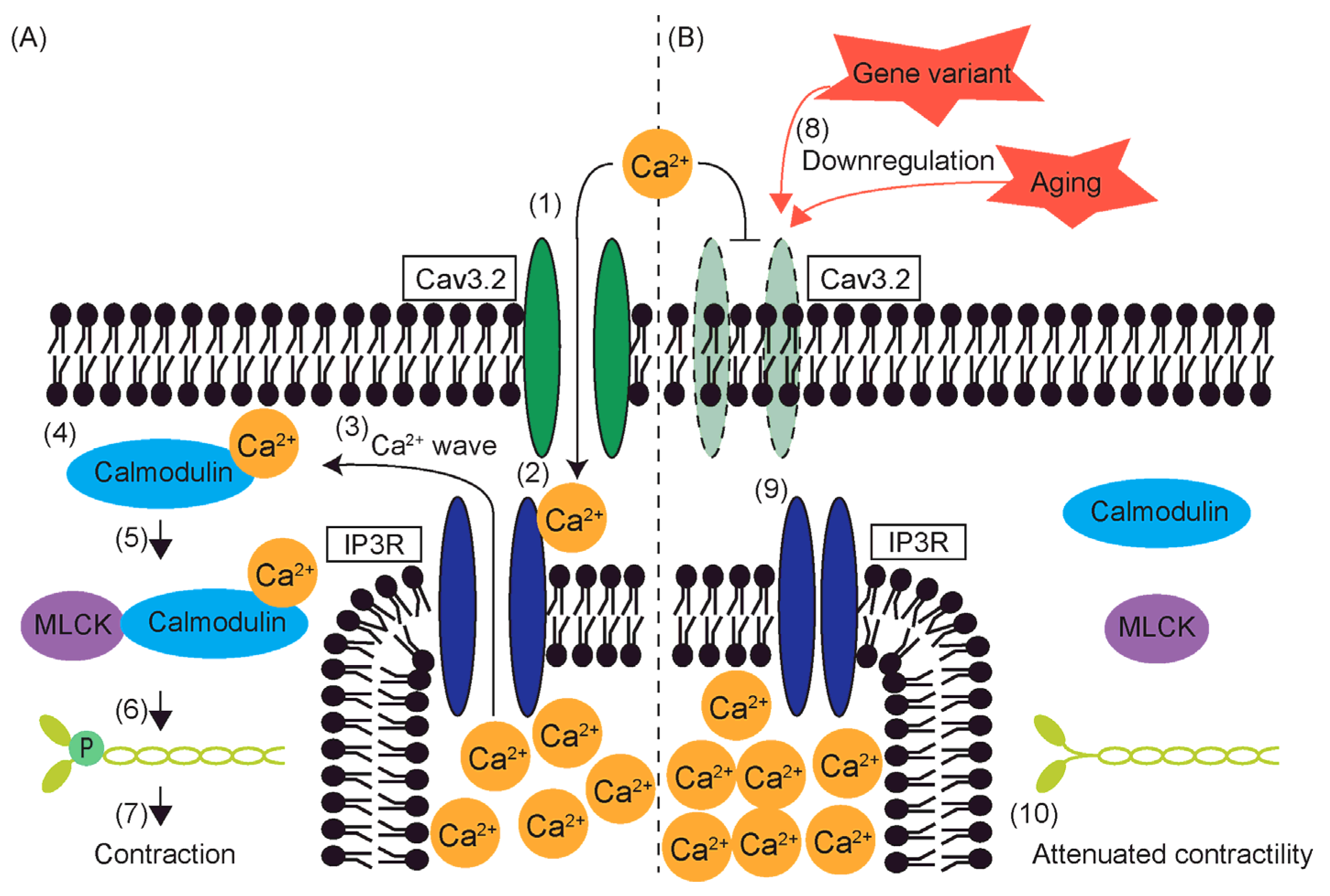
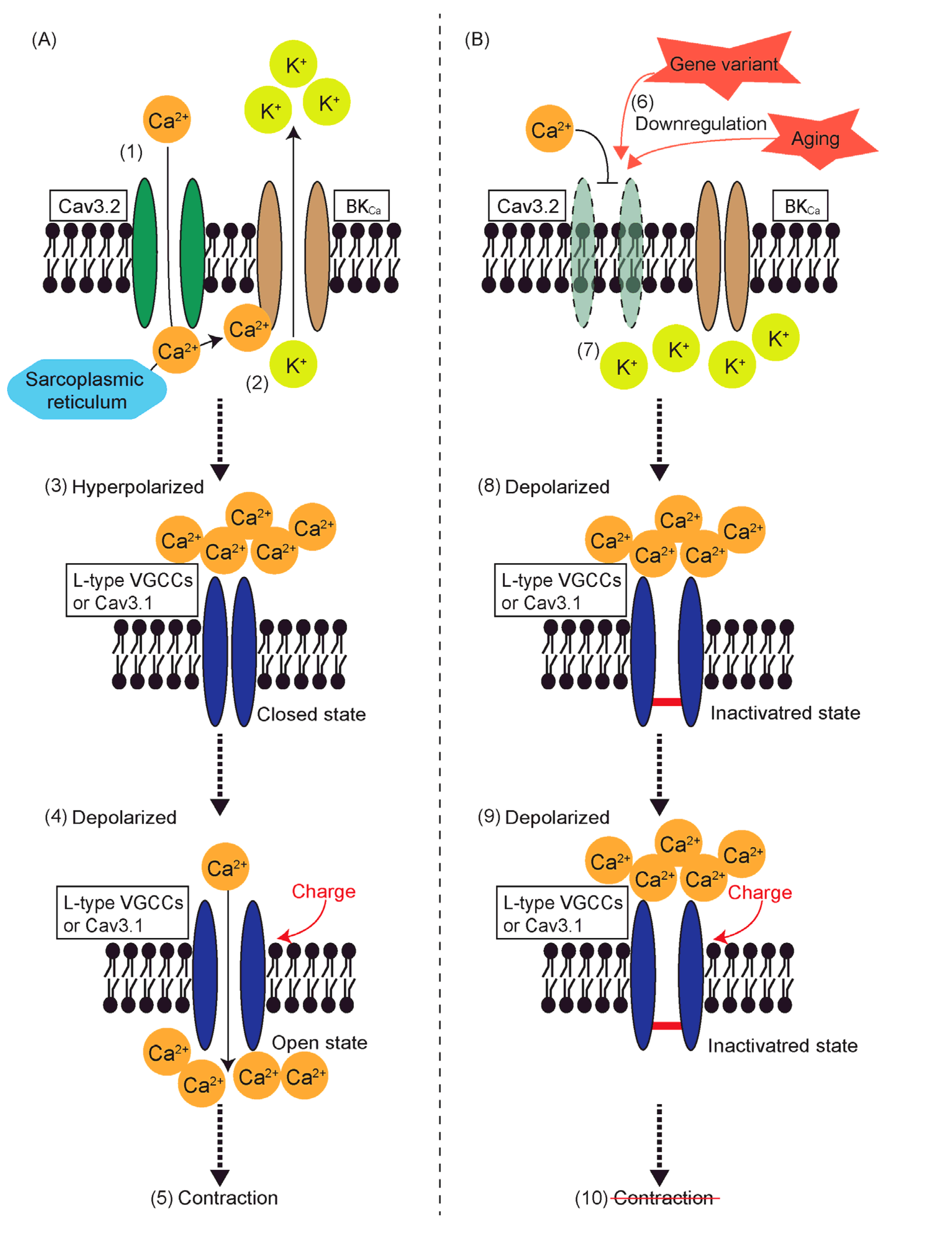
6.4. Aging
7. Functions of T-Type VGCCs Other than Contraction Induction
7.1. Proliferation
7.2. Apoptosis
7.3. T Cell Differentiation and Cytokine Production
8. Reduced Contractility and T-Type VGCCs
9. Multi-Omics Analysis of Smooth Muscle Cells
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tomida, S.; Aizawa, K.; Nishida, N.; Aoki, H.; Imai, Y.; Nagai, R.; Suzuki, T. Indomethacin reduces rates of aortic dissection and rupture of the abdominal aorta by inhibiting monocyte/macrophage accumulation in a murine model. Sci. Rep. 2019, 9, 10751. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Malak, M.; Eshak, I.B. Aortic Dissection: A Review of the Pathophysiology, Management and Prospective Advances. Curr. Cardiol. Rev. 2021, 17, 12–26. [Google Scholar]
- Rombouts, K.B.; van Merrienboer, T.A.R.; Ket, J.C.F.; Bogunovic, N.; van der Velden, J.; Yeung, K.K. The role of vascular smooth muscle cells in the development of aortic aneurysms and dissections. Eur. J. Clin. Investig. 2022, 52, e13697. [Google Scholar] [CrossRef] [PubMed]
- Karimi, A.; Milewicz, D.M. Structure of the Elastin-Contractile Units in the Thoracic Aorta and How Genes That Cause Thoracic Aortic Aneurysms and Dissections Disrupt This Structure. Can. J. Cardiol. 2016, 32, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.H.; LeMaire, S.A. Molecular pathogenesis of genetic and sporadic aortic aneurysms and dissections. Curr. Probl. Surg. 2017, 54, 95–155. [Google Scholar] [CrossRef]
- Negishi, K.; Aizawa, K.; Shindo, T.; Suzuki, T.; Sakurai, T.; Saito, Y.; Miyakawa, T.; Tanokura, M.; Kataoka, Y.; Maeda, M.; et al. An Myh11 single lysine deletion causes aortic dissection by reducing aortic structural integrity and contractility. Sci. Rep. 2022, 12, 8844. [Google Scholar] [CrossRef]
- Tomida, S.; Ishima, T.; Sawaki, D.; Imai, Y.; Nagai, R.; Aizawa, K. Multi-Omics of Familial Thoracic Aortic Aneurysm and Dissection: Calcium Transport Impairment Predisposes Aortas to Dissection. Int. J. Mol. Sci. 2023, 24, 15213. [Google Scholar] [CrossRef]
- Huang, J.; Fan, X.; Jin, X.; Lyu, C.; Guo, Q.; Liu, T.; Chen, J.; Davakan, A.; Lory, P.; Yan, N. Structural basis for human Cav3.2 inhibition by selective antagonists. Cell Res. 2024, 34, 440–450. [Google Scholar] [CrossRef]
- Andreasen, D.; Friis, U.G.; Uhrenholt, T.R.; Jensen, B.L.; Skøtt, O.; Hansen, P.B. Coexpression of Voltage-Dependent Calcium Channels Cav1.2, 2.1a, and 2.1b in Vascular Myocytes. Hypertension 2006, 47, 735–741. [Google Scholar] [CrossRef]
- Mironova, G.Y.; Haghbin, N.; Welsh, D.G. Functional tuning of Vascular L-type Ca2+ channels. Front. Physiol. 2022, 13, 1058744. [Google Scholar] [CrossRef]
- Akin, E.J.; Aoun, J.; Jimenez, C.; Mayne, K.; Baeck, J.; Young, M.D.; Sullivan, B.; Sanders, K.M.; Ward, S.M.; Bulley, S.; et al. ANO1, CaV1.2, and IP3R form a localized unit of EC-coupling in mouse pulmonary arterial smooth muscle. J. General. Physiol. 2023, 155, e202213217. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, F.; Andreasen, D.; Salomonsson, M.; Jensen, B.L.; Holstein-Rathlou, N.-H. Conducted vasoconstriction in rat mesenteric arterioles: Role for dihydropyridine-insensitive Ca2+ channels. Am. J. Physiol.-Heart Circ. Physiol. 2001, 280, H582–H590. [Google Scholar] [CrossRef] [PubMed]
- Webb, R.C. Smooth muscle contraction and relaxation. Adv. Physiol. Educ. 2003, 27, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Rufenach, B.; Van Petegem, F. Structure and function of STAC proteins: Calcium channel modulators and critical components of muscle excitation–contraction coupling. J. Biol. Chem. 2021, 297, 100874. [Google Scholar] [CrossRef] [PubMed]
- Jahan, K.S.; Shi, J.; Greenberg, H.Z.E.; Khavandi, S.; Baudel, M.M.-A.; Barrese, V.; Greenwood, I.A.; Albert, A.P. MARCKS mediates vascular contractility through regulating interactions between voltage-gated Ca2+ channels and PIP2. Vasc. Pharmacol. 2020, 132, 106776. [Google Scholar] [CrossRef]
- El-Rahman, R.R.A.; Harraz, O.F.; Brett, S.E.; Anfinogenova, Y.; Mufti, R.E.; Goldman, D.; Welsh, D.G. Identification of L- and T-type Ca2+ channels in rat cerebral arteries: Role in myogenic tone development. Am. J. Physiol.-Heart Circ. Physiol. 2013, 304, H58–H71. [Google Scholar] [CrossRef]
- Lory, P.; Bidaud, I.; Chemin, J. T-type calcium channels in differentiation and proliferation. Cell Calcium 2006, 40, 135–146. [Google Scholar] [CrossRef]
- Yao, X.; Gao, S.; Yan, N. Structural biology of voltage-gated calcium channels. Channels 2024, 18, 2290807. [Google Scholar] [CrossRef]
- Klöckner, U.; Lee, J.-H.; Cribbs, L.L.; Daud, A.; Hescheler, J.; Pereverzev, A.; Perez-Reyes, E.; Schneider, T. Comparison of the Ca2+ currents induced by expression of three cloned α1 subunits, α1G, α1H and α1I, of low-voltage-activated T-type Ca2+ channels. Eur. J. Neurosci. 1999, 11, 4171–4178. [Google Scholar] [CrossRef]
- Lee, J.-H.; Gomora, J.C.; Cribbs, L.L.; Perez-Reyes, E. Nickel Block of Three Cloned T-Type Calcium Channels: Low Concentrations Selectively Block α1H. Biophys. J. 1999, 77, 3034–3042. [Google Scholar] [CrossRef]
- Perez-Reyes, E. Molecular Physiology of Low-Voltage-Activated T-type Calcium Channels. Physiol. Rev. 2003, 83, 117–161. [Google Scholar] [CrossRef] [PubMed]
- Iftinca, M.C.; Zamponi, G.W. Regulation of neuronal T-type calcium channels. Trends Pharmacol. Sci. 2009, 30, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Guidelli, R.; Becucci, L. Deterministic model of Cav3.1 Ca2+ channel and a proposed sequence of its conformations. Bioelectrochemistry 2020, 136, 107618. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef]
- He, L.; Yu, Z.; Geng, Z.; Huang, Z.; Zhang, C.; Dong, Y.; Gao, Y.; Wang, Y.; Chen, Q.; Sun, L.; et al. Structure, gating, and pharmacology of human CaV3.3 channel. Nat. Commun. 2022, 13, 2084. [Google Scholar] [CrossRef]
- Weiss, N.; Zamponi, G.W. T-type calcium channels: From molecule to therapeutic opportunities. Int. J. Biochem. Cell Biol. 2019, 108, 34–39. [Google Scholar] [CrossRef]
- Kuriyama, H.; Kitamura, K.; Itoh, T.; Inoue, R. Physiological Features of Visceral Smooth Muscle Cells, with Special Reference to Receptors and Ion Channels. Physiol. Rev. 1998, 78, 811–920. [Google Scholar] [CrossRef]
- Huguenard, J.R. Low-threshold calcium currents in central nervous system neurons. Annu. Rev. Physiol. 1996, 58, 329–348. [Google Scholar] [CrossRef]
- Staes, M.; Talavera, K.; Klugbauer, N.; Prenen, J.; Lacinová, L.; Droogmans, G.; Hofmann, F.; Nilius, B. The amino side of the C-terminus determines fast inactivation of the T-type calcium channel α1G. J. Physiol. 2001, 530, 35–45. [Google Scholar] [CrossRef]
- Hering, J.; Feltz, A.; Lambert, R.C. Slow inactivation of the CaV3.1 isotype of T-type calcium channels. J. Physiol. 2004, 555 Pt 2, 331–344. [Google Scholar] [CrossRef]
- Chemin, J.; Taiakina, V.; Monteil, A.; Piazza, M.; Guan, W.; Stephens, R.F.; Kitmitto, A.; Pang, Z.P.; Dolphin, A.C.; Perez-Reyes, E.; et al. Calmodulin regulates Cav3 T-type channels at their gating brake. J. Biol. Chem. 2017, 292, 20010–20031. [Google Scholar] [CrossRef] [PubMed]
- Vitko, I.; Bidaud, I.; Arias, J.M.; Mezghrani, A.; Lory, P.; Perez-Reyes, E. The I–II Loop Controls Plasma Membrane Expression and Gating of Cav3.2 T-Type Ca2+ Channels: A Paradigm for Childhood Absence Epilepsy Mutations. J. Neurosci. 2007, 27, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, J.P.; Vitko, I.; Bidaud, I.; Kondratskyi, A.; Lory, P.; Perez-Reyes, E. I–II Loop Structural Determinants in the Gating and Surface Expression of Low Voltage-Activated Calcium Channels. PLoS ONE 2008, 3, e2976. [Google Scholar] [CrossRef]
- Chemin, J.; Stamenic, T.T.; Cazade, M.; Llinares, J.; Blesneac, I.; Todorovic, S.M.; Lory, P. A novel phospho-modulatory mechanism contributes to the calcium-dependent regulation of T-type Ca2+ channels. Sci. Rep. 2019, 9, 15642. [Google Scholar] [CrossRef] [PubMed]
- Tamang, H.K.; Yang, R.B.; Song, Z.H.; Hsu, S.C.; Peng, C.C.; Tung, Y.C.; Tzeng, B.H.; Chen, C.C. CaV 3.2 T-type calcium channel regulates mouse platelet activation and arterial thrombosis. J. Thromb. Haemost. 2022, 20, 1887–1899. [Google Scholar] [CrossRef] [PubMed]
- Pandi, S.P.S.; Shattock, M.J.; Hendry, B.M.; Sharpe, C.C. Stimulated phosphorylation of ERK in mouse kidney mesangial cells is dependent upon expression of Cav3.1. BMC Nephrol. 2022, 23, 211. [Google Scholar] [CrossRef]
- Lin, S.-S.; Tzeng, B.-H.; Lee, K.-R.; Smith, R.J.H.; Campbell, K.P.; Chen, C.-C. Cav3.2 T-type calcium channel is required for the NFAT-dependent Sox9 expression in tracheal cartilage. Proc. Natl. Acad. Sci. USA 2014, 111, E1990–E1998. [Google Scholar] [CrossRef]
- Meng, Y.; Toledo-Rodriguez, M.; Fedorenko, O.; Smith, P.A. Sex and age affect depot expression of Ca2+ channels in rat white fat adipocytes. J. Mol. Endocrinol. 2024, 72, e230108. [Google Scholar] [CrossRef]
- Liu, Q.; Lu, Z.; Ren, H.; Fu, L.; Wang, Y.; Bu, H.; Ma, M.; Ma, L.; Huang, C.; Wang, J.; et al. Cav3.2 T-Type calcium channels downregulation attenuates bone cancer pain induced by inhibiting IGF-1/HIF-1α signaling pathway in the rat spinal cord. J. Bone Oncol. 2023, 42, 100495. [Google Scholar] [CrossRef]
- Mehregan, A.; Ardestani, G.; Akizawa, H.; Carvacho, I.; Fissore, R. Deletion of TRPV3 and CaV3.2 T-type channels in mice undermines fertility and Ca2+ homeostasis in oocytes and eggs. J. Cell Sci. 2021, 134, jcs257956. [Google Scholar] [CrossRef]
- Cai, H.; Chen, S.; Sun, Y.; Zheng, T.; Liu, Y.; Tao, J.; Zhang, Y. Interleukin-22 receptor 1-mediated stimulation of T-type Ca2+ channels enhances sensory neuronal excitability through the tyrosine-protein kinase Lyn-dependent PKA pathway. Cell Commun. Signal. 2024, 22, 307. [Google Scholar] [CrossRef] [PubMed]
- Leandrou, E.; Chalatsa, I.; Anagnostou, D.; Machalia, C.; Semitekolou, M.; Filippa, V.; Makridakis, M.; Vlahou, A.; Anastasiadou, E.; Vekrellis, K.; et al. α-Synuclein oligomers potentiate neuroinflammatory NF-κB activity and induce CaV3.2 calcium signaling in astrocytes. Transl. Neurodegener. 2024, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.W.; Kim, S.E.; Kim, D.Y.; Jeong, I.; Kim, S.; Chung, S.; Lee, S.E. Cav3.2 T-Type Calcium Channel Mediates Acute Itch and Contributes to Chronic Itch and Inflammation in Experimental Atopic Dermatitis. J. Investig. Dermatol. 2024, 144, 612–620.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, X.; Xue, L.; Xing, J.; Jouvin, M.H.; Putney, J.W.; Anderson, M.P.; Trebak, M.; Kinet, J.P. Low-Voltage-Activated CaV3.1 Calcium Channels Shape T Helper Cell Cytokine Profiles. Immunity 2016, 44, 782–794. [Google Scholar] [CrossRef]
- Shao, J.; Gao, D.-S.; Liu, Y.-H.; Chen, S.-P.; Liu, N.; Zhang, L.; Zhou, X.-Y.; Xiao, Q.; Wang, L.-P.; Hu, H.-L.; et al. Cav3.1-driven bursting firing in ventromedial hypothalamic neurons exerts dual control of anxiety-like behavior and energy expenditure. Mol. Psychiatry 2022, 27, 2901–2913. [Google Scholar] [CrossRef]
- Zöphel, D.; Hof, C.; Lis, A. Altered Ca2+ Homeostasis in Immune Cells during Aging: Role of Ion Channels. Int. J. Mol. Sci. 2020, 22, 110. [Google Scholar] [CrossRef]
- Mayadali, Ü.S.; Chertes, C.A.M.; Sinicina, I.; Shaikh, A.G.; Horn, A.K.E. Ion channel profiles of extraocular motoneurons and internuclear neurons in human abducens and trochlear nuclei. Front. Neuroanat. 2024, 18, 1411154. [Google Scholar] [CrossRef]
- Maksemous, N.; Blayney, C.D.; Sutherland, H.G.; Smith, R.A.; Lea, R.A.; Tran, K.N.; Ibrahim, O.; McArthur, J.R.; Haupt, L.M.; Cader, M.Z.; et al. Investigation of CACNA1I Cav3.3 Dysfunction in Hemiplegic Migraine. Front. Mol. Neurosci. 2022, 15, 892820. [Google Scholar] [CrossRef]
- Eickhoff, A.; Tjaden, J.; Stahlke, S.; Vorgerd, M.; Theis, V.; Matschke, V.; Theiss, C. Effects of progesterone on T-type-Ca2+-channel expression in Purkinje cells. Neural Regen. Res. 2022, 17, 2465–2471. [Google Scholar]
- Cmarko, L.; Weiss, N. Selective inhibition of neuronal Cav3.3 T-type calcium channels by TAT-based channel peptide. Mol. Brain 2020, 13, 95. [Google Scholar] [CrossRef]
- Bertolesi, G.E.; Jollimore, C.A.B.; Shi, C.; Elbaum, L.; Denovan-Wright, E.M.; Barnes, S.; Kelly, M.E.M. Regulation of α1G T-type calcium channel gene (CACNA1G) expression during neuronal differentiation. Eur. J. Neurosci. 2003, 17, 1802–1810. [Google Scholar] [CrossRef] [PubMed]
- González-Ramírez, R.; Martínez-Hernández, E.; Sandoval, A.; Felix, R. Transcription Factor Sp1 Regulates T-Type Ca2+ Channel CaV3.1 Gene Expression. J. Cell. Physiol. 2014, 229, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Banderali, U.; Jain, M.; Thakur, S.; Jayanthan, A.; Belke, D.D.; Giles, W.R.; Narendran, A. The T-type Calcium Channel Cav3.1 in Y79 Retinoblastoma Cells is Regulated by the Epidermal Growth Factor Receptor via the MAPK Signaling Pathway. Curr. Eye Res. 2022, 47, 426–435. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, C.; Liu, Y.; Ren, L.; Qi, J.; Yang, Y.; Chen, W.; Yao, Y.; Cai, X.; Liu, Z.; et al. NRF-2/HO-1 Pathway-Mediated SHOX2 Activation Is a Key Switch for Heart Rate Acceleration by Yixin-Fumai Granules. Oxidative Med. Cell. Longev. 2022, 2022, 8488269. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, Y.; Han, S.; Zi, Y.; Zhang, Y.; Kong, R.; Liu, Z.; Cai, Z.; Zhong, C.; Liu, W.; et al. Nkx2-5 Regulates the Proliferation and Migration of H9c2 Cells. Med. Sci. Monit. 2020, 26, e925388. [Google Scholar] [CrossRef] [PubMed]
- Tjaden, J.; Eickhoff, A.; Stahlke, S.; Gehmeyr, J.; Vorgerd, M.; Theis, V.; Matschke, V.; Theiss, C. Expression Pattern of T-Type Ca2+ Channels in Cerebellar Purkinje Cells after VEGF Treatment. Cells 2021, 10, 2277. [Google Scholar] [CrossRef]
- Ito, J.; Minemura, T.; Wälchli, S.; Niimi, T.; Fujihara, Y.; Kuroda, S.I.; Takimoto, K.; Maturana, A.D. Id2 Represses Aldosterone-Stimulated Cardiac T-Type Calcium Channels Expression. Int. J. Mol. Sci. 2021, 22, 3561. [Google Scholar] [CrossRef]
- Stroedecke, K.; Meinel, S.; Markwardt, F.; Kloeckner, U.; Straetz, N.; Quarch, K.; Schreier, B.; Kopf, M.; Gekle, M.; Grossmann, C. The mineralocorticoid receptor leads to increased expression of EGFR and T-type calcium channels that support HL-1 cell hypertrophy. Sci. Rep. 2021, 11, 13229. [Google Scholar] [CrossRef]
- Mohan, R.A.; Bosada, F.M.; van Weerd, J.H.; van Duijvenboden, K.; Wang, J.; Mommersteeg, M.T.M.; Hooijkaas, I.B.; Wakker, V.; de Gier-de Vries, C.; Coronel, R.; et al. T-box transcription factor 3 governs a transcriptional program for the function of the mouse atrioventricular conduction system. Proc. Natl. Acad. Sci. USA 2020, 117, 18617–18626. [Google Scholar] [CrossRef]
- Trainito, A.; Muscarà, C.; Gugliandolo, A.; Chiricosta, L.; Salamone, S.; Pollastro, F.; Mazzon, E.; D’Angiolini, S. Cannabinol (CBN) Influences the Ion Channels and Synaptic-Related Genes in NSC-34 Cell Line: A Transcriptomic Study. Cells 2024, 13, 1573. [Google Scholar] [CrossRef]
- Markandeya, Y.S.; Fahey, J.M.; Pluteanu, F.; Cribbs, L.L.; Balijepalli, R.C. Caveolin-3 Regulates Protein Kinase A Modulation of the CaV3.2 (α1H) T-type Ca2+ Channels. J. Biol. Chem. 2011, 286, 2433–2444. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, K.; Saito, Y.; Takano, M.; Arai, Y.; Yasuno, S.; Nakagawa, Y.; Takahashi, N.; Adachi, Y.; Takemura, G.; Horie, M.; et al. NRSF regulates the fetal cardiac gene program and maintains normal cardiac structure and function. EMBO J. 2003, 22, 6310–6321. [Google Scholar] [CrossRef] [PubMed]
- Somekawa, S.; Imagawa, K.; Naya, N.; Takemoto, Y.; Onoue, K.; Okayama, S.; Takeda, Y.; Kawata, H.; Horii, M.; Nakajima, T.; et al. Regulation of Aldosterone and Cortisol Production by the Transcriptional Repressor Neuron Restrictive Silencer Factor. Endocrinology 2009, 150, 3110–3117. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, L.; Zhou, H.; Zhang, K.; Zhao, M. Identification of miRNA-mediated gene regulatory networks in L-methionine exposure counteracts cocaine-conditioned place preference in mice. Front. Genet. 2023, 13, 1076156. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, L.; Bai, Y.-G.; Song, J.-B.; Cheng, J.-H.; Ma, H.-Z.; Ma, J.; Xie, M.-J. miR-137 and its target T-type CaV3.1 channel modulate dedifferentiation and proliferation of cerebrovascular smooth muscle cells in simulated microgravity rats by regulating calcineurin/NFAT pathway. Cell Prolif. 2020, 53, e12774. [Google Scholar] [CrossRef]
- Zheng, Q.; Li, X.; Xu, X.; Tang, X.; Hammad, B.; Xing, J.; Zhang, D. The mmu_circ_003062, hsa_circ_0075663/miR-490-3p/CACNA1H axis mediates apoptosis in renal tubular cells in association with endoplasmic reticulum stress following ischemic acute kidney injury. Int. Immunopharmacol. 2024, 132, 111956. [Google Scholar] [CrossRef]
- Koyama, R.; Mannic, T.; Ito, J.; Amar, L.; Zennaro, M.-C.; Rossier, M.F.; Maturana, A.D. MicroRNA-204 Is Necessary for Aldosterone-Stimulated T-Type Calcium Channel Expression in Cardiomyocytes. Int. J. Mol. Sci. 2018, 19, 2941. [Google Scholar] [CrossRef]
- Qi, R.; Cao, J.; Sun, Y.; Li, Y.; Huang, Z.; Jiang, D.; Jiang, X.-H.; Snutch, T.P.; Zhang, Y.; Tao, J. Histone methylation-mediated microRNA-32-5p down-regulation in sensory neurons regulates pain behaviors via targeting Cav3.2 channels. Proc. Natl. Acad. Sci. USA 2022, 119, e2117209119. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, M.; Wang, Y.; Liu, X.; Zhou, L.; Zhang, C.; Shao, L. Histone deacetylase inhibition by MS-275 potentiates glucose-stimulated insulin secretion without affecting glucose oxidation. Life Sci. 2020, 257, 118073. [Google Scholar] [CrossRef]
- Kim, J.W.; Oh, H.A.; Kim, S.R.; Ko, M.J.; Seung, H.; Lee, S.H.; Shin, C.Y. Epigenetically Upregulated T-Type Calcium Channels Contribute to Abnormal Proliferation of Embryonic Neural Progenitor Cells Exposed to Valproic Acid. Biomol. Ther. 2020, 28, 389–396. [Google Scholar] [CrossRef]
- Stein, A.B.; Goonewardena, S.N.; Jones, T.A.; Prusick, P.J.; Bazzi, A.A.; Belyavskaya, J.M.; McCoskey, M.M.; Dandar, R.A. The PTIP-Associated Histone Methyltransferase Complex Prevents Stress-Induced Maladaptive Cardiac Remodeling. PLoS ONE 2015, 10, e0127839. [Google Scholar] [CrossRef] [PubMed]
- Toyota, M.; Ho, C.; Ohe-Toyota, M.; Baylin, S.B.; Issa, J.-P.J. Inactivation of CACNA1G, a T-Type Calcium Channel Gene, by Aberrant Methylation of Its 5′ CpG Island in Human Tumors1. Cancer Res. 1999, 59, 4535–4541. [Google Scholar] [PubMed]
- Svahn, F.; Solhusløkk Höse, K.; Stenman, A.; Liu, Y.; Calissendorff, J.; Tham, E.; Végvári, Á.; Zubarev, R.A.; Wang, N.; Korah, R.; et al. Genetic variants and down-regulation of CACNA1H in pheochromocytoma. Endocr. Relat. Cancer 2024, 31, e230061. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Kaßmann, M.; Cui, Y.; Matthaeus, C.; Kunz, S.; Zhong, C.; Zhu, S.; Xie, Y.; Tsvetkov, D.; Daumke, O.; et al. Age attenuates the T-type Ca(V) 3.2-RyR axis in vascular smooth muscle. Aging Cell 2020, 19, e13134. [Google Scholar] [CrossRef]
- Yabuki, Y.; Matsuo, K.; Yu, M.; Xu, J.; Sakimura, K.; Shioda, N.; Fukunaga, K. Cav3.1 t-type calcium channel is critical for cell proliferation and survival in newly generated cells of the adult hippocampus. Acta Physiol. 2021, 232, e13613. [Google Scholar] [CrossRef]
- Rodman, D.M.; Reese, K.; Harral, J.; Fouty, B.; Wu, S.; West, J.; Hoedt-Miller, M.; Tada, Y.; Li, K.-X.; Cool, C.; et al. Low-Voltage-Activated (T-Type) Calcium Channels Control Proliferation of Human Pulmonary Artery Myocytes. Circ. Res. 2005, 96, 864–872. [Google Scholar] [CrossRef]
- Tzeng, B.-H.; Chen, Y.-H.; Huang, C.-H.; Lin, S.-S.; Lee, K.-R.; Chen, C.-C. The Cav3.1 T-type calcium channel is required for neointimal formation in response to vascular injury in mice. Cardiovasc. Res. 2012, 96, 533–542. [Google Scholar] [CrossRef]
- Duckles, H.; Boycott, H.E.; Al-Owais, M.M.; Elies, J.; Johnson, E.; Dallas, M.L.; Porter, K.E.; Giuntini, F.; Boyle, J.P.; Scragg, J.L.; et al. Heme oxygenase-1 regulates cell proliferation via carbon monoxide-mediated inhibition of T-type Ca2+ channels. Pflügers Arch.-Eur. J. Physiol. 2015, 467, 415–427. [Google Scholar] [CrossRef]
- Dziegielewska, B.; Brautigan, D.L.; Larner, J.M.; Dziegielewski, J. T-Type Ca2+ Channel Inhibition Induces p53-Dependent Cell Growth Arrest and Apoptosis through Activation of p38-MAPK in Colon Cancer Cells. Mol. Cancer Res. 2014, 12, 348–358. [Google Scholar] [CrossRef]
- Du, N.H.; Ngoc, T.T.B.; Cang, H.Q.; Luyen, N.T.T.; Thuoc, T.L.; Le Quan, T.; Thao, D.T.P. KTt-45, a T-type calcium channel blocker, acts as an anticancer agent by inducing apoptosis on HeLa cervical cancer cell line. Sci. Rep. 2023, 13, 22092. [Google Scholar] [CrossRef]
- Sedeeq, M.; Maklad, A.; Dutta, T.; Feng, Z.; Wilson, R.; Gueven, N.; Azimi, I. T-Type Calcium Channel Inhibitors Induce Apoptosis in Medulloblastoma Cells Associated with Altered Metabolic Activity. Mol. Neurobiol. 2022, 59, 2932–2945. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, T.; Yamazaki, J. T-type voltage-activated calcium channel Cav3.1, but not Cav3.2, is involved in the inhibition of proliferation and apoptosis in MCF-7 human breast cancer cells. Int. J. Oncol. 2012, 41, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Li, S.; Hao, M.; Chen, J.; Zhao, Z.; Hong, S.; Min, J.; Tang, J.; Hu, M.; Hong, L. T-type calcium channel blockade induces apoptosis in C2C12 myotubes and skeletal muscle via endoplasmic reticulum stress activation. FEBS Open Bio 2020, 10, 2122–2136. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Oh, H.A.; Lee, S.H.; Kim, K.C.; Eun, P.H.; Ko, M.J.; Gonzales, E.L.T.; Seung, H.; Kim, S.; Bahn, G.H.; et al. T-Type Calcium Channels Are Required to Maintain Viability of Neural Progenitor Cells. Biomol. Ther. 2018, 26, 439–445. [Google Scholar] [CrossRef]
- Ferreira, A.F.F.; Britto, L.R.G. The transient receptor potential melastatin 2: A new therapeutical target for Parkinson’s disease? Neural Regen. Res. 2023, 18, 1652–1656. [Google Scholar] [CrossRef]
- Dai, F.; Hu, C.; Li, X.; Zhang, Z.; Wang, H.; Zhou, W.; Wang, J.; Geng, Q.; Dong, Y.; Tang, C. Cav3.2 channel regulates cerebral ischemia/reperfusion injury: A promising target for intervention. Neural Regen. Res. 2024, 19, 2480–2487. [Google Scholar] [CrossRef]
- Yang, F.; Zhou, L.; Shen, Y.; Wang, X.; Fan, X.; Yang, L. Multi-omics approaches for drug-response characterization in primary biliary cholangitis and autoimmune hepatitis variant syndrome. J. Transl. Med. 2024, 22, 214. [Google Scholar] [CrossRef]
- Erdogmus, S.; Concepcion, A.R.; Yamashita, M.; Sidhu, I.; Tao, A.Y.; Li, W.; Rocha, P.P.; Huang, B.; Garippa, R.; Lee, B.; et al. Cavβ1 regulates T cell expansion and apoptosis independently of voltage-gated Ca2+ channel function. Nat. Commun. 2022, 13, 2033. [Google Scholar] [CrossRef]
- Capone, A.; Volpe, E. Transcriptional Regulators of T Helper 17 Cell Differentiation in Health and Autoimmune Diseases. Front. Immunol. 2020, 11, 348. [Google Scholar] [CrossRef]
- Johnson, B.V.; Bert, A.G.; Ryan, G.R.; Condina, A.; Cockerill, P.N. Granulocyte-Macrophage Colony-Stimulating Factor Enhancer Activation Requires Cooperation between NFAT and AP-1 Elements and Is Associated with Extensive Nucleosome Reorganization. Mol. Cell. Biol. 2004, 24, 7914–7930. [Google Scholar] [CrossRef]
- El-Lakany, M.A.; Haghbin, N.; Arora, N.; Hashad, A.M.; Mironova, G.Y.; Sancho, M.; Gros, R.; Welsh, D.G. CaV3.1 channels facilitate calcium wave generation and myogenic tone development in mouse mesenteric arteries. Sci. Rep. 2023, 13, 20407. [Google Scholar] [CrossRef] [PubMed]
- Paknejad, N.; Sapuru, V.; Hite, R.K. Structural titration reveals Ca2+-dependent conformational landscape of the IP3 receptor. Nat. Commun. 2023, 14, 6897. [Google Scholar] [CrossRef] [PubMed]
- Hashad, A.M.; Harraz, O.F.; Brett, S.E.; Romero, M.; Kassmann, M.; Puglisi, J.L.; Wilson, S.M.; Gollasch, M.; Welsh, D.G. Caveolae Link Ca(V)3.2 Channels to BK(Ca)-Mediated Feedback in Vascular Smooth Muscle. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Harraz, O.F.; Jensen, L.J. Aging, calcium channel signaling and vascular tone. Mech. Ageing Dev. 2020, 191, 111336. [Google Scholar] [CrossRef]
- Helle, F.; Hultström, M.; Kavvadas, P.; Iversen, B.; Chadjichristos, C.E.; Chatziantoniou, C. Deletion of Notch3 Impairs Contractility of Renal Resistance Vessels Due to Deficient Ca2+ Entry. Int. J. Mol. Sci. 2022, 23, 16068. [Google Scholar] [CrossRef]
- Plante, A.E.; Whitt, J.P.; Meredith, A.L. BK channel activation by L-type Ca2+ channels CaV1.2 and CaV1.3 during the subthreshold phase of an action potential. J. Neurophysiol. 2021, 126, 427–439. [Google Scholar] [CrossRef]
- Vivas, O.; Moreno, C.M.; Santana, L.F.; Hille, B. Proximal clustering between BK and CaV1.3 channels promotes functional coupling and BK channel activation at low voltage. eLife 2017, 6, e28029. [Google Scholar] [CrossRef]
- Numata, T.; Sato-Numata, K.; Yoshino, M. BK Channels Are Activated by Functional Coupling With L-Type Ca2+ Channels in Cricket Myocytes. Front. Insect Sci. 2021, 1, 662414. [Google Scholar] [CrossRef]
- Schmitz, E.A.; Takahashi, H.; Karakas, E. Structural basis for activation and gating of IP3 receptors. Nat. Commun. 2022, 13, 1408. [Google Scholar] [CrossRef]
- Feng, M.-G.; Li, M.; Navar, L.G. T-type calcium channels in the regulation of afferent and efferent arterioles in rats. Am. J. Physiol.-Ren. Physiol. 2004, 286, F331–F337. [Google Scholar] [CrossRef]
- Pérez, G.J.; Bonev, A.D.; Patlak, J.B.; Nelson, M.T. Functional Coupling of Ryanodine Receptors to KCa Channels in Smooth Muscle Cells from Rat Cerebral Arteries. J. Gen. Physiol. 1999, 113, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Findlay, I. β-Adrenergic stimulation modulates Ca2+- and voltage-dependent inactivation of L-type Ca2+ channel currents in guinea-pig ventricular myocytes. J. Physiol. 2002, 541, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lu, C.; Ma, L.; Li, C.; Luo, H.; Liu, Y.; Liu, X.; Li, H.; Cui, Y.; Zeng, J.; et al. The T-Type Calcium Channel CACNA1H is Required for Smooth Muscle Cytoskeletal Organization During Tracheal Tubulogenesis. Adv. Sci. 2024, e2308622. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Iijima, T. Cardiac T-type Ca2+ channels in the heart. J. Mol. Cell. Cardiol. 2010, 48, 65–70. [Google Scholar] [CrossRef]
- Yu, P.; Cai, X.; Liang, Y.; Wang, M.; Yang, W. Roles of NAD+ and Its Metabolites Regulated Calcium Channels in Cancer. Molecules 2020, 25, 4826. [Google Scholar] [CrossRef]
- Kanemaru, K.; Cranley, J.; Muraro, D.; Miranda, A.M.A.; Ho, S.Y.; Wilbrey-Clark, A.; Patrick Pett, J.; Polanski, K.; Richardson, L.; Litvinukova, M.; et al. Spatially resolved multiomics of human cardiac niches. Nature 2023, 619, 801–810. [Google Scholar] [CrossRef]
- Qian, Y.; Xiong, S.; Li, L.; Sun, Z.; Zhang, L.; Yuan, W.; Cai, H.; Feng, G.; Wang, X.; Yao, H.; et al. Spatial multiomics atlas reveals smooth muscle phenotypic transformation and metabolic reprogramming in diabetic macroangiopathy. Cardiovasc. Diabetol. 2024, 23, 358. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Property | Cav3.1 | Cav3.2 | Cav3.3 |
|---|---|---|---|
| Gene [17,18] | CACNA1G | CACNA1H | CACNA1I |
| Activation [19] | Fastest | Slower | Slower |
| Inactivation [19] | Fastest | Slower | Slower |
| Deactivation [19] | Slower | Slower | Fastest |
| IC50 for Ni2+ [20] | 13 µM | 250 µM | 216 µM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomida, S.; Ishima, T.; Nagai, R.; Aizawa, K. T-Type Voltage-Gated Calcium Channels: Potential Regulators of Smooth Muscle Contractility. Int. J. Mol. Sci. 2024, 25, 12420. https://doi.org/10.3390/ijms252212420
Tomida S, Ishima T, Nagai R, Aizawa K. T-Type Voltage-Gated Calcium Channels: Potential Regulators of Smooth Muscle Contractility. International Journal of Molecular Sciences. 2024; 25(22):12420. https://doi.org/10.3390/ijms252212420
Chicago/Turabian StyleTomida, Shota, Tamaki Ishima, Ryozo Nagai, and Kenichi Aizawa. 2024. "T-Type Voltage-Gated Calcium Channels: Potential Regulators of Smooth Muscle Contractility" International Journal of Molecular Sciences 25, no. 22: 12420. https://doi.org/10.3390/ijms252212420
APA StyleTomida, S., Ishima, T., Nagai, R., & Aizawa, K. (2024). T-Type Voltage-Gated Calcium Channels: Potential Regulators of Smooth Muscle Contractility. International Journal of Molecular Sciences, 25(22), 12420. https://doi.org/10.3390/ijms252212420