
Molecular Modeling of Vasodilatory Activity: Unveiling Novel Candidates Through Density Functional Theory, QSAR, and Molecular Dynamics

 , , , , , ,
, , , , , ,  ,
,  and
and
Abstract
:1. Introduction
2. Results
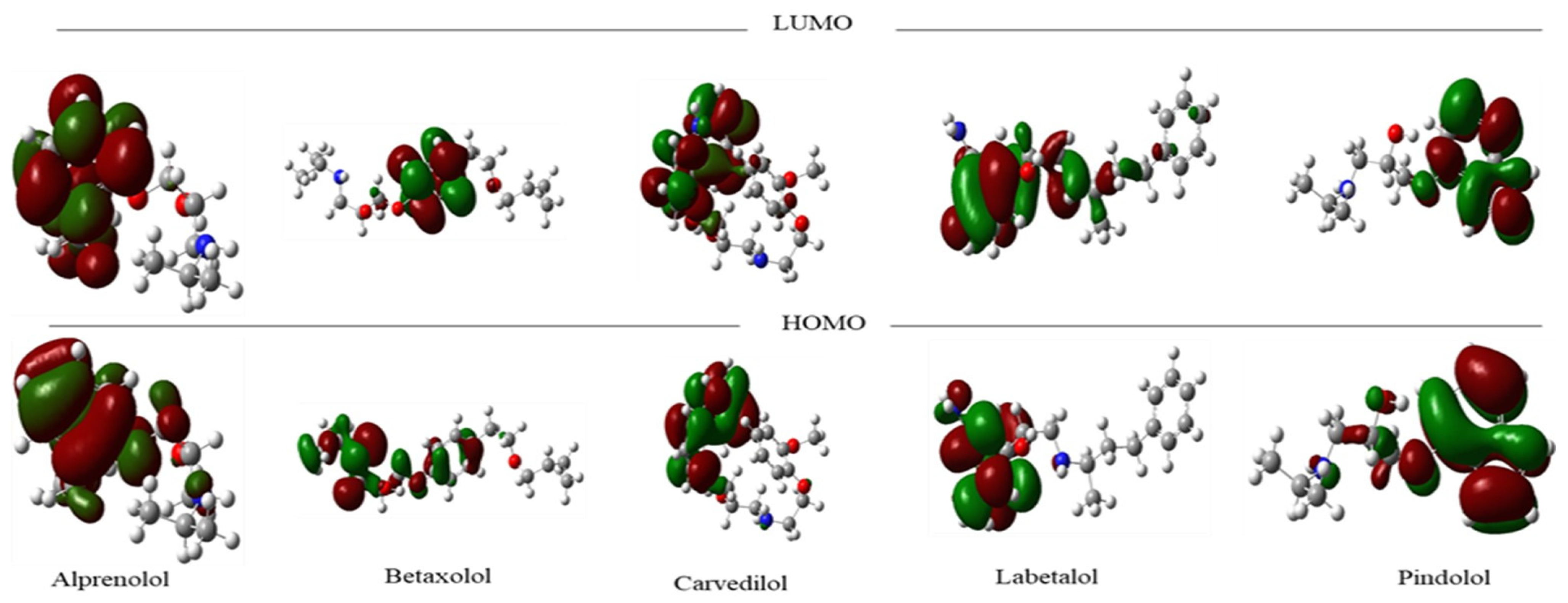
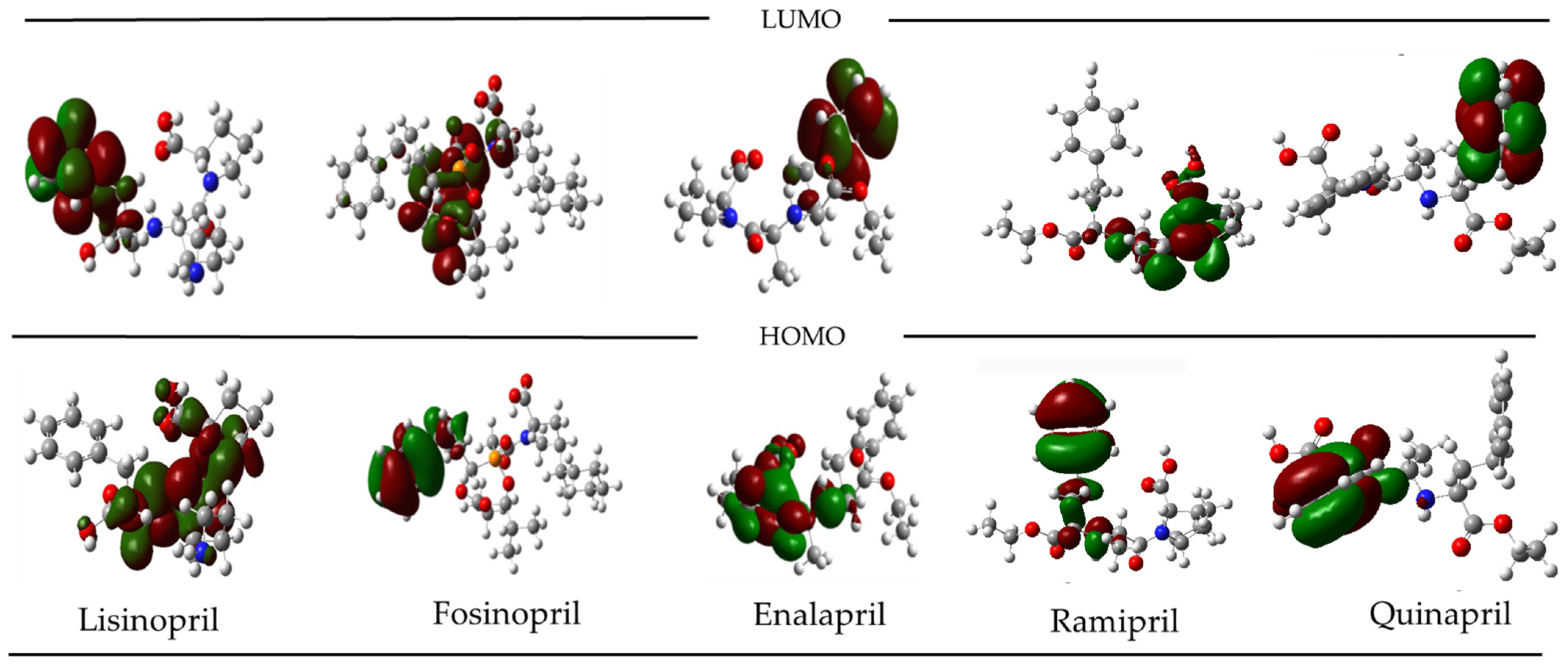
2.1. Molecular Frontier Orbitals Analysis
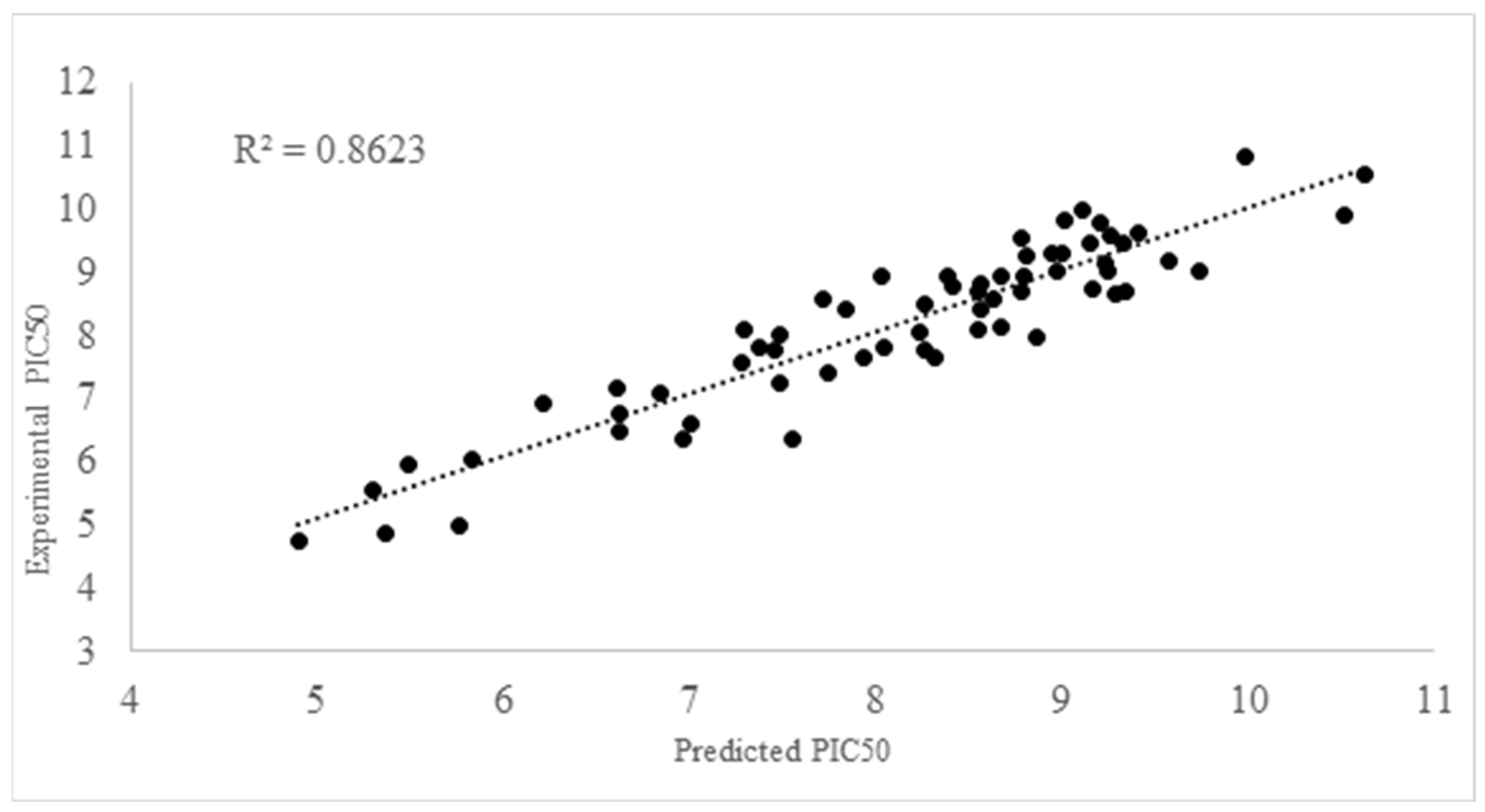
2.2. QSAR Modelling
- pKI50 = 4.2275 + 2.1251A + 3.5830E − 05B − 10.1982C + 1.18754D − 1.0493E + 8.1419F + 0.6900G − 3.8395H; R2 = 0.8623, F = 33.088; s = 0.5846; N = 72; Q2loo = 0.7886; RMSE = 0.9377; MAE = 0.7941
- pKI50 = 3.2838 +2.3428A + 0.0001B−11.0363C + 1.0286D−0.9335 E + 8.8309F + 0.8707G; R2 = 0.8307, F = 32.2440; s = 0.6241; N = 72; Q2loo = 0.7581; RMSE = 0.6741; MAE = 0.5431
- pKI50 = 2.5933 + 2.3755A+ 0.0001B−0.3186C + 0.9599D + 8.3454E + 0.8515G R2 = 0.7876, F = 29.0428; s = 0.6916; N = 72; Q2loo = 0.7190; RMSE = 0.7421; MAE = 0.6035
2.3. Drug Bank 5.1.7 Screening
2.4. Molecular Orbital Analysis for the Best Candidate from Drug Bank Repurpose
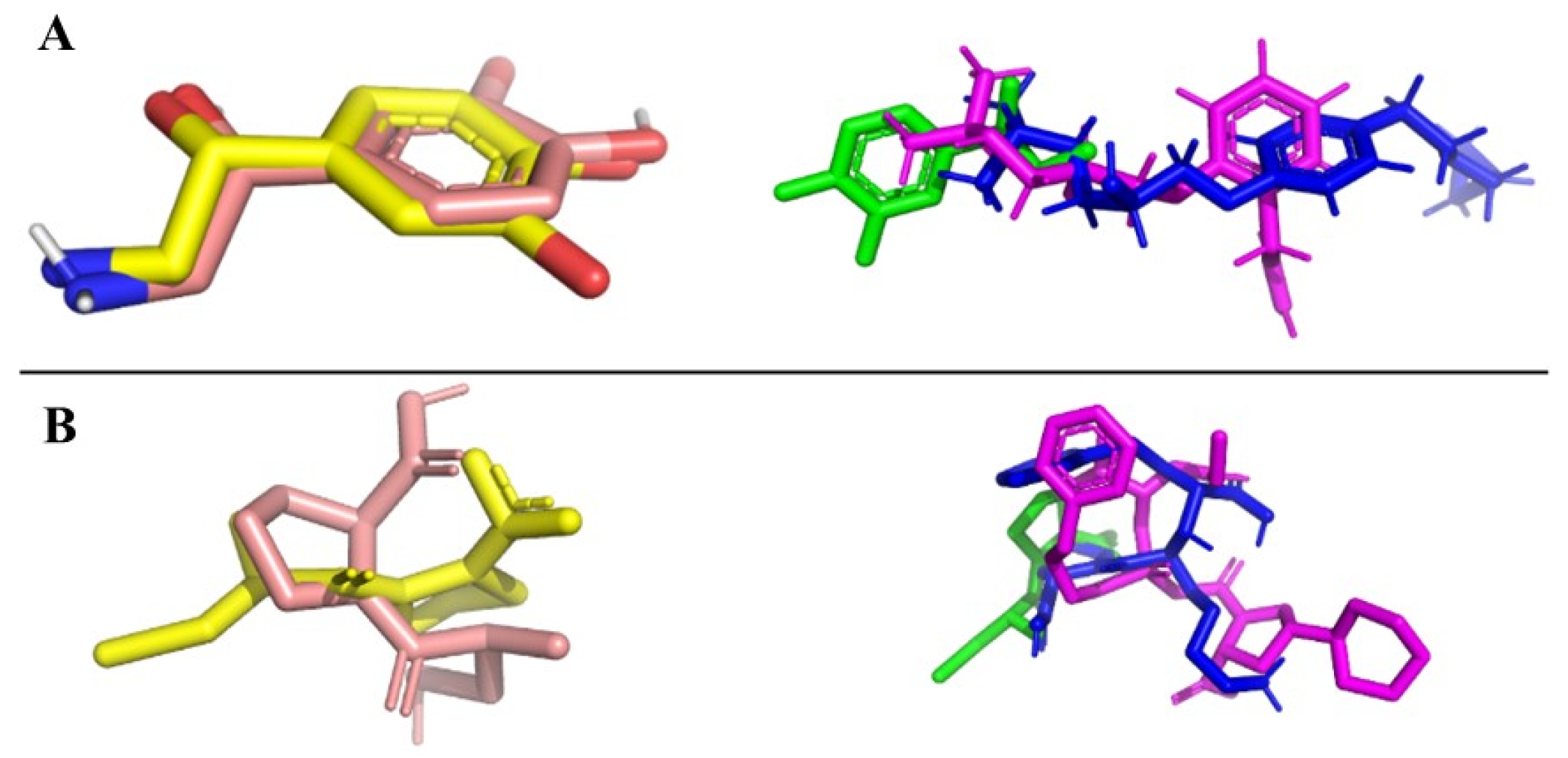
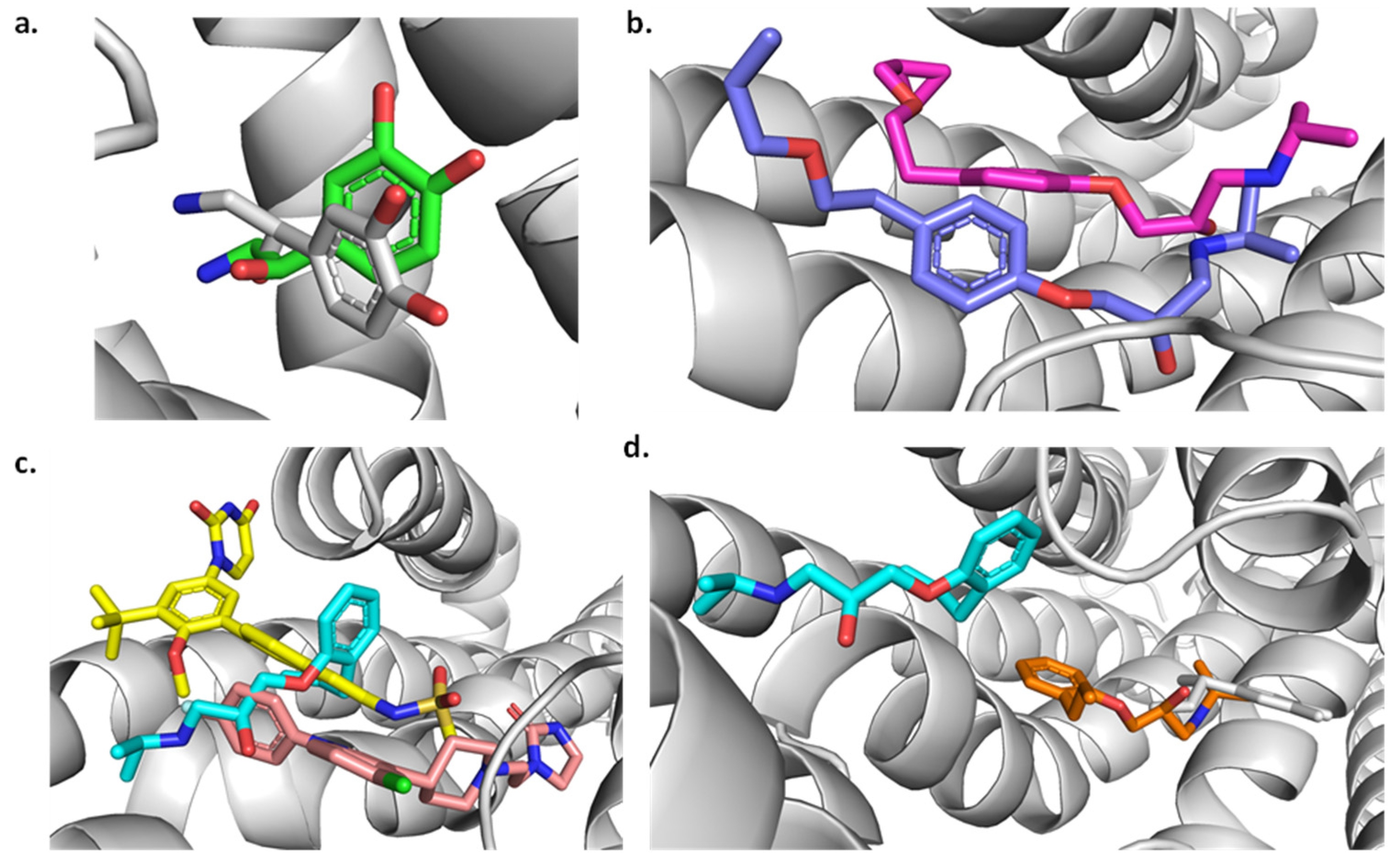
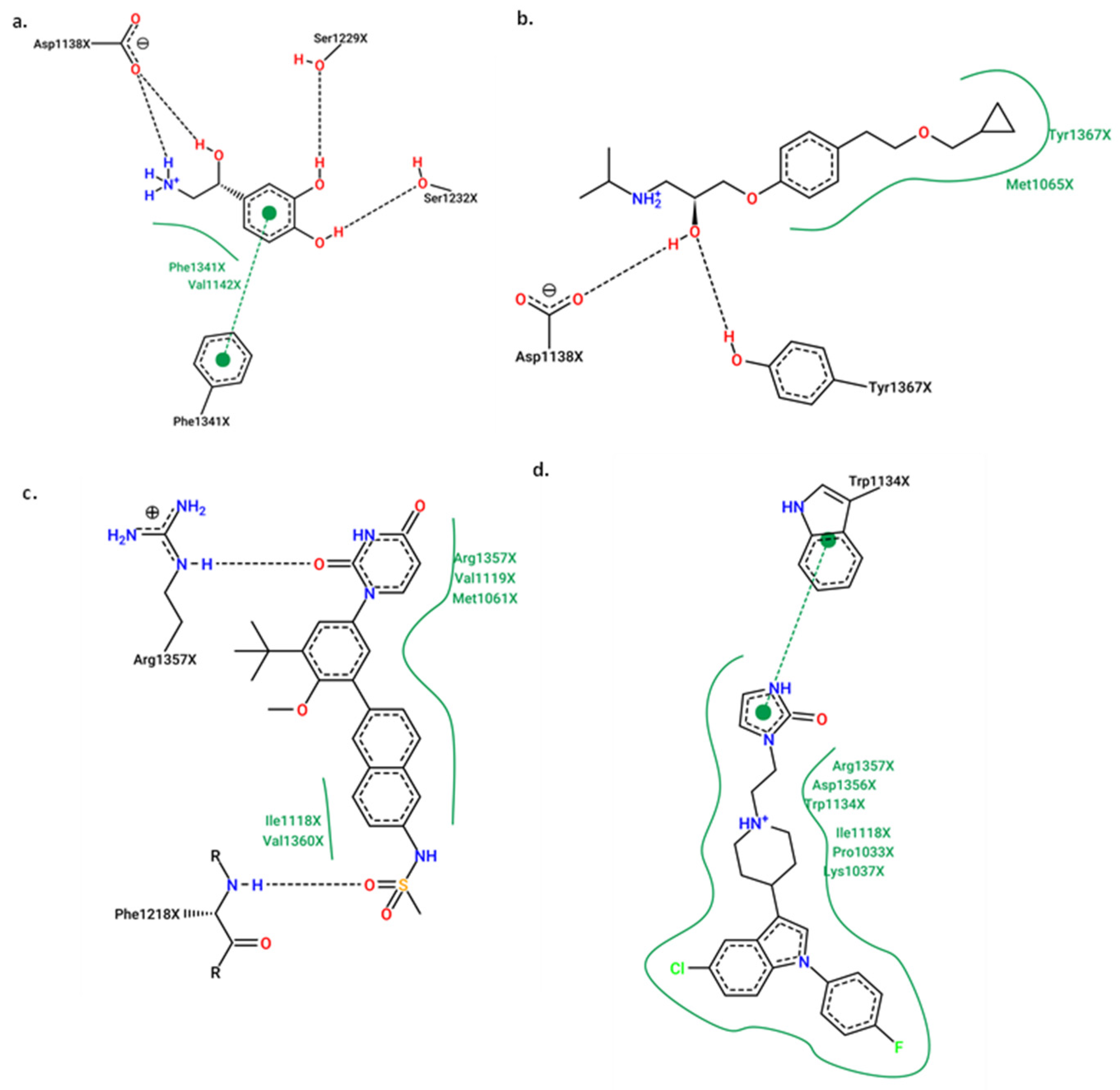
2.5. Molecular Docking
Redocking
2.6. Molecular Docking for the Best Compounds Repurposing from the FDA Database
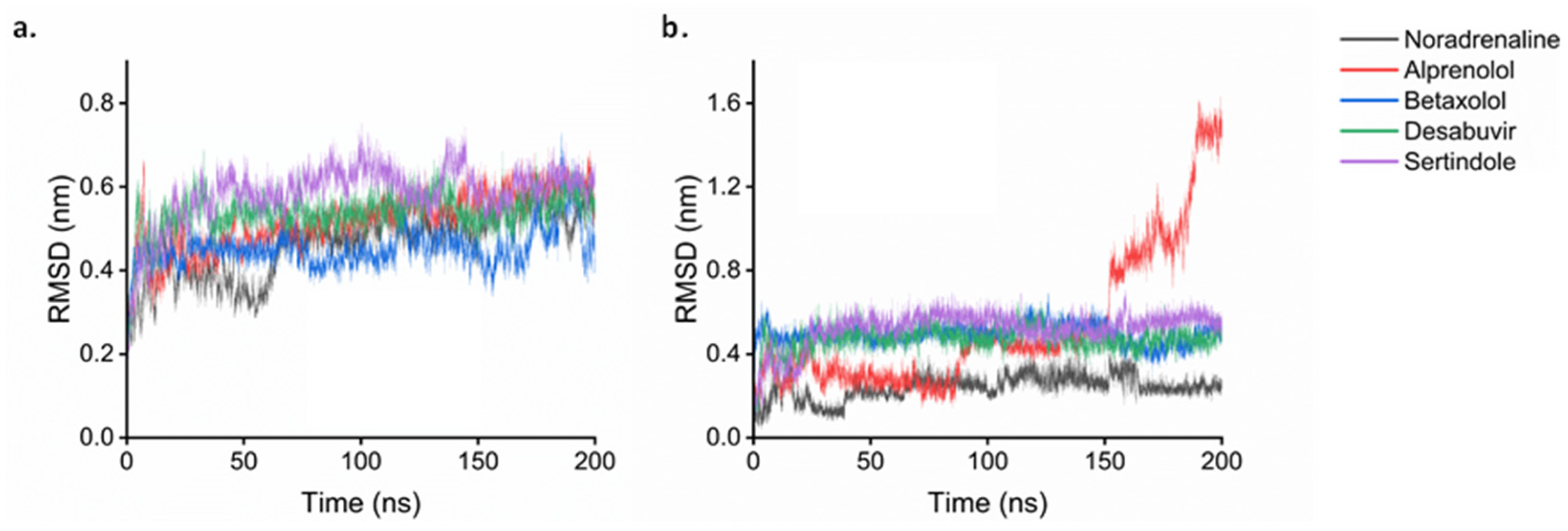
2.7. Molecular Dynamics
3. Materials and Methods
3.1. Data Selection
3.2. Molecular Descriptors Calculation
3.3. QSAR Modeling Building
3.4. Applicability Domain
3.5. Model Performance
3.6. Molecular Docking
3.6.1. Preparation of Protein Structures
3.6.2. Ligand Preparation
3.6.3. Redocking Procedure
3.6.4. Docking Simulations
3.7. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, Regional, and National Age-Sex-Specific Mortality for 282 Causes of Death in 195 Countries and Territories, 1980–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2021 Update. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.; Leong, D.; McKee, M.; Anand, S.S.; Schwalm, J.-D.; Teo, K.; Mente, A.; Yusuf, S. Reducing the Global Burden of Cardiovascular Disease, Part 1. Circ. Res. 2017, 121, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, Y.; Morimoto, A.; Asayama, K.; Sonoda, N.; Miyamatsu, N.; Ohno, Y.; Miyamoto, Y.; Izawa, S.; Ohkubo, T. Fasting Blood Glucose Predicts Incidence of Hypertension Independent of HbA1c Levels and Insulin Resistance in Middle-Aged Japanese: The Saku Study. Am. J. Hypertens. 2019, 32, 1178–1185. [Google Scholar] [CrossRef]
- Vranckx, P.; White, H.D.; Huang, Z.; Mahaffey, K.W.; Armstrong, P.W.; Van de Werf, F.; Moliterno, D.J.; Wallentin, L.; Held, C.; Aylward, P.E.; et al. Validation of BARC Bleeding Criteria in Patients With Acute Coronary Syndromes. J. Am. Coll. Cardiol. 2016, 67, 2135–2144. [Google Scholar] [CrossRef]
- Pan, S.; Lai, H.; Shen, Y.; Breeze, C.; Beck, S.; Hong, T.; Wang, C.; Teschendorff, A.E. DNA Methylome Analysis Reveals Distinct Epigenetic Patterns of Ascending Aortic Dissection and Bicuspid Aortic Valve. Cardiovasc. Res. 2017, 113, 692–704. [Google Scholar] [CrossRef]
- Bhanu, C.; Nimmons, D.; Petersen, I.; Orlu, M.; Davis, D.; Hussain, H.; Magammanage, S.; Walters, K. Drug-Induced Orthostatic Hypotension: A Systematic Review and Meta-Analysis of Randomised Controlled Trials. PLoS Med. 2021, 18, e10038212021. [Google Scholar] [CrossRef]
- Rivasi, G.; Rafanelli, M.; Mossello, E.; Brignole, M.; Ungar, A. Drug-Related Orthostatic Hypotension: Beyond Anti-Hypertensive Medications. Drugs Aging 2020, 37, 725–738. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, S.; Zhao, J.; Wu, K.; Xu, B.; Wang, W. Increased Plasma Renin by Vasodilators Promotes the Progression of Abdominal Aortic Aneurysm. Front. Pharmacol. 2023, 14, 1174278. [Google Scholar] [CrossRef]
- Armario, P.; del Rey, R.H.; Pardell, H. Adverse Effects of Direct-Acting Vasodilators. Drug Saf. 1994, 11, 80–85. [Google Scholar] [CrossRef]
- Tropsha, A.; Isayev, O.; Varnek, A.; Schneider, G.; Cherkasov, A. Integrating QSAR Modelling and Deep Learning in Drug Discovery: The Emergence of Deep QSAR. Nat. Rev. Drug Discov. 2024, 23, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Srour, A.M.; Dawood, D.H.; Saleh, D.O. Synthesis, 3D-Pharmacophore Modelling and 2D-QSAR Study of New Pyridine-3-Carbonitriles as Vasorelaxant Active Agents. New J. Chem. 2021, 45, 7731–7740. [Google Scholar] [CrossRef]
- Tsopka, I.-C.; Hadjipavlou-Litina, D. QSAR Studies of Nitric Oxide Synthase Inhibitors. Curr. Top. Med. Chem. 2023, 23, 2723–2734. [Google Scholar] [CrossRef]
- Sherikar, A.S.; Bhatia, M.S.; Dhavale, R.P. Identification and Investigation of Chalcone Derivatives as Calcium Channel Blockers: Pharmacophore Modeling, Docking Studies, In Vitro Screening, and 3D-QSAR Analysis. Curr. Comput. Aided Drug Des. 2021, 17, 676–686. [Google Scholar] [CrossRef]
- Luna-Vázquez, F.J.; Ibarra-Alvarado, C.; Camacho-Corona, M.d.R.; Rojas-Molina, A.; Rojas-Molina, J.I.; García, A.; Bah, M. Vasodilator Activity of Compounds Isolated from Plants Used in Mexican Traditional Medicine. Molecules 2018, 23, 1474. [Google Scholar] [CrossRef]
- Alagarsamy, V.; Chitra, K.; Saravanan, G.; Solomon, V.R.; Sulthana, M.; Narendhar, B. An Overview of Quinazolines: Pharmacological Significance and Recent Developments. Eur. J. Med. Chem. 2018, 151, 628–685. [Google Scholar] [CrossRef] [PubMed]
- Cañizares-Carmenate, Y.; Mena-Ulecia, K.; Perera-Sardiña, Y.; Torrens, F.; Castillo-Garit, J.A. An Approach to Identify New Antihypertensive Agents Using Thermolysin as Model: In Silico Study Based on QSARINS and Docking. Arab. J. Chem. 2019, 12, 4861–4877. [Google Scholar] [CrossRef]
- You, H.; Su, X.; Su, G. Novel Thiazole–Pyrazolone Hybrids as Potent ACE Inhibitors and Their Cardioprotective Effect on Isoproterenol-induced Myocardial Infarction. Arch. Pharm. 2020, 353, 140. [Google Scholar] [CrossRef] [PubMed]
- Soyka, R.; Guth, B.D.; Weisenberger, H.M.; Luger, P.; Müller, T.H. Guanidine Derivatives as Combined Thromboxane A2 Receptor Antagonists and Synthase Inhibitors. J. Med. Chem. 1999, 42, 1235–1249. [Google Scholar] [CrossRef]
- Isert, C.; Kromann, J.C.; Stiefl, N.; Schneider, G.; Lewis, R.A. Machine Learning for Fast, Quantum Mechanics-Based Approximation of Drug Lipophilicity. ACS Omega 2023, 8, 2046–2056. [Google Scholar] [CrossRef]
- Priya, S.; Tripathi, G.; Singh, D.B.; Jain, P.; Kumar, A. Machine Learning Approaches and Their Applications in Drug Discovery and Design. Chem. Biol. Drug Des. 2022, 100, 136–153. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Fatima, A.; Singh, M.; Verma, I.; Khanum, G.; Muthu, S.; Althubeiti, K.; Abualnaja, K.M.; Ahmad, M.; Siddiqui, N.; et al. Experimental Spectroscopic, Quantum Computational, Hirshfeld Surface, Molecular Docking, and Electronic Excitation Studies on an Antibiotic Agent: SDZ. Polycycl. Aromat. Compd. 2023, 43, 3122–3146. [Google Scholar] [CrossRef]
- Fatima, A.; Singh, M.; Singh, N.; Savita, S.; Verma, I.; Siddiqui, N.; Javed, S. Investigations on Experimental, Theoretical Spectroscopic, Electronic Excitations, Molecular Docking of Sulfaguanidine (SG): An Antibiotic Drug. Chem. Phys. Lett. 2021, 783, 139049. [Google Scholar] [CrossRef]
- Bureš, F. Fundamental Aspects of Property Tuning in Push–Pull Molecules. RSC Adv. 2014, 4, 58826–58851. [Google Scholar] [CrossRef]
- Panja, S.K.; Dwivedi, N.; Saha, S. Tuning the Intramolecular Charge Transfer (ICT) Process in Push–Pull Systems: Effect of Nitro Groups. RSC Adv. 2016, 6, 105786–105794. [Google Scholar] [CrossRef]
- Verbitskiy, E.V.; le Poul, P.; Bureš, F.; Achelle, S.; Barsella, A.; Kvashnin, Y.A.; Rusinov, G.L.; Charushin, V.N. Push–Pull Derivatives Based on 2,4′-Biphenylene Linker with Quinoxaline, [1,2,5]Oxadiazolo [3,4-B]Pyrazine and [1,2,5]Thiadiazolo [3,4-B]Pyrazine Electron Withdrawing Parts. Molecules 2022, 27, 4250. [Google Scholar] [CrossRef]
- Ali, D.C.; Naveed, M.; Gordon, A.; Majeed, F.; Saeed, M.; Ogbuke, M.I.; Atif, M.; Zubair, H.M.; Changxing, L. β-Adrenergic Receptor, an Essential Target in Cardiovascular Diseases. Heart Fail. Rev. 2020, 25, 343–354. [Google Scholar] [CrossRef]
- Boyce, T.G.; Ballone, N.T.; Certa, K.M.; Becker, M.A. The Use of β-Adrenergic Receptor Antagonists in Psychiatry: A Review. J. Acad. Consult. Liaison Psychiatry 2021, 62, 404–412. [Google Scholar] [CrossRef]
- Archer, C.; Wiles, N.; Kessler, D.; Turner, K.; Caldwell, D.M. Beta-Blockers for the Treatment of Anxiety Disorders: A Systematic Review and Meta-Analysis. J. Affect. Disord. 2024, 368, 90–99. [Google Scholar] [CrossRef]
- Archer, C.; Kessler, D.; Wiles, N.; A Chew-Graham, C.; Turner, K. Exploring GPs’ Views on Beta-Blocker Prescribing for People with Anxiety Disorders: A Qualitative Study. Br. J. General. Pract. 2024, 74, e735–e741. [Google Scholar] [CrossRef]
- Chrysant, S.G.; Chrysant, G.S. Antihypertensive and Cardioprotective Effects of Three Generations of Beta-Adrenergic Blockers: An Historical Perspective. Hosp. Pract. 2022, 50, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Almudayni, H.K.; Alhowaish, R.K.; Alotaibi, B.M.; Alshehri, A.M.; Alqahtani, A.M.; Tmraz, S.F.; Alotaibi, S.M. An Overview on Hyperthyroidism, Evaluation and Management Approach in Primary Health Care Centre. Arch. Pharm. Pract. 2021, 12, 134–139. [Google Scholar] [CrossRef]
- Izzo, J.L.; Weir, M.R. Angiotensin-Converting Enzyme Inhibitors. J. Clin. Hypertens. 2011, 13, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, P. From Density Functional Theory to Conceptual Density Functional Theory and Biosystems. Pharmaceuticals 2022, 15, 1112. [Google Scholar] [CrossRef]
- Zanni, R.; Galvez-Llompart, M.; García-Domenech, R.; Galvez, J. Latest Advances in Molecular Topology Applications for Drug Discovery. Expert. Opin. Drug Discov. 2015, 10, 945–957. [Google Scholar] [CrossRef]
- Zanni, R.; Galvez-Llompart, M.; Garcia-Domenech, R.; Galvez, J. What Place Does Molecular Topology Have in Today’s Drug Discovery? Expert. Opin. Drug Discov. 2020, 15, 1133–1144. [Google Scholar] [CrossRef]
- Yoshimori, A. Prediction of Molecular Properties Using Molecular Topographic Map. Molecules 2021, 26, 4475. [Google Scholar] [CrossRef]
- Pinzi, L.; Bisi, N.; Rastelli, G. How Drug Repurposing Can Advance Drug Discovery: Challenges and Opportunities. Front. Drug Discov. 2024, 4, 1460100. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Agnihotry, S.; Pathak, R.K.; Srivastav, A.; Shukla, P.K.; Gautam, B. Molecular Docking and Structure-Based Drug Design. In Computer-Aided Drug Design; Springer: Singapore, 2020; pp. 115–131. [Google Scholar]
- Smith, A.J.T.; Zhang, X.; Leach, A.G.; Houk, K.N. Beyond Picomolar Affinities: Quantitative Aspects of Noncovalent and Covalent Binding of Drugs to Proteins. J. Med. Chem. 2009, 52, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Natesh, R.; Schwager, S.L.U.; Evans, H.R.; Sturrock, E.D.; Acharya, K.R. Structural Details on the Binding of Antihypertensive Drugs Captopril and Enalaprilat to Human Testicular Angiotensin I-Converting Enzyme. Biochemistry 2004, 43, 8718–8724. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; A Shoemaker, B.; A Thiessen, P.; Yu, B.; et al. PubChem 2023 Update. Nucleic Acids Res 2023, 51, D1373–D1380. [Google Scholar] [CrossRef] [PubMed]
- Pence, H.E.; Williams, A. ChemSpider: An Online Chemical Information Resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. ChemInf. 2012, 4, 17. [Google Scholar] [CrossRef]
- Márquez, E.; Mora, J.R.; Flores-Morales, V.; Insuasty, D.; Calle, L. Modeling the Antileukemia Activity of Ellipticine-Related Compounds: QSAR and Molecular Docking Study. Molecules 2020, 25, 24. [Google Scholar] [CrossRef]
- Anchique, L.; Alcázar, J.J.; Ramos-Hernandez, A.; Méndez-López, M.; Mora, J.R.; Rangel, N.; Paz, J.L.; Márquez, E. Predicting the Adsorption of Amoxicillin and Ibuprofen on Chitosan and Graphene Oxide Materials: A Density Functional Theory Study. Polymers 2021, 13, 1620. [Google Scholar] [CrossRef]
- García-Jacas, C.R.; Marrero-Ponce, Y.; Vivas-Reyes, R.; Suárez-Lezcano, J.; Martinez-Rios, F.; Terán, J.E.; Aguilera-Mendoza, L. Distributed and Multicore QuBiLS-MIDAS Software v2.0: Computing Chiral, Fuzzy, Weighted and Truncated Geometrical Molecular Descriptors Based on Tensor Algebra. J. Comput. Chem. 2020, 41, 1209–1227. [Google Scholar] [CrossRef]
- González, J.; Ortega, J.; Damas, M.; Martín-Smith, P.; Gan, J.Q. A New Multi-Objective Wrapper Method for Feature Selection—Accuracy and Stability Analysis for BCI. Neurocomputing 2019, 333, 407–418. [Google Scholar] [CrossRef]
- Frank, E.; Hall, M.; Holmes, G.; Kirkby, R.; Pfahringer, B.; Witten, I.H.; Trigg, L. Weka-A Machine Learning Workbench for Data Mining. In Data Mining and Knowledge Discovery Handbook; Springer: Boston, MA, USA, 2009; pp. 1269–1277. [Google Scholar]
- Sahigara, F.; Mansouri, K.; Ballabio, D.; Mauri, A.; Consonni, V.; Todeschini, R. Comparison of Different Approaches to Define the Applicability Domain of QSAR Models. Molecules 2012, 17, 4791–4810. [Google Scholar] [CrossRef]
- Roy, K.; Kar, S.; Ambure, P. On a Simple Approach for Determining Applicability Domain of QSAR Models. Chemom. Intell. Lab. Syst. 2015, 145, 22–29. [Google Scholar] [CrossRef]
- Xu, X.; Kaindl, J.; Clark, M.J.; Hübner, H.; Hirata, K.; Sunahara, R.K.; Gmeiner, P.; Kobilka, B.K.; Liu, X. Binding Pathway Determines Norepinephrine Selectivity for the Human Β1AR over Β2AR. Cell Res. 2021, 31, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved Side-chain Torsion Potentials for the Amber Ff99SB Protein Force Field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- da Silva, A.W.S.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser InterfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving Physical Realism, Stereochemistry, and Side-chain Accuracy in Homology Modeling: Four Approaches That Performed Well in CASP8. Proteins Struct. Funct. Bioinform. 2009, 77, 114–122. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G-Mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Criterion | Cross-Validation | External Validation | ||
|---|---|---|---|---|
| Result | Assessment | Result | Assessment | |
| R2 > 0.6 | 0.803 | PASS | 0.803 | PASS |
| Q2Val > 0.5 | 0.730 | PASS | 0.724 | PASS |
| (Q2Val − R02)/Q2Val < 0.1 | 0.004 | PASS | 0.087 | PASS |
| (Q2Val − R0′2)/Q2Val < 0.1 | 0.077 | PASS | 0.004 | PASS |
| abs(R02 − R0′2) < 0.1 | 0.054 | PASS | 0.060 | PASS |
| 0.85 < k < 1.15 | 0.997 | PASS | 0.996 | PASS |
| 0.85 < km < 1.15 | 0.994 | PASS | 0.999 | PASS |
| Compound | Predicted PKI50 | Compound | Predicted PKI50 |
|---|---|---|---|
| Astemizole | 12.17 | Rimonabant | 11.04 |
| Ozanimod | 11.36 | Idarubicin | 10.96 |
| Fluphenazine | 11.23 | Topotecan | 10.92 |
| Propericiazine | 11.12 | Sarecycline | 10.86 |
| Lasofoxifene | 11.11 | Dasabuvir | 10.81 |
| Methylergometrine | 10.51 | Irbesartan | 10.48 |
| Ursodeoxycholic acid | 10.47 | Tedizolid | 10.38 |
| Oxandrolone | 10.34 | Deoxycholic acid | 10.30 |
| Gemifloxacin | 10.30 | Nalfurafine | 10.23 |
| Quetiapine | 10.23 | Nicergoline | 10.19 |
| Ethynyl estradiol | 10.14 | Niraparib | 10.14 |
| Megestrol | 10.05 | Domperidone | 10.04 |
| Linagliptin | 9.98 | Sertindole | 9.97 |
| Ribociclib | 9.96 | Flupentixol | 9.96 |
| Cholic acid | 9.95 | Lidoflazine | 9.95 |
| Molecule | Angiotensin-I- Converting | β-Adrenergic Receptor | Molecule | Angiotensin-I- Converting | β-Adrenergic Receptor |
|---|---|---|---|---|---|
| Captopril | −6.6 | - | Megestrol | −9.2 | −8.7 |
| Cyclazocine | - | −8.5 | Methylergometrine | −9.2 | −9.1 |
| Astemizole | −9.4 | −9.9 | Nalfurafine | −8.9 | −8.9 |
| Cholic Acid | −9.6 | −8.9 | Nicergoline | −9.6 | −9.8 |
| Dasabuvir | −10.6 | −10.7 | Niraparib | −9.2 | −10.2 |
| Flupentixol | −9.1 | −9.7 | Oxandrolone | −9.0 | −9.5 |
| Fluphenazine | −9.3 | −9.1 | Ozanimod | −9.4 | −9.7 |
| Irbesartan | −9.8 | −9.6 | Propericiazine | −9.3 | −9.3 |
| Lidoflazine | −9.8 | −10.5 | Ribociclib | −9.7 | −9.4 |
| Linagliptin | −10.3 | −9.5 | Rimonabant | −10.1 | −9.5 |
| Idarubicin | −10.5 | −9.7 | Sarecycline | −8.8 | −9.3 |
| Sertindole | −9.7 | −10.2 |
| Compound | van der Waals Energy | Electrostatic Energy | SASA Energy | Binding Energy |
|---|---|---|---|---|
| Noradrenaline | −22.91 | −19.31 | −2.66 | −12.07 |
| Betaxolol | −47.54 | −12.72 | −5.14 | −29.96 |
| Alprenolol | −29.69 | −10.62 | −4.00 | −12.46 |
| Dasabuvir | −53.62 | −14.63 | −5.76 | −27.42 |
| Sertindole | −54.71 | −4.86 | −5.53 | −28.95 |
| Captopril | −94.67 | −59.49 | −12.21 | −25.14 |
| Idarubicin | −36.70 | −9.32 | −4.38 | −15.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernal, A.; Márquez, E.A.; Flores-Sumoza, M.; Cuesta, S.A.; Mora, J.R.; Paz, J.L.; Mendoza-Mendoza, A.; Rodríguez-Macías, J.; Salazar, F.; Insuasty, D.; et al. Molecular Modeling of Vasodilatory Activity: Unveiling Novel Candidates Through Density Functional Theory, QSAR, and Molecular Dynamics. Int. J. Mol. Sci. 2024, 25, 12649. https://doi.org/10.3390/ijms252312649
Bernal A, Márquez EA, Flores-Sumoza M, Cuesta SA, Mora JR, Paz JL, Mendoza-Mendoza A, Rodríguez-Macías J, Salazar F, Insuasty D, et al. Molecular Modeling of Vasodilatory Activity: Unveiling Novel Candidates Through Density Functional Theory, QSAR, and Molecular Dynamics. International Journal of Molecular Sciences. 2024; 25(23):12649. https://doi.org/10.3390/ijms252312649
Chicago/Turabian StyleBernal, Anthony, Edgar A. Márquez, Máryury Flores-Sumoza, Sebastián A. Cuesta, José Ramón Mora, José L. Paz, Adel Mendoza-Mendoza, Juan Rodríguez-Macías, Franklin Salazar, Daniel Insuasty, and et al. 2024. "Molecular Modeling of Vasodilatory Activity: Unveiling Novel Candidates Through Density Functional Theory, QSAR, and Molecular Dynamics" International Journal of Molecular Sciences 25, no. 23: 12649. https://doi.org/10.3390/ijms252312649
APA StyleBernal, A., Márquez, E. A., Flores-Sumoza, M., Cuesta, S. A., Mora, J. R., Paz, J. L., Mendoza-Mendoza, A., Rodríguez-Macías, J., Salazar, F., Insuasty, D., Marrero-Ponce, Y., Agüero-Chapin, G., Flores-Morales, V., & Carrascal-Hernández, D. C. (2024). Molecular Modeling of Vasodilatory Activity: Unveiling Novel Candidates Through Density Functional Theory, QSAR, and Molecular Dynamics. International Journal of Molecular Sciences, 25(23), 12649. https://doi.org/10.3390/ijms252312649