
Advancing Therapeutic and Vaccine Proteins: Switching from Recombinant to Ribosomal Delivery—A Humanitarian Cause



Abstract
:
1. Introduction
2. Recombinant Proteins (RPs)
Manufacturing Setup Cost of Recombinant Proteins
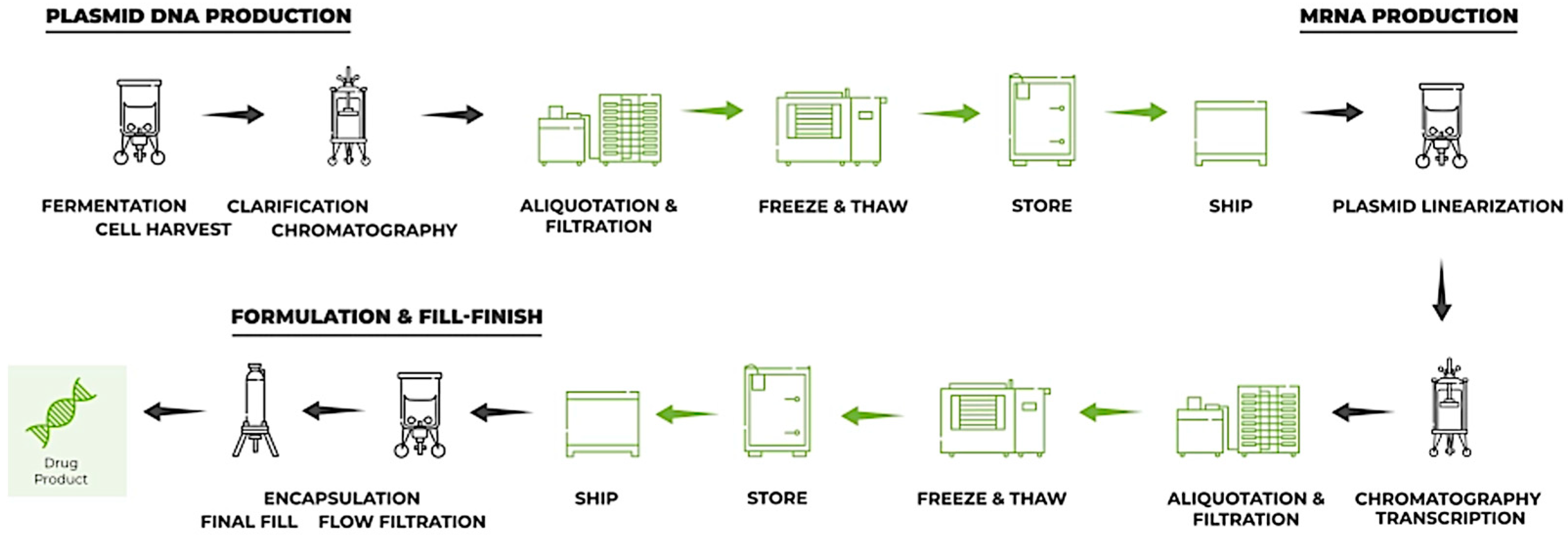
3. Ex Vivo mRNA Proteins (EMP)
4. In Vivo mRNA Proteins (IMPs)
- It is faster and more efficient than RP technology and EMP. This means that development and production can happen more quickly. Complex and time-consuming cloning and expression in host cells are prerequisites for conventional recombinant technologies. On the other hand, IMP technology employs in vivo expression, removing all constraints of structural variability of recombinant proteins, which adds extensive cost to establish a reproducible process.
- mRNAs are smaller than plasmid DNA and never cross the nuclear membrane, staying in the cytoplasm for expression, eliminating genetic manipulation risks.
- They are more adaptable to producing complicated proteins, even ones that defy recombinant expression such as modification to make novel proteins, conjugates such as binding with transferrin protein, or expression of only minor parts of antibodies such as their scFv.
- In the future, the cell-based manufacturing systems used in mRNA and polymerase chain reaction (PCR) will be less susceptible to contamination from endotoxins and adventitious agents, reducing the safety risks.
- A short manufacturing cycle reduces costs significantly, and release testing is much simpler.
- IMP technology provides scalability and productivity benefits. One mRNA molecule can produce hundreds or even thousands of target protein molecules, making it highly efficient and reducing dosing, which brings better safety and lower costs.
4.1. Vaccines
4.2. Manufacturing Setup Costs of mRNA
5. Intellectual Property
6. Talent Access
7. Regulatory
7.1. Biosimilars and Generics
7.2. Copy of Licensed mRNA Product
- Infectious diseases: Moderna is advancing mRNA vaccines for influenza (mRNA-1010), respiratory syncytial virus (RSV) with mRNA-1345, and cytomegalovirus (CMV) with mRNA-1647, all of which are in phase 2 or 3 trials. Other early-phase products target Zika, herpes, Lyme disease, and Nipah virus.
- Cancer: There is significant work in cancer vaccines, such as Moderna’s individualized neoantigen therapies (e.g., mRNA-4157), which target melanoma, non-small cell lung cancer, and renal cell carcinoma. These therapies are based on patient-specific tumor antigens and are in late-stage clinical development.
- Rare diseases: mRNA therapeutics are also being explored for metabolic and rare genetic diseases, including treatments for propionic acidemia (mRNA-3927) and methylmalonic acidemia (mRNA-3705). These therapies aim to replace or enhance deficient enzymes; a novel application for mRNA beyond traditional vaccines.
- Combination vaccines: Moderna and Pfizer are developing combination mRNA vaccines to simultaneously address multiple viruses, such as the flu and COVID-19 vaccine (mRNA-1083), which are progressing through phase 3 trials.
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Requena, J.R. The protean prion protein. PLOS Biol. 2020, 18, e3000754. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F. The Arrangement of Amino Acids in Proteins. In Advances in Protein Chemistry; Anson, M.L., Bailey, K., Edsall, J.T., Eds.; Academic Press: Cambridge, MA, USA, 1952; pp. 1–67. [Google Scholar]
- Pauling, L.; Corey, R.B.; Branson, H.R. The structure of proteins: Two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA 1951, 37, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Kendrew, J.C.; Bodo, G.; Dintzis, H.M.; Parrish, R.G.; Wyckoff, H.; Phillips, D.C. A Three-Dimensional Model of the Myoglobin Molecule Obtained by X-Ray Analysis. Nature 1958, 181, 662–666. [Google Scholar] [CrossRef]
- Chaffey, N. Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K. and Walter, P. Molecular biology of the cell. 4th edn. Ann. Bot. 2003, 91, 401. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Palade, G.E. A small particulate component of the cytoplasm. J. Biophys. Biochem. Cytol. 1955, 1, 59–68. [Google Scholar] [CrossRef]
- Jacob, F.; Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961, 3, 318–356. [Google Scholar] [CrossRef]
- Meselson, M.; Stahl, F.W. The replication of dna in Escherichia coli. Proc. Natl. Acad. Sci. USA 1958, 44, 671–682. [Google Scholar] [CrossRef]
- Nirenberg, M.W.; Matthaei, J.H. The dependence of cell-free protein synthesis in E. coli upon naturally occurring or synthetic polyribonucleotides. Proc. Natl. Acad. Sci. USA 1961, 47, 1588–1602. [Google Scholar] [CrossRef]
- Rathore, A.S.; Gardner, P.J.; Chhabra, H.; Raman, R. Global outlook on the affordability of biotherapeutic drugs. Ann. N. Y. Acad. Sci. 2024, 1537, 168–178. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, M.; Lin, S.; Jian, R.; Li, X.; Chan, J.; Dong, G.; Fang, H.; Robinson, A.E.; Snyder, M.P.; et al. A Quantitative Proteome Map of the Human Body. Cell 2020, 183, 269–283.e19. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, I.; Tornali, C.; Bragazzi, N.L.; Martini, M. The Discovery of Insulin: An Important Milestone in the History of Medicine. Front. Endocrinol. 2018, 9, 613. [Google Scholar] [CrossRef]
- Ayyar, V.S. History of growth hormone therapy. Indian. J. Endocrinol. Metab. 2011, 15 (Suppl. S3), S162–S165. [Google Scholar] [CrossRef]
- Mannucci, P.M.; Tuddenham, E.G. The hemophilias--from royal genes to gene therapy. N. Engl. J. Med. 2001, 344, 1773–1779. [Google Scholar] [CrossRef] [PubMed]
- Greenwalt, T.J. A short history of transfusion medicine. Transfusion 1997, 37, 550–563. [Google Scholar] [CrossRef]
- Bergman, S.J.; Ferguson, M.C.; Santanello, C. Interferons as therapeutic agents for infectious diseases. Infect. Dis. Clin. N. Am. 2011, 25, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Leão Rde, B.; Esteves, S.C. Gonadotropin therapy in assisted reproduction: An evolutionary perspective from biologics to biotech. Clinics 2014, 69, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Szmuness, W.; Stevens, C.E.; Harley, E.J.; Zang, E.A.; Oleszko, W.R.; William, D.C.; Sadovsky, R.; Morrison, J.M.; Kellner, A. Hepatitis B vaccine: Demonstration of efficacy in a controlled clinical trial in a high-risk population in the United States. N. Engl. J. Med. 1980, 303, 833–841. [Google Scholar] [CrossRef]
- Berg, P.; Mertz, J.E. Personal reflections on the origins and emergence of recombinant DNA technology. Genetics 2010, 184, 9–17. [Google Scholar] [CrossRef]
- Walsh, G. Biopharmaceutical benchmarks 2010. Nat. Biotechnol. 2010, 28, 917–924. [Google Scholar] [CrossRef]
- Jackson, D.A.; Symons, R.H.; Berg, P. Biochemical method for inserting new genetic information into DNA of Simian Virus 40: Circular SV40 DNA molecules containing lambda phage genes and the galactose operon of Escherichia coli. Proc. Natl. Acad. Sci. USA 1972, 69, 2904–2909. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.N.; Chang, A.C.Y.; Boyer, H.W.; Helling, R.B. Construction of biologically functional bacterial plasmids in vitro. Proc. Natl. Acad. Sci. USA 1973, 70, 3240–3244. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.S. Making dollars out of DNA: The first major patent in biotechnology and the commercialization of molecular biology, 1974–1980. Isis 2001, 92, 541–575. [Google Scholar] [CrossRef] [PubMed]
- Quianzon, C.C.; Cheikh, I. History of insulin. J. Community Hosp. Intern. Med. Perspect. 2012, 2, 18701. [Google Scholar] [CrossRef]
- Niazi, S.K. Biosimilars Adoption: Recognizing and Removing the RoadBlocks. Clin. Outcomes Res. 2023, 15, 281–294. [Google Scholar] [CrossRef]
- Niazi, S.K. Biosimilars: Harmonizing the Approval Guidelines. Biologics 2022, 2, 171–195. [Google Scholar] [CrossRef]
- Niazi, S.K. Biosimilars: A futuristic fast-to-market advice to developers. Expert Opin. Biol. Ther. 2022, 22, 149–155. [Google Scholar] [CrossRef]
- Morin, S.; Segafredo, G.; Piccolis, M.; Das, A.; Das, M.; Loffredi, N.; Larbi, A.; Mwamelo, K.; Villanueva, E.; Nobre, S.; et al. Expanding access to biotherapeutics in low-income and middle-income countries through public health non-exclusive voluntary intellectual property licensing: Considerations, requirements, and opportunities. Lancet Glob. Health 2023, 11, e145–e154. [Google Scholar] [CrossRef]
- WHO. Roadmap for Access to Medicines, Vaccines, and Health Product 2019–2023: Comprehensive Support for Access to Medicines, Vaccines, and Other Health Products. Available online: https://www.who.int/publications/i/item/9789241517034 (accessed on 1 October 2024).
- Geng, S.L.; Zhao, X.J.; Zhang, X.; Zhang, J.H.; Mi, C.L.; Wang, T.Y. Recombinant therapeutic proteins degradation and overcoming strategies in CHO cells. Appl. Microbiol. Biotechnol. 2024, 108, 182. [Google Scholar] [CrossRef]
- Markets, R. Recombinant Proteins Market. Available online: https://www.researchandmarkets.com/report/protein-drugs#tag-pos-12024 (accessed on 1 October 2024).
- Purdue University. Clinical Drug Experience Knowledgebase: FDA Approvals|Recombinant Protein. 2024. Available online: https://cdek.pharmacy.purdue.edu/fda-approvals/Recombinant%20protein/ (accessed on 1 October 2024).
- Aggarwal, S.R. What’s fueling the biotech engine-2011 to 2012. Nat. Biotechnol. 2012, 30, 1191–1197. [Google Scholar] [CrossRef]
- Sampathkumar, K.; Kerwin, B.A. Roadmap for Drug Product Development and Manufacturing of Biologics. J. Pharm. Sci. 2024, 113, 314–331. [Google Scholar] [CrossRef] [PubMed]
- Vivek Arora, D.K.; Patel, P.; Rajendran, R. Reimagining the Future of Biopharma Manufacturing. Available online: https://www.mckinsey.com/industries/life-sciences/our-insights/reimagining-the-future-of-biopharma-manufacturing. (accessed on 1 October 2024).
- Zhu, M.M.; Mollet, M.; Hubert, R.S.; Kyung, Y.S.; Zhang, G.G. Industrial Production of Therapeutic Proteins: Cell Lines, Cell Culture, and Purification. In Handbook of Industrial Chemistry and Biotechnology; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Harrison, R.G.; Todd, P.W.; Rudge, S.R.; Petrides, D.P. Bioseparations Science and Engineering, 2nd ed.; Oxford University Press: New York, NY, USA, 2015. [Google Scholar]
- Khanal, O.; Lenhoff, A.M. Developments and opportunities in continuous biopharmaceutical manufacturing. mAbs 2021, 13, 1903664. [Google Scholar] [CrossRef] [PubMed]
- FDA. Facts About the Current Good Manufacturing Practice (CGMP). 2024. Available online: https://www.fda.gov/drugs/pharmaceutical-quality-resources/facts-about-current-good-manufacturing-practice-cgmp (accessed on 1 October 2024).
- Müller, D.; Klein, L.; Lemke, J.; Schulze, M.; Kruse, T.; Saballus, M.; Matuszczyk, J.; Kampmann, M.; Zijlstra, G. Process intensification in the biopharma industry: Improving efficiency of protein manufacturing processes from development to production scale using synergistic approaches. Chem. Eng. Process.—Process Intensif. 2022, 171, 108727. [Google Scholar] [CrossRef]
- Mckinsey. R&D Biosimilars. 2022. Available online: https://www.mckinsey.com/industries/life-sciences/our-insights/three-imperatives-for-r-and-d-in-biosimilars (accessed on 1 October 2024).
- Forster, A.C.; Church, G.M. Towards synthesis of a minimal cell. Mol. Syst. Biol. 2006, 2, 45. [Google Scholar] [CrossRef] [PubMed]
- Katzen, F.; Chang, G.; Kudlicki, W. The past, present and future of cell-free protein synthesis. Trends Biotechnol. 2005, 23, 150–156. [Google Scholar] [CrossRef]
- Shimizu, Y.; Kanamori, T.; Ueda, T. Protein synthesis by pure translation systems. Methods 2005, 36, 299–304. [Google Scholar] [CrossRef]
- Hodgman, C.E.; Jewett, M.C. Cell-free synthetic biology: Thinking outside the cell. Metab. Eng. 2012, 14, 261–269. [Google Scholar] [CrossRef]
- Caschera, F.; Noireaux, V. Synthesis of 2.3 mg/mL of protein with an all Escherichia coli cell-free transcription–translation system. Biochimie 2014, 99, 162–168. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Niazi, S.K. Making COVID-19 mRNA vaccines accessible: Challenges resolved. Expert Rev. Vaccines 2022, 21, 1163–1176. [Google Scholar] [CrossRef]
- Niazi, S.K.; Magoola, M. mRNA and Synthesis-Based Therapeutic Proteins: A Non-Recombinant Affordable Option. Biologics 2023, 3, 355–379. [Google Scholar] [CrossRef]
- Niazi, S.K. RNA Therapeutics: A Healthcare Paradigm Shift. Biomedicines 2023, 11, 1275. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- FDA. Vaccines Licensed for Use in the United States. 2024. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/vaccines-licensed-use-united-states (accessed on 1 October 2024).
- Niazi, S.K. Anti-Idiotypic mRNA Vaccine to Treat Autoimmune Disorders. Vaccines 2024, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- CYTIVA. Get a Better Grip on mRNA Therapeutics. 2024. Available online: https://www.cytivalifesciences.com/en/us/solutions/bioprocessing/products-and-solutions/mrna-manufacturing?srsltid=AfmBOoqjJZoY1pM9_qGTBHIR8fFyuDI6T_3EqZ-APJfLkaPrnOiSNLAS (accessed on 1 October 2024).
- Omojuyigbe, J.O.; Ade-Adekunle, O.A.; Atobatele, I.R.; Adekunle, F.O. How the African vaccine manufacturing accelerator can assist in strengthening Africa’s response to global health challenges. Vaccine X 2024, 19, 100499. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A. The mRNA vaccine revolution is the dividend from decades of basic science research. J. Clin. Investig. 2021, 131, e153721. [Google Scholar] [CrossRef]
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J.B.; Yu, D. mRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol. Ther. 2019, 27, 757–772. [Google Scholar] [CrossRef]
- Fang, E.; Liu, X.; Li, M.; Zhang, Z.; Song, L.; Zhu, B.; Wu, X.; Liu, J.; Zhao, D.; Li, Y. Advances in COVID-19 mRNA vaccine development. Signal Transduct. Target. Ther. 2022, 7, 94. [Google Scholar] [CrossRef]
- Schlake, T.; Thess, A.; Fotin-Mleczek, M.; Kallen, K.J. Developing mRNA-vaccine technologies. RNA Biol. 2012, 9, 1319–1330. [Google Scholar] [CrossRef]
- Whitley, J.; Zwolinski, C.; Denis, C.; Maughan, M.; Hayles, L.; Clarke, D.; Snare, M.; Liao, H.; Chiou, S.; Marmura, T.; et al. Development of mRNA manufacturing for vaccines and therapeutics: mRNA platform requirements and development of a scalable production process to support early phase clinical trials. Transl. Res. 2022, 242, 38–55. [Google Scholar] [CrossRef]
- Kohli, M.A.; Maschio, M.; Joshi, K.; Lee, A.; Fust, K.; Beck, E.; Van de Velde, N.; Weinstein, M.C. The potential clinical impact and cost-effectiveness of the updated COVID-19 mRNA fall 2023 vaccines in the United States. J. Med. Econ. 2023, 26, 1532–1545. [Google Scholar] [CrossRef] [PubMed]
- Rosa, S.S.; Prazeres, D.M.F.; Azevedo, A.M.; Marques, M.P.C. mRNA vaccines manufacturing: Challenges and bottlenecks. Vaccine 2021, 39, 2190–2200. [Google Scholar] [CrossRef]
- Citizen. How to Make Enough Vaccine for the World in One Year. 2022. Available online: https://www.citizen.org/article/how-to-make-enough-vaccine-for-the-world-in-one-year/ (accessed on 1 October 2024).
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Precision. RNA-LNPs: Navigating the Regulatory Challenges. 2023. Available online: https://www.pharmaceutical-technology.com/sponsored/rna-lnps-navigating-the-regulatory-challenges/ (accessed on 1 October 2024).
- IPWATCHDOG. The mRNA Patent and Competitive Landscape: Pioneers, Litigation Outlook and Big Pharma’s Next Moves. 2021. Available online: https://ipwatchdog.com/2021/04/30/mrna-patent-competitive-landscape-pioneers-litigation-outlook-big-pharmas-next-moves-part-iii/id=132936/ (accessed on 1 October 2024).
- FBRICE. Lipid Nanoparticles for RNA Vaccines—Even More Layers of Patents. 2023. Available online: https://www.fbrice.com.au/ip-news-insights/beyond-the-surface-delving-into-even-more-patent-layers-for-rna-vaccine-lipid-nanoparticles (accessed on 1 October 2024).
- THIRD WORLD ECONOMICS. Exemption from Pharmaceutical Patents Agreed for LDCs. 2015. Available online: https://www.twn.my/title2/twe/2015/603/8.htm (accessed on 1 October 2024).
- UNCTAD. Least Developed Countries. 2024. Available online: https://unctad.org/topic/least-developed-countries/list (accessed on 1 October 2024).
- World Trade Organization. WTO Members Agree to Extend TRIPS Transition Period for LDCs Until 1 July 2034. 2021. Available online: https://www.wto.org/english/news_e/news21_e/trip_30jun21_e.htm (accessed on 1 October 2024).
- Naik, R.; Peden, K. Regulatory Considerations on the Development of mRNA Vaccines. Curr. Top. Microbiol. Immunol. 2022, 440, 187–205. [Google Scholar]
- ICH. CH Q6A Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances—Scientific Guideline. Available online: https://www.ema.europa.eu/en/ich-q6a-specifications-test-procedures-acceptance-criteria-new-drug-substances-new-drug-products-chemical-substances-scientific-guideline (accessed on 1 October 2024).
- Guerriaud, M.; Kohli, E. RNA-based drugs and regulation: Toward a necessary evolution of the definitions issued from the European union legislation. Front. Med. 2022, 9, 1012497. [Google Scholar] [CrossRef] [PubMed]
- FDA. Guidance for Human Somatic Cell Therapy and Gene Therapy. Available online: https://www.fda.gov/regulatoryinformation/ (accessed on 1 October 2024).
- FDA. Biosimilars Product Information. 2024. Available online: https://www.fda.gov/drugs/biosimilars/biosimilar-product-information (accessed on 1 October 2024).
- McKinsey. An Inflection Point for Biosimilars. 2021. Available online: https://www.mckinsey.com/industries/life-sciences/our-insights/an-inflection-point-for-biosimilars (accessed on 1 October 2024).
- Wang, Y.-S.; Kumari, M.; Chen, G.-H.; Hong, M.-H.; Yuan, J.P.-Y.; Tsai, J.-L.; Wu, H.C. mRNA-based vaccines and therapeutics: An in-depth survey of current and upcoming clinical applications. J. Biomed. Sci. 2023, 30, 84. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.K. The FDA’s New Guideline “Generally Accepted Scientific Knowledge” (GASK): An Opportunity to Expedite the Approval of Biosimilars. Pharmaceuticals 2023, 16, 1517. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Therapeutic RP | Brand Example | Application | Mechanism/Function |
|---|---|---|---|
| Adalimumab | Humira | Rheumatoid arthritis, psoriasis | TNF-alpha inhibitor |
| Aflibercept | Eylea | Age-related macular degeneration | VEGF inhibitor |
| Albutrepenonacog Alfa | Idelvion | Hemophilia B | Recombinant factor IX |
| Aldesleukin | Proleukin | Metastatic renal cell carcinoma | Interleukin-2 analog |
| Alglucosidase Alfa | Myozyme, Lumizyme | Pompe disease | Enzyme replacement therapy |
| Alirocumab | Praluent | Hypercholesterolemia | PCSK9 inhibitor |
| Anakinra | Kineret | Rheumatoid arthritis | IL-1 receptor antagonist |
| Asparaginase | Elspar | Acute lymphoblastic leukemia | An enzyme that depletes asparagine |
| Atezolizumab | Tecentriq | Various cancers | PD-L1 inhibitor |
| Avalglucosidase Alfa | Nexviazyme | Pompe disease | Enzyme replacement therapy |
| Avatrombopag | Doptelet | Thrombocytopenia | Thrombopoietin receptor agonist |
| Belatacept | Nulojix | Organ transplant rejection | Selective T cell co-stimulation blocker |
| Belimumab | Benlysta | Systemic lupus erythematosus | BAFF inhibitor |
| Bevacizumab | Avastin | Various cancers (e.g., colorectal, lung) | VEGF inhibitor |
| Blinatumomab | Blincyto | Acute lymphoblastic leukemia | BiTE targeting CD19 and CD3 |
| Blisibimod | A-623 | Systemic lupus erythematosus | BAFF inhibitor |
| Bremelanotide | Vyleesi | Hypoactive sexual desire disorder | Melanocortin receptor agonist |
| Brodalumab | Siliq | Psoriasis | IL-17 receptor antagonist |
| Burosumab | Crysvita | X-linked hypophosphatemia | FGF23 inhibitor |
| Canakinumab | Ilaris | Periodic fever syndromes | IL-1 beta inhibitor |
| Caplacizumab | Cablivi | Thrombotic thrombocytopenic purpura | von Willebrand factor inhibitor |
| Cenegermin | Oxervate | Neurotrophic keratitis | Recombinant human nerve growth factor |
| Crizanlizumab | Adakveo | Sickle cell disease | P-selectin inhibitor |
| Darbepoetin Alfa | Aranesp | Anemia (chronic kidney disease) | Stimulates red blood cell production |
| Denosumab | Prolia, Xgeva | Osteoporosis, bone cancer | RANKL inhibitor |
| Dornase Alfa | Pulmozyme | Cystic fibrosis | Mucolytic agent |
| Dostarlimab | Jemperli | Mismatch repair-deficient cancers | PD-1 inhibitor |
| Dupilumab | Dupixent | Atopic dermatitis, asthma | IL-4 receptor alpha antagonist |
| Durvalumab | Imfinzi | Lung cancer | PD-L1 inhibitor |
| Eculizumab | Soliris | Paroxysmal nocturnal hemoglobinuria | Complement inhibitor |
| Efgartigimod | Vyvgart | Myasthenia gravis | FcRn antagonist |
| Eltrombopag | Promacta | Thrombocytopenia | Thrombopoietin receptor agonist |
| Emicizumab | Hemlibra | Hemophilia A | Bispecific factor IXa- and X-directed antibody |
| Erenumab | Aimovig | Migraine prevention | CGRP receptor antagonist |
| Erythropoietin | Epogen, Procrit | Anemia (chronic kidney disease) | Stimulates red blood cell production |
| Etanercept | Enbrel | Autoimmune diseases | TNF receptor fusion protein |
| Etrolizumab | In development | Ulcerative colitis | Anti-beta7 integrin antibody |
| Evolocumab | Repatha | Hypercholesterolemia | PCSK9 inhibitor |
| Factor IX | BeneFIX, Idelvion | Hemophilia B | Clotting factor replacement |
| Factor VIII | Advate, Eloctate | Hemophilia A | Clotting factor replacement |
| Filgrastim | Neupogen | Neutropenia | Stimulates white blood cell growth |
| Fremanezumab | Ajovy | Migraine prevention | CGRP inhibitor |
| Galcanezumab | Emgality | Migraine prevention | CGRP inhibitor |
| Givosiran | Givlaari | Acute hepatic porphyria | RNA interference (RNAi) agent targeting ALAS1 |
| Golimumab | Simponi | Rheumatoid arthritis, psoriatic arthritis | TNF-alpha inhibitor |
| Ibalizumab-uiyk | Trogarzo | HIV-1 infection | CD4-directed post-attachment inhibitor |
| Idursulfase | Elaprase | Hunter syndrome | Enzyme replacement therapy |
| Imiglucerase | Cerezyme | Gaucher disease | Enzyme replacement therapy |
| Imlifidase | Idefirix | Desensitization in kidney transplantation | IgG-degrading enzyme |
| Inclisiran | Leqvio | Hypercholesterolemia | siRNA targeting PCSK9 |
| Infliximab | Remicade | Crohn’s disease, rheumatoid arthritis | TNF-alpha inhibitor |
| Insulin | Humulin, Novolin | Diabetes | Blood sugar regulation |
| Interferon Beta-1a | Avonex, Rebif | Multiple sclerosis | Immune modulation |
| Ipilimumab | Yervoy | Melanoma | CTLA-4 inhibitor |
| Lanadelumab | Takhzyro | Hereditary angioedema | Kallikrein inhibitor |
| Lanreotide | Somatuline Depot | Acromegaly | Somatostatin analog |
| Laronidase | Aldurazyme | Mucopolysaccharidosis I | Enzyme replacement therapy |
| Leronlimab | In development | HIV, cancer | CCR5 antagonist |
| Luspatercept | Reblozyl | Anemia in beta-thalassemia | Erythroid maturation agent |
| Maribavir | Livtencity | CMV infection post-transplant | UL97 protein kinase inhibitor |
| Natalizumab | Tysabri | Multiple sclerosis, Crohn’s disease | Integrin inhibitor |
| Niraparib | Zejula | Ovarian cancer | PARP inhibitor |
| Nivolumab | Opdivo | Melanoma, lung cancer | PD-1 inhibitor |
| Ocrelizumab | Ocrevus | Multiple sclerosis | Anti-CD20 monoclonal antibody |
| Olaparib | Lynparza | BRCA-mutated cancers | PARP inhibitor |
| Omalizumab | Xolair | Asthma | IgE inhibitor |
| Palivizumab | Synagis | Respiratory syncytial virus (RSV) | RSV-specific monoclonal antibody |
| Pegloticase | Krystexxa | Chronic gout | Uric acid breakdown enzyme |
| Pegzilarginase | AEB1102 | Arginase deficiency | Recombinant human arginase |
| Pembrolizumab | Keytruda | Melanoma, lung cancer | PD-1 inhibitor |
| Plerixafor | Mozobil | Stem cell mobilization | CXCR4 antagonist |
| Ranibizumab | Lucentis | Diabetic macular edema | VEGF inhibitor |
| Raxibacumab | Abthrax | Anthrax infection | Anthrax toxin inhibitor |
| Rilonacept | Arcalyst | Cryopyrin-associated periodic syndromes | IL-1 inhibitor |
| Risankizumab | Skyrizi | Psoriasis | IL-23 inhibitor |
| Risdiplam | Evrysdi | Spinal muscular atrophy | SMN2 splicing modifier |
| Rituximab | Rituxan | Non-Hodgkin’s lymphoma | CD20-targeted antibody |
| Romiplostim | Nplate | Thrombocytopenia | Thrombopoietin receptor agonist |
| Romosozumab | Evenity | Osteoporosis | Sclerostin inhibitor |
| Satralizumab | Enspryng | Neuromyelitis optica | IL-6 receptor antagonist |
| Secukinumab | Cosentyx | Psoriasis, ankylosing spondylitis | IL-17A inhibitor |
| Sutimlimab | Enjaymo | Cold agglutinin disease | Complement C1s inhibitor |
| Teplizumab | Tzield | Type 1 diabetes (delay onset) | Anti-CD3 antibody |
| Teprotumumab | Tepezza | Thyroid eye disease | IGF-1 receptor antagonist |
| Tildrakizumab | Ilumya | Psoriasis | IL-23 inhibitor |
| Tocilizumab | Actemra | Rheumatoid arthritis | IL-6 receptor antagonist |
| Tralokinumab | Adtralza | Atopic dermatitis | IL-13 inhibitor |
| Trastuzumab | Herceptin | HER2-positive breast cancer | Targets HER2 receptor |
| Ustekinumab | Stelara | Psoriasis, Crohn’s disease | IL-12 and IL-23 inhibitor |
| Vedolizumab | Entyvio | Ulcerative colitis, Crohn’s disease | Integrin antagonist |
| Vibecotolimab | In development | Cancer immunotherapy | TIM-3 inhibitor |
| Vilobelimab | Vilova | Sepsis | C5a inhibitor |
| Vaccine | Brand Example | Target Protein | Prevention of |
|---|---|---|---|
| Hepatitis B | Engerix-B, Recombivax HB | Hepatitis B surface antigen (HBsAg) | Hepatitis B virus infection |
| Human Papillomavirus (HPV) | Gardasil, Cervarix | L1 protein from various HPV types (e.g., 6, 11, 16, and 18) | HPV-related cancers and genital warts |
| Influenza (Flu) | Flublok Quadrivalent | Hemagglutinin protein from multiple influenza strains | Seasonal influenza |
| SARS-CoV-2 (COVID-19) | NVX-CoV2373 (Novavax) | Spike (S) protein of SARS-CoV-2 | COVID-19 |
| Meningococcal B | Trumenba, Bexsero | Factor H binding protein (fHbp), NHBA, and others | Neisseria meningitidis serogroup B infection |
| Malaria | Mosquirix (RTS, S) | Circumsporozoite protein (CSP) of Plasmodium falciparum | Malaria in children |
| Lyme Disease | LYMErix (discontinued) | Outer surface protein A (OspA) of Borrelia burgdorferi | Lyme disease (was discontinued) |
| Rabies | Rabivax-S | Glycoprotein G of rabies virus | Rabies, typically for post-exposure prophylaxis |
| Ebola Virus | Ervebo (primarily viral vector) | Glycoprotein from the Ebola virus | Ebola virus disease |
| Herpes Zoster (Shingles) | Shingrix | Glycoprotein E of the varicella-zoster virus | Shingles (herpes zoster) in older adults |
| Dengue Virus | Dengvaxia | Envelope and membrane proteins from multiple dengue virus serotypes | Dengue virus in endemic areas |
| Pertussis Component of DTaP | Infanrix, Daptacel | Pertussis toxin, filamentous hemagglutinin, pertactin (in acellular vaccines) | Pertussis (whooping cough), especially in children |
| RSV (Respiratory Syncytial Virus) | In development | F protein (fusion protein) of RSV | RSV infection, especially in infants and the elderly |
| Yellow Fever | In development | Envelope proteins of the yellow fever virus | Yellow fever |
| HIV | In development | Envelope glycoprotein (gp120) or gp160 of HIV | HIV infection |
| Leishmaniasis | In development | Kinetoplastid membrane protein (KMP-11) and others for Leishmania species | Leishmaniasis, particularly in endemic areas |
| Chikungunya Virus | In development | Envelope protein of chikungunya virus | Chikungunya virus infection |
| Step | Recombinant Protein Engineering | mRNA Technology | Comparison |
|---|---|---|---|
| 1. Research and development | High: Extensive work on gene design, host cell selection, and vector optimization. | Moderate: mRNA sequence design and modification (e.g., codon optimization, UTR engineering). | Recombinant costs higher due to host cell studies. |
| 2. Plasmid construction | High: Stable plasmid generation for host cells (e.g., CHO cells). | Moderate: Plasmid or template synthesis for IVT (in vitro transcription). | Recombinant cost higher for stable plasmids. |
| 3. Cell line development | Very high: Creation of stable expression systems (e.g., CHO, E. coli). | None: mRNA avoids the need for cell lines. | Recombinant is far more expensive. |
| 4. Upstream production | High: Large-scale bioreactors for fermentation and cell culture. | Low: Scalable IVT reactions in cell-free systems. | Recombinant is costlier due to cell culture. |
| 5. Downstream purification | Very high: Protein purification steps like chromatography, filtration, and refolding. | Moderate: RNA purification (e.g., chromatography, ultrafiltration). | Recombinant is more intensive and costly. |
| 6. Formulation development | Moderate: Stabilizing proteins (e.g., lyophilization, additives). | Moderate: Stabilization of mRNA (e.g., lipid nanoparticle [LNP] formulation). | Similar costs. |
| 7. Scalability | High: Requires optimized bioreactor and downstream processes. | Low: IVT is inherently scalable with fewer bottlenecks. | mRNA is more scalable and cost-effective. |
| 8. Quality control and testing | Very high: Protein structure/function analysis, glycosylation profiling, etc. | Moderate: Sequencing, purity checks, and potency assays for mRNA. | Recombinant involves complex QC steps. |
| 9. Regulatory approval costs | High: Extensive CMC data and biosimilar comparability studies. | High: Emerging guidelines but fewer comparability studies are needed. | Comparable costs. |
| 10. Manufacturing cost per dose | Very high: Dependent on yields, process efficiency, and scalability. | Low: Cost-efficient once IVT and LNP formulation are optimized. | mRNA is cheaper per dose. |
| 11. Time to market | Long: Several years (5–10 years) due to cell line development and process optimization. | Short: 1–3 years due to faster development and more straightforward production. | mRNA faster to market. |
| Total cost estimate | High cost per unit, especially for low-yield, complex proteins (can exceed hundreds of millions in total development costs for a commercial product). | Lower overall costs due to reduced infrastructure needs and simpler production processes (~30–50% less expensive than recombinant protein production). | An affordable option for the majority of the world. |
| Company Name | Headquarters | Key mRNA Products/Research Areas | Notable Collaborations |
|---|---|---|---|
| Arcturus Therapeutics | San Diego, CA, USA | mRNA-based vaccines and therapeutics, including COVID-19 vaccine candidates | Duke–NUS Medical School (COVID-19 vaccine development) |
| BioNTech | Mainz, Germany | COVID-19 vaccine (Comirnaty), cancer immunotherapies, infectious disease vaccines | Pfizer (COVID-19 vaccine development) |
| Chimeron Bio | Philadelphia, PA, USA | mRNA-based vaccines and therapeutics using the proprietary ChaESAR platform | None specified |
| CureVac | Tübingen, Germany | mRNA-based vaccines and therapeutics for infectious diseases and cancer | GSK (influenza and COVID-19 vaccines) |
| eTheRNA Immunotherapies | Niel, Belgium | mRNA-based immunotherapies for cancer and infectious diseases | None specified |
| Ethris | Planegg, Germany | mRNA-based therapeutics for respiratory diseases | None specified |
| GSK (GlaxoSmithKline) | Brentford, UK | mRNA vaccines for influenza and COVID-19 | CureVac (mRNA vaccine development) |
| Laronde | Cambridge, MA, USA | Endless RNA™ (eRNA) therapeutics for various diseases | None specified |
| Moderna | Cambridge, MA, USA | COVID-19 vaccine (Spikevax), RSV vaccine (Mresvia), cancer vaccines, rare disease therapeutics | None specified |
| Nutcracker Therapeutics | Emeryville, CA, USA | mRNA-based cancer treatments | None specified |
| Omega Therapeutics | Cambridge, MA, USA | mRNA-based epigenomic programming for various diseases | None specified |
| Pfizer | New York, NY, USA | mRNA-based COVID-19 vaccine (Comirnaty) | BioNTech (COVID-19 vaccine development) |
| RaNA Therapeutics | Cambridge, MA, USA | mRNA-targeted therapies for genetic diseases | None specified |
| Replicate Bioscience | San Diego, CA, USA | Self-replicating RNA therapeutics for cancer and autoimmune diseases | None specified |
| RNAimmune | Gaithersburg, MD, USA | mRNA-based therapies for cancer, rare diseases, and prophylactic vaccines | None specified |
| Sanofi | Paris, France | mRNA vaccines and therapeutics for infectious diseases | Acquired Translate Bio |
| Strand Therapeutics | Cambridge, MA, USA | mRNA-based therapeutics with programmable control for cancer and other diseases | None specified |
| Tiba Biotech | Cambridge, MA, USA | mRNA vaccines and therapeutics using novel nanoparticle delivery systems | None specified |
| Translate Bio | Lexington, MA, USA | mRNA therapeutics for various diseases, including cystic fibrosis and infectious diseases | Sanofi (acquired Translate Bio) |
| Vaxart | South San Francisco, CA, USA | Oral recombinant vaccines, including mRNA-based candidates for COVID-19 and other viruses | None specified |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niazi, S.K.; Magoola, M. Advancing Therapeutic and Vaccine Proteins: Switching from Recombinant to Ribosomal Delivery—A Humanitarian Cause. Int. J. Mol. Sci. 2024, 25, 12797. https://doi.org/10.3390/ijms252312797
Niazi SK, Magoola M. Advancing Therapeutic and Vaccine Proteins: Switching from Recombinant to Ribosomal Delivery—A Humanitarian Cause. International Journal of Molecular Sciences. 2024; 25(23):12797. https://doi.org/10.3390/ijms252312797
Chicago/Turabian StyleNiazi, Sarfaraz K., and Matthias Magoola. 2024. "Advancing Therapeutic and Vaccine Proteins: Switching from Recombinant to Ribosomal Delivery—A Humanitarian Cause" International Journal of Molecular Sciences 25, no. 23: 12797. https://doi.org/10.3390/ijms252312797
APA StyleNiazi, S. K., & Magoola, M. (2024). Advancing Therapeutic and Vaccine Proteins: Switching from Recombinant to Ribosomal Delivery—A Humanitarian Cause. International Journal of Molecular Sciences, 25(23), 12797. https://doi.org/10.3390/ijms252312797