
Functional Analysis of TAAR1 Expression in the Intestine Wall and the Effect of Its Gene Knockout on the Gut Microbiota in Mice

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Results
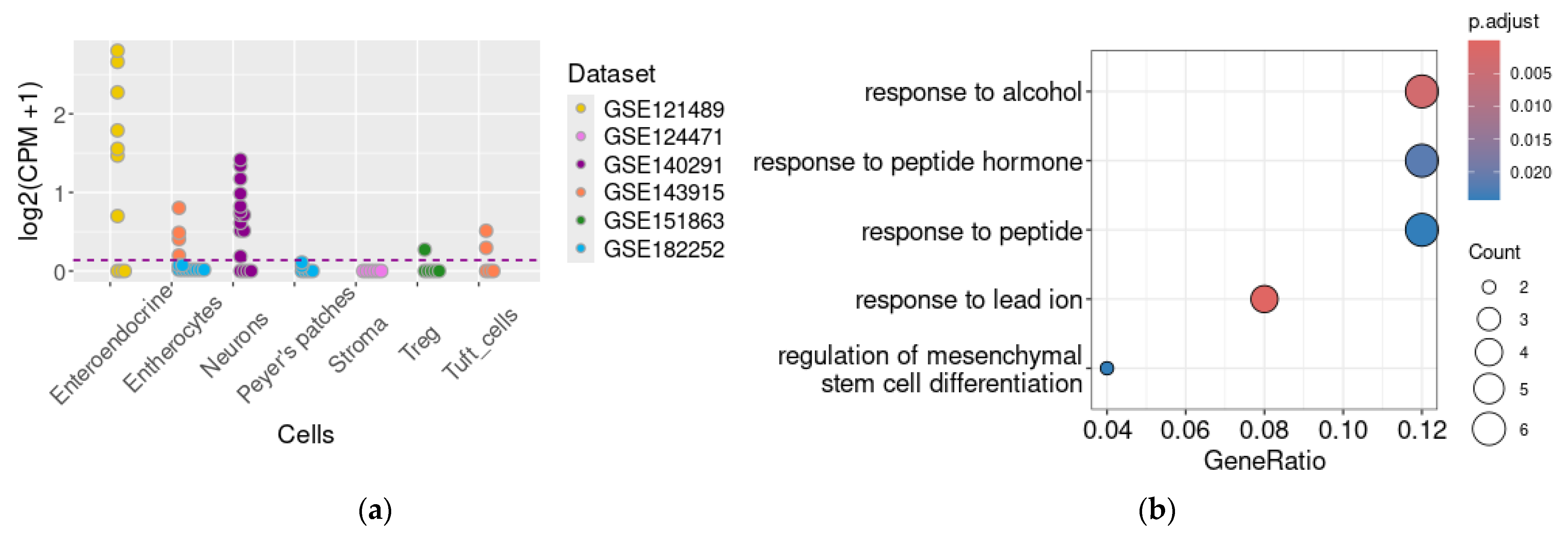
2.1. TAAR1 Expression Is Heterogeneously Distributed in Colon Epithelium Cell Population
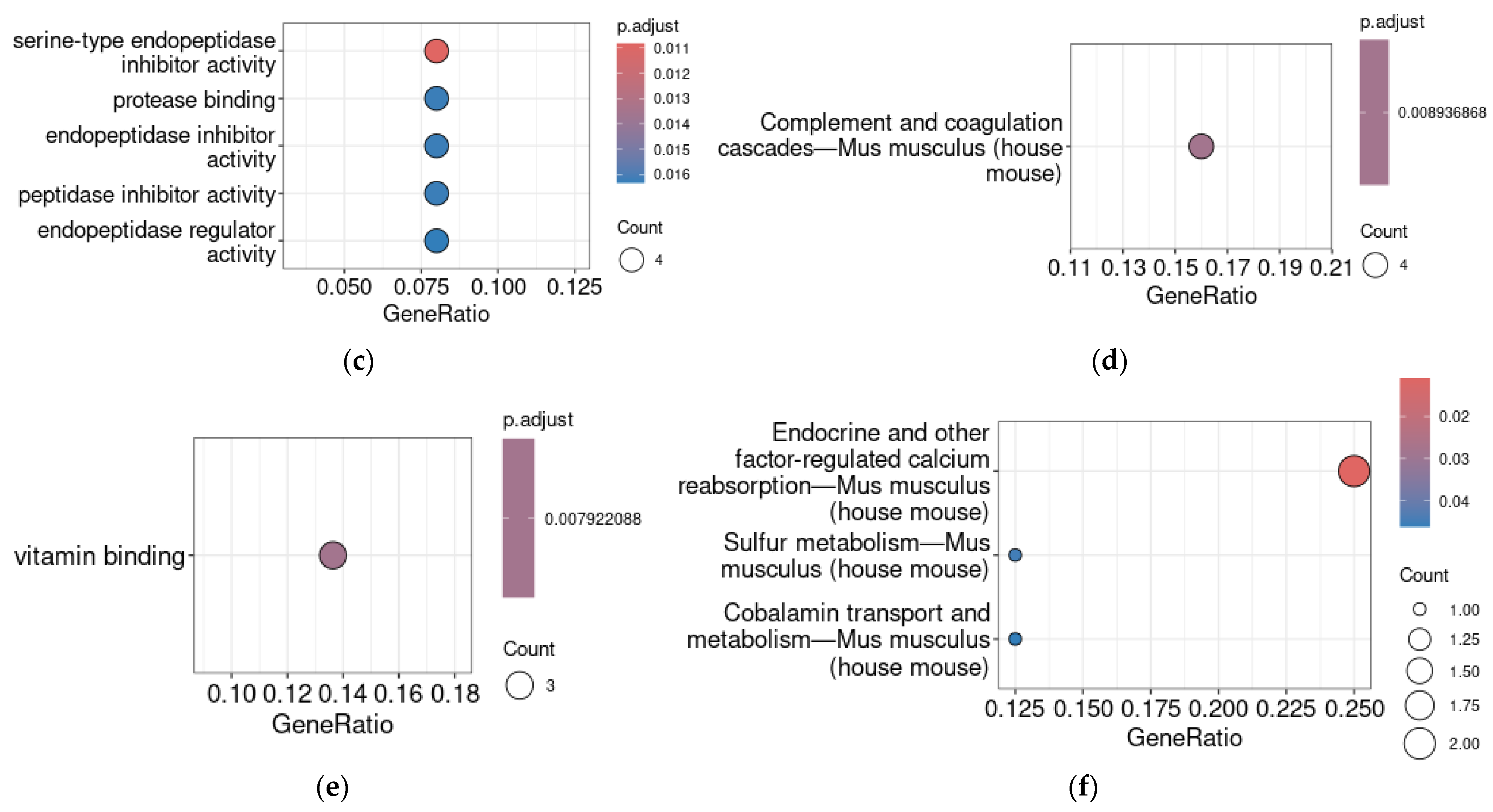
2.2. Genes Co-Expressed with TAAR1 in the Enteric Secretory Cells
2.3. Genes Co-Expressed with TAAR1 in the Myenteric Neural Cells
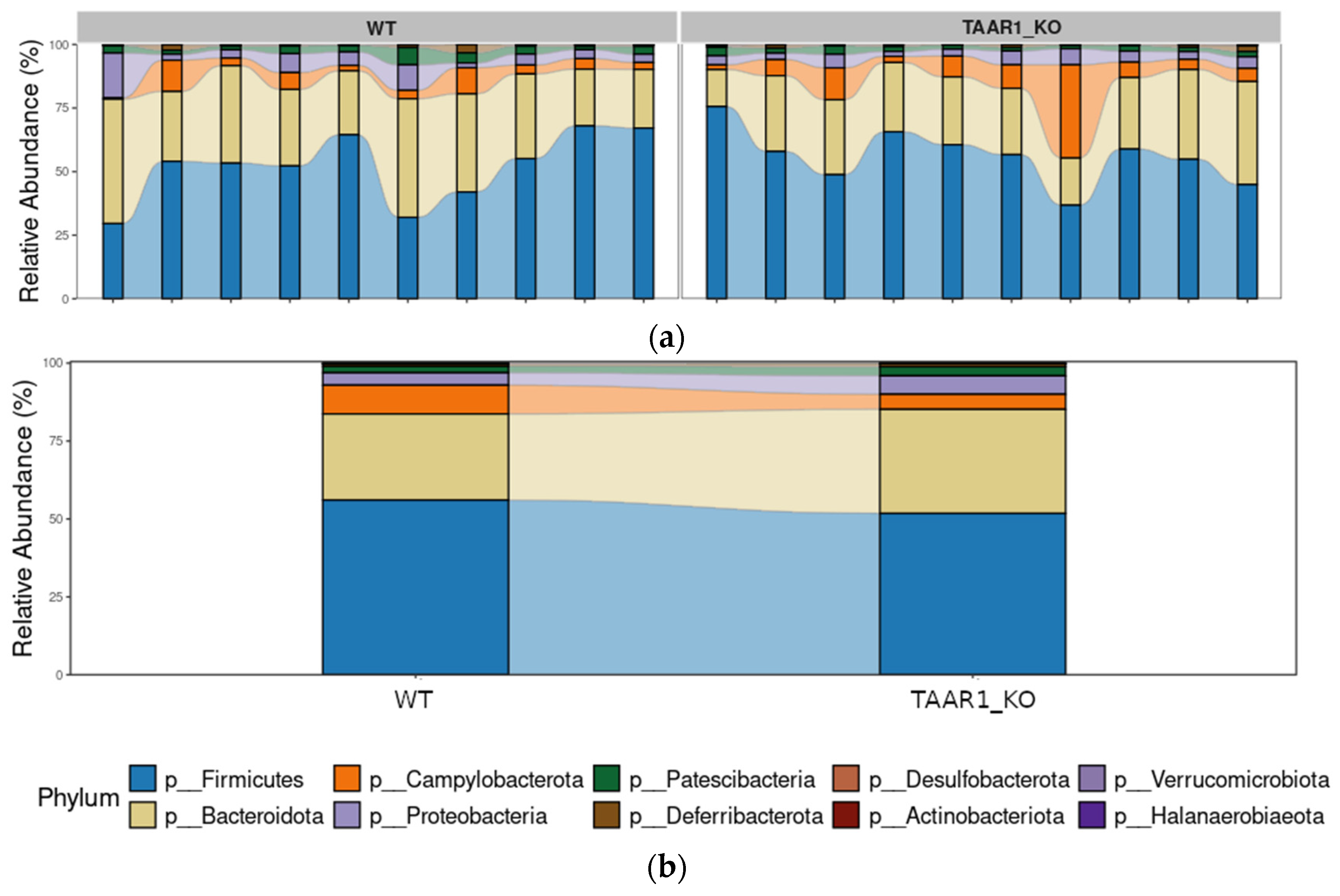
2.4. Gut Microbiota Composition in TAAR1 Knockout Mice
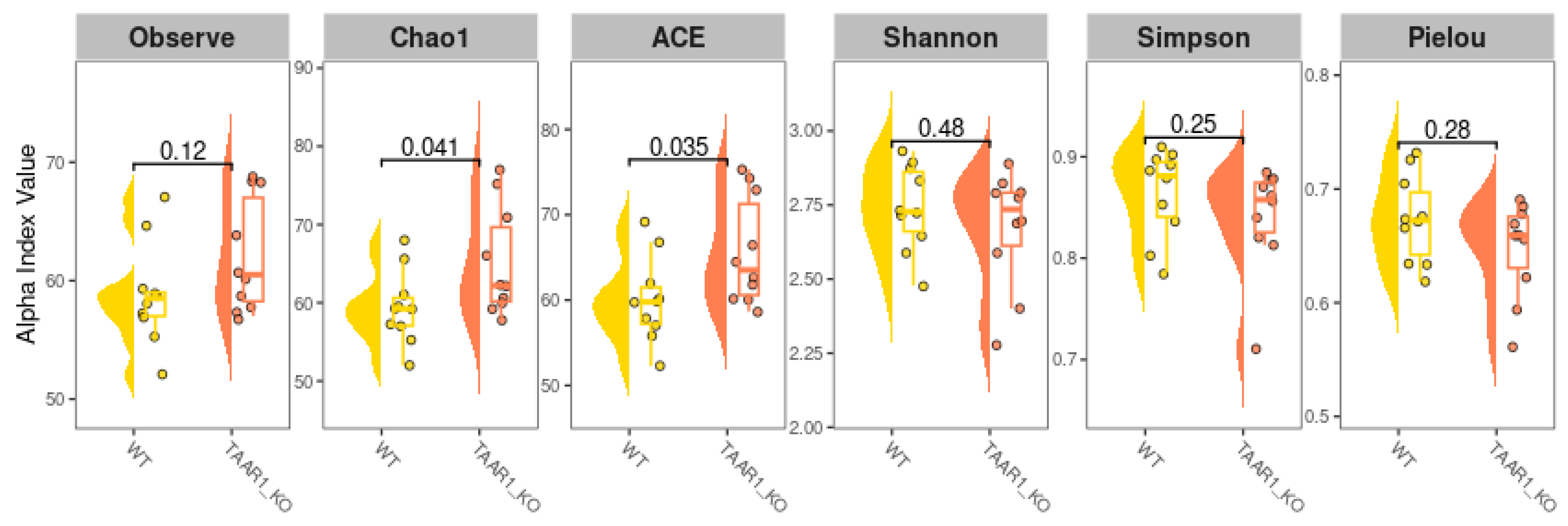
2.5. Alterations in Fecal Microbiota α-Diversity in TAAR1-KO Mice
2.6. The TAAR1-KO Genotype Is Associated with More Stable Fecal Microbiome Composition and Slight Reorganization of Bacterial Interactions in the Gut Microbial Community
3. Discussion
4. Materials and Methods
4.1. Data Collection and Inclusion Criteria for Datasets
4.2. Data Normalization and Statistical Analysis
4.3. Gene Co-Expression Measurement and Pathway Enrichment Analysis
4.4. Animals and Sample Collection
4.5. Gut Bacterial DNA Extraction and Sequencing
4.6. Sequencing Data Processing
4.7. Statistical Analysis of 16S rRNA Sequencing Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berry, M.D.; Gainetdinov, R.R.; Hoener, M.C.; Shahid, M. Pharmacology of human trace amine-associated receptors: Therapeutic opportunities and challenges. Pharmacol. Ther. 2017, 180, 161–180. [Google Scholar] [CrossRef] [PubMed]
- Gainetdinov, R.R.; Hoener, M.C.; Berry, M.D. Trace Amines and Their Receptors. Pharmacol. Rev. 2018, 70, 549–620. [Google Scholar] [CrossRef] [PubMed]
- Amato, A.; Terzo, S.; Marchesa, P.; Maffongelli, A.; Martorana, M.; Scoglio, S.; Mulè, F. Spasmolytic Effects of Aphanizomenon Flos Aquae (AFA) Extract on the Human Colon Contractility. Nutrients 2021, 13, 3445. [Google Scholar] [CrossRef] [PubMed]
- Bugda Gwilt, K.; González, D.P.; Olliffe, N.; Oller, H.; Hoffing, R.; Puzan, M.; El Aidy, S.; Miller, G.M. Actions of Trace Amines in the Brain-Gut-Microbiome Axis via Trace Amine-Associated Receptor-1 (TAAR1). Cell Mol. Neurobiol. 2020, 40, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Sudo, N. Biogenic Amines: Signals Between Commensal Microbiota and Gut Physiology. Front. Endocrinol. 2019, 10, 504. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, Y.; Mori, Y.; Nara, M.; Kotani, Y.; Nagai, E.; Kawada, H.; Kitamura, M.; Hirano, R.; Shimokawa, H.; Nakagawa, A.; et al. Gut Bacterial Aromatic Amine Production: Aromatic Amino Acid Decarboxylase and Its Effects on Peripheral Serotonin Production. Gut Microbes 2022, 14, 2128605. [Google Scholar] [CrossRef]
- Zhai, L.; Huang, C.; Ning, Z.; Zhang, Y.; Zhuang, M.; Yang, W.; Wang, X.; Wang, J.; Zhang, L.; Xiao, H.; et al. Ruminococcus Gnavus Plays a Pathogenic Role in Diarrhea-Predominant Irritable Bowel Syndrome by Increasing Serotonin Biosynthesis. Cell Host Microbe 2023, 31, 33–44.e5. [Google Scholar] [CrossRef]
- Wei, Y.-X.; Zheng, K.-Y.; Wang, Y.-G. Gut Microbiota-Derived Metabolites as Key Mucosal Barrier Modulators in Obesity. World J. Gastroenterol. 2021, 27, 5555–5565. [Google Scholar] [CrossRef]
- Kuvarzin, S.R.; Sukhanov, I.; Onokhin, K.; Zakharov, K.; Gainetdinov, R.R. Unlocking the Therapeutic Potential of Ulotaront as a Trace Amine-Associated Receptor 1 Agonist for Neuropsychiatric Disorders. Biomedicines 2023, 11, 1977. [Google Scholar] [CrossRef]
- Kantrowitz, J.T. Trace Amine-Associated Receptor 1 as a Target for the Development of New Antipsychotics: Current Status of Research and Future Directions. CNS Drugs 2021, 35, 1153–1161. [Google Scholar] [CrossRef]
- Le, G.H.; Gillissie, E.S.; Rhee, T.G.; Cao, B.; Alnefeesi, Y.; Guo, Z.; Di Vincenzo, J.D.; Jawad, M.Y.; March, A.M.; Ramachandra, R.; et al. Efficacy, Safety, and Tolerability of Ulotaront (SEP-363856, a Trace Amine-Associated Receptor 1 Agonist) for the Treatment of Schizophrenia and Other Mental Disorders: A Systematic Review of Preclinical and Clinical Trials. Expert. Opin. Investig. Drugs 2023, 32, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Raab, S.; Wang, H.; Uhles, S.; Cole, N.; Alvarez-Sanchez, R.; Künnecke, B.; Ullmer, C.; Matile, H.; Bedoucha, M.; Norcross, R.D.; et al. Incretin-like Effects of Small Molecule Trace Amine-Associated Receptor 1 Agonists. Mol. Metab. 2016, 5, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Dedic, N.; Wang, L.; Hajos-Korcsok, E.; Hecksher-Sørensen, J.; Roostalu, U.; Vickers, S.P.; Wu, S.; Anacker, C.; Synan, C.; Jones, P.G.; et al. TAAR1 Agonists Improve Glycemic Control, Reduce Body Weight and Modulate Neurocircuits Governing Energy Balance and Feeding. Mol. Metab. 2024, 80, 101883. [Google Scholar] [CrossRef] [PubMed]
- Zhukov, I.S.; Ptukha, M.A.; Zolotoverkhaja, E.A.; Sinitca, E.L.; Tissen, I.Y.; Karpova, I.V.; Volnova, A.B.; Gainetdinov, R.R. Evaluation of Approach to a Conspecific and Blood Biochemical Parameters in TAAR1 Knockout Mice. Brain Sci. 2022, 12, 614. [Google Scholar] [CrossRef]
- Vaganova, A.N.; Kuvarzin, S.R.; Sycheva, A.M.; Gainetdinov, R.R. Deregulation of Trace Amine-Associated Receptors (TAAR) Expression and Signaling Mode in Melanoma. Biomolecules 2022, 12, 114. [Google Scholar] [CrossRef]
- Vaganova, A.N.; Maslennikova, D.D.; Konstantinova, V.V.; Kanov, E.V.; Gainetdinov, R.R. The Expression of Trace Amine-Associated Receptors (TAARs) in Breast Cancer Is Coincident with the Expression of Neuroactive Ligand-Receptor Systems and Depends on Tumor Intrinsic Subtype. Biomolecules 2023, 13, 1361. [Google Scholar] [CrossRef]
- Zhai, L.; Xiao, H.; Lin, C.; Wong, H.L.X.; Lam, Y.Y.; Gong, M.; Wu, G.; Ning, Z.; Huang, C.; Zhang, Y.; et al. Gut Microbiota-Derived Tryptamine and Phenethylamine Impair Insulin Sensitivity in Metabolic Syndrome and Irritable Bowel Syndrome. Nat. Commun. 2023, 14, 4986. [Google Scholar] [CrossRef]
- Broadley, K.J.; Anwar, M.A.; Herbert, A.A.; Fehler, M.; Jones, E.M.; Davies, W.E.; Kidd, E.J.; Ford, W.R. Effects of Dietary Amines on the Gut and Its Vasculature. Br. J. Nutr. 2008, 101, 1645–1652. [Google Scholar] [CrossRef]
- Ohta, H.; Takebe, Y.; Murakami, Y.; Takahama, Y.; Morimura, S. Tyramine and β-Phenylethylamine, from Fermented Food Products, as Agonists for the Human Trace Amine-Associated Receptor 1 (hTAAR1) in the Stomach. Biosci. Biotechnol. Biochem. 2017, 81, 1002–1006. [Google Scholar] [CrossRef]
- Sánchez, M.; Suárez, L.; Andrés, M.T.; Flórez, B.H.; Bordallo, J.; Riestra, S.; Cantabrana, B. Modulatory Effect of Intestinal Polyamines and Trace Amines on the Spontaneous Phasic Contractions of the Isolated Ileum and Colon Rings of Mice. Food Nutr. Res. 2017, 61, 1321948. [Google Scholar] [CrossRef]
- Pretorius, L.; Van Staden, A.D.P.; Van der Merwe, J.J.; Henning, N.; Smith, C. Alterations to Microbial Secretome by Estrogen May Contribute to Sex Bias in Irritable Bowel Syndrome. Inflammopharmacol 2022, 30, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, L.; Smith, C. The Trace Aminergic System: A Gender-Sensitive Therapeutic Target for IBS? J. Biomed. Sci. 2020, 27, 95. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, J.; Chen, Y. Regulation of Neurotransmitters by the Gut Microbiota and Effects on Cognition in Neurological Disorders. Nutrients 2021, 13, 2099. [Google Scholar] [CrossRef] [PubMed]
- Heimroth, R.D.; Casadei, E.; Salinas, I. Molecular Drivers of Lymphocyte Organization in Vertebrate Mucosal Surfaces: Revisiting the TNF Superfamily Hypothesis. J. Immunol. 2020, 204, 2697–2711. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous Bacteria from the Gut Microbiota Regulate Host Serotonin Biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef]
- Nataf, S.; Pays, L. Molecular Insights into SARS-CoV2-Induced Alterations of the Gut/Brain Axis. Int. J. Mol. Sci. 2021, 22, 10440. [Google Scholar] [CrossRef]
- Bienenstock, J.; Kunze, W.A.; Forsythe, P. Disruptive Physiology: Olfaction and the Microbiome–Gut–Brain Axis. Biol. Rev. 2018, 93, 390–403. [Google Scholar] [CrossRef]
- Fernandez-Cantos, M.V.; Babu, A.F.; Hanhineva, K.; Kuipers, O.P. Identification of Metabolites Produced by Six Gut Commensal Bacteroidales Strains Using Non-Targeted LC-MS/MS Metabolite Profiling. Microbiol. Res. 2024, 283, 127700. [Google Scholar] [CrossRef]
- Luqman, A.; Muttaqin, M.Z.; Yulaipi, S.; Ebner, P.; Matsuo, M.; Zabel, S.; Tribelli, P.M.; Nieselt, K.; Hidayati, D.; Götz, F. Trace Amines Produced by Skin Bacteria Accelerate Wound Healing in Mice. Commun. Biol. 2020, 3, 277. [Google Scholar] [CrossRef]
- Liu, M.; Nieuwdorp, M.; de Vos, W.M.; Rampanelli, E. Microbial Tryptophan Metabolism Tunes Host Immunity, Metabolism, and Extraintestinal Disorders. Metabolites 2022, 12, 834. [Google Scholar] [CrossRef]
- Dicks, L.M.T. Our Mental Health Is Determined by an Intrinsic Interplay between the Central Nervous System, Enteric Nerves, and Gut Microbiota. Int. J. Mol. Sci. 2024, 25, 38. [Google Scholar] [CrossRef] [PubMed]
- Dicks, L.M.T.; Hurn, D.; Hermanus, D. Gut Bacteria and Neuropsychiatric Disorders. Microorganisms 2021, 9, 2583. [Google Scholar] [CrossRef] [PubMed]
- MacFarland, S.; Bagatell, R. Advances in Neuroblastoma Therapy. Curr. Opin. Pediatr. 2019, 31, 14. [Google Scholar] [CrossRef] [PubMed]
- Salahpour, A.; Espinoza, S.; Masri, B.; Lam, V.; Barak, L.S.; Gainetdinov, R.R. BRET Biosensors to Study GPCR Biology, Pharmacology, and Signal Transduction. Front. Endocrinol. 2012, 3, 105. [Google Scholar] [CrossRef] [PubMed]
- Santoru, M.L.; Piras, C.; Murgia, A.; Palmas, V.; Camboni, T.; Liggi, S.; Ibba, I.; Lai, M.A.; Orrù, S.; Blois, S.; et al. Cross Sectional Evaluation of the Gut-Microbiome Metabolome Axis in an Italian Cohort of IBD Patients. Sci. Rep. 2017, 7, 9523. [Google Scholar] [CrossRef]
- Hendel, S.K.; Kellermann, L.; Hausmann, A.; Bindslev, N.; Jensen, K.B.; Nielsen, O.H. Tuft Cells and Their Role in Intestinal Diseases. Front. Immunol. 2022, 13, 822867. [Google Scholar] [CrossRef]
- McCann, J.R.; Rawls, J.F. Essential Amino Acid Metabolites as Chemical Mediators of Host-Microbe Interaction in the Gut. Annu. Rev. Microbiol. 2023, 77, 479–497. [Google Scholar] [CrossRef]
- Yang, Z.; Cheng, J.; Shang, P.; Sun, J.-P.; Yu, X. Emerging Roles of Olfactory Receptors in Glucose Metabolism. Trends Cell Biol. 2023, 33, 463–476. [Google Scholar] [CrossRef]
- Vaganova, A.N.; Shemyakova, T.S.; Lenskaia, K.V.; Rodionov, R.N.; Steenblock, C.; Gainetdinov, R.R. Trace Amine-Associated Receptors and Monoamine-Mediated Regulation of Insulin Secretion in Pancreatic Islets. Biomolecules 2023, 13, 1618. [Google Scholar] [CrossRef]
- Mkaouar, H.; Mariaule, V.; Rhimi, S.; Hernandez, J.; Kriaa, A.; Jablaoui, A.; Akermi, N.; Maguin, E.; Lesner, A.; Korkmaz, B.; et al. Gut Serpinome: Emerging Evidence in IBD. Int. J. Mol. Sci. 2021, 22, 6088. [Google Scholar] [CrossRef]
- Sánchez-Navarro, A.; González-Soria, I.; Caldiño-Bohn, R.; Bobadilla, N.A. An Integrative View of Serpins in Health and Disease: The Contribution of SerpinA3. Am. J. Physiol. Cell Physiol. 2021, 320, C106–C118. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, U.; Saxena, M.; O’Neill, N.K.; Saadatpour, A.; Yuan, G.-C.; Herbert, Z.; Murata, K.; Shivdasani, R.A. Dynamic Reorganization of Chromatin Accessibility Signatures during Dedifferentiation of Secretory Precursors into Lgr5+ Intestinal Stem Cells. Cell Stem Cell 2017, 21, 65–77.e5. [Google Scholar] [CrossRef]
- Singh, P.N.P.; Gu, W.; Madha, S.; Lynch, A.W.; Cejas, P.; He, R.; Bhattacharya, S.; Gomez, M.M.; Oser, M.G.; Brown, M.; et al. Transcription Factor Dynamics, Oscillation, and Functions in Human Enteroendocrine Cell Differentiation. bioRxiv 2024. [Google Scholar] [CrossRef]
- Brehmer, A. Classification of Human Enteric Neurons. Histochem. Cell Biol. 2021, 156, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Zetzmann, K.; Strehl, J.; Geppert, C.; Kuerten, S.; Jabari, S.; Brehmer, A. Calbindin D28k-Immunoreactivity in Human Enteric Neurons. Int. J. Mol. Sci. 2018, 19, 194. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.Y.; Parathan, P.; Boonma, P.; Wu, Q.; Wang, Y.; Haag, A.; Luna, R.A.; Bornstein, J.C.; Savidge, T.C.; Foong, J.P.P. Antibiotic Exposure Postweaning Disrupts the Neurochemistry and Function of Enteric Neurons Mediating Colonic Motor Activity. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G1042–G1053. [Google Scholar] [CrossRef]
- Obata, Y.; Castaño, Á.; Boeing, S.; Bon-Frauches, A.C.; Fung, C.; Fallesen, T.; de Agüero, M.G.; Yilmaz, B.; Lopes, R.; Huseynova, A.; et al. Neuronal Programming by Microbiota Regulates Intestinal Physiology. Nature 2020, 578, 284–289. [Google Scholar] [CrossRef]
- Habib, A.M.; Nagi, K.; Thillaiappan, N.B.; Sukumaran, V.; Akhtar, S. Vitamin D and Its Potential Interplay With Pain Signaling Pathways. Front. Immunol. 2020, 11, 820. [Google Scholar] [CrossRef]
- Larsson, S.; Voss, U. Neuroprotective Effects of Vitamin D on High Fat Diet- and Palmitic Acid-Induced Enteric Neuronal Loss in Mice. BMC Gastroenterol. 2018, 18, 175. [Google Scholar] [CrossRef]
- Li, N.; Seetharam, S.; Rosenblatt, D.S.; Seetharam, B. Expression of Transcobalamin II mRNA in Human Tissues and Cultured Fibroblasts from Normal and Transcobalamin II-Deficient Patients. Biochem. J. 1994, 301 Pt 2, 585–590. [Google Scholar] [CrossRef]
- Kurashima, Y.; Yamamoto, D.; Nelson, S.; Uematsu, S.; Ernst, P.B.; Nakayama, T.; Kiyono, H. Mucosal Mesenchymal Cells: Secondary Barrier and Peripheral Educator for the Gut Immune System. Front. Immunol. 2017, 8, 01787. [Google Scholar] [CrossRef]
- Takahashi, K.; Morita, N.; Tamano, R.; Gao, P.; Iida, N.; Andoh, A.; Imaeda, H.; Kurokawa, K.; Tsuboi, M.; Hayakawa, Y.; et al. Mouse IgA Modulates Human Gut Microbiota with Inflammatory Bowel Disease Patients. J. Gastroenterol. 2024, 59, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Tsai, C.-Y.; McAdams, Z.L.; Oo, M.; Hansen, M.; Dougher, M.; Sansano, A.; Watson, A.; LoMauro, K.; Antilus-Sainte, R.; et al. Wild Mouse Gut Microbiota Limits Initial Tuberculosis Infection in BALB/c Mice. PLoS ONE 2023, 18, e0288290. [Google Scholar] [CrossRef] [PubMed]
- Oren, A.; Garrity, G.M. Valid Publication of the Names of Forty-Two Phyla of Prokaryotes. Int. J. Syst. Evol. Microbiol. 2021, 71, 005056. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, M.; Xue, J.; Huang, J.; Zhuang, R.; Zhou, X.; Zhang, H.; Fu, Q.; Hao, Y. Body Mass Index Differences in the Gut Microbiota Are Gender Specific. Front. Microbiol. 2018, 9, 012510. [Google Scholar] [CrossRef]
- Ke, S.; Guimond, A.-J.; Tworoger, S.S.; Huang, T.; Chan, A.T.; Liu, Y.-Y.; Kubzansky, L.D. Gut Feelings: Associations of Emotions and Emotion Regulation with the Gut Microbiome in Women. Psychol. Med. 2023, 53, 7151–7160. [Google Scholar] [CrossRef]
- Moroishi, Y.; Gui, J.; Hoen, A.G.; Morrison, H.G.; Baker, E.R.; Nadeau, K.C.; Li, H.; Li, Z.; Madan, J.C.; Karagas, M.R. The Relationship between the Gut Microbiome and the Risk of Respiratory Infections among Newborns. Commun. Med. 2022, 2, 87. [Google Scholar] [CrossRef]
- Petrak, F.; Herpertz, S.; Hirsch, J.; Röhrig, B.; Donati-Hirsch, I.; Juckel, G.; Meier, J.J.; Gatermann, S. Gut Microbiota Differs in Composition between Adults with Type 1 Diabetes with or without Depression and Healthy Control Participants: A Case-Control Study. BMC Microbiol. 2022, 22, 169. [Google Scholar] [CrossRef]
- Barandouzi, Z.A.; Lee, J.; Maas, K.; Starkweather, A.R.; Cong, X.S. Altered Gut Microbiota in Irritable Bowel Syndrome and Its Association with Food Components. J. Pers. Med. 2021, 11, 35. [Google Scholar] [CrossRef]
- Zhukov, I.S.; Vaganova, A.N.; Murtazina, R.Z.; Alferova, L.S.; Ermolenko, E.I.; Gainetdinov, R.R. Gut Microbiota Alterations in Trace Amine-Associated Receptor 9 (TAAR9) Knockout Rats. Biomolecules 2022, 12, 1823. [Google Scholar] [CrossRef]
- Li, Z.; Liu, B.; Cui, H.; Ding, J.; Li, H.; Xie, G.; Ren, N.; Xing, D. The complete genome sequence of Ethanoligenens harbinense reveals the metabolic pathway of acetate-ethanol fermentation: A novel understanding of the principles of anaerobic biotechnology. Environ. Int. 2019, 131, 105053. [Google Scholar] [CrossRef]
- Chen, S.; Dong, X. Acetanaerobacterium elongatum gen. nov., sp. nov., from paper mill waste water. Int. J. Syst. Evol. Microbiol. 2004, 54, 2257–2262. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S. Eggerthellaceae. In Bergey’s Manual of Systematics of Archaea and Bacteria; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2021; pp. 1–11. [Google Scholar] [CrossRef]
- Huus, K.E.; Ley, R.E. Gut bacterial metabolism produces neuroactive steroids in pregnant women. Life Metab. 2024, 3, loae030. [Google Scholar] [CrossRef]
- Lkhagva, E.; Chung, H.-J.; Hong, J.; Tang, W.H.W.; Lee, S.-I.; Hong, S.-T.; Lee, S. The Regional Diversity of Gut Microbiome along the GI Tract of Male C57BL/6 Mice. BMC Microbiol. 2021, 21, 44. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.-S.; Lkhagva, E.; Jung, S.; Kim, H.-J.; Chung, H.-J.; Hong, S.-T. Fecal Microbiome Does Not Represent Whole Gut Microbiome. Cell. Microbiol. 2023, 2023, 6868417. [Google Scholar] [CrossRef]
- Otaru, N.; Greppi, A.; Plüss, S.; Zünd, J.; Mujezinovic, D.; Baur, J.; Koleva, E.; Lacroix, C.; Pugin, B. Intestinal Bacteria-Derived Tryptamine and Its Impact on Human Gut Microbiota. Front. Microbiomes 2024, 3, 1373335. [Google Scholar] [CrossRef]
- Pugin, B.; Barcik, W.; Westermann, P.; Heider, A.; Wawrzyniak, M.; Hellings, P.; Akdis, C.A.; O’Mahony, L. A Wide Diversity of Bacteria from the Human Gut Produces and Degrades Biogenic Amines. Microb. Ecol. Health Dis. 2017, 28, 1353881. [Google Scholar] [CrossRef]
- Marcobal, A.; De Las Rivas, B.; Landete, J.M.; Tabera, L.; Muñoz, R. Tyramine and Phenylethylamine Biosynthesis by Food Bacteria. Crit. Rev. Food Sci. Nutr. 2012, 52, 448–467. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets--Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Wolinsky, T.D.; Swanson, C.J.; Smith, K.E.; Zhong, H.; Borowsky, B.; Seeman, P.; Branchek, T.; Gerald, C.P. The Trace Amine 1 receptor knockout mouse: An animal model with relevance to schizophrenia. Genes Brain Behav. 2007, 6, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Peplies, J.; Glöckner, F.O. SINA: Accurate High-Throughput Multiple Sequence Alignment of Ribosomal RNA Genes. Bioinformatics 2012, 28, 1823–1829. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhan, L.; Tang, W.; Wang, Q.; Dai, Z.; Zhou, L.; Feng, T.; Chen, M.; Wu, T.; Hu, E.; et al. Microbiota Process: A Comprehensive R Package for Deep Mining Microbiome. Innovation 2023, 4, 100388. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Griffith, D.M.; Veech, J.A.; Marsh, C.J. Cooccur: Probabilistic Species Co-Occurrence Analysis in R. J. Stat. Softw. Code Snippets 2016, 69, 1–17. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal Complex Syst. 2006, 1695, 1–9. Available online: https://igraph.org/ (accessed on 29 November 2024).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Title | Cell Type | n * |
|---|---|---|---|
| GSE121489 | Transcriptomics analysis of enteroendocrine cells after vertical sleeve gastrectomy | Enteroendocrine cells form ileum and colon | 5 (ileum) and 5 (colon) |
| GSE124471 | Bulk RNA-sequencing and Single-cell RNA-sequencing of adult mice intestinal stromal cells and lacteal LECs with PDGFRb-specific deletion of LATS1/2 [RNA-seq] | Intestinal stromal cell | 6 |
| GSE140291 | Expression analysis of enteric neurons from the mice housed in different animal facilities (the Francis Crick Institute and the University of Bern) | Neurons | 16 |
| GSE143915 | Transcriptomic and Proteomic Signatures of Stemness and Differentiation in the Colon Crypt | Tuft cells and enterocytes | 5 tuft cells and 5 enterocyte samples |
| GSE151863 | Impaired estrogen signaling underlies regulatory T cell loss-of-function in the chronically-inflamed intestine | Regulatory T cells | 6 |
| GSE182252 | Transcriptomic analysis of the follicle associated epithelium of Peyer’s Patches, intestinal villous epithelium and ileum from young and aged mice | Enterocytes and epithelium of Peyer’s Patches | Follicle associated epithelium (n = 3 mice), intestinal and ileum epithelium (n = 4–6 mice) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaganova, A.N.; Zhukov, I.S.; Shemiakova, T.S.; Rozhkov, K.A.; Alferova, L.S.; Karaseva, A.B.; Ermolenko, E.I.; Gainetdinov, R.R. Functional Analysis of TAAR1 Expression in the Intestine Wall and the Effect of Its Gene Knockout on the Gut Microbiota in Mice. Int. J. Mol. Sci. 2024, 25, 13216. https://doi.org/10.3390/ijms252313216
Vaganova AN, Zhukov IS, Shemiakova TS, Rozhkov KA, Alferova LS, Karaseva AB, Ermolenko EI, Gainetdinov RR. Functional Analysis of TAAR1 Expression in the Intestine Wall and the Effect of Its Gene Knockout on the Gut Microbiota in Mice. International Journal of Molecular Sciences. 2024; 25(23):13216. https://doi.org/10.3390/ijms252313216
Chicago/Turabian StyleVaganova, Anastasia N., Ilya S. Zhukov, Taisiia S. Shemiakova, Konstantin A. Rozhkov, Lyubov S. Alferova, Alena B. Karaseva, Elena I. Ermolenko, and Raul R. Gainetdinov. 2024. "Functional Analysis of TAAR1 Expression in the Intestine Wall and the Effect of Its Gene Knockout on the Gut Microbiota in Mice" International Journal of Molecular Sciences 25, no. 23: 13216. https://doi.org/10.3390/ijms252313216
APA StyleVaganova, A. N., Zhukov, I. S., Shemiakova, T. S., Rozhkov, K. A., Alferova, L. S., Karaseva, A. B., Ermolenko, E. I., & Gainetdinov, R. R. (2024). Functional Analysis of TAAR1 Expression in the Intestine Wall and the Effect of Its Gene Knockout on the Gut Microbiota in Mice. International Journal of Molecular Sciences, 25(23), 13216. https://doi.org/10.3390/ijms252313216