
Semisynthesis and Antitumour Evaluation of Natural Derivatives from ent-Kaurene ent-15α-Angeloyloxykaur-l6-en-3β-ol Isolated from Distichoselinum tenuifolium

 , , , , ,
, , , , ,  , , and
, , and
Abstract

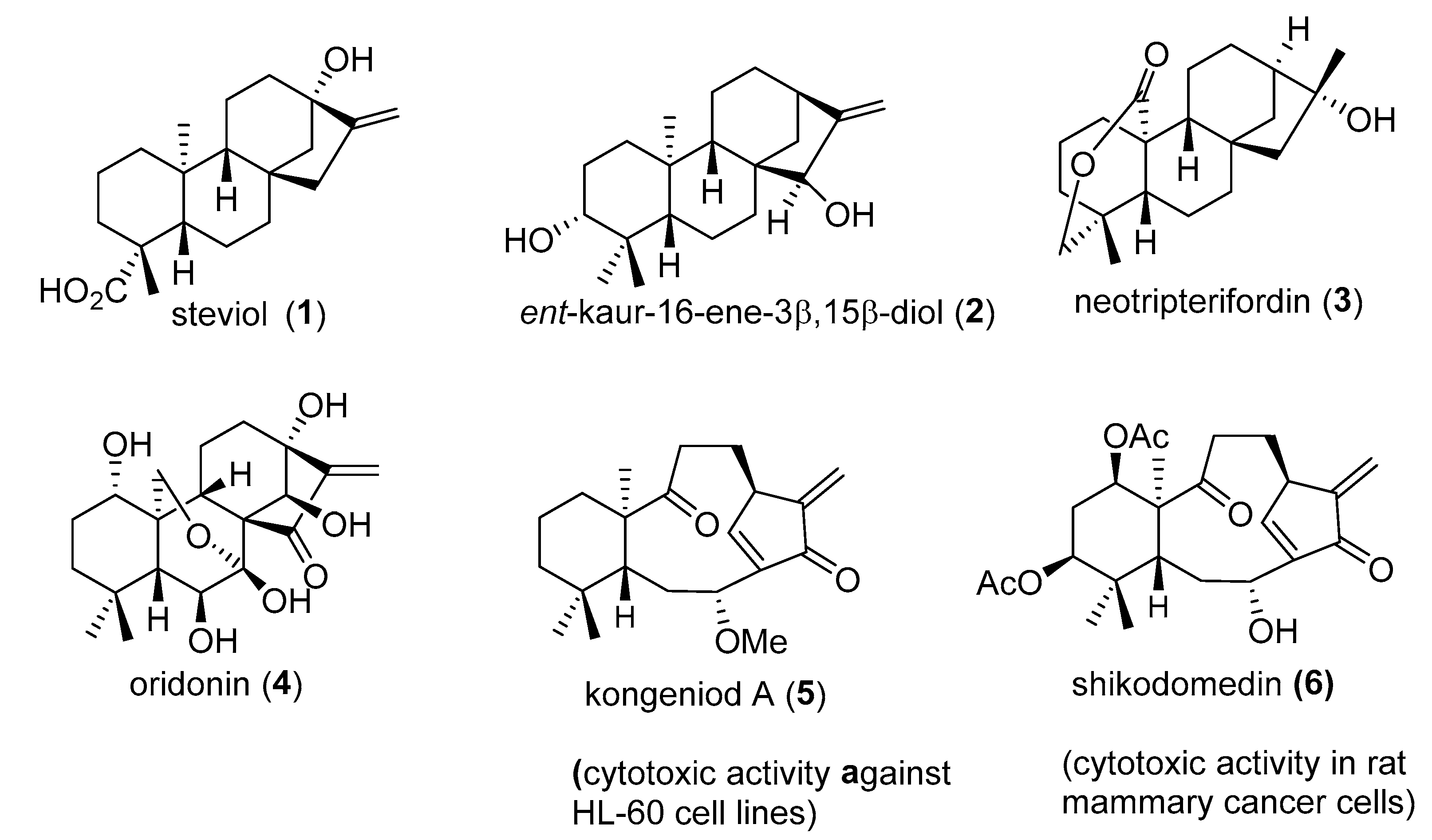
1. Introduction
2. Results
2.1. Chemistry
2.2. Anticancer Activity
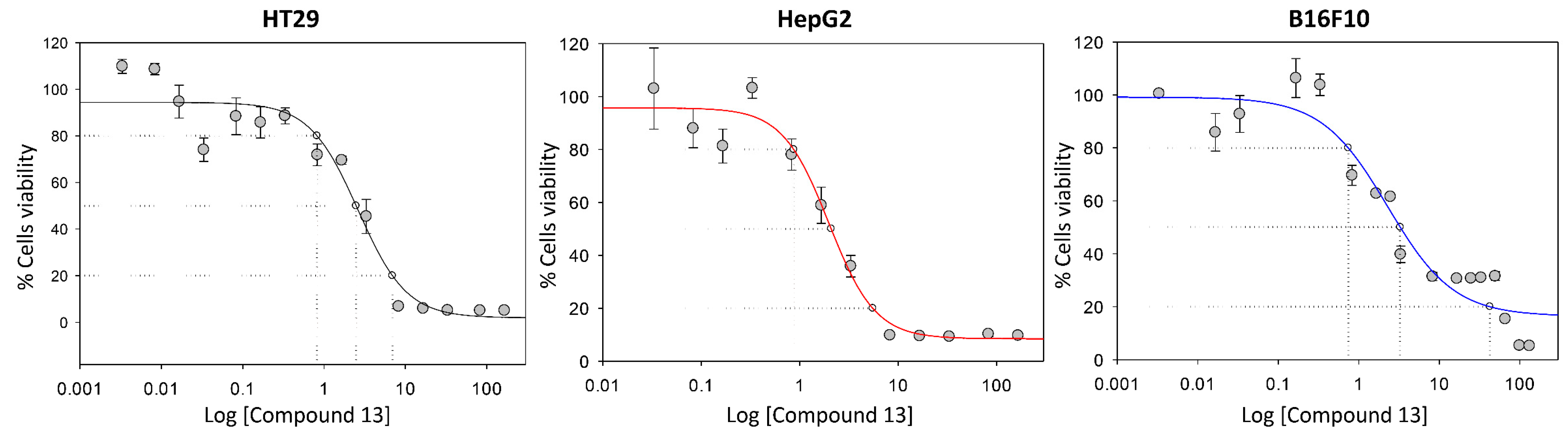
2.2.1. Cell Viability
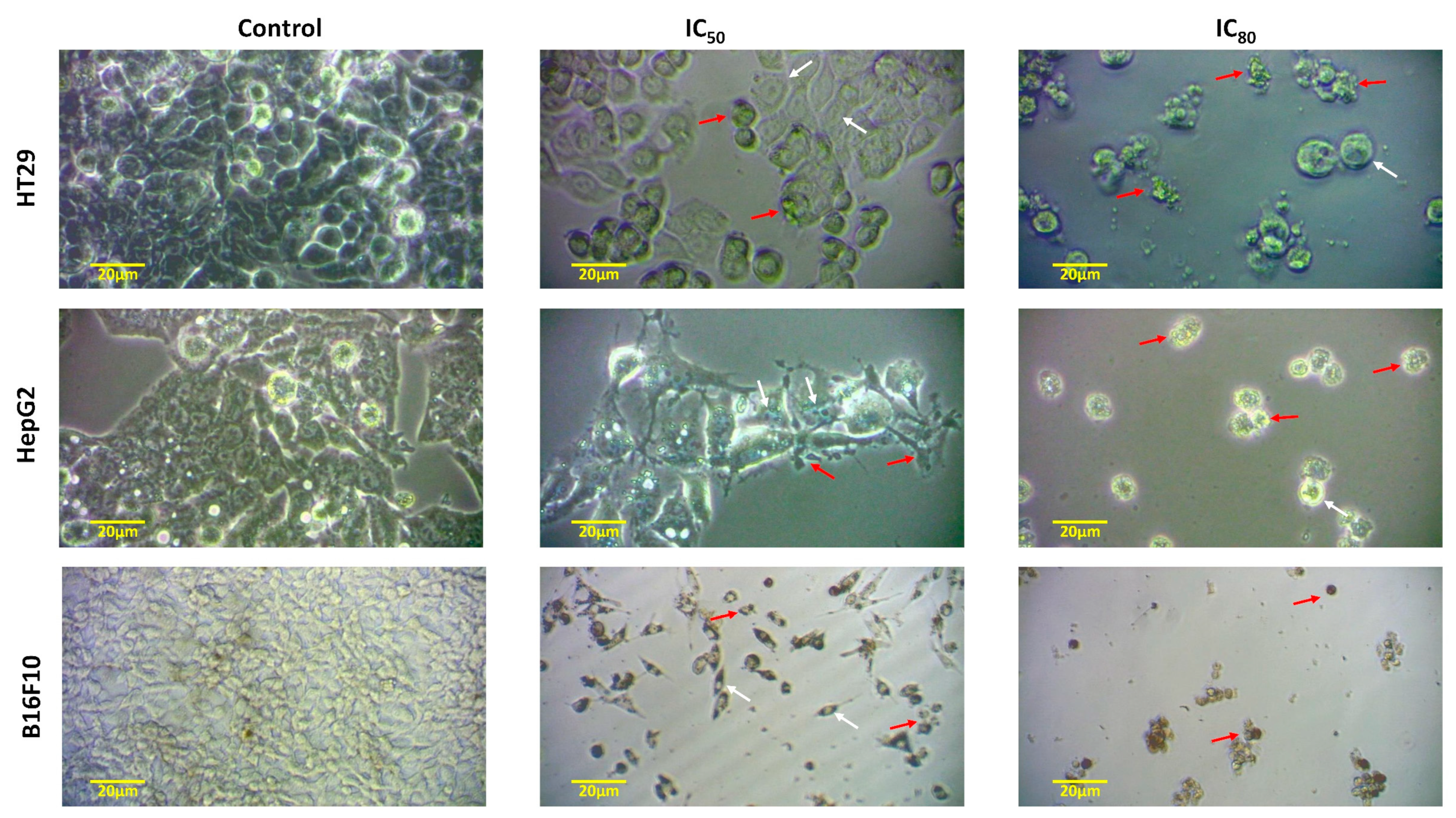
2.2.2. Cell Morphology
2.2.3. Induction of Apoptosis
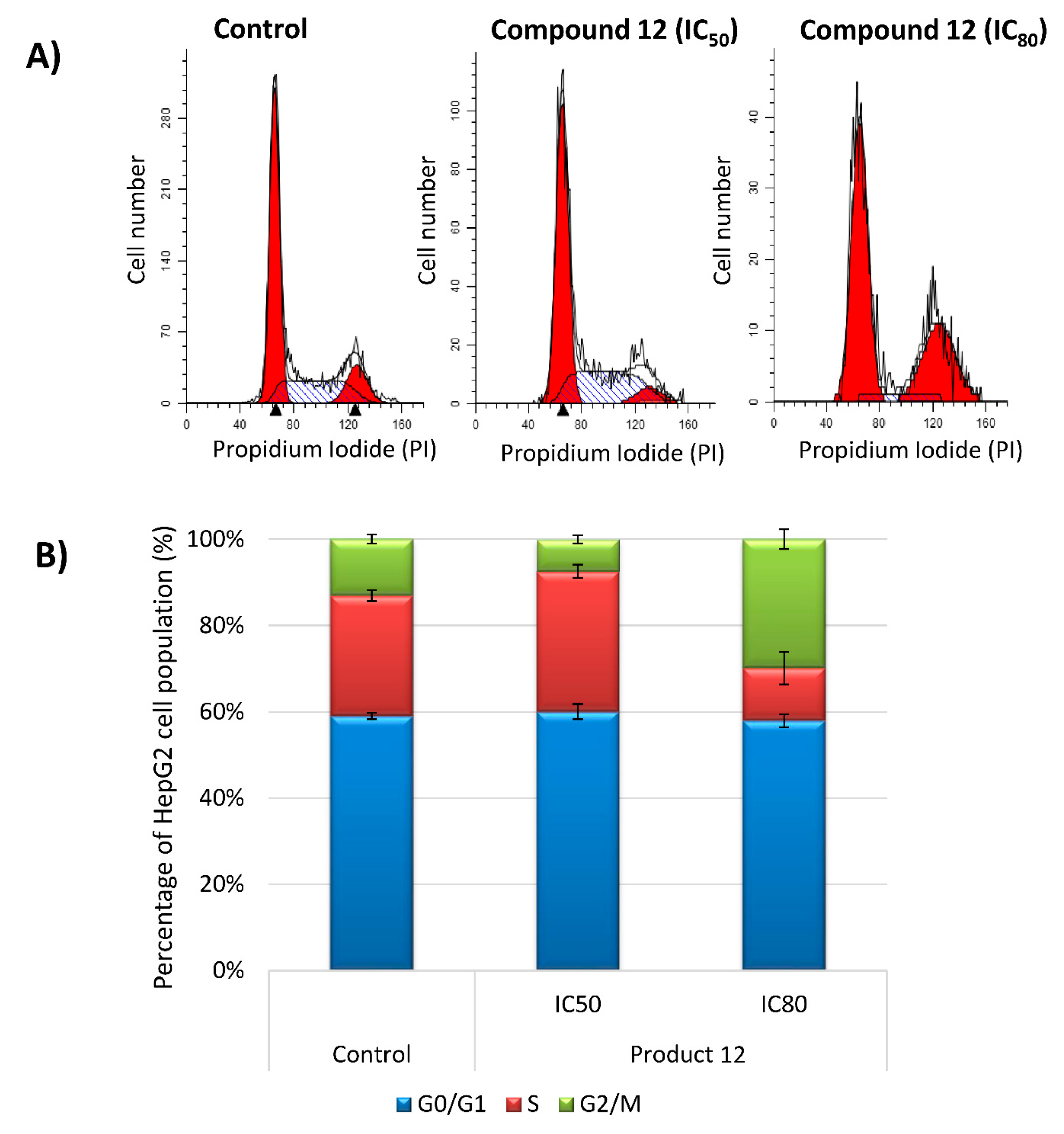
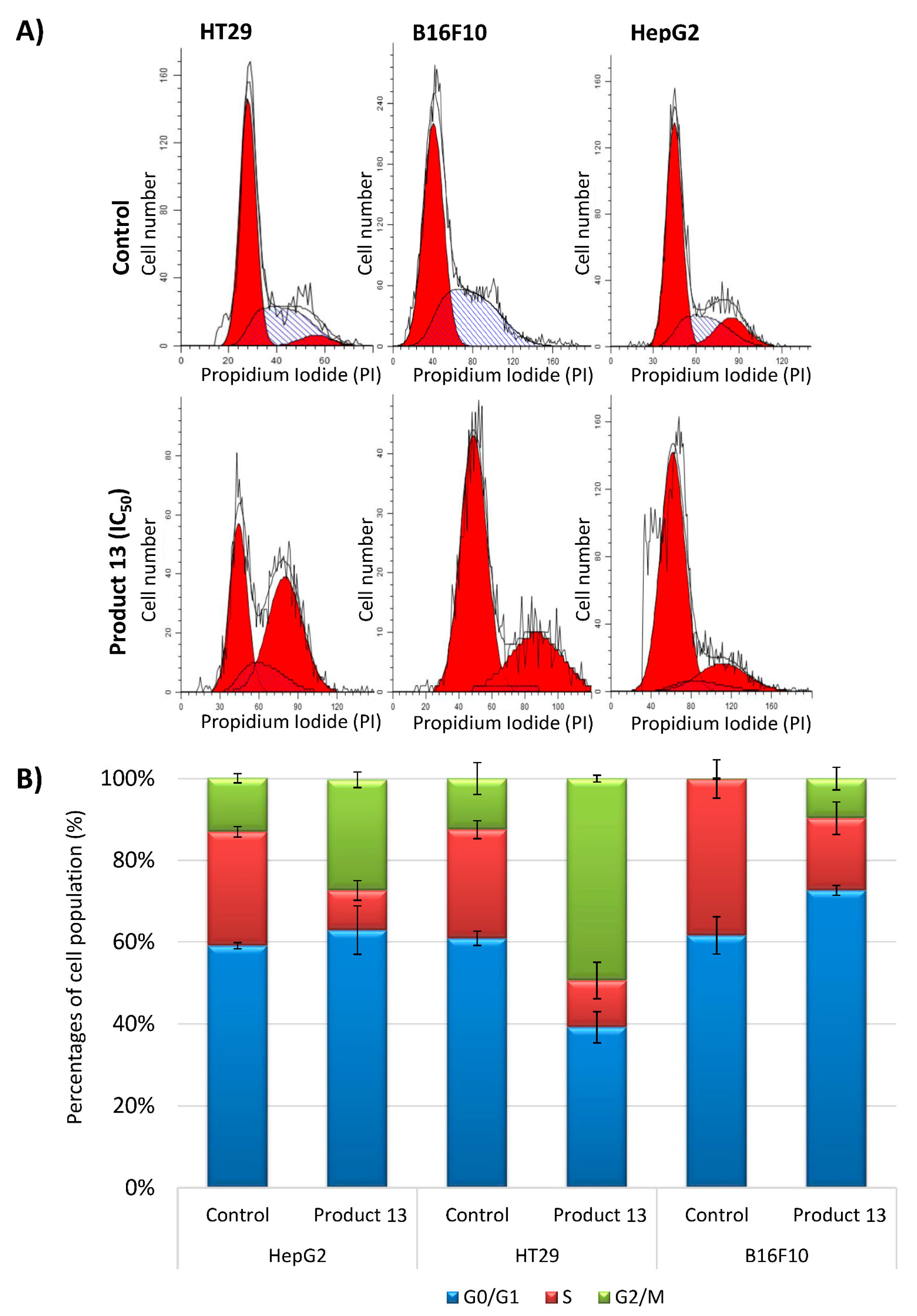
2.2.4. Analysis of Cell Cycle
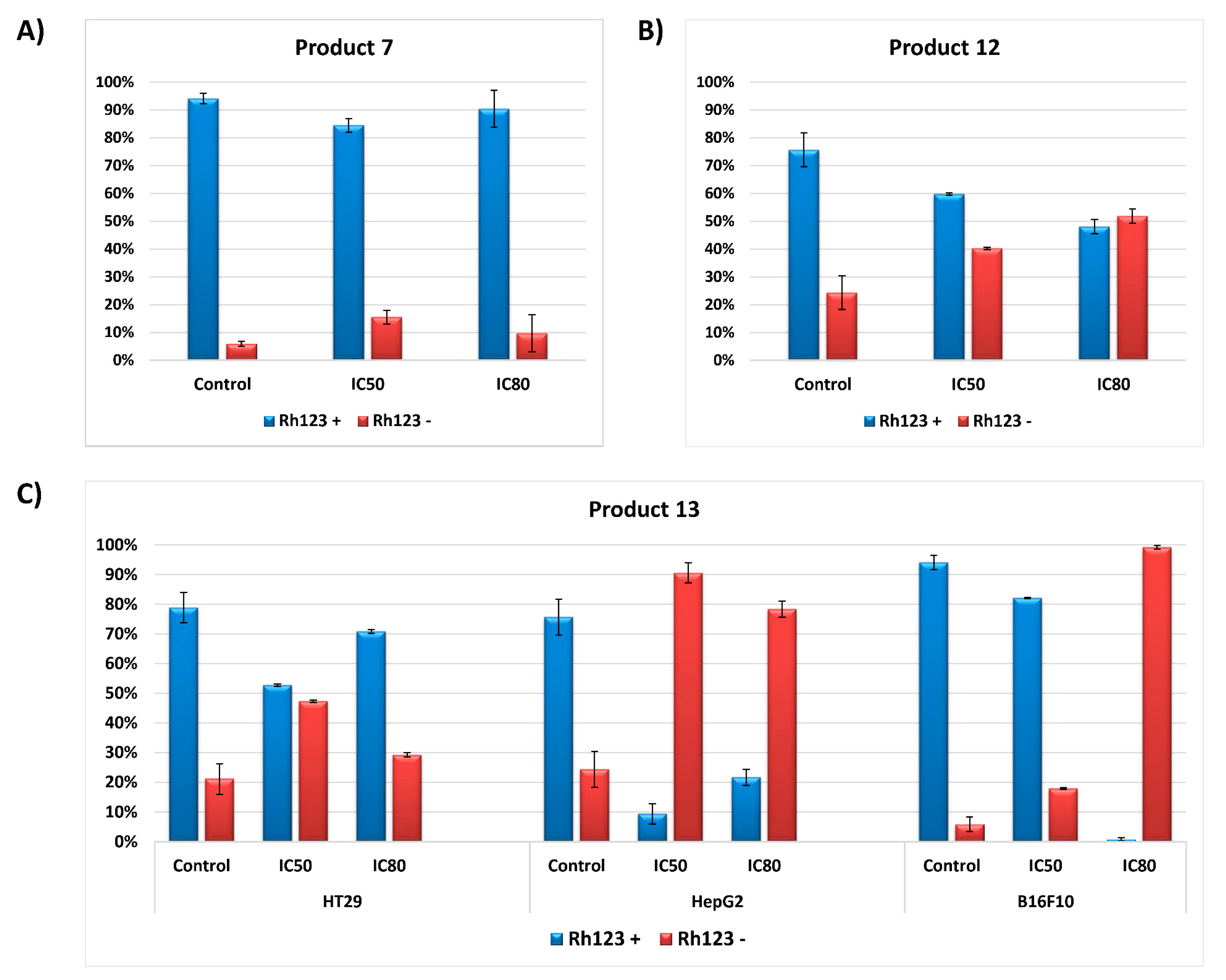
2.2.5. Analysis of Mitochondrial Membrane Potential
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Experimental Procedures
4.1.2. Extraction and Isolation
4.1.3. Synthesis and Structural Characterisation
Structural Characterisation of Compounds (7) and (8)
Synthesis of Acetate 9
Synthesis of Ketone 10
Synthesis of Diketone 11
Synthesis of Hydroxyketone 12
Synthesis of Diol 2
Synthesis of Hydroxyenone 13
4.2. Biological Experimental Procedures
4.2.1. Materials Used
4.2.2. Test Compounds
4.2.3. Cell Culture and Viability Assay
4.2.4. Cell Cycle Analysis
4.2.5. Apoptosis Analysis
4.2.6. Flow Cytometric Analysis of the Mitochondrial Membrane Potential
4.2.7. Statistical ANALYSIS
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fujita, E.; Nagao, Y.; Kaneko, K.; Nakazawa, S.; Kuroda, H. Antitumor and Antibacterial Activity of Isodon Diterpenoids. Chem. Pharm. Bull. 1976, 24, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Riehl, P.S.; DePorre, Y.C.; Armaly, A.M.; Groso, E.J.; Schindler, C.S. New avenues for the synthesis of ent-kaurene diterpenoids. Tetrahedron 2015, 71, 6629–6650. [Google Scholar] [CrossRef]
- Ding, C.; Ding, Y.; Chen, H.; Zhou, J. Chemistry and Bioactivity of ent-Kaurene Diterpenoids. Stud. Nat. Prod. Chem. 2017, 54, 141–197. [Google Scholar]
- Li, J.L.; Chen, Q.Q.; Jin, Q.P.; Gao, J.; Zhao, P.J.; Lu, S.; Zeng, Y. IeCPS2 is potentially involved in the biosynthesis pharmacologically active Isodon diterpenoids rather than gibberellin. Phytochemistry 2012, 76, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.D.; Huang, S.X.; Han, Q.B. Diterpenoids from Isodon species and their biological activities. Nat. Prod. Rep. 2006, 23, 673–698. [Google Scholar] [CrossRef]
- Nagashima, F.T.M.; Asakawa, Y. Terpenoids from some Japonese Liverworts. Phytochemistry 1990, 29, 673–698. [Google Scholar] [CrossRef]
- Grande, M.; Macias, M.J.; Mancheno, B.; Segura, M.; Zarzo, A. New Kaurane Diterpenoids from the Aerial Parts of Distichoselinum-Tenuifolium. J. Nat. Prod. 1991, 54, 866–869. [Google Scholar] [CrossRef]
- Das, B.; Chakravarty, A.K.; Masuda, K.; Suzuki, H.; Ageta, H. A Diterpenoid from Roots of Gelonium-Multiflorum. Phytochemistry 1994, 37, 1363–1366. [Google Scholar] [CrossRef]
- Brito, L.S.; Batista, A.; Santos, F.A.; de Lima, R.P.; Ayala, A.P.; Canuto, K.M.; Silveira, E.R.; Pessoa, O.D.L. Anti-inflammatory kaurane diterpenoids of Erythroxylum bezerrae. Fitoterapia 2023, 165, 105424. [Google Scholar] [CrossRef]
- Chen, K.; Shi, Q.A.; Fujioka, T.; Nakano, T.; Hu, C.Q.; Jin, J.Q.; Kilkuskie, R.E.; Lee, K.H. Anti-Aids Agents.19. Neotripterifordin, a Novel Anti-Hiv Principle from Tripterygium-Wilfordii-Isolation and Structural Elucidation. Bioorgan. Med. Chem. 1995, 3, 1345–1348. [Google Scholar] [CrossRef]
- Corey, E.J.; Liu, K. Enantioselective total synthesis of the potent anti-HIV agent neotripterifordin. Reassignment of stereochemistry at C(16). J. Am. Chem. Soc. 1997, 119, 9929–9930. [Google Scholar] [CrossRef]
- Meade-Tollin, L.C.; Wijeratne, E.M.K.; Cooper, D.; Guild, M.; Jon, E.; Fritz, A.; Zhou, G.X.; Whitesell, L.; Liang, J.Y.; Gunatilaka, A.A.L. Ponicidin and oridonin are responsible for the antiangiogenic activity of Rabdosia rubescens, a constituent of the herbal supplement PCSPES. J. Nat. Prod. 2004, 67, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Huang, R.W.; Lin, D.J.; Peng, J.; Wu, X.Y.; Pan, X.L.; Li, M.Q.; Lin, Q. Anti-proliferative effects of oridonin on SPC-A-1 cells and its mechanism of action. J. Int. Med. Res. 2004, 32, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Wu, X.Y.; Peng, J.; Pan, X.L.; Lu, H.L. Antiproliferation effects of oridonin on HL-60 cells. Ann. Hematol. 2004, 83, 691–695. [Google Scholar] [CrossRef]
- Shi, S.Q.; Fan, Y.Y.; Xu, C.H.; Ding, J.; Wang, G.W.; Yue, J.M. Cytotoxic 8,9-seco-ent-kaurane diterpenoids from Croton kongensis. J. Asian Nat. Prod. Res. 2018, 20, 920–927. [Google Scholar] [CrossRef]
- Fujita, T.T.Y.; Shingu, T.; Kido, M.; Taira, Z. Structures of shikodomedin (X-ray analysis) and shikokiamedin: New cytotoxic 8,9-seco-ent-kaurenoids from Rabdosia shikokiana var. Intermedia. J. Chem. Soc. Chem. Commun. 1982, 162. [Google Scholar] [CrossRef]
- Wang, M.J.; Wang, M.; Zhan, X.Q.; Liu, L.; Wu, Q.; An, F.L.; Lu, Y.B.; Guo, L.L.; Zhang, Z.X.; Fei, D.Q. Antimicrobial diterpenoids from the leaves and twigs of Croton kongensis Gagnepain. Fitoterapia 2023, 164, 105350. [Google Scholar] [CrossRef]
- Li, Y.Q.; Hou, B.L.; Wang, M.J.; Wang, R.Y.; Chen, X.H.; Liu, X.; Fei, D.Q.; Zhang, Z.X.; Li, E.W. Diterpenoids and C(13) Nor-Isoprenoid Identified From the Leaves and Twigs of Croton yanhuii Activating Apoptosis and Pyroptosis. Front. Chem. 2022, 10, 861278. [Google Scholar] [CrossRef]
- Grande, M.; Segura, M.; Mancheno, B. New Kaurane Diterpenoids from the Roots of Elaeoselinum-Tenuifolium. J. Nat. Prod. 1986, 49, 259–264. [Google Scholar] [CrossRef]
- Xu, H.Z.; Huang, Y.; Wu, Y.L.; Zhao, Y.; Xiao, W.L.; Lin, Q.S.; Sun, H.D.; Dai, W.; Chen, G.Q. Pharicin A, a novel natural ent-kaurene diterpenoid, induces mitotic arrest and mitotic catastrophe of cancer cells by interfering with BubR1 function. Cell Cycle 2010, 9, 2897–2907. [Google Scholar] [CrossRef]
- Da Costa, R.M.; Bastos, J.K.; Costa, M.C.A.; Ferreira, M.M.C.; Mizuno, C.S.; Caramori, G.F.; Nagurniak, G.R.; Simao, M.R.; Dos Santos, R.A.; Veneziani, R.C.S.; et al. In vitro cytotoxicity and structure-activity relationship approaches of ent-kaurenoic acid derivatives against human breast carcinoma cell line. Phytochemistry 2018, 156, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Goes, L.D.M.C.H.O.; Duarte, J.L.; Salgueiro, L.R.; Cavaleiro, C.; Perazzo, F.F.; Carvalho, J.C.T. Effect of Distichoselinum tenuifolium (Lag.) Garcia Martin & Silvestre essential oil on analgesic and behavioral assays. Afr. J. Pharm. Pharmacol. 2015, 9, 460–467. [Google Scholar]
- Grande, M.; Moran, J.R.; Macias, M.J.; Mancheno, B. C-13 Nuclear-Magnetic-Resonance Spectra of Some Tetracyclic Diterpenoids Isolated from Elaeoselinum Species. Phytochem. Anal. 1993, 4, 19–24. [Google Scholar] [CrossRef]
- Villalobos, N.; Martin, L.; Macias, M.J.; Mancheno, B.; Grande, M. Gibberellin-Like Activity of Some Tetracyclic Diterpenoids from Elaeoselinum Species and Their Derivatives. Phytochemistry 1994, 37, 635–639. [Google Scholar] [CrossRef]
- Jannus, F.; Medina-O’Donnell, M.; Rivas, F.; Díaz-Ruiz, L.; Rufino-Palomares, E.E.; Lupiáñez, J.A.; Parra, A.; Reyes-Zurita, F.J. A Diamine-PEGylated Oleanolic Acid Derivative Induced Efficient Apoptosis through a Death Receptor and Mitochondrial Apoptotic Pathway in HepG2 Human Hepatoma Cells. Biomolecules 2020, 10, 1375. [Google Scholar] [CrossRef]
- Zentar, H.; Jannus, F.; Medina-O’Donnell, M.; El Mansouri, A.E.; Fernández, A.; Justicia, J.; Alvarez-Manzaneda, E.; Reyes-Zurita, F.J.; Chahboun, R. Synthesis of Tricyclic Pterolobirin H Analogue: Evaluation of Anticancer and Anti-Inflammatory Activities and Molecular Docking Investigations. Molecules 2023, 28, 6208. [Google Scholar] [CrossRef]
- Hossain, M.; Das, U.; Dimmock, J.R. Recent advances in α,β-unsaturated carbonyl compounds as mitochondrial toxins. Eur. J. Med. Chem. 2019, 183, 111687. [Google Scholar] [CrossRef]
- Ding, Y.; Ding, C.Y.; Ye, N.; Liu, Z.Q.; Wold, E.A.; Chen, H.Y.; Wild, C.; Shen, Q.; Zhou, J. Discovery and development of natural product oridonin-inspired anticancer agents. Eur. J. Med. Chem. 2016, 122, 102–117. [Google Scholar] [CrossRef]
- Sarwar, M.S.; Xia, Y.X.; Liang, Z.M.; Tsang, S.W.; Zhang, H.J. Mechanistic Pathways and Molecular Targets of Plant-Derived Anticancer ent-Kaurane Diterpenes. Biomolecules 2020, 10, 144. [Google Scholar] [CrossRef]
- Du, J.; Chen, C.Y.; Sun, Y.Q.; Zheng, L.; Wang, W.C. Ponicidin suppresses HT29 cell growth via the induction of G1 cell cycle arrest and apoptosis. Mol. Med. Rep. 2015, 12, 5816–5820. [Google Scholar] [CrossRef]
- Liu, J.J.; Zhang, Y.; Guang, W.B.; Yang, H.Z.; Lin, D.J.; Xiao, R.Z. Ponicidin Inhibits Monocytic Leukemia Cell Growth by Induction of Apoptosis. Int. J. Mol. Sci. 2008, 9, 2265–2277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; Han, Q.B.; Zhao, A.H.; Sun, H.D. Diterpenoids from Isodon japonica. Fitoterapia 2003, 74, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, M.; He, P.C.; Zhao, J.J.; Chen, Y.; Qi, J.; Wang, Y. Proteomic analysis of oridonin-induced apoptosis in multiple myeloma cells. Mol. Med. Rep. 2017, 15, 1807–1815. [Google Scholar] [CrossRef]
- Kibet, S.; Kimani, N.M.; Mwanza, S.S.; Mudalungu, C.M.; Santos, C.B.R.; Tanga, C.M. Unveiling the Potential of ent-Kaurane Diterpenoids: Multifaceted Natural Products for Drug Discovery. Pharmaceuticals 2024, 17, 510. [Google Scholar] [CrossRef]
- Zentar, H.; Jannus, F.; Medina-O’Donnell, M.; Lupiáñez, J.A.; Justicia, J.; Alvarez-Manzaneda, R.; Reyes-Zurita, F.J.; Alvarez-Manzaneda, E.; Chahboun, R. Synthesis and Biological Evaluation of Cassane Diterpene (5α)-Vuacapane-8(14), 9(11)-Diene and of Some Related Compounds. Molecules 2022, 27, 5705. [Google Scholar] [CrossRef]
- Zentar, H.; Jannus, F.; Gutierrez, P.; Medina-O’Donnell, M.; Lupiáñez, J.A.; Reyes-Zurita, F.J.; Alvarez-Manzaneda, E.; Chahboun, R. Semisynthesis and Evaluation of Anti-Inflammatory Activity of the Cassane-Type Diterpenoid Taepeenin F and of Some Synthetic Intermediates. J. Nat. Prod. 2022, 85, 2372–2384. [Google Scholar] [CrossRef]
- Li, D.; Han, T.; Liao, J.; Hu, X.; Xu, S.; Tian, K.; Gu, X.; Cheng, K.; Li, Z.; Hua, H.; et al. Oridonin, a Promising ent-Kaurane Diterpenoid Lead Compound. Int. J. Mol. Sci. 2016, 17, 1395. [Google Scholar] [CrossRef]
- Liu, X.; Kang, J.; Wang, H.; Huang, T. Mitochondrial ROS contribute to oridonin-induced HepG2 apoptosis through PARP activation. Oncol. Lett. 2018, 15, 2881–2888. [Google Scholar] [CrossRef]
- Santagata, S.; Xu, Y.M.; Wijeratne, E.M.; Kontnik, R.; Rooney, C.; Perley, C.C.; Kwon, H.; Clardy, J.; Kesari, S.; Whitesell, L.; et al. Using the heat-shock response to discover anticancer compounds that target protein homeostasis. ACS Chem. Biol. 2012, 7, 340–349. [Google Scholar] [CrossRef]
- You, L.T.; Dong, X.X.; Ni, B.R.; Fu, J.; Yang, C.J.; Yin, X.B.; Leng, X.; Ni, J. Triptolide Induces Apoptosis Through Fas Death and Mitochondrial Pathways in HepaRG Cell Line. Front. Pharmacol. 2018, 9, 813. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Compound | IC20 (µM) | IC50 (µM) | IC80 (µM) | IC50 7/IC50 # |
|---|---|---|---|---|---|
| HT29 | 2 | 1.90 ± 1.12 | 10.08 ± 2.69 | 50.55 ± 4.37 | 7.46 |
| 7 | 71.82 ± 0.27 | 75.27 ± 0.90 | 79.47 ± 2.53 | 1.00 | |
| 8 | 83.57 ± 4.89 | 111.72 ± 3.39 | 148.32 ± 2.67 | 0.67 | |
| 9 | 92.42 ± 2.27 | 153.83 ± 0.71 | 202.16 ± 1.70 | 0.49 | |
| 11 | 49.71 ± 3.29 | 74.96 ± 5.91 | 114.91 ± 11.43 | 1.00 | |
| 12 | 27.28 ± 8.36 | 45.37 ± 5.34 | 85.58 ± 27.54 | 1.66 | |
| 13 | 0.73 ± 0.08 | 2.71 ± 0.23 | 8.43 ± 2.61 | 27.76 | |
| HepG2 | 2 | 39.15 ± 15.37 | 70.22 ± 9.92 | 125.00 ± 4.66 | 1.04 |
| 7 | 71.38 ± 0.66 | 72.84 ± 0.29 | 74.30 ± 0.15 | 1.00 | |
| 8 | 66.06 ± 12.57 | 107.69 ± 6.08 | 166.59 ± 7.72 | 0.68 | |
| 9 | 48.02 ± 7.57 | 101.49 ± 7.65 | 171.30 ± 6.00 | 0.72 | |
| 11 | 31.76 ± 4.10 | 47.26 ± 5.78 | 72.90 ± 10.87 | 1.54 | |
| 12 | 8.79 ± 9.30 | 24.43 ± 16.35 | 81.75 ± 13.64 | 2.98 | |
| 13 | 0.79 ± 0.30 | 2.12 ± 0.23 | 5.36 ± 0.86 | 34.42 | |
| B16-F10 | 2 | 52.32 ± 3.74 | 79.58 ± 3.58 | 125.56 ± 5.55 | 0.41 |
| 7 | 27.40 ± 4.62 | 32.43 ± 3.28 | 37.93 ± 2.94 | 1.00 | |
| 8 | 128.36 ± 2.81 | 145.32 ± 2.76 | 168.42 ± 3.60 | 0.22 | |
| 9 | 83.33 ± 4.50 | 107.13 ± 3.95 | 138.94 ± 4.68 | 0.30 | |
| 11 | 25.04 ± 2.28 | 42.04 ± 1.55 | 66.97 ± 1.05 | 0.77 | |
| 12 | 64.26 ± 6.65 | 94.54 ± 8.33 | 143.50 ± 9.36 | 0.34 | |
| 13 | 0.79 ± 0.13 | 2.65 ± 0.13 | 54.29 ± 4.00 | 12.26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasser, Y.K.; Gil, D.; Zentar, H.; Durán-Peña, M.J.; Prados-Lopez, B.; Juárez-Moreno, J.; Botubol-Ares, J.M.; Haidour, A.; Sainz, J.; Fernández, A.; et al. Semisynthesis and Antitumour Evaluation of Natural Derivatives from ent-Kaurene ent-15α-Angeloyloxykaur-l6-en-3β-ol Isolated from Distichoselinum tenuifolium. Int. J. Mol. Sci. 2024, 25, 13222. https://doi.org/10.3390/ijms252313222
Yasser YK, Gil D, Zentar H, Durán-Peña MJ, Prados-Lopez B, Juárez-Moreno J, Botubol-Ares JM, Haidour A, Sainz J, Fernández A, et al. Semisynthesis and Antitumour Evaluation of Natural Derivatives from ent-Kaurene ent-15α-Angeloyloxykaur-l6-en-3β-ol Isolated from Distichoselinum tenuifolium. International Journal of Molecular Sciences. 2024; 25(23):13222. https://doi.org/10.3390/ijms252313222
Chicago/Turabian StyleYasser, Yass K., Daniel Gil, Houda Zentar, María Jesús Durán-Peña, Belen Prados-Lopez, Jorge Juárez-Moreno, José Manuel Botubol-Ares, Ali Haidour, Juan Sainz, Antonio Fernández, and et al. 2024. "Semisynthesis and Antitumour Evaluation of Natural Derivatives from ent-Kaurene ent-15α-Angeloyloxykaur-l6-en-3β-ol Isolated from Distichoselinum tenuifolium" International Journal of Molecular Sciences 25, no. 23: 13222. https://doi.org/10.3390/ijms252313222
APA StyleYasser, Y. K., Gil, D., Zentar, H., Durán-Peña, M. J., Prados-Lopez, B., Juárez-Moreno, J., Botubol-Ares, J. M., Haidour, A., Sainz, J., Fernández, A., Alvarez-Manzaneda, R., Chahboun, R., & Reyes-Zurita, F. J. (2024). Semisynthesis and Antitumour Evaluation of Natural Derivatives from ent-Kaurene ent-15α-Angeloyloxykaur-l6-en-3β-ol Isolated from Distichoselinum tenuifolium. International Journal of Molecular Sciences, 25(23), 13222. https://doi.org/10.3390/ijms252313222