

Genome-Wide Identification and Characterization of GRAS Transcription Factor Family in Cultivated Hybrid Sugarcane ZZ1 (Saccharum officinarum) and Their Role in Development and Stress

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Identification and Localization of the GRAS TFs
2.2. Molecular Weight, Isoelectric Point
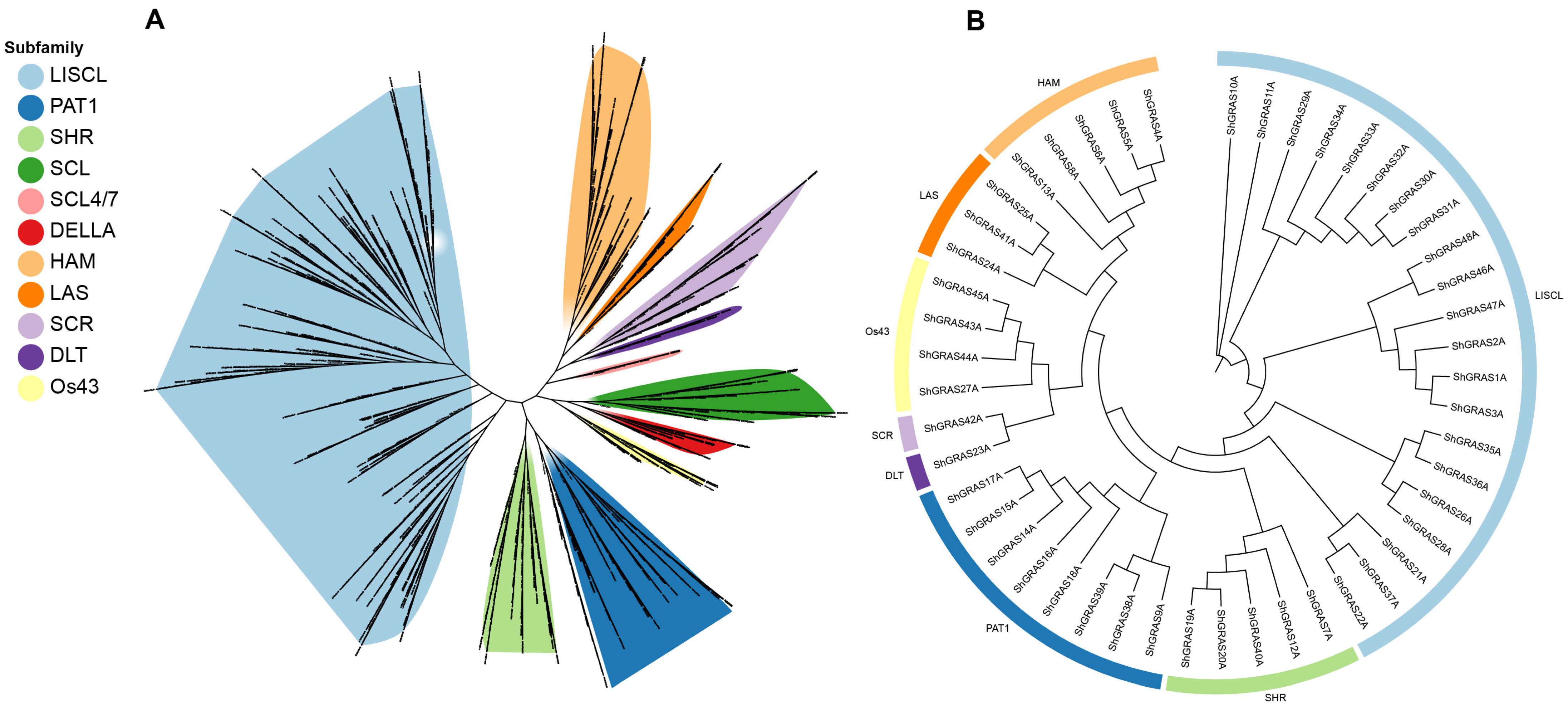
2.3. Phylogenetic and Clade Analyses
2.4. Protein Motif and Gene Structure Analysis
2.5. Analysis of Cis-Regulatory Elements in Promoter Regions
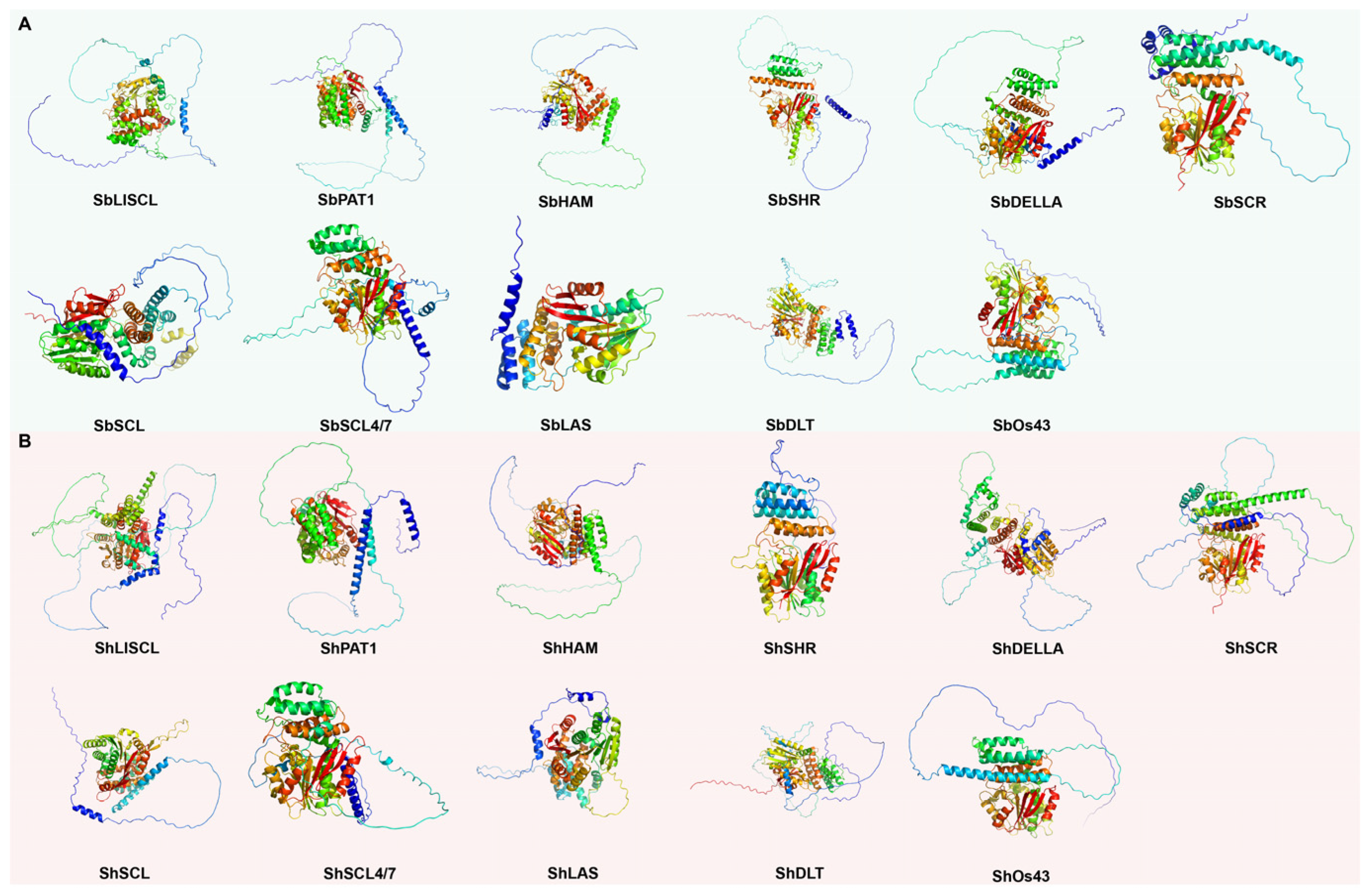
2.6. Tertiary Structure
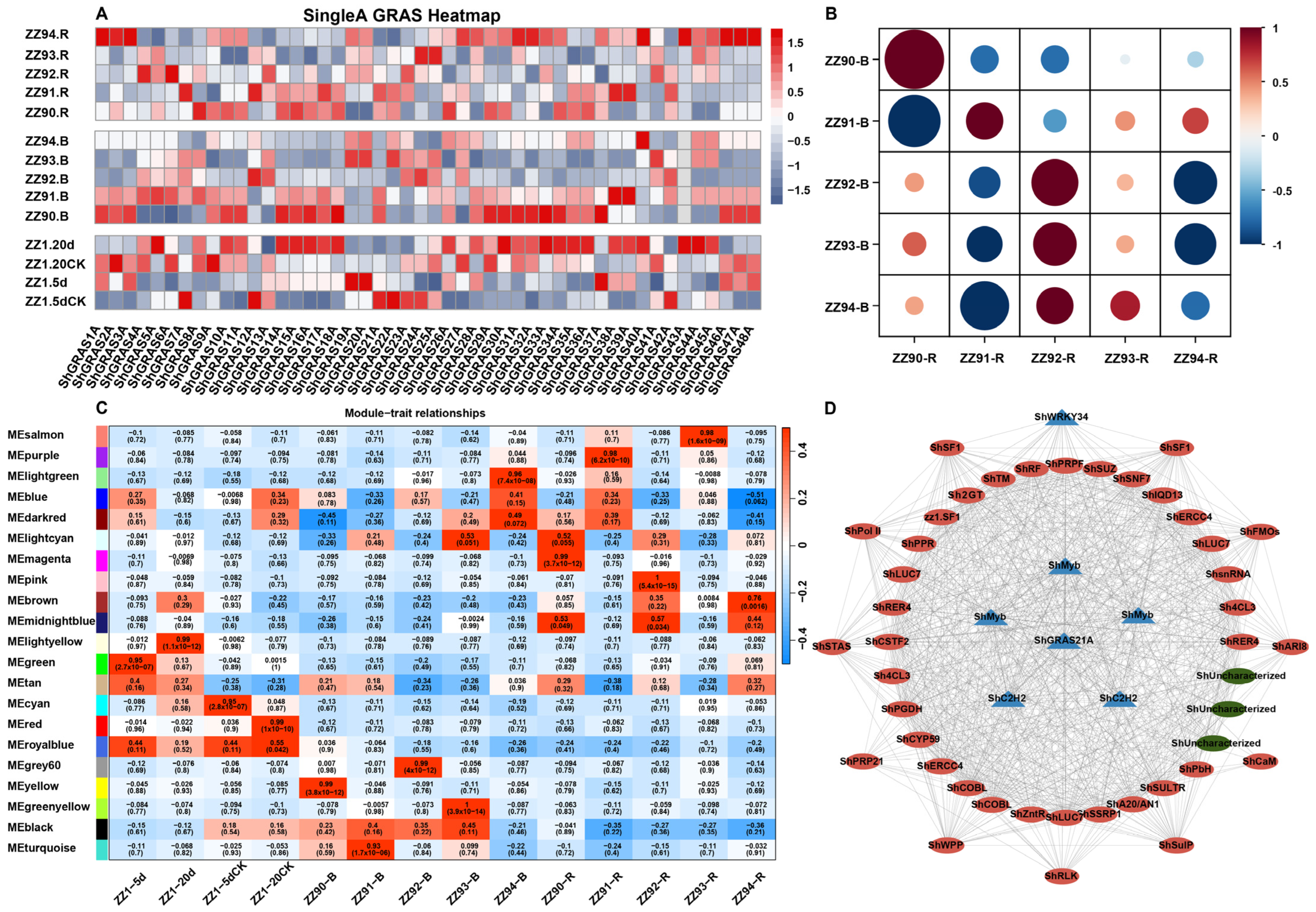
2.7. Expression and Network Analysis
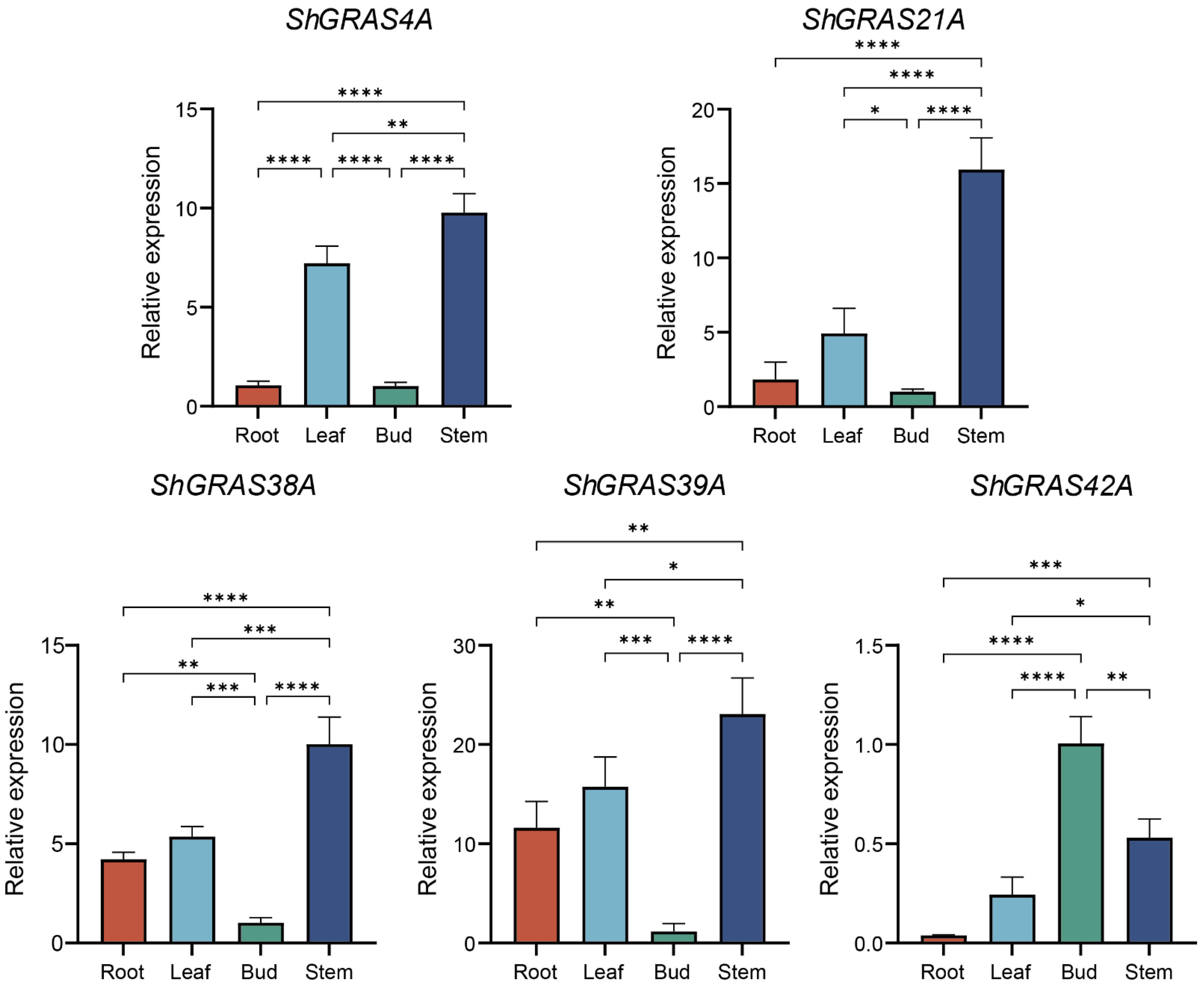
2.8. Expression Pattern Analysis of ShGRASs by RT-qPCR
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Data Resources
5.2. GRAS TFs Identification and Chromosome Distribution
5.3. Molecular Weight, Isoelectric Point Analysis
5.4. Phylogenetic and Class Analyses
5.5. Gene Structure and Conserved Motif Analysis
5.6. Promoter Analysis
5.7. Tertiary Structure Analysis
5.8. Transcriptome and Expression Analysis
5.9. RNA Isolation and RT-qPCR Expression Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Karin, M. Too Many Transcription Factors: Positive and Negative Interactions. New Biol. 1990, 2, 126–131. [Google Scholar] [PubMed]
- Latchman, D.S. Transcription Factors: An Overview. Int. J. Exp. Pathol. 1993, 74, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Xian, Z.; Kang, X.; Tang, N.; Li, Z. Genome-Wide Identification, Phylogeny and Expression Analysis of GRAS Gene Family in Tomato. BMC Plant Biol. 2015, 15, 209. [Google Scholar] [CrossRef]
- Paz-Ares, J.; Ghosal, D.; Wienand, U.; Peterson, P.A.; Saedler, H. The Regulatory C1 Locus of Zea Mays Encodes a Protein with Homology to Myb Proto-Oncogene Products and with Structural Similarities to Transcriptional Activators. EMBO J. 1987, 6, 3553–3558. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Liu, H.; Zhu, K.; Cheng, Z.-M. Evolution and Functional Analysis of the GRAS Family Genes in Six Rosaceae Species. BMC Plant Biol. 2022, 22, 569. [Google Scholar] [CrossRef]
- Fan, Y.; Yan, J.; Lai, D.; Yang, H.; Xue, G.; He, A.; Guo, T.; Chen, L.; Cheng, X.; Xiang, D.; et al. Genome-Wide Identification, Expression Analysis, and Functional Study of the GRAS Transcription Factor Family and Its Response to Abiotic Stress in Sorghum [Sorghum Bicolor (L.) Moench]. BMC Genom. 2021, 22, 509. [Google Scholar] [CrossRef]
- Peng, J.; Carol, P.; Richards, D.E.; King, K.E.; Cowling, R.J.; Murphy, G.P.; Harberd, N.P. The Arabidopsis GAI Gene Defines a Signaling Pathway That Negatively Regulates Gibberellin Responses. Genes Dev. 1997, 11, 3194–3205. [Google Scholar] [CrossRef]
- Pysh, L.D.; Wysocka-Diller, J.W.; Camilleri, C.; Bouchez, D.; Benfey, P.N. The GRAS Gene Family in Arabidopsis: Sequence Characterization and Basic Expression Analysis of the SCARECROW-LIKE Genes. Plant J. 1999, 18, 111–119. [Google Scholar] [CrossRef]
- Rana, D.; Sharma, P.; Arpita, K.; Srivastava, H.; Sharma, S.; Gaikwad, K. Genome-Wide Identification and Characterization of GRAS Gene Family in Pigeonpea (Cajanus cajan (L.) Millspaugh). 3 Biotech 2023, 13, 363. [Google Scholar] [CrossRef]
- Silverstone, A.L.; Ciampaglio, C.N.; Sun, T. The Arabidopsis RGA Gene Encodes a Transcriptional Regulator Repressing the Gibberellin Signal Transduction Pathway. Plant Cell 1998, 10, 155–169. [Google Scholar] [CrossRef]
- Huang, X.; Zentella, R.; Park, J.; Reser, L.; Bai, D.L.; Ross, M.M.; Shabanowitz, J.; Hunt, D.F.; Sun, T. Phosphorylation Activates Master Growth Regulator DELLA by Promoting Histone H2A Binding at Chromatin in Arabidopsis. Nat. Commun. 2024, 15, 7694. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, N.R. A Structure for Plant-Specific Transcription Factors: The GRAS Domain Revealed. Plant Cell 2016, 28, 993–994. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Widmer, A.; Li, S.; Zhao, Y.; Zhao, Z.; Wu, X.; Sun, L.; Liu, Q.; Wu, Y. Crystal Structure of the GRAS Domain of SCARECROW-LIKE7 in Oryza Sativa. Plant Cell 2016, 28, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Hakoshima, T. Structural Basis of the Specific Interactions of GRAS Family Proteins. FEBS Lett. 2018, 592, 489–501. [Google Scholar] [CrossRef]
- Liu, X.; Widmer, A. Genome-Wide Comparative Analysis of the GRAS Gene Family in Populus, Arabidopsis and Rice. Plant Mol. Biol. Report. 2014, 32, 1129–1145. [Google Scholar] [CrossRef]
- Tian, C.; Wan, P.; Sun, S.; Li, J.; Chen, M. Genome-Wide Analysis of the GRAS Gene Family in Rice and Arabidopsis. Plant Mol. Biol. 2004, 54, 519–532. [Google Scholar] [CrossRef]
- Hirsch, S.; Kim, J.; Muñoz, A.; Heckmann, A.B.; Downie, J.A.; Oldroyd, G.E.D. GRAS Proteins Form a DNA Binding Complex to Induce Gene Expression during Nodulation Signaling in Medicago truncatula. Nat. Genet. 2009, 21, 545–557. [Google Scholar] [CrossRef]
- To, V.-T.; Shi, Q.; Zhang, Y.; Shi, J.; Shen, C.; Zhang, D.; Cai, W. Genome-Wide Analysis of the GRAS Gene Family in Barley (Hordeum Vulgare L.). Genes 2020, 11, 553. [Google Scholar] [CrossRef]
- Song, X.-M.; Liu, T.-K.; Duan, W.-K.; Ma, Q.-H.; Ren, J.; Wang, Z.; Li, Y.; Hou, X.-L. Genome-Wide Analysis of the GRAS Gene Family in Chinese Cabbage (Brassica rapa ssp. Pekinensis). Genomics 2014, 103, 135–146. [Google Scholar] [CrossRef]
- Zhao, X.; Xu, Y.; He, G.; He, K.; Xiao, L.; Hu, R.; Li, S. Genome-Wide Characterization and Expression Profiling of the GRAS Gene Family in Salt and Alkali Stresses in Miscanthus Sinensis. Int. J. Mol. Sci. 2022, 23, 14521. [Google Scholar] [CrossRef]
- Tong, N.; Li, D.; Zhang, S.; Tang, M.; Chen, Y.; Zhang, Z.; Huang, Y.; Lin, Y.; Cheng, Z.; Lai, Z. Genome-Wide Identification and Expression Analysis of the GRAS Family under Low-Temperature Stress in Bananas. Front. Plant Sci. 2023, 14, 1216070. [Google Scholar] [CrossRef] [PubMed]
- Zaman, Q.U.; Hussain, M.A.; Khan, L.U.; Cui, J.-P.; Hui, L.; Khan, D.; Lv, W.; Wang, H.-F. Genome-Wide Identification and Expression Pattern of the GRAS Gene Family in Pitaya (Selenicereus undatus L.). Biology 2022, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Mi, L.; Xu, L.; Yu, C.; Li, C.; Chen, C. Genome-Wide Identification, Characterization, Interaction Network and Expression Profile of GRAS Gene Family in Sweet Orange (Citrus sinensis). Sci. Rep. 2019, 9, 2156. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Bhalothia, P.; Chen, J.; Yan, Q.; Li, J.; Feng, L.; Zhang, Y.; Xu, J.; Xia, R.; Zeng, Z.; et al. The GRAS Gene Family and Its Roles in Seed Development in Litchi (Litchi chinensis Sonn). BMC Plant Biol. 2021, 21, 423. [Google Scholar] [CrossRef]
- Jing, C.-Y.; Zhang, F.-M.; Wang, X.-H.; Wang, M.-X.; Zhou, L.; Cai, Z.; Han, J.-D.; Geng, M.-F.; Yu, W.-H.; Jiao, Z.-H.; et al. Genomic Insights into the Recent Chromosome Reduction of Autopolyploid Sugarcane Saccharum spontaneum. Nat. Genet. 2022, 54, 885–896. [Google Scholar] [CrossRef]
- Yu, F.; Wang, P.; Li, X.; Huang, Y.; Wang, Q.; Luo, L.; Jing, Y.; Liu, X.; Deng, Z.; Wu, J.; et al. Characterization of Chromosome Composition of Sugarcane in Nobilization by Using Genomic in Situ Hybridization. Mol. Cytogenet. 2018, 11, 35. [Google Scholar] [CrossRef]
- Bao, Y.; Zhang, Q.; Huang, J.; Zhang, S.; Yao, W.; Yu, Z.; Deng, Z.; Yu, J.; Kong, W.; Yu, X.; et al. A Chromosomal-Scale Genome Assembly of Modern Cultivated Hybrid Sugarcane Provides Insights into Origination and Evolution. Nat. Commun. 2024, 15, 3041. [Google Scholar] [CrossRef]
- Cenci, A.; Rouard, M. Evolutionary Analyses of GRAS Transcription Factors in Angiosperms. Front. Plant Sci. 2017, 8, 273. [Google Scholar] [CrossRef]
- Liu, B.; Sun, Y.; Xue, J.; Jia, X.; Li, R. Genome-Wide Characterization and Expression Analysis of GRAS Gene Family in Pepper (Capsicum Annuum L.). PeerJ 2018, 6, e4796. [Google Scholar] [CrossRef]
- Wang, X.; Li, G.; Sun, Y.; Qin, Z.; Feng, P. Genome-Wide Analysis and Characterization of GRAS Family in Switchgrass. Bioengineered 2021, 12, 6096–6114. [Google Scholar] [CrossRef]
- Sun, X.; Jones, W.T.; Rikkerink, E.H.A. GRAS Proteins: The Versatile Roles of Intrinsically Disordered Proteins in Plant Signalling. Biochem. J. 2012, 442, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dobrev, V.S.; Fred, L.M.; Gerhart, K.P.; Metallo, S.J. Characterization of the Binding of Small Molecules to Intrinsically Disordered Proteins. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 611, ISBN 978-0-12-815649-0. [Google Scholar]
- Lee, R.; van der Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of Intrinsically Disordered Regions and Proteins. Available online: https://pubs.acs.org/doi/full/10.1021/cr400525m (accessed on 12 October 2024).
- Sun, X.; Xue, B.; Jones, W.T.; Rikkerink, E.; Dunker, A.K.; Uversky, V.N. A Functionally Required Unfoldome from the Plant Kingdom: Intrinsically Disordered N-Terminal Domains of GRAS Proteins Are Involved in Molecular Recognition during Plant Development. Plant Mol. Biol. 2011, 77, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Diao, Z.; Yang, D.; Wang, X.; Zheng, X.; Xiang, X.; Xiao, Y.; Chen, Z.; Wang, W.; Wu, Y.; et al. The 14-3-3 Protein GF14c Positively Regulates Immunity by Modulating the Protein Homoeostasis of the GRAS Protein OsSCL7 in Rice. Plant Cell Environ. 2022, 45, 1065–1081. [Google Scholar] [CrossRef] [PubMed]
- De Vleesschauwer, D.; Seifi, H.S.; Filipe, O.; Haeck, A.; Huu, S.N.; Demeestere, K.; Höfte, M. The DELLA Protein SLR1 Integrates and Amplifies Salicylic Acid- and Jasmonic Acid-Dependent Innate Immunity in Rice. Plant Physiol. 2016, 170, 1831–1847. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Xiao, H.; Yao, P.; Ma, X.; Shi, Q.; Yang, J.; Hou, H.; Li, L. Unveiling the Mechanism of Broad-spectrum Blast Resistance in Rice: The Collaborative Role of Transcription Factor OsGRAS30 and Histone Deacetylase OsHDAC1. Plant Biotechnol. J. 2024, 22, 1740–1756. [Google Scholar] [CrossRef]
- Gullerova, M.; Barta, A.; Lorković, Z.J. AtCyp59 Is a Multidomain Cyclophilin from Arabidopsis thaliana That Interacts with SR Proteins and the C-Terminal Domain of the RNA Polymerase II. RNA 2006, 12, 631–643. [Google Scholar] [CrossRef]
- Sharma, A.; Sharma, D.; Verma, S.K. Zinc Binding Proteome of a Phytopathogen Xanthomonas Translucens Pv. Undulosa. Plant Physiol. Biochem. 2019, 6, 190369. [Google Scholar] [CrossRef]
- Sharma, A.; Sharma, D.; Verma, S.K.; Saha, D.; Prasad, A.M.; Srinivasan, R. Pentatricopeptide Repeat Proteins and Their Emerging Roles in Plants. Plant Physiol. Biochem. 2007, 45, 521–534. [Google Scholar] [CrossRef]
- Zeiner, A.; Colina, F.J.; Citterico, M.; Wrzaczek, M. CYSTEINE-RICH RECEPTOR-LIKE PROTEIN KINASES: Their Evolution, Structure, and Roles in Stress Response and Development. J. Exp. Bot. 2023, 74, 4910–4927. [Google Scholar] [CrossRef]
- Craig, A.; Ewan, R.; Mesmar, J.; Gudipati, V.; Sadanandom, A. E3 Ubiquitin Ligases and Plant Innate Immunity. J. Exp. Bot. 2009, 60, 1123–1132. [Google Scholar] [CrossRef]
- Snedden, W.A.; Fromm, H. Calmodulin, Calmodulin-Related Proteins and Plant Responses to the Environment. Trends Plant Sci. 1998, 3, 299–304. [Google Scholar] [CrossRef]
- Vij, S.; Tyagi, A.K. A20/AN1 Zinc-Finger Domain-Containing Proteins in Plants and Animals Represent Common Elements in Stress Response. Funct. Integr. Genom. 2008, 8, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Wani, S.H.; Anand, S.; Singh, B.; Bohra, A.; Joshi, R. WRKY Transcription Factors and Plant Defense Responses: Latest Discoveries and Future Prospects. Plant Cell Rep. 2021, 40, 1071–1085. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER Web Server: Interactive Sequence Similarity Searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Gel, B.; Serra, E. karyoploteR: An R/Bioconductor Package to Plot Customizable Genomes Displaying Arbitrary Data. Bioinformatics 2017, 33, 3088–3090. [Google Scholar] [CrossRef]
- Osorio, D.; Rondón-Villarreal, P.; Torres, R. Peptides: A Package for Data Mining of Antimicrobial Peptides. R J. 2015, 7, 4. [Google Scholar] [CrossRef]
- Edgar, R.C.; Li, W.-Y.; Wang, X.; Li, R.; Li, W.-Q.; Chen, K.-M. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Dutta, M.; Saha, A.; Moin, M.; Kirti, P.B. Genome-Wide Identification, Transcript Profiling and Bioinformatic Analyses of GRAS Transcription Factor Genes in Rice. Front. Plant Sci. 2021, 12, 777285. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M. PlantCARE, a Database of Plant Cis-Acting Regulatory Elements and a Portal to Tools for in Silico Analysis of Promoter Sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Bramucci, E.; Paiardini, A.; Bossa, F.; Pascarella, S. PyMod: Sequence Similarity Searches, Multiple Sequence-Structure Alignments, and Homology Modeling within PyMOL. BMC Bioinform. 2012, 13, S2. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Zhang, Y.; Park, C.; Bennett, C.; Thornton, M.; Kim, D. Rapid and Accurate Alignment of Nucleotide Conversion Sequencing Reads with HISAT-3N. Genome Res. 2021, 31, 1290–1295. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, L.; Lun, A.T.L.; Baldoni, P.L.; Smyth, G.K. edgeR 4.0: Powerful Differential Analysis of Sequencing Data with Expanded Functionality and Improved Support for Small Counts and Larger Datasets. bioRxiv 2024. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The R Package Rsubread Is Easier, Faster, Cheaper and Better for Alignment and Quantification of RNA Sequencing Reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GRAS Subfamilies | Fraction Disordered | RS | GRAS Subfamilies | Fraction Disordered | RS | RMSD | MatchAlign Score |
|---|---|---|---|---|---|---|---|
| ShLISCL | 0.49 | 0.82 | SbLISCL | 0.38 | 0.83 | 1.626 | 1180.0 |
| ShPAT1 | 0.37 | 0.82 | SbPAT1 | 0.40 | 0.81 | 1.930 | 1744.5 |
| ShHAM | 0.41 | 0.81 | SbHAM | 0.40 | 0.80 | 2.570 | 3164.5 |
| ShSHR | 0.13 | 0.91 | SbSHR | 0.36 | 0.85 | 1.524 | 554.5 |
| ShDELLA | 0.49 | 0.81 | SbDELLA | 0.30 | 0.80 | 28.276 | 837.0 |
| ShSCR | 0.36 | 0.80 | SbSCR | 0.17 | 0.82 | 0.226 | 2547.5 |
| ShSCL | 0.25 | 0.84 | SbSCL | 0.27 | 0.85 | 1.201 | 536.0 |
| ShSCL4/7 | 0.27 | 0.82 | SbSCL4/7 | 0.27 | 0.84 | 4.022 | 2794.5 |
| ShLAS | 0.24 | 0.86 | SbLAS | 0.05 | 0.89 | 0.498 | 1290.0 |
| ShDLT | 0.37 | 0.84 | SbDLT | 0.39 | 0.83 | 0.288 | 3037.5 |
| ShOs43 | 0.26 | 0.84 | SbOs43 | 0.25 | 0.84 | 0.568 | 2655.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, H.; Wang, L.; Gong, Y.; Zhang, Y.; Zhao, T.; Feng, C.; Wang, J.; Lin, J. Genome-Wide Identification and Characterization of GRAS Transcription Factor Family in Cultivated Hybrid Sugarcane ZZ1 (Saccharum officinarum) and Their Role in Development and Stress. Int. J. Mol. Sci. 2024, 25, 13470. https://doi.org/10.3390/ijms252413470
Wen H, Wang L, Gong Y, Zhang Y, Zhao T, Feng C, Wang J, Lin J. Genome-Wide Identification and Characterization of GRAS Transcription Factor Family in Cultivated Hybrid Sugarcane ZZ1 (Saccharum officinarum) and Their Role in Development and Stress. International Journal of Molecular Sciences. 2024; 25(24):13470. https://doi.org/10.3390/ijms252413470
Chicago/Turabian StyleWen, Hao, Lidan Wang, Yuqing Gong, Yu Zhang, Tingting Zhao, Cuilian Feng, Jungang Wang, and Jishan Lin. 2024. "Genome-Wide Identification and Characterization of GRAS Transcription Factor Family in Cultivated Hybrid Sugarcane ZZ1 (Saccharum officinarum) and Their Role in Development and Stress" International Journal of Molecular Sciences 25, no. 24: 13470. https://doi.org/10.3390/ijms252413470
APA StyleWen, H., Wang, L., Gong, Y., Zhang, Y., Zhao, T., Feng, C., Wang, J., & Lin, J. (2024). Genome-Wide Identification and Characterization of GRAS Transcription Factor Family in Cultivated Hybrid Sugarcane ZZ1 (Saccharum officinarum) and Their Role in Development and Stress. International Journal of Molecular Sciences, 25(24), 13470. https://doi.org/10.3390/ijms252413470