Alterations in Striatal Architecture and Biochemical Markers’ Levels During Postnatal Development in the Rat Model of an Attention Deficit/Hyperactivity Disorder (ADHD)


 and
and 
Abstract
:1. Introduction
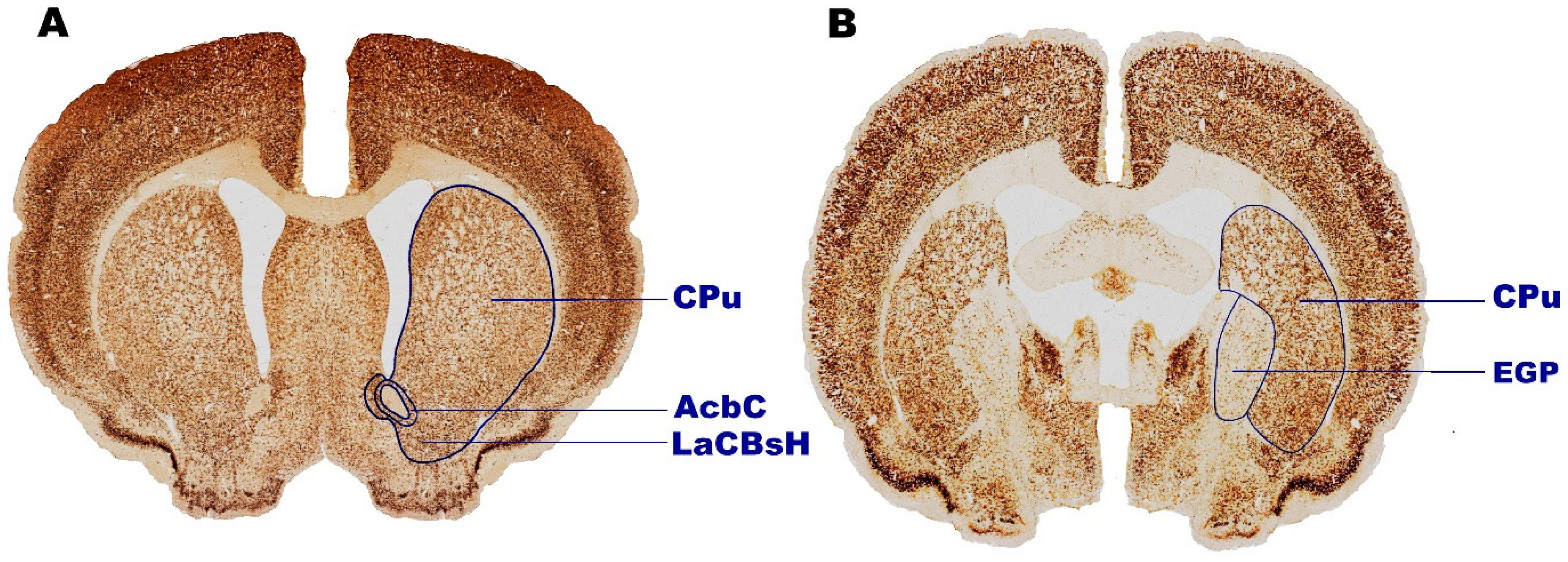
2. Results
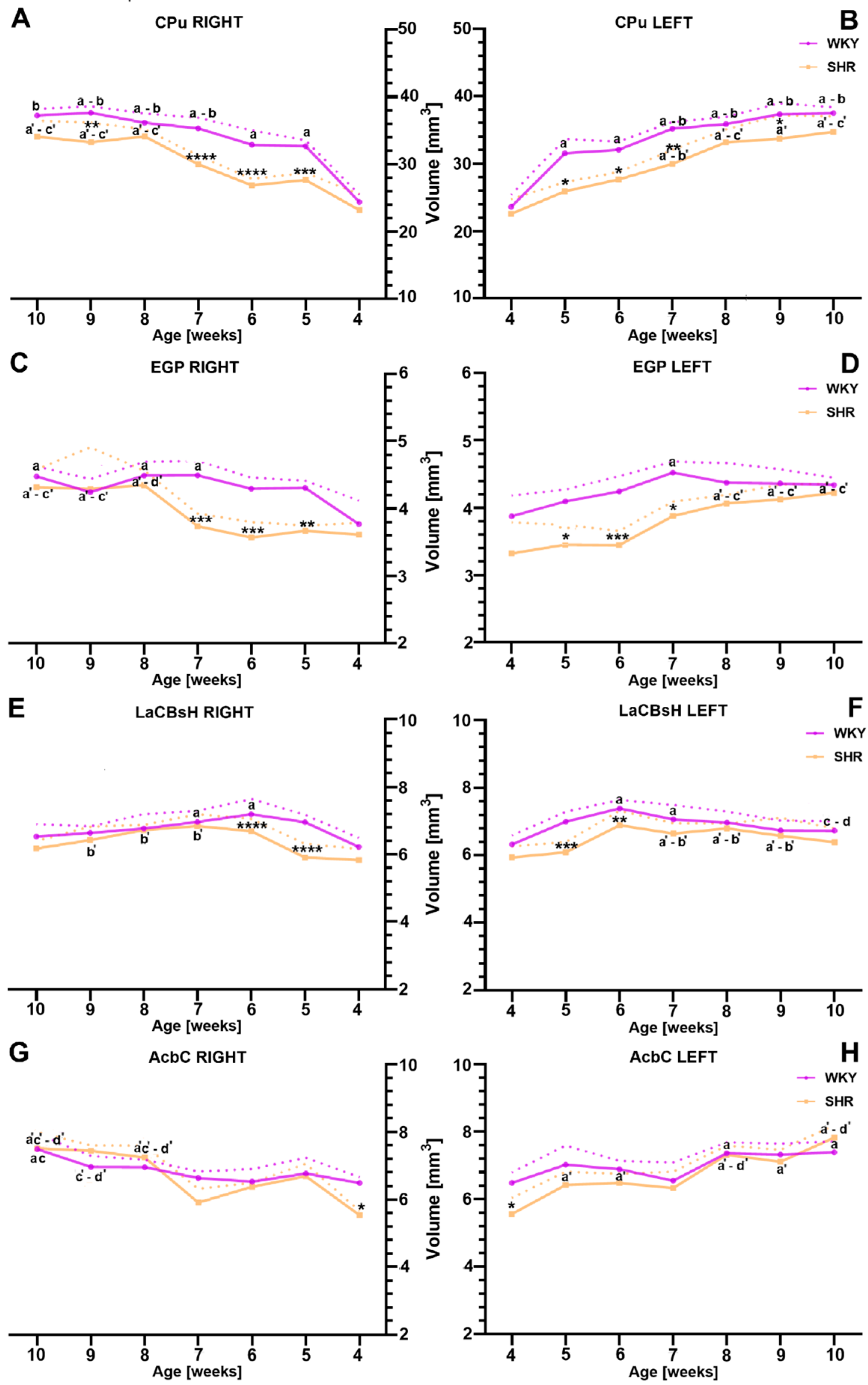
2.1. Volumetric Alternations
2.1.1. Caudate Putamen (CPu)
2.1.2. External Globus Pallidus (EGP)
2.1.3. Lateral Accumbens Shell (LaCBsH)
2.1.4. Nucleus Accumbens Core (AcbC)
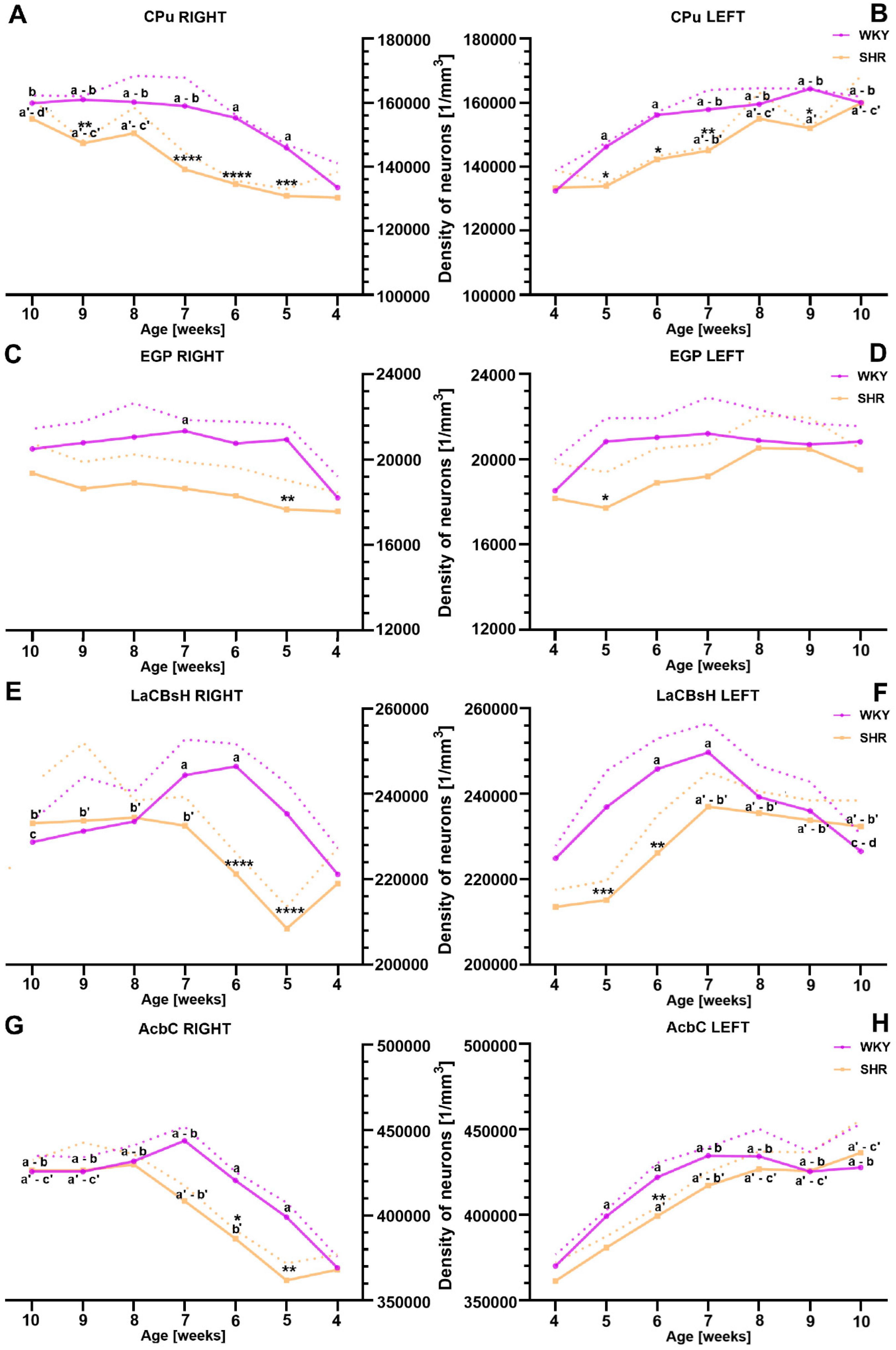
2.2. Alternations in Neuronal Density Across Distinct Striatal Regions
2.2.1. Caudate Putamen (CPu)
2.2.2. External Globus Pallidus (EGP)
2.2.3. Lateral Accumbens Shell (LaCBsH)
2.2.4. Nucleus Accumbens Core (AcbC)
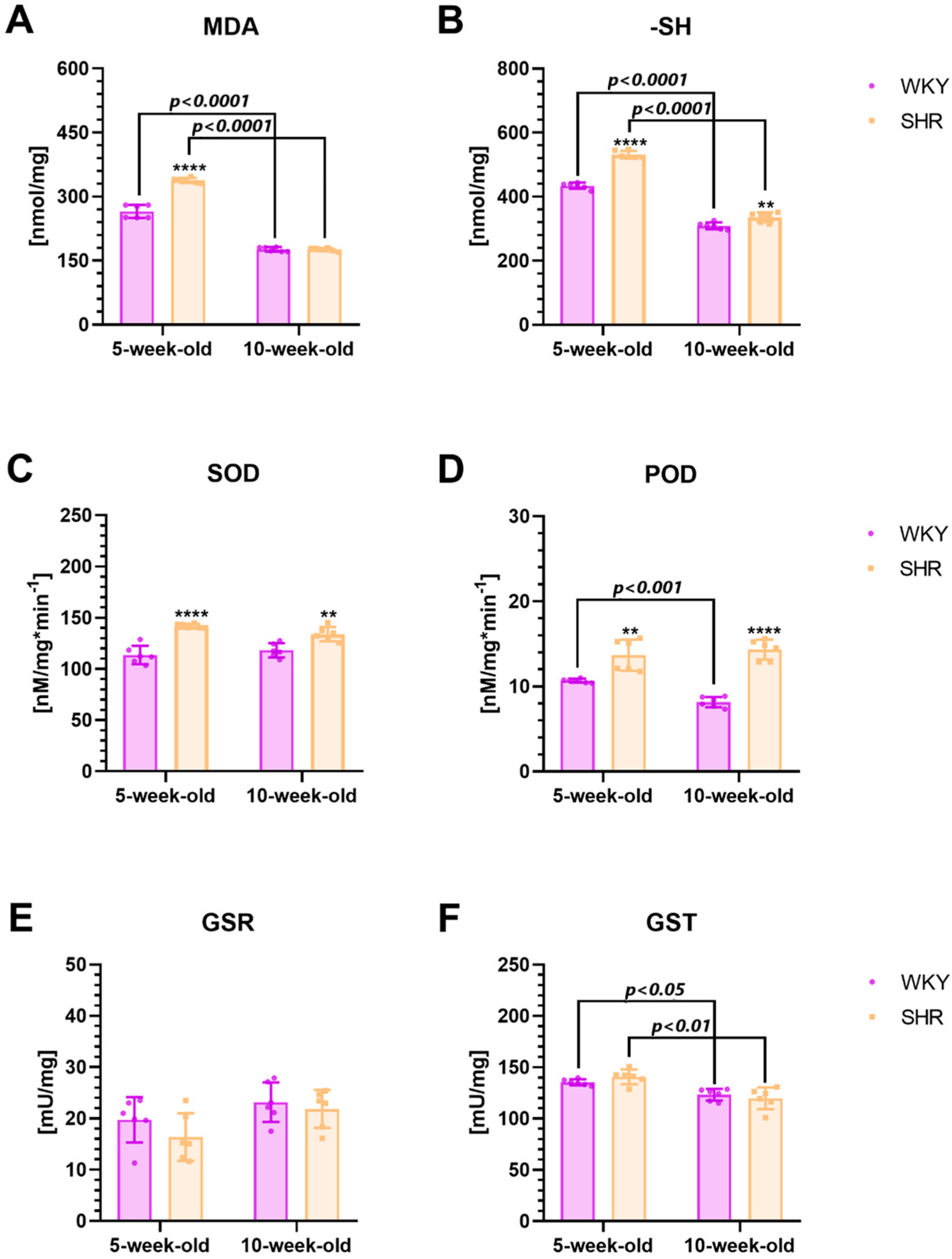
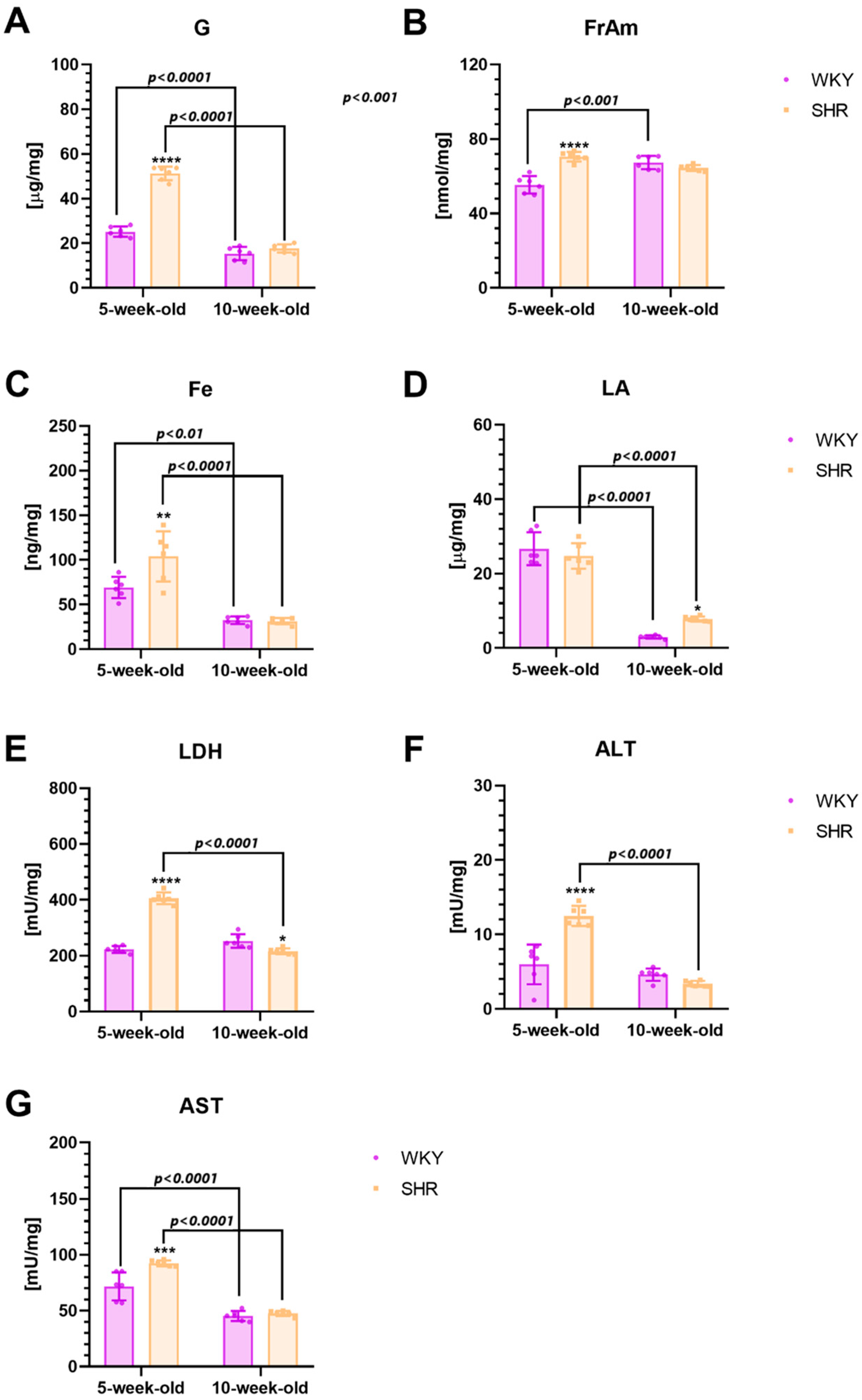
2.3. Biochemical Alternationss in the Striatum
2.3.1. Inflammatory Markers
2.3.2. Oxidative Stress Markers
2.3.3. Metabolic Markers
3. Discussion
3.1. Striatal Volume and Neuron Density Alternations
3.2. Inflammatory Markers
3.3. Oxidative Stress Markers
3.4. Metabolism Markers
4. Materials and Methods
4.1. Animals
4.1.1. Animals with Transcardial Perfusion
4.1.2. Animals Without Transcardial Perfusion
4.2. Preparation of Brains
4.2.1. Preparation of Brains for Neuron-Specific Nuclear Protein (NeuN) Staining
4.2.2. Neuron-Specific Nuclear Protein (NeuN) Staining
4.2.3. Preparation of Brains for Measuring the Content of Inflammatory, Oxidative Stress, and Metabolic Markers
4.3. Morphometric Analysis of Motor Cortex
4.3.1. Volumetric Analyses
4.3.2. The Density Quantification
4.4. Assessment of Immune, Oxidative Stress, and Metabolic Markers
Preparation of Test Material and Determination of Immune, Oxidative Stress, and Metabolic Markers/Enzymes
4.5. Statistics
5. Limitations of a Study
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilens, T.E.; Spencer, T.J. Understanding Attention-Deficit/Hyperactivity Disorder from Childhood to Adulthood. Postgrad. Med. 2010, 122, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Nijmeijer, J.S.; Minderaa, R.B.; Buitelaar, J.K.; Mulligan, A.; Hartman, C.A.; Hoekstra, P.J. Attention-deficit/hyperactivity disorder and social dysfunctioning. Clin. Psychol. Rev. 2008, 28, 692–708. [Google Scholar] [CrossRef] [PubMed]
- Danielson, M.L.; Claussen, A.H.; Bitsko, R.H.; Katz, S.M.; Newsome, K.; Blumberg, S.J.; Kogan, M.D.; Ghandour, R. ADHD Prevalence Among U.S. Children and Adolescents in 2022: Diagnosis, Severity, Co-Occurring Disorders, and Treatment. J. Clin. Child Adolesc. Psychol. 2024, 53, 343–360. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Zha, M.; Yang, Q.; Zhang, Y.; Li, X.; Rudan, I. The prevalence of adult attention-deficit hyperactivity disorder: A global systematic review and meta-analysis. J. Glob. Health 2021, 11, 04009. [Google Scholar] [CrossRef]
- Kates, N. Attention deficit disorder in adults. Management in primary care. Can. Fam. Physician 2005, 51, 53–59. [Google Scholar]
- Magnus, W.; Nazir, S.; Anilkumar, A.C.; Shaban, K. Attention Deficit Hyperactivity Disorder. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024; Available online: http://www.ncbi.nlm.nih.gov/books/NBK441838/ (accessed on 23 August 2024).
- Almeida, L.G.; Ricardo-Garcell, J.; Prado, H.; Barajas, L.; Fernández-Bouzas, A.; Avila, D.; Martínez, R.B. Reduced right frontal cortical thickness in children, adolescents and adults with ADHD and its correlation to clinical variables: A cross-sectional study. J. Psychiatr. Res. 2010, 44, 1214–1223. [Google Scholar] [CrossRef]
- Cupertino, R.B.; Soheili-Nezhad, S.; Grevet, E.H.; Bandeira, C.E.; Picon, F.A.; Tavares, M.E.d.A.; Naaijen, J.; van Rooij, D.; Akkermans, S.; Vitola, E.S.; et al. Reduced fronto-striatal volume in attention-deficit/hyperactivity disorder in two cohorts across the lifespan. NeuroImage Clin. 2020, 28, 102403. [Google Scholar] [CrossRef]
- Castellanos, F.X. Quantitative Brain Magnetic Resonance Imaging in Attention-Deficit Hyperactivity Disorder. Arch. Gen. Psychiatry 1996, 53, 607. [Google Scholar] [CrossRef]
- Castellanos, F.X.; Lee, P.P.; Sharp, W.; Jeffries, N.O.; Greenstein, D.K.; Clasen, L.S.; Blumenthal, J.D.; James, R.S.; Ebens, C.L.; Walter, J.M.; et al. Developmental trajectories of brain volume abnormalities in children and adolescents with attention-deficit/hyperactivity disorder. JAMA 2002, 288, 1740–1748. [Google Scholar] [CrossRef]
- Castellanos, F.X.; Giedd, J.N.; Eckburg, P.; Marsh, W.L.; Vaituzis, A.C.; Kaysen, D.; Hamburger, S.D.; Rapoport, J.L. Quantitative morphology of the caudate nucleus in attention deficit hyperactivity disorder. Am. J. Psychiatry 1994, 151, 1791–1796. [Google Scholar] [CrossRef]
- Filipek, P.A.; Semrud-Clikeman, M.; Steingard, R.J.; Renshaw, P.F.; Kennedy, D.N.; Biederman, J. Volumetric MRI analysis comparing subjects having attention-deficit hyperactivity disorder with normal controls. Neurology 1997, 48, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Hynd, G.W.; Hern, K.L.; Novey, E.S.; Eliopulos, D.; Marshall, R.; Gonzalez, J.J.; Voeller, K.K. Attention deficit-hyperactivity disorder and asymmetry of the caudate nucleus. J. Child Neurol. 1993, 8, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Mataró, M.; Garcia-Sánchez, C.; Junqué, C.; Estévez-González, A.; Pujol, J. Magnetic resonance imaging measurement of the caudate nucleus in adolescents with attention-deficit hyperactivity disorder and its relationship with neuropsychological and behavioral measures. Arch. Neurol. 1997, 54, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Pineda, D.A.; Restrepo, M.A.; Sarmiento, R.J.; Gutierrez, J.E.; Vargas, S.A.; Quiroz, Y.T.; Hynd, G.W. Statistical analyses of structural magnetic resonance imaging of the head of the caudate nucleus in Colombian children with attention-deficit hyperactivity disorder. J. Child Neurol. 2002, 17, 97–105. [Google Scholar] [CrossRef]
- Semrud-Clikeman, M.; Steingard, R.J.; Filipek, P.; Biederman, J.; Bekken, K.; Renshaw, P.F. Using MRI to examine brain-behavior relationships in males with attention deficit disorder with hyperactivity. J. Am. Acad. Child Adolesc. Psychiatry 2000, 39, 477–484. [Google Scholar] [CrossRef]
- Utter, A.A.; Basso, M.A. The basal ganglia: An overview of circuits and function. Neurosci. Biobehav. Rev. 2008, 32, 333–342. [Google Scholar] [CrossRef]
- Haber, S.N. The primate basal ganglia: Parallel and integrative networks. J. Chem. Neuroanat. 2003, 26, 317–330. [Google Scholar] [CrossRef]
- Plichta, M.M.; Scheres, A. Ventral-striatal responsiveness during reward anticipation in ADHD and its relation to trait impulsivity in the healthy population: A meta-analytic review of the fMRI literature. Neurosci. Biobehav. Rev. 2014, 38, 125–134. [Google Scholar] [CrossRef]
- Long, Y.; Pan, N.; Ji, S.; Qin, K.; Chen, Y.; Zhang, X.; He, M.; Suo, X.; Yu, Y.; Wang, S.; et al. Distinct brain structural abnormalities in attention-deficit/hyperactivity disorder and substance use disorders: A comparative meta-analysis. Transl. Psychiatry 2022, 12, 368. [Google Scholar] [CrossRef]
- Brennan, A.R.; Arnsten, A.F.T. Neuronal Mechanisms Underlying Attention Deficit Hyperactivity Disorder. Ann. N. Y. Acad. Sci. 2008, 1129, 236–245. [Google Scholar] [CrossRef]
- Castellanos, F.X.; Sharp, W.S.; Gottesman, R.F.; Greenstein, D.K.; Giedd, J.N.; Rapoport, J.L. Anatomic brain abnormalities in monozygotic twins discordant for attention deficit hyperactivity disorder. Am. J. Psychiatry 2003, 160, 1693–1696. [Google Scholar] [CrossRef] [PubMed]
- Castellanos, F.X.; Giedd, J.N.; Berquin, P.C.; Walter, J.M.; Sharp, W.; Tran, T.; Vaituzis, A.C.; Blumenthal, J.D.; Nelson, J.; Bastain, T.M.; et al. Quantitative brain magnetic resonance imaging in girls with attention-deficit/hyperactivity disorder. Arch. Gen. Psychiatry 2001, 58, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Hoogman, M.; Bralten, J.; Hibar, D.P.; Mennes, M.; Zwiers, M.P.; Schweren, L.S.J.; van Hulzen, K.J.E.; Medland, S.E.; Shumskaya, E.; Jahanshad, N.; et al. Subcortical brain volume differences in participants with attention deficit hyperactivity disorder in children and adults: A cross-sectional mega-analysis. Lancet Psychiatry 2017, 4, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.; Fekete, T.; Siciliano, F.; Biezonski, D.; Greenhill, L.; Pliszka, S.R.; Blader, J.C.; Roy, A.K.; Leibenluft, E.; Posner, J. Neural Correlates of Aggression in Medication-Naive Children with ADHD: Multivariate Analysis of Morphometry and Tractography. Neuropsychopharmacology 2015, 40, 1717–1725. [Google Scholar] [CrossRef]
- Mills, K.L.; Bathula, D.; Dias, T.G.C.; Iyer, S.P.; Fenesy, M.C.; Musser, E.D.; Stevens, C.A.; Thurlow, B.L.; Carpenter, S.D.; Nagel, B.J.; et al. Altered Cortico-Striatal–Thalamic Connectivity in Relation to Spatial Working Memory Capacity in Children with ADHD. Front. Psychiatry 2012, 3, 2. [Google Scholar] [CrossRef]
- Kumar, U.; Arya, A.; Agarwal, V. Altered functional connectivity in children with ADHD while performing cognitive control task. Psychiatry Res. Neuroimaging 2022, 326, 111531. [Google Scholar] [CrossRef]
- Oldehinkel, M.; Beckmann, C.F.; Pruim, R.H.R.; van Oort, E.S.B.; Franke, B.; Hartman, C.A.; Hoekstra, P.J.; Oosterlaan, J.; Heslenfeld, D.; Buitelaar, J.K.; et al. Attention-Deficit/Hyperactivity Disorder symptoms coincide with altered striatal connectivity. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2016, 1, 353. [Google Scholar] [CrossRef]
- Kozłowska, A.; Wojtacha, P.; Równiak, M.; Kolenkiewicz, M.; Huang, A.C.W. ADHD pathogenesis in the immune, endocrine and nervous systems of juvenile and maturating SHR and WKY rats. Psychopharmacology 2019, 236, 2937–2958. [Google Scholar] [CrossRef]
- Bogdańska-Chomczyk, E.; Równiak, M.; Wojtacha, P.; Tsai, M.-L.; Kozłowska, A. Age-related changes in the architecture and biochemical markers levels in motor-related cortical areas of SHR Ratsan ADHD animal model. Front. Mol. Neurosci. 2024, 17, 1414457. [Google Scholar] [CrossRef]
- Hsu, J.-W.; Lee, L.-C.; Chen, R.-F.; Yen, C.-T.; Chen, Y.-S.; Tsai, M.-L. Striatal volume changes in a rat model of childhood attention-deficit/hyperactivity disorder. Psychiatry Res. 2010, 179, 338–341. [Google Scholar] [CrossRef]
- Tarver, J.; Daley, D.; Sayal, K. Attention-deficit hyperactivity disorder (ADHD): An updated review of the essential facts. Child Care Health Dev. 2014, 40, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Altabella, L.; Zoratto, F.; Adriani, W.; Canese, R. MR imaging-detectable metabolic alterations in attention deficit/hyperactivity disorder: From preclinical to clinical studies. AJNR Am. J. Neuroradiol. 2014, 35, S55–S63. [Google Scholar] [CrossRef]
- Joseph, N.; Zhang-James, Y.; Perl, A.; Faraone, S.V. Oxidative Stress and ADHD: A Meta-Analysis. J. Atten. Disord. 2015, 19, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Kozłowska, A.; Wojtacha, P.; Równiak, M.; Kolenkiewicz, M.; Tsai, M.-L. Differences in serum steroid hormones concentrations in Spontaneously Hypertensive Rats (SHR)—An animal model of Attention-Deficit/Hyperactivity Disorder (ADHD). Physiol. Res. 2019, 68, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Corona, J.C. Role of Oxidative Stress and Neuroinflammation in Attention-Deficit/Hyperactivity Disorder. Antioxidants 2020, 9, 1039. [Google Scholar] [CrossRef] [PubMed]
- Cortese, S.; Angriman, M.; Comencini, E.; Vincenzi, B.; Maffeis, C. Association between inflammatory cytokines and ADHD symptoms in children and adolescents with obesity: A pilot study. Psychiatry Res. 2019, 278, 7–11. [Google Scholar] [CrossRef]
- Donfrancesco, R.; Nativio, P.; Di Benedetto, A.; Villa, M.P.; Andriola, E.; Melegari, M.G.; Cipriano, E.; Di Trani, M. Anti-Yo Antibodies in Children With ADHD: First Results About Serum Cytokines. J. Atten. Disord. 2020, 24, 1497–1502. [Google Scholar] [CrossRef]
- Oades, R.D.; Myint, A.-M.; Dauvermann, M.R.; Schimmelmann, B.G.; Schwarz, M.J. Attention-deficit hyperactivity disorder (ADHD) and glial integrity: An exploration of associations of cytokines and kynurenine metabolites with symptoms and attention. Behav. Brain Funct. 2010, 6, 32. [Google Scholar] [CrossRef]
- Chang, S.-J.; Kuo, H.-C.; Chou, W.-J.; Tsai, C.-S.; Lee, S.-Y.; Wang, L.-J. Cytokine Levels and Neuropsychological Function among Patients with Attention-Deficit/Hyperactivity Disorder and Atopic Diseases. J. Pers. Med. 2022, 12, 1155. [Google Scholar] [CrossRef]
- Darwish, A.H. Posterior Reversible Encephalopathy Syndrome in Children: A Prospective Follow-up Study. J. Child Neurol. 2020, 35, 55–62. [Google Scholar] [CrossRef]
- O’Shea, T.M.; Joseph, R.M.; Kuban, K.C.K.; Allred, E.N.; Ware, J.; Coster, T.; Fichorova, R.N.; Dammann, O.; Leviton, A. ELGAN Study Investigators Elevated blood levels of inflammation-related proteins are associated with an attention problem at age 24 mo in extremely preterm infants. Pediatr. Res. 2014, 75, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Felger, J.C.; Miller, A.H. Cytokine Effects on the Basal Ganglia and Dopamine Function: The Subcortical Source of Inflammatory Malaise. Front. Neuroendocrinol. 2012, 33, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Oades, R.D.; Dauvermann, M.R.; Schimmelmann, B.G.; Schwarz, M.J.; Myint, A.-M. Attention-deficit hyperactivity disorder (ADHD) and glial integrity: S100B, cytokines and kynurenine metabolism--effects of medication. Behav. Brain Funct. 2010, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- di Penta, A.; Moreno, B.; Reix, S.; Fernandez-Diez, B.; Villanueva, M.; Errea, O.; Escala, N.; Vandenbroeck, K.; Comella, J.X.; Villoslada, P. Oxidative Stress and Proinflammatory Cytokines Contribute to Demyelination and Axonal Damage in a Cerebellar Culture Model of Neuroinflammation. PLoS ONE 2013, 8, e54722. [Google Scholar] [CrossRef] [PubMed]
- Taherkhani, S.; Suzuki, K.; Castell, L. A Short Overview of Changes in Inflammatory Cytokines and Oxidative Stress in Response to Physical Activity and Antioxidant Supplementation. Antioxidants 2020, 9, 886. [Google Scholar] [CrossRef]
- Yang, D.; Elner, S.G.; Bian, Z.-M.; Till, G.O.; Petty, H.R.; Elner, V.M. Pro-inflammatory Cytokines Increase Reactive Oxygen Species through Mitochondria and NADPH Oxidase in Cultured RPE Cells. Exp. Eye Res. 2007, 85, 462–472. [Google Scholar] [CrossRef]
- Liu, X.; Lin, J.; Zhang, H.; Khan, N.U.; Zhang, J.; Tang, X.; Cao, X.; Shen, L. Oxidative Stress in Autism Spectrum Disorder—Current Progress of Mechanisms and Biomarkers. Front. Psychiatry 2022, 13, 813304. Available online: https://www.frontiersin.org/articles/10.3389/fpsyt.2022.813304 (accessed on 25 October 2023). [CrossRef]
- Nishimura, Y.; Kanda, Y.; Sone, H.; Aoyama, H. Oxidative Stress as a Common Key Event in Developmental Neurotoxicity. Oxid. Med. Cell. Longev. 2021, 2021, 6685204. [Google Scholar] [CrossRef]
- Oztop, D.; Altun, H.; Baskol, G.; Ozsoy, S. Oxidative stress in children with attention deficit hyperactivity disorder. Clin. Biochem. 2012, 45, 745–748. [Google Scholar] [CrossRef]
- Usui, N.; Kobayashi, H.; Shimada, S. Neuroinflammation and Oxidative Stress in the Pathogenesis of Autism Spectrum Disorder. Int. J. Mol. Sci. 2023, 24, 5487. [Google Scholar] [CrossRef]
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural Regen. Res. 2012, 7, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Amen, D.G.; Carmichael, B.D. High-resolution brain SPECT imaging in ADHD. Ann. Clin. Psychiatry 1997, 9, 81–86. [Google Scholar] [CrossRef]
- Sieg, K.G.; Gaffney, G.R.; Preston, D.F.; Hellings, J.A. SPECT brain imaging abnormalities in attention deficit hyperactivity disorder. Clin. Nucl. Med. 1995, 20, 55–60. [Google Scholar] [CrossRef]
- Zametkin, A.J.; Liebenauer, L.L.; Fitzgerald, G.A.; King, A.C.; Minkunas, D.V.; Herscovitch, P.; Yamada, E.M.; Cohen, R.M. Brain metabolism in teenagers with attention-deficit hyperactivity disorder. Arch. Gen. Psychiatry 1993, 50, 333–340. [Google Scholar] [CrossRef]
- Zametkin, A.J.; Nordahl, T.E.; Gross, M.; King, A.C.; Semple, W.E.; Rumsey, J.; Hamburger, S.; Cohen, R.M. Cerebral glucose metabolism in adults with hyperactivity of childhood onset. N. Engl. J. Med. 1990, 323, 1361–1366. [Google Scholar] [CrossRef]
- Lou, H.C. Etiology and pathogenesis of attention-deficit hyperactivity disorder (ADHD): Significance of prematurity and perinatal hypoxic-haemodynamic encephalopathy. Acta Paediatr. 1996, 85, 1266–1271. [Google Scholar] [CrossRef]
- Lou, H.C.; Henriksen, L.; Bruhn, P. Focal cerebral dysfunction in developmental learning disabilities. Lancet Lond. Engl. 1990, 335, 8–11. [Google Scholar] [CrossRef]
- Lou, H.C.; Henriksen, L.; Bruhn, P.; Børner, H.; Nielsen, J.B. Striatal dysfunction in attention deficit and hyperkinetic disorder. Arch. Neurol. 1989, 46, 48–52. [Google Scholar] [CrossRef]
- Lou, H.C.; Henriksen, L.; Bruhn, P. Focal cerebral hypoperfusion in children with dysphasia and/or attention deficit disorder. Arch. Neurol. 1984, 41, 825–829. [Google Scholar] [CrossRef]
- Kim, D.; Yadav, D.; Song, M. An updated review on animal models to study attention-deficit hyperactivity disorder. Transl. Psychiatry 2024, 14, 187. [Google Scholar] [CrossRef] [PubMed]
- Johansen, E.B.; Sagvolden, T.; Kvande, G. Effects of delayed reinforcers on the behavior of an animal model of attention-deficit/hyperactivity disorder (ADHD). Behav. Brain Res. 2005, 162, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Johansen, E.B.; Killeen, P.R.; Sagvolden, T. Behavioral variability, elimination of responses, and delay-of-reinforcement gradients in SHR and WKY rats. Behav. Brain Funct. 2007, 3, 60. [Google Scholar] [CrossRef] [PubMed]
- Sontag, T.A.; Tucha, O.; Walitza, S.; Lange, K.W. Animal models of attention deficit/hyperactivity disorder (ADHD): A critical review. Atten. Deficit Hyperact. Disord. 2010, 2, 1–20. [Google Scholar] [CrossRef]
- Bogdańska-Chomczyk, E.; Równiak, M.; Huang, A.C.-W.; Kozłowska, A. Parvalbumin interneuron deficiency in the prefrontal and motor cortices of spontaneously hypertensive rats: An attention-deficit hyperactivity disorder animal model insight. Front. Psychiatry 2024, 15, 1359237. [Google Scholar] [CrossRef]
- Watanabe, Y.; Fujita, M.; Ito, Y.; Okada, T.; Kusuoka, H.; Nishimura, T. Brain dopamine transporter in spontaneously hypertensive rats. J. Nucl. Med. 1997, 38, 470–474. [Google Scholar]
- Simchon, Y.; Weizman, A.; Rehavi, M. The effect of chronic methylphenidate administration on presynaptic dopaminergic parameters in a rat model for ADHD. Eur. Neuropsychopharmacol. 2010, 20, 714–720. [Google Scholar] [CrossRef]
- Coelho-Santos, V.; Cardoso, F.L.; Leitão, R.A.; Fontes-Ribeiro, C.A.; Silva, A.P. Impact of developmental exposure to methylphenidate on rat brain’s immune privilege and behavior: Control versus ADHD model. Brain. Behav. Immun. 2018, 68, 169–182. [Google Scholar] [CrossRef]
- Park, S.; Shin, J.; Hong, Y.; Kim, S.; Lee, S.; Park, K.; Lkhagvasuren, T.; Lee, S.-R.; Chang, K.-T.; Hong, Y. Forced Exercise Enhances Functional Recovery after Focal Cerebral Ischemia in Spontaneously Hypertensive Rats. Brain Sci. 2012, 2, 483. [Google Scholar] [CrossRef]
- França, A.P.; Schamne, M.G.; De Souza, B.S.; Da Luz Scheffer, D.; Bernardelli, A.K.; Corrêa, T.; De Souza Izídio, G.; Latini, A.; Da Silva-Santos, J.E.; Canas, P.M.; et al. Caffeine Consumption plus Physical Exercise Improves Behavioral Impairments and Stimulates Neuroplasticity in Spontaneously Hypertensive Rats (SHR): An Animal Model of Attention Deficit Hyperactivity Disorder. Mol. Neurobiol. 2020, 57, 3902–3919. [Google Scholar] [CrossRef]
- Wender, P.H.; Wolf, L.E.; Wasserstein, J. Adults with ADHD: An Overview. Ann. N. Y. Acad. Sci. 2001, 931, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sobel, L.J.; Bansal, R.; Maia, T.V.; Sanchez, J.; Mazzone, L.; Durkin, K.; Liu, J.; Hao, X.; Ivanov, I.; Miller, A.; et al. Basal Ganglia Surface Morphology and the Effects of Stimulant Medications in Youth With Attention Deficit Hyperactivity Disorder. Am. J. Psychiatry 2010, 167, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Brain Development Cooperative Group. Total and regional brain volumes in a population-based normative sample from 4 to 18 years: The NIH MRI Study of Normal Brain Development. Cereb. Cortex 2012, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Greven, C.U.; Bralten, J.; Mennes, M.; O’Dwyer, L.; van Hulzen, K.J.E.; Rommelse, N.; Schweren, L.J.S.; Hoekstra, P.J.; Hartman, C.A.; Heslenfeld, D.; et al. Developmentally Stable Whole-Brain Volume Reductions and Developmentally Sensitive Caudate and Putamen Volume Alterations in Those With Attention-Deficit/Hyperactivity Disorder and Their Unaffected Siblings. JAMA Psychiatry 2015, 72, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Nakao, T.; Radua, J.; Rubia, K.; Mataix-Cols, D. Gray matter volume abnormalities in ADHD: Voxel-based meta-analysis exploring the effects of age and stimulant medication. Am. J. Psychiatry 2011, 168, 1154–1163. [Google Scholar] [CrossRef]
- Frodl, T.; Skokauskas, N. Meta-analysis of structural MRI studies in children and adults with attention deficit hyperactivity disorder indicates treatment effects. Acta Psychiatr. Scand. 2012, 125, 114–126. [Google Scholar] [CrossRef]
- Ahrendts, J.; Rüsch, N.; Wilke, M.; Philipsen, A.; Eickhoff, S.B.; Glauche, V.; Perlov, E.; Ebert, D.; Hennig, J.; van Elst, L.T. Visual cortex abnormalities in adults with ADHD: A structural MRI study. World J. Biol. Psychiatry 2011, 12, 260–270. [Google Scholar] [CrossRef]
- Depue, B.E.; Burgess, G.C.; Bidwell, L.C.; Willcutt, E.G.; Banich, M.T. Behavioral performance predicts grey matter reductions in the right inferior frontal gyrus in young adults with combined type ADHD. Psychiatry Res. 2010, 182, 231–237. [Google Scholar] [CrossRef]
- Seidman, L.J.; Valera, E.M.; Makris, N.; Monuteaux, M.C.; Boriel, D.L.; Kelkar, K.; Kennedy, D.N.; Caviness, V.S.; Bush, G.; Aleardi, M.; et al. Dorsolateral prefrontal and anterior cingulate cortex volumetric abnormalities in adults with attention-deficit/hyperactivity disorder identified by magnetic resonance imaging. Biol. Psychiatry 2006, 60, 1071–1080. [Google Scholar] [CrossRef]
- Onnink, A.M.H.; Zwiers, M.P.; Hoogman, M.; Mostert, J.C.; Kan, C.C.; Buitelaar, J.; Franke, B. Brain alterations in adult ADHD: Effects of gender, treatment and comorbid depression. Eur. Neuropsychopharmacol. 2014, 24, 397–409. [Google Scholar] [CrossRef]
- Biezonski, D.; Shah, R.; Krivko, A.; Cha, J.; Guilfoyle, D.N.; Hrabe, J.; Gerum, S.; Xie, S.; Duan, Y.; Bansal, R.; et al. Longitudinal magnetic resonance imaging reveals striatal hypertrophy in a rat model of long-term stimulant treatment. Transl. Psychiatry 2016, 6, e884. [Google Scholar] [CrossRef] [PubMed]
- Lolansen, S.D.; Barbuskaite, D.; Ye, F.; Xiang, J.; Keep, R.F.; MacAulay, N. Spontaneously hypertensive rats can become hydrocephalic despite undisturbed secretion and drainage of cerebrospinal fluid. Fluids Barriers CNS 2023, 20, 53. [Google Scholar] [CrossRef] [PubMed]
- Douglas, P.K.; Gutman, B.; Anderson, A.; Larios, C.; Lawrence, K.E.; Narr, K.; Sengupta, B.; Cooray, G.; Douglas, D.B.; Thompson, P.M.; et al. Hemispheric Brain Asymmetry Differences in Youths with Attention-Deficit/Hyperactivity Disorder. Neuroimage Clin. 2018, 18, 744–752. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5988460/ (accessed on 10 March 2024). [CrossRef] [PubMed]
- Ellison-Wright, I.; Ellison-Wright, Z.; Bullmore, E. Structural brain change in Attention Deficit Hyperactivity Disorder identified by meta-analysis. BMC Psychiatry 2008, 8, 51. [Google Scholar] [CrossRef]
- Valera, E.M.; Faraone, S.V.; Murray, K.E.; Seidman, L.J. Meta-analysis of structural imaging findings in attention-deficit/hyperactivity disorder. Biol. Psychiatry 2007, 61, 1361–1369. [Google Scholar] [CrossRef]
- Schrimsher, G.W.; Billingsley, R.L.; Jackson, E.F.; Moore, B.D. Caudate nucleus volume asymmetry predicts attention-deficit hyperactivity disorder (ADHD) symptomatology in children. J. Child Neurol. 2002, 17, 877–884. [Google Scholar] [CrossRef]
- Dang, L.C.; Samanez-Larkin, G.R.; Young, J.S.; Cowan, R.L.; Kessler, R.M.; Zald, D.H. Caudate asymmetry is related to attentional impulsivity and an objective measure of ADHD-like attentional problems in healthy adults. Brain Struct. Funct. 2016, 221, 277–286. [Google Scholar] [CrossRef]
- Sun, L.; Jin, Z.; Zang, Y.; Zeng, Y.-W.; Liu, G.; Li, Y.; Seidman, L.J.; Faraone, S.V.; Wang, Y. Differences between attention-deficit disorder with and without hyperactivity: A 1H-magnetic resonance spectroscopy study. Brain Dev. 2005, 27, 340–344. [Google Scholar] [CrossRef]
- Ross Brian, E.R.D. (Ed.) Magnetic Resonance Spectroscopy Diagnosis of Neurological Diseases; CRC Press: Boca Raton, FL, USA, 2014; ISBN 978-0-429-17720-0. [Google Scholar]
- Vermathen, P.; Laxer, K.D.; Matson, G.B.; Weiner, M.W. Hippocampal structures: Anteroposterior N-acetylaspartate differences in patients with epilepsy and control subjects as shown with proton MR spectroscopic imaging. Radiology 2000, 214, 403–410. [Google Scholar] [CrossRef]
- Dautry, C.; Vaufrey, F.; Brouillet, E.; Bizat, N.; Henry, P.G.; Condé, F.; Bloch, G.; Hantraye, P. Early N-acetylaspartate depletion is a marker of neuronal dysfunction in rats and primates chronically treated with the mitochondrial toxin 3-nitropropionic acid. J. Cereb. Blood Flow Metab. 2000, 20, 789–799. [Google Scholar] [CrossRef]
- Jin, Z.; Zang, Y.F.; Zeng, Y.W.; Zhang, L.; Wang, Y.F. Striatal neuronal loss or dysfunction and choline rise in children with attention-deficit hyperactivity disorder: A 1H-magnetic resonance spectroscopy study. Neurosci. Lett. 2001, 315, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Ben Amor, L. 1H-Magnetic resonance spectroscopy study of stimulant medication effect on brain metabolites in French Canadian children with attention deficit hyperactivity disorder. Neuropsychiatr. Dis. Treat. 2014, 2014, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Fayed, N.; Modrego, P.J.; Castillo, J.; Dávila, J. Evidence of Brain Dysfunction in Attention Deficit-Hyperactivity Disorder: A Controlled Study with Proton Magnetic Resonance Spectroscopy. Acad. Radiol. 2007, 14, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.E.M.S.; Palmini, A.; Bau, C.H.D.; Grevet, E.H.; Hoefel, J.R.; Rohde, L.A.; Anés, M.; Ferreira, E.E.; Belmonte-de-Abreu, P. Differentiating attention-deficit/hyperactivity disorder inattentive and combined types: A 1H-magnetic resonance spectroscopy study of fronto-striato-thalamic regions. J. Neural Transm. 2009, 116, 623–629. [Google Scholar] [CrossRef]
- Alexander, A.L.; Lee, J.E.; Lazar, M.; Field, A.S. Diffusion tensor imaging of the brain. Neurotherapeutics 2007, 4, 316–329. [Google Scholar] [CrossRef]
- Peters, J.M.; Sahin, M.; Vogel-Farley, V.K.; Jeste, S.S.; Nelson, C.A.; Gregas, M.C.; Prabhu, S.P.; Scherrer, B.; Warfield, S.K. Loss of white matter microstructural integrity is associated with adverse neurological outcome in Tuberous Sclerosis Complex. Acad. Radiol. 2012, 19, 17–25. [Google Scholar] [CrossRef]
- Ashtari, M.; Kumra, S.; Bhaskar, S.L.; Clarke, T.; Thaden, E.; Cervellione, K.L.; Rhinewine, J.; Kane, J.M.; Adesman, A.; Milanaik, R.; et al. Attention-deficit/hyperactivity disorder: A preliminary diffusion tensor imaging study. Biol. Psychiatry 2005, 57, 448–455. [Google Scholar] [CrossRef]
- de Zeeuw, P.; Mandl, R.C.W.; Hulshoff Pol, H.E.; van Engeland, H.; Durston, S. Decreased frontostriatal microstructural organization in attention deficit/hyperactivity disorder. Hum. Brain Mapp. 2011, 33, 1941–1951. [Google Scholar] [CrossRef]
- Liston, C.; Watts, R.; Tottenham, N.; Davidson, M.C.; Niogi, S.; Ulug, A.M.; Casey, B.J. Frontostriatal microstructure modulates efficient recruitment of cognitive control. Cereb. Cortex 2006, 16, 553–560. [Google Scholar] [CrossRef]
- Casey, B.J.; Epstein, J.N.; Buhle, J.; Liston, C.; Davidson, M.C.; Tonev, S.T.; Spicer, J.; Niogi, S.; Millner, A.J.; Reiss, A.; et al. Frontostriatal connectivity and its role in cognitive control in parent-child dyads with ADHD. Am. J. Psychiatry 2007, 164, 1729–1736. [Google Scholar] [CrossRef]
- Durston, S.; de Zeeuw, P.; Staal, W.G. Imaging genetics in ADHD: A focus on cognitive control. Neurosci. Biobehav. Rev. 2009, 33, 674–689. [Google Scholar] [CrossRef] [PubMed]
- Booth, J.R.; Burman, D.D.; Meyer, J.R.; Lei, Z.; Trommer, B.L.; Davenport, N.D.; Li, W.; Parrish, T.B.; Gitelman, D.R.; Mesulam, M.M. Larger deficits in brain networks for response inhibition than for visual selective attention in attention deficit hyperactivity disorder (ADHD). J. Child Psychol. Psychiatry 2005, 46, 94–111. [Google Scholar] [CrossRef] [PubMed]
- Konrad, K.; Neufang, S.; Hanisch, C.; Fink, G.R.; Herpertz-Dahlmann, B. Dysfunctional attentional networks in children with attention deficit/hyperactivity disorder: Evidence from an event-related functional magnetic resonance imaging study. Biol. Psychiatry 2006, 59, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Paloyelis, Y.; Mehta, M.A.; Kuntsi, J.; Asherson, P. Functional magnetic resonance imaging in attention deficit hyperactivity disorder (ADHD): A systematic literature review. Expert Rev. Neurother. 2007, 7, 1337–1356. [Google Scholar] [CrossRef]
- Tsai, M.-L.; Kozłowska, A.; Li, Y.-S.; Shen, W.-L.; Huang, A.C.W. Social factors affect motor and anxiety behaviors in the animal model of attention-deficit hyperactivity disorders: A housing-style factor. Psychiatry Res. 2017, 254, 290–300. [Google Scholar] [CrossRef]
- Hsieh, Y.-L.; Yang, C.-C. Age-series characteristics of locomotor activities in spontaneously hypertensive rats: A comparison with the Wistar-Kyoto strain. Physiol. Behav. 2008, 93, 777–782. [Google Scholar] [CrossRef]
- Schreiber, J.E.; Possin, K.L.; Girard, J.M.; Rey-Casserly, C. Executive Function in Children with Attention Deficit/Hyperactivity Disorder: The NIH EXAMINER battery. J. Int. Neuropsychol. Soc. 2014, 20, 41–51. [Google Scholar] [CrossRef]
- Kollins, S.H.; Adcock, R.A. ADHD, altered dopamine neurotransmission, and disrupted reinforcement processes: Implications for smoking and nicotine dependence. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 52, 70–78. [Google Scholar] [CrossRef]
- Yoo, H.K.; Huh, H.; Lee, S.; Jung, K.; Kim, B. Development of the Attention-Deficit Hyperactivity Disorder-After School Checklist. J. Korean Acad. Child Adolesc. Psychiatry 2018, 29, 47–53. [Google Scholar] [CrossRef]
- Forero, D.A.; Arboleda, G.H.; Vasquez, R.; Arboleda, H. Candidate genes involved in neural plasticity and the risk for attention-deficit hyperactivity disorder: A meta-analysis of 8 common variants. J. Psychiatry Neurosci. 2009, 34, 361–366. [Google Scholar]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Quinn, R. Comparing rat’s to human’s age: How old is my rat in people years? Nutrition 2005, 21, 775–777. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.; Eckstrand, K.; Sharp, W.; Blumenthal, J.; Lerch, J.P.; Greenstein, D.; Clasen, L.; Evans, A.; Giedd, J.; Rapoport, J.L. Attention-deficit/hyperactivity disorder is characterized by a delay in cortical maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19649–19654. [Google Scholar] [CrossRef] [PubMed]
- Qiu, A.; Crocetti, D.; Adler, M.; Mahone, E.M.; Denckla, M.B.; Miller, M.I.; Mostofsky, S.H. Basal ganglia volume and shape in children with attention deficit hyperactivity disorder. Am. J. Psychiatry 2009, 166, 74–82. [Google Scholar] [CrossRef]
- Sagvolden, T.; Russell, V.A.; Aase, H.; Johansen, E.B.; Farshbaf, M. Rodent Models of Attention-Deficit/Hyperactivity Disorder. Biol. Psychiatry 2005, 57, 1239–1247. [Google Scholar] [CrossRef]
- Abikoff, H.B.; Jensen, P.S.; Arnold, L.L.E.; Hoza, B.; Hechtman, L.; Pollack, S.; Martin, D.; Alvir, J.; March, J.S.; Hinshaw, S.; et al. Observed classroom behavior of children with ADHD: Relationship to gender and comorbidity. J. Abnorm. Child Psychol. 2002, 30, 349–359. [Google Scholar] [CrossRef]
- Song, Y.; Yuan, H.; Chen, T.; Lu, M.; Lei, S.; Han, X. An Shen Ding Zhi Ling Alleviates Symptoms of Attention Deficit Hyperactivity Disorder via Anti-Inflammatory Effects in Spontaneous Hypertensive Rats. Front. Pharmacol. 2021, 11, 617581. Available online: https://www.frontiersin.org/articles/10.3389/fphar.2020.617581 (accessed on 22 March 2023). [CrossRef]
- Leffa, D.T.; Bellaver, B.; de Oliveira, C.; de Macedo, I.C.; de Freitas, J.S.; Grevet, E.H.; Caumo, W.; Rohde, L.A.; Quincozes-Santos, A.; Torres, I.L.S. Increased Oxidative Parameters and Decreased Cytokine Levels in an Animal Model of Attention-Deficit/Hyperactivity Disorder. Neurochem. Res. 2017, 42, 3084–3092. [Google Scholar] [CrossRef]
- Barchowsky, A.; Frleta, D.; Vincenti, M.P. Integration of the NF-κB and mitogen-activated protein kinase/AP-1 pathways at the Collagenase-1 promoter: Divergence of IL-1 and TNF-dependent signal transduction in rabbit primary synovial fibroblasts. Cytokine 2000, 12, 1469–1479. [Google Scholar] [CrossRef]
- Mattson, M.P.; Camandola, S. NF-κB in neuronal plasticity and neurodegenerative disorders. J. Clin. Investig. 2001, 107, 247–254. [Google Scholar] [CrossRef]
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Gonçalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef] [PubMed]
- Pua, L.J.W.; Mai, C.-W.; Chung, F.F.-L.; Khoo, A.S.-B.; Leong, C.-O.; Lim, W.-M.; Hii, L.-W. Functional Roles of JNK and p38 MAPK Signaling in Nasopharyngeal Carcinoma. Int. J. Mol. Sci. 2022, 23, 1108. [Google Scholar] [CrossRef] [PubMed]
- Mancini, M.; Natoli, S.; Gardoni, F.; Di Luca, M.; Pisani, A. Dopamine Transmission Imbalance in Neuroinflammation: Perspectives on Long-Term COVID-19. Int. J. Mol. Sci. 2023, 24, 5618. [Google Scholar] [CrossRef] [PubMed]
- Channer, B.; Matt, S.M.; Nickoloff-Bybel, E.A.; Pappa, V.; Agarwal, Y.; Wickman, J.; Gaskill, P.J. Dopamine, Immunity, and Disease. Pharmacol. Rev. 2023, 75, 62–158. [Google Scholar] [CrossRef]
- Russell, V.A.; de Villiers, A.S.; Sagvolden, T.; Lamm, M.C.; Taljaard, J.J. Methylphenidate affects striatal dopamine differently in an animal model for attention-deficit/hyperactivity disorder--the spontaneously hypertensive rat. Brain Res. Bull. 2000, 53, 187–192. [Google Scholar] [CrossRef]
- Miller, E.M.; Pomerleau, F.; Huettl, P.; Russell, V.A.; Gerhardt, G.A.; Glaser, P.E.A. The spontaneously hypertensive and Wistar Kyoto rat models of ADHD exhibit sub-regional differences in dopamine release and uptake in the striatum and nucleus accumbens. Neuropharmacology 2012, 63, 1327–1334. [Google Scholar] [CrossRef]
- Aster, H.-C.; Romanos, M.; Walitza, S.; Gerlach, M.; Mühlberger, A.; Rizzo, A.; Andreatta, M.; Hasenauer, N.; Hartrampf, P.E.; Nerlich, K.; et al. Responsivity of the Striatal Dopamine System to Methylphenidate—A Within-Subject I-123-β-CIT-SPECT Study in Male Children and Adolescents With Attention-Deficit/Hyperactivity Disorder. Front. Psychiatry 2022, 13, 804730. [Google Scholar] [CrossRef]
- Sagvolden, T.; Johansen, E.B.; Aase, H.; Russell, V.A. A dynamic developmental theory of attention-deficit/hyperactivity disorder (ADHD) predominantly hyperactive/impulsive and combined subtypes. Behav. Brain Sci. 2005, 28, 397–419; discussion 419–468. [Google Scholar] [CrossRef]
- Lyu, D.; Yu, W.; Tang, N.; Wang, R.; Zhao, Z.; Xie, F.; He, Y.; Du, H.; Chen, J. The mTOR Signaling Pathway Regulates Pain-Related Synaptic Plasticity in Rat Entorhinal-Hippocampal Pathways. Mol. Pain 2013, 9, 1744-8069-9-64. [Google Scholar] [CrossRef]
- Lee, D.Y. Roles of mTOR Signaling in Brain Development. Exp. Neurobiol. 2015, 24, 177–185. [Google Scholar] [CrossRef]
- Liu, X.; Li, Y.; Yu, L.; Vickstrom, C.R.; Liu, Q. VTA mTOR Signaling Regulates Dopamine Dynamics, Cocaine-Induced Synaptic Alterations, and Reward. Neuropsychopharmacology 2018, 43, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- LaSarge, C.L.; Pun, R.Y.K.; Gu, Z.; Santos, V.R.; Danzer, S.C. Impact of mTOR hyperactive neurons on the morphology and physiology of adjacent neurons: Do PTEN KO cells make bad neighbors? Exp. Neurol. 2019, 321, 113029. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.X.; Timbang, L.; Kim, J.-I.; Shang, Y.; Sandoval, K.; Tang, A.A.; Whistler, J.L.; Ding, J.B.; Huang, E.J. TGF-β Signaling in Dopaminergic Neurons Regulates Dendritic Growth, Excitatory-Inhibitory Synaptic Balance, and Reversal Learning. Cell Rep. 2016, 17, 3233–3245. [Google Scholar] [CrossRef]
- Meyers, E.A.; Kessler, J.A. TGF-β Family Signaling in Neural and Neuronal Differentiation, Development, and Function. Cold Spring Harb. Perspect. Biol. 2017, 9, a022244. [Google Scholar] [CrossRef]
- van der Meer, D.; Hoekstra, P.J.; Bralten, J.; van Donkelaar, M.; Heslenfeld, D.J.; Oosterlaan, J.; Faraone, S.V.; Franke, B.; Buitelaar, J.K.; Hartman, C.A. Interplay between stress response genes associated with attention-deficit hyperactivity disorder and brain volume. Genes Brain Behav. 2016, 15, 627–636. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Li, F.; Maiese, K. Activating Akt and the brain’s resources to drive cellular survival and prevent inflammatory injury. Histol. Histopathol. 2005, 20, 299–315. [Google Scholar]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef]
- Garabedian, M.J.; Harris, C.A.; Jeanneteau, F. Glucocorticoid receptor action in metabolic and neuronal function. F1000Research 2017, 6, 1208. [Google Scholar] [CrossRef]
- Li, X.; Hu, S.; Cai, Y.; Liu, X.; Luo, J.; Wu, T. Revving the engine: PKB/AKT as a key regulator of cellular glucose metabolism. Front. Physiol. 2024, 14, 1320964. [Google Scholar] [CrossRef]
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell. Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Pieczenik, S.R.; Neustadt, J. Mitochondrial dysfunction and molecular pathways of disease. Exp. Mol. Pathol. 2007, 83, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Bulut, M.; Selek, S.; Gergerlioglu, H.S.; Savas, H.A.; Yilmaz, H.R.; Yuce, M.; Ekici, G. Malondialdehyde levels in adult attention-deficit hyperactivity disorder. J. Psychiatry Neurosci. 2007, 32, 435–438. [Google Scholar] [PubMed]
- Verlaet, A.A.J.; Breynaert, A.; Ceulemans, B.; De Bruyne, T.; Fransen, E.; Pieters, L.; Savelkoul, H.F.J.; Hermans, N. Oxidative stress and immune aberrancies in attention-deficit/hyperactivity disorder (ADHD): A case-control comparison. Eur. Child Adolesc. Psychiatry 2019, 28, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Ashour, F.A.; Elshafie, M.K.; Ameen, O.; Abdelnabi, S.A.; Naguib, Y.M. Early detection of attention deficit hyperactivity disorder and/or epilepsy by oxidative stress biomarkers. Menoufia Med. J. 2016, 29, 954–960. [Google Scholar]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Avila-Costa, M.R.; Colín-Barenque, L.; Fortoul, T.I.; Machado-Salas, J.P.; Espinosa-Villanueva, J.; Rugerio-Vargas, C.; Borgonio, G.; Dorado, C.; Rivas-Arancibia, S. Motor impairments in an oxidative stress model and its correlation with cytological changes on rat striatum and prefrontal cortex. Int. J. Neurosci. 2001, 108, 193–200. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Archana, E.; Pai, P.; Prabhu, B.K.; Shenoy, R.P.; Prabhu, K.; Rao, A. Altered biochemical parameters in saliva of pediatric attention deficit hyperactivity disorder. Neurochem. Res. 2012, 37, 330–334. [Google Scholar] [CrossRef]
- Öğütlü, H.; Mertoğlu, C.; Gök, G.; Neşelioğlu, S. Thiols and ceruloplasmin levels in serum of children with attention deficit hyperactivity disorder: A cross-sectional study. Psychiatry Res. 2020, 294, 113546. [Google Scholar] [CrossRef]
- Baba, S.P.; Bhatnagar, A. Role of Thiols in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Otal, Y.; Kahraman, F.A.; Haydar, F.G.; Erel, Ö. Dynamic thiol/disulfide homeostasis as oxidative stress marker in diabetic ketoacidosis. Turk. J. Med. Sci. 2021, 51, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Ertürk, C.; Altay, M.A.; Büyükdoğan, H.; Çalışkan, G.; Erel, Ö. Thiol/disulfide homeostasis as a novel indicator of oxidative stress during the treatment process of patients with septic arthritis. Jt. Dis. Relat. Surg. 2020, 31, 502–508. [Google Scholar] [CrossRef]
- Zinellu, E.; Zinellu, A.; Pau, M.C.; Piras, B.; Fois, A.G.; Mellino, S.; Carru, C.; Mangoni, A.A.; Pirina, P. Glutathione Peroxidase in Stable Chronic Obstructive Pulmonary Disease: A Systematic Review and Meta-analysis. Antioxidants 2021, 10, 1745. [Google Scholar] [CrossRef]
- Selek, S.; Bulut, M.; Ocak, A.R.; Kalenderoğlu, A.; Savaş, H.A. Evaluation of total oxidative status in adult attention deficit hyperactivity disorder and its diagnostic implications. J. Psychiatr. Res. 2012, 46, 451–455. [Google Scholar] [CrossRef]
- Ceylan, M.; Sener, S.; Bayraktar, A.C.; Kavutcu, M. Oxidative imbalance in child and adolescent patients with attention-deficit/hyperactivity disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2010, 34, 1491–1494. [Google Scholar] [CrossRef]
- Simsek, S.; Gencoglan, S.; Ozaner, S.; Kaplan, I.; Kaya, M.C. Antioxidant Status and DNA Damage in Children with Attention Deficit Hyperactivity Disorder with or without Comorbid Disruptive Behavioral Disorders. Klin. Psikofarmakol. Bül.-Bull. Clin. Psychopharmacol. 2016, 26, 119–125. [Google Scholar] [CrossRef]
- Boriskin, P.; Gulenko, O.; Deviatkin, A.; Pavlova, O.; Toropovskiy, A. Correlation of superoxide dismutase activity distribution in serum and tissues of small experimental animals. IOP Conf. Ser. Earth Environ. Sci. 2019, 403, 012112. [Google Scholar] [CrossRef]
- Karababa, İ.F.; Savas, S.N.; Selek, S.; Cicek, E.; Cicek, E.I.; Asoglu, M.; Bayazit, H.; Kandemir, H.; Kati, M.; Ulas, T. Homocysteine Levels and Oxidative Stress Parameters in Patients With Adult ADHD. J. Atten. Disord. 2017, 21, 487–493. [Google Scholar] [CrossRef]
- Cannon Homaei, S.; Barone, H.; Kleppe, R.; Betari, N.; Reif, A.; Haavik, J. ADHD symptoms in neurometabolic diseases: Underlying mechanisms and clinical implications. Neurosci. Biobehav. Rev. 2022, 132, 838–856. [Google Scholar] [CrossRef]
- Guney, E.; Cetin, F.H.; Alisik, M.; Tunca, H.; Tas Torun, Y.; Iseri, E.; Isik Taner, Y.; Cayci, B.; Erel, O. Attention Deficit Hyperactivity Disorder and oxidative stress: A short term follow up study. Psychiatry Res. 2015, 229, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.J.; Puri, B.K. A randomized double-blind, placebo-controlled study of the effects of supplementation with highly unsaturated fatty acids on ADHD-related symptoms in children with specific learning difficulties. Prog. Neuropsychopharmacol. Biol. Psychiatry 2002, 26, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Manor, I.; Magen, A.; Keidar, D.; Rosen, S.; Tasker, H.; Cohen, T.; Richter, Y.; Zaaroor-Regev, D.; Manor, Y.; Weizman, A. The effect of phosphatidylserine containing Omega3 fatty-acids on attention-deficit hyperactivity disorder symptoms in children: A double-blind placebo-controlled trial, followed by an open-label extension. Eur. Psychiatry 2012, 27, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Perera, H.; Jeewandara, K.C.; Seneviratne, S.; Guruge, C. Combined ω3 and ω6 supplementation in children with attention-deficit hyperactivity disorder (ADHD) refractory to methylphenidate treatment: A double-blind, placebo-controlled study. J. Child Neurol. 2012, 27, 747–753. [Google Scholar] [CrossRef]
- Veluthakal, R.; Esparza, D.; Hoolachan, J.M.; Balakrishnan, R.; Ahn, M.; Oh, E.; Jayasena, C.S.; Thurmond, D.C. Mitochondrial Dysfunction, Oxidative Stress, and Inter-Organ Miscommunications in T2D Progression. Int. J. Mol. Sci. 2024, 25, 1504. [Google Scholar] [CrossRef]
- Tangvarasittichai, S. Oxidative stress, insulin resistance, dyslipidemia and type 2 diabetes mellitus. World J. Diabetes 2015, 6, 456–480. [Google Scholar] [CrossRef]
- Ernst, M.; Liebenauer, L.L.; King, A.C.; Fitzgerald, G.A.; Cohen, R.M.; Zametkin, A.J. Reduced brain metabolism in hyperactive girls. J. Am. Acad. Child Adolesc. Psychiatry 1994, 33, 858–868. [Google Scholar] [CrossRef]
- Verma, P.; Singh, A.; Nthenge-Ngumbau, D.N.; Rajamma, U.; Sinha, S.; Mukhopadhyay, K.; Mohanakumar, K.P. Attention deficit-hyperactivity disorder suffers from mitochondrial dysfunction. BBA Clin. 2016, 6, 153–158. [Google Scholar] [CrossRef]
- Siddiqui, M.F.; Elwell, C.; Johnson, M.H. Mitochondrial Dysfunction in Autism Spectrum Disorders. Autism-Open Access 2016, 6, 1000190. [Google Scholar] [CrossRef]
- Welsh, K.J.; Kirkman, M.S.; Sacks, D.B. Role of Glycated Proteins in the Diagnosis and Management of Diabetes: Research Gaps and Future Directions. Diabetes Care 2016, 39, 1299–1306. [Google Scholar] [CrossRef]
- Sauerzopf, U.; Weidenauer, A.; Dajic, I.; Bauer, M.; Bartova, L.; Meyer, B.; Nics, L.; Philippe, C.; Pfaff, S.; Pichler, V.; et al. Disrupted relationship between blood glucose and brain dopamine D2/3 receptor binding in patients with first-episode schizophrenia. NeuroImage Clin. 2021, 32, 102813. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, G.; Maillard, P.; Himali, J.J.; Beiser, A.S.; Au, R.; Wolf, P.A.; Seshadri, S.; DeCarli, C. Glucose indices are associated with cognitive and structural brain measures in young adults. Neurology 2015, 84, 2329–2337. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Gammon, S.T.; Sutton, M.N.; Zacharias, N.M.; Bhattacharya, P.; Piwnica-Worms, D. Excess exogenous pyruvate inhibits lactate dehydrogenase activity in live cells in an MCT1-dependent manner. J. Biol. Chem. 2021, 297, 100775. [Google Scholar] [CrossRef] [PubMed]
- Medin, T.; Medin, H.; Hefte, M.B.; Storm-Mathisen, J.; Bergersen, L.H. Upregulation of the lactate transporter monocarboxylate transporter 1 at the blood-brain barrier in a rat model of attention-deficit/hyperactivity disorder suggests hyperactivity could be a form of self-treatment. Behav. Brain Res. 2019, 360, 279–285. [Google Scholar] [CrossRef]
- Li, C.; Liu, F.-Y.; Shen, Y.; Tian, Y.; Han, F.-J. Research progress on the mechanism of glycolysis in ovarian cancer. Front. Immunol. 2023, 14, 1284853. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Y.; Wang, H.; Liu, L.; Li, W.; Xie, P. The regulatory effects of lactic acid on neuropsychiatric disorders. Discov. Ment. Health 2022, 2, 8. [Google Scholar] [CrossRef]
- Edden, R.A.E.; Crocetti, D.; Zhu, H.; Gilbert, D.L.; Mostofsky, S.H. Reduced GABA Concentration in Attention-Deficit/Hyperactivity Disorder. Arch. Gen. Psychiatry 2012, 69, 750–753. [Google Scholar] [CrossRef]
- Bollmann, S.; Ghisleni, C.; Poil, S.-S.; Martin, E.; Ball, J.; Eich-Höchli, D.; Edden, R.A.E.; Klaver, P.; Michels, L.; Brandeis, D.; et al. Developmental changes in gamma-aminobutyric acid levels in attention-deficit/hyperactivity disorder. Transl. Psychiatry 2015, 5, e589. [Google Scholar] [CrossRef]
- Ende, G.; Cackowski, S.; Van Eijk, J.; Sack, M.; Demirakca, T.; Kleindienst, N.; Bohus, M.; Sobanski, E.; Krause-Utz, A.; Schmahl, C. Impulsivity and Aggression in Female BPD and ADHD Patients: Association with ACC Glutamate and GABA Concentrations. Neuropsychopharmacology 2016, 41, 410–418. [Google Scholar] [CrossRef]
- Schür, R.R.; Draisma, L.W.R.; Wijnen, J.P.; Boks, M.P.; Koevoets, M.G.J.C.; Joëls, M.; Klomp, D.W.; Kahn, R.S.; Vinkers, C.H. Brain GABA levels across psychiatric disorders: A systematic literature review and meta-analysis of (1) H-MRS studies. Hum. Brain Mapp. 2016, 37, 3337–3352. [Google Scholar] [CrossRef]
- Ross, J.M.; Öberg, J.; Brené, S.; Coppotelli, G.; Terzioglu, M.; Pernold, K.; Goiny, M.; Sitnikov, R.; Kehr, J.; Trifunovic, A.; et al. High brain lactate is a hallmark of aging and caused by a shift in the lactate dehydrogenase A/B ratio. Proc. Natl. Acad. Sci. USA 2010, 107, 20087–20092. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, P.; Žorić, L.; Stefanović, I.; Džunić, B.; Djordjević-Jocić, J.; Radenković, M.; Jovanović, M. Lactate dehydrogenase and oxidative stress activity in primary open-angle glaucoma aqueous humour. Bosn. J. Basic Med. Sci. 2010, 10, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, T.; Wang, C.; Wang, S.; Wang, W.; Ma, D.; Li, Y.; Zhao, H.; Chen, J. Effects of Tianshu Capsule on Spontaneously Hypertensive Rats as Revealed by 1H-NMR-Based Metabolic Profiling. Front. Pharmacol. 2019, 10, 989. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef]
- McGill, M.R. The past and present of serum aminotransferases and the future of liver injury biomarkers. EXCLI J. 2016, 15, 817–828. [Google Scholar] [CrossRef]
- Skalny, A.V.; Mazaletskaya, A.L.; Zaitseva, I.P.; Skalny, A.A.; Spandidos, D.A.; Tsatsakis, A.; Lobanova, Y.N.; Skalnaya, M.G.; Aschner, M.; Tinkov, A.A. Alterations in serum amino acid profiles in children with attention deficit/hyperactivity disorder. Biomed. Rep. 2021, 14, 47. [Google Scholar] [CrossRef]
- Kim, S.L.; Shin, S.; Yang, S.J. Iron Homeostasis and Energy Metabolism in Obesity. Clin. Nutr. Res. 2022, 11, 316–330. [Google Scholar] [CrossRef]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef]
- Long, H.; Zhu, W.; Wei, L.; Zhao, J. Iron homeostasis imbalance and ferroptosis in brain diseases. MedComm 2023, 4, e298. [Google Scholar] [CrossRef]
- McBean, G.J.; López, M.G.; Wallner, F.K. Redox-based therapeutics in neurodegenerative disease. Br. J. Pharmacol. 2017, 174, 1750–1770. [Google Scholar] [CrossRef]
- Everett, J.; Céspedes, E.; Shelford, L.R.; Exley, C.; Collingwood, J.F.; Dobson, J.; van der Laan, G.; Jenkins, C.A.; Arenholz, E.; Telling, N.D. Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer’s disease peptide β-amyloid (1-42). J. R. Soc. Interface 2014, 11, 20140165. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Zhang, J. Iron Metabolism, Ferroptosis, and the Links With Alzheimer’s Disease. Front. Neurosci. 2019, 13, 1443. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guo, X.; Zeng, Y.; Mo, X.; Hong, S.; He, H.; Li, J.; Fatima, S.; Liu, Q. Oxidative stress induces mitochondrial iron overload and ferroptotic cell death. Sci. Rep. 2023, 13, 15515. [Google Scholar] [CrossRef]
- García-Yébenes, I.; Sobrado, M.; Moraga, A.; Zarruk, J.G.; Romera, V.G.; Pradillo, J.M.; Perez de la Ossa, N.; Moro, M.A.; Dávalos, A.; Lizasoain, I. Iron overload, measured as serum ferritin, increases brain damage induced by focal ischemia and early reperfusion. Neurochem. Int. 2012, 61, 1364–1369. [Google Scholar] [CrossRef]
- Yang, J.; Wang, M.; Wang, S.; Li, G.; Gao, Y. Study on ferroptosis pathway that operates in hypertensive brain damage. Clin. Exp. Hypertens. 2020, 42, 748–752. [Google Scholar] [CrossRef]
- Chen, J.-R.; Hsu, S.-F.; Hsu, C.-D.; Hwang, L.-H.; Yang, S.-C. Dietary patterns and blood fatty acid composition in children with attention-deficit hyperactivity disorder in Taiwan. J. Nutr. Biochem. 2004, 15, 467–472. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, L.; Zhang, L.; Qu, Y.; Mu, D. Iron Status in Attention-Deficit/Hyperactivity Disorder: A Systematic Review and Meta-Analysis. PLoS ONE 2017, 12, e0169145. [Google Scholar] [CrossRef]
- Tseng, P.-T.; Cheng, Y.-S.; Yen, C.-F.; Chen, Y.-W.; Stubbs, B.; Whiteley, P.; Carvalho, A.F.; Li, D.-J.; Chen, T.-Y.; Yang, W.-C.; et al. Peripheral iron levels in children with attention-deficit hyperactivity disorder: A systematic review and meta-analysis. Sci. Rep. 2018, 8, 788. [Google Scholar] [CrossRef]
- Collings, R.; Harvey, L.J.; Hooper, L.; Hurst, R.; Brown, T.J.; Ansett, J.; King, M.; Fairweather-Tait, S.J. The absorption of iron from whole diets: A systematic review. Am. J. Clin. Nutr. 2013, 98, 65–81. [Google Scholar] [CrossRef]
- Navas, F.J.; Córdova, A. Iron distribution in different tissues in rats following exercise. Biol. Trace Elem. Res. 2000, 73, 259–268. [Google Scholar] [CrossRef]
- Glancy, B.; Kane, D.A.; Kavazis, A.N.; Goodwin, M.L.; Willis, W.T.; Gladden, L.B. Mitochondrial lactate metabolism: History and implications for exercise and disease. J. Physiol. 2022, 599, 863–888. Available online: https://pmc.ncbi.nlm.nih.gov/articles/PMC8439166/ (accessed on 27 November 2024). [CrossRef] [PubMed]
- Kozłowska, A.; Wojtacha, P.; Majewski, M.; Równiak, M. The cytokine alterations/abnormalities and oxidative damage in the pancreas during hypertension development. Pflugers Arch. 2019, 471, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Mullen, R.J.; Buck, C.R.; Smith, A.M. NeuN, a neuronal specific nuclear protein in vertebrates. Dev. Camb. Engl. 1992, 116, 201–211. [Google Scholar] [CrossRef]
- Páleníček, T.; Fujáková, M.; Brunovský, M.; Balíková, M.; Horáček, J.; Gorman, I.; Tylš, F.; Tišlerová, B.; Soš, P.; Bubeníková-Valešová, V.; et al. Electroencephalographic spectral and coherence analysis of ketamine in rats: Correlation with behavioral effects and pharmacokinetics. Neuropsychobiology 2011, 63, 202–218. [Google Scholar] [CrossRef]
- Yang, C.; Hu, Y.-M.; Zhou, Z.-Q.; Zhang, G.-F.; Yang, J.-J. Acute administration of ketamine in rats increases hippocampal BDNF and mTOR levels during forced swimming test. Ups. J. Med. Sci. 2013, 118, 3–8. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 5th ed.; Elsevier Academic Press: Amsterdam, The Netherlands, 2005; ISBN 978-0-12-088472-8. Available online: http://catdir.loc.gov/catdir/description/els051/2004303983.html (accessed on 25 October 2023).
- Oorschot, D.E. Total number of neurons in the neostriatal, pallidal, subthalamic, and substantia nigral nuclei of the rat basal ganglia: A stereological study using the cavalieri and optical disector methods. J. Comp. Neurol. 1996, 366, 580–599. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open Source platform for biological image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- DeVito, J.L.; Graham, J.; Sackett, G.P. Volumetric growth of the major brain divisions in fetal Macaca nemestrina. J. Hirnforsch. 1989, 30, 479–487. [Google Scholar]
- West, M.J.; Gundersen, H.J. Unbiased stereological estimation of the number of neurons in the human hippocampus. J. Comp. Neurol. 1990, 296, 1–22. [Google Scholar] [CrossRef]
- Weitner, T.; Inić, S.; Jablan, J.; Gabričević, M.; Domijan, A.-M. Spectrophotometric Determination of Malondialdehyde in Urine Suitable for Epidemiological Studies. Croat. Chem. Acta 2016, 89, 133–139. [Google Scholar] [CrossRef]
- Chan, K.-Y.; Wasserman, B.P. Direct Colorimetric Assay of Free Thiol Groups and Disulfide Bonds in Suspensions of Solubilized and Particulate Cereal Proteins’. Cereal Chem. 1993, 70, 22. [Google Scholar]
- Tkachenko, H.; Kurhaluk, N.; Grudniewska, J. Oxidative stress biomarkers in different tissues of rainbow trout (Oncorhynchus mykiss) exposed to Disinfectant-CIP formulated with peracetic acid and hydrogen peroxide. Fish. Aquat. Life 2014, 22, 207–219. [Google Scholar] [CrossRef]
- Bovaird, J.H.; Ngo, T.T.; Lenhoff, H.M. Optimizing the o-phenylenediamine assay for horseradish peroxidase: Effects of phosphate and pH, substrate and enzyme concentrations, and stopping reagents. Clin. Chem. 1982, 28, 2423–2426. [Google Scholar] [CrossRef] [PubMed]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974, 249, 7130–7139. [Google Scholar] [CrossRef]
- Calipari, E.S.; Huggins, K.N.; Mathews, T.A.; Jones, S.R. Conserved dorsal–ventral gradient of dopamine release and uptake rate in mice, rats and rhesus macaques. Neurochem. Int. 2012, 61, 986–991. [Google Scholar] [CrossRef]
- Bocarsly, M.E.; Silva, D.d.S.e.; Kolb, V.; Luderman, K.D.; Shashikiran, S.; Rubinstein, M.; Sibley, D.R.; Dobbs, L.K.; Alvarez, V.A. A Mechanism Linking Two Known Vulnerability Factors for Alcohol Abuse: Heightened Alcohol Stimulation and Low Striatal Dopamine D2 Receptors. Cell Rep. 2019, 29, 1147–1163.e5. [Google Scholar] [CrossRef]
- Zhang-James, Y.; Middleton, F.A.; Faraone, S.V. Genetic architecture of Wistar-Kyoto rat and spontaneously hypertensive rat substrains from different sources. Physiol. Genomics 2013, 45, 528–538. [Google Scholar] [CrossRef]
- Nam, H.; Clinton, S.M.; Jackson, N.L.; Kerman, I.A. Learned helplessness and social avoidance in the Wistar-Kyoto rat. Front. Behav. Neurosci. 2014, 8, 109. [Google Scholar] [CrossRef]
- Dunn, G.A.; Nigg, J.T.; Sullivan, E.L. Neuroinflammation as a risk factor for attention deficit hyperactivity disorder. Pharmacol. Biochem. Behav. 2019, 182, 22–34. [Google Scholar] [CrossRef]
- Leffa, D.T.; Torres, I.L.S.; Rohde, L.A. A Review on the Role of Inflammation in Attention-Deficit/Hyperactivity Disorder. Neuroimmunomodulation 2018, 25, 328–333. [Google Scholar] [CrossRef]
- Koç, S.; Güler, E.M.; Derin, S.; Gültekin, F.; Aktaş, S. Oxidative and Inflammatory Parameters in Children and Adolescents with ADHD. J. Atten. Disord. 2023, 27, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Biehl, S.C.; Gschwendtner, K.M.; Guhn, A.; Müller, L.D.; Reichert, S.; Heupel, J.; Reif, A.; Deckert, J.; Herrmann, M.J.; Jacob, C.P. Does adult ADHD interact with COMTval158met genotype to influence working memory performance? ADHD Atten. Deficit Hyperact. Disord. 2015, 7, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Wang, G.-J.; Tomasi, D.; Kollins, S.H.; Wigal, T.L.; Newcorn, J.H.; Telang, F.W.; Fowler, J.S.; Logan, J.; Wong, C.T.; et al. Methylphenidate-Elicited Dopamine Increases in Ventral Striatum Are Associated with Long-Term Symptom Improvement in Adults with Attention Deficit Hyperactivity Disorder. J. Neurosci. 2012, 32, 841–849. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age (Weeks) | SHRs Body Weight (g) | WKYs Body Weight (g) |
|---|---|---|
| 4 | 89.80 ± 1.90 | 84.30 ± 0.09 |
| 5 | 115.08 ± 6.31 | 120.08 ± 0.08 |
| 6 | 154.86 ± 6.13 | 159.28 ± 0.05 |
| 7 | 200.78 ± 5.37 | 191.13 ± 6.06 |
| 8 | 220.35 ± 10.76 | 226.95 ± 0.04 |
| 9 | 249.79 ± 9.86 | 242.67 ± 9.09 |
| 10 | 269.58 ± 7.92 | 264.95 ± 10.02 |
| Marker/ Enzyme | Determination Method | ||
|---|---|---|---|
| Immune | IL-1α | The ELISA test plate’s absorbance was quantified using a TECAN Infinite M200 PRO plate reader (Austria) at a wavelength of λ = 450 nm. The outcomes were expressed relative to the amount of protein, presented per milligram. | ELISA Kit for Rat IL-1 alfa (E0071r; EIAab; Wuhan, China) |
| IL-1β | ELISA Kit for Rat IL-1 beta (E0563r; EIAab; Wuhan, China) | ||
| IL-6 | ELISA Kit for Rat IL-6 (E0079r; EIAab; Wuhan, China) | ||
| mTOR | ELISA kit for Rat Serine/threonine-protein mTOR (ER1520; Wuhan Fine Biotech Co, Ltd.; Wuhan, China) | ||
| TGF-β | TGF beta-1 Multispecies Matched An-tibody Pair, (CHC1683, ThermoFisher Scientific, Waltham, MA, USA) | ||
| AKT-1 | ELISA Kit FOR rat RAC-alpha serine/threonine-protein kinase (E0382r; EIAab; Wuhan, China) | ||
| GCsRβ | ELISA kit for rat Glucocorticoid receptor β (E1608r; EIAab; Wuhan, China) | ||
| Oxidative Stress | MDA | Markers’ levels/activity were assessed following the following: | Weitner et al. procedure [213] |
| -SH | Ellman method [214] | ||
| SOD | Tkachenko and Grudniewska protocol [215] | ||
| POD | Kinetic method [216] | ||
| GSR | Tkachenko and Grudniewska method [215] | ||
| GST | Spectrophotometric assay procedure [217] | ||
| Metabolic | G | Markers’ content/activity was measured employing specific commercial reagent sets | Glucose oxidase reagent set (G7521; Pointe Scientific, Inc.; Poland) |
| FrAm | Fructosamine reagent set (F7546; Pointe Scientific, Inc.; Poland) | ||
| Fe | Total iron reagent set (17505; Pointe Scientific, Inc.; Poland) | ||
| LA | Lactate acid reagent set (L7596; Pointe Scientific, Inc.; Poland) | ||
| LDH | Lactate dehydrogenase reagent set (L7572; Pointe Scientific, Inc.; Poland) | ||
| ALT | Alanine transaminase (SGPT) reagent set (A7526; Pointe Scientific, Inc.; Poland) | ||
| AST | Aspartate aminotransferase (SGOT) reagent set; (A7561; Pointe Scientific, Inc.; Poland) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogdańska-Chomczyk, E.; Wojtacha, P.; Tsai, M.-L.; Huang, A.C.W.; Kozłowska, A. Alterations in Striatal Architecture and Biochemical Markers’ Levels During Postnatal Development in the Rat Model of an Attention Deficit/Hyperactivity Disorder (ADHD). Int. J. Mol. Sci. 2024, 25, 13652. https://doi.org/10.3390/ijms252413652
Bogdańska-Chomczyk E, Wojtacha P, Tsai M-L, Huang ACW, Kozłowska A. Alterations in Striatal Architecture and Biochemical Markers’ Levels During Postnatal Development in the Rat Model of an Attention Deficit/Hyperactivity Disorder (ADHD). International Journal of Molecular Sciences. 2024; 25(24):13652. https://doi.org/10.3390/ijms252413652
Chicago/Turabian StyleBogdańska-Chomczyk, Ewelina, Paweł Wojtacha, Meng-Li Tsai, Andrew Chih Wei Huang, and Anna Kozłowska. 2024. "Alterations in Striatal Architecture and Biochemical Markers’ Levels During Postnatal Development in the Rat Model of an Attention Deficit/Hyperactivity Disorder (ADHD)" International Journal of Molecular Sciences 25, no. 24: 13652. https://doi.org/10.3390/ijms252413652
APA StyleBogdańska-Chomczyk, E., Wojtacha, P., Tsai, M.-L., Huang, A. C. W., & Kozłowska, A. (2024). Alterations in Striatal Architecture and Biochemical Markers’ Levels During Postnatal Development in the Rat Model of an Attention Deficit/Hyperactivity Disorder (ADHD). International Journal of Molecular Sciences, 25(24), 13652. https://doi.org/10.3390/ijms252413652