

Beyond NMDA Receptors: A Narrative Review of Ketamine’s Rapid and Multifaceted Mechanisms in Depression Treatment

Abstract
:1. Introduction
2. Materials and Methods
- What molecular mechanisms underlie the rapid antidepressant effects of ketamine?
- What changes in neuronal networks are induced by ketamine, and how do they contribute to its therapeutic efficacy in depression?
- What molecular mechanisms influence therapeutic approaches to using ketamine in MDD, including its impact on treatment personalization and efficacy optimization?
3. Pharmacology Profile of Ketamine
3.1. Structure
3.2. Routes of Administration and Bioavailability
3.3. Dosages and Time of Action
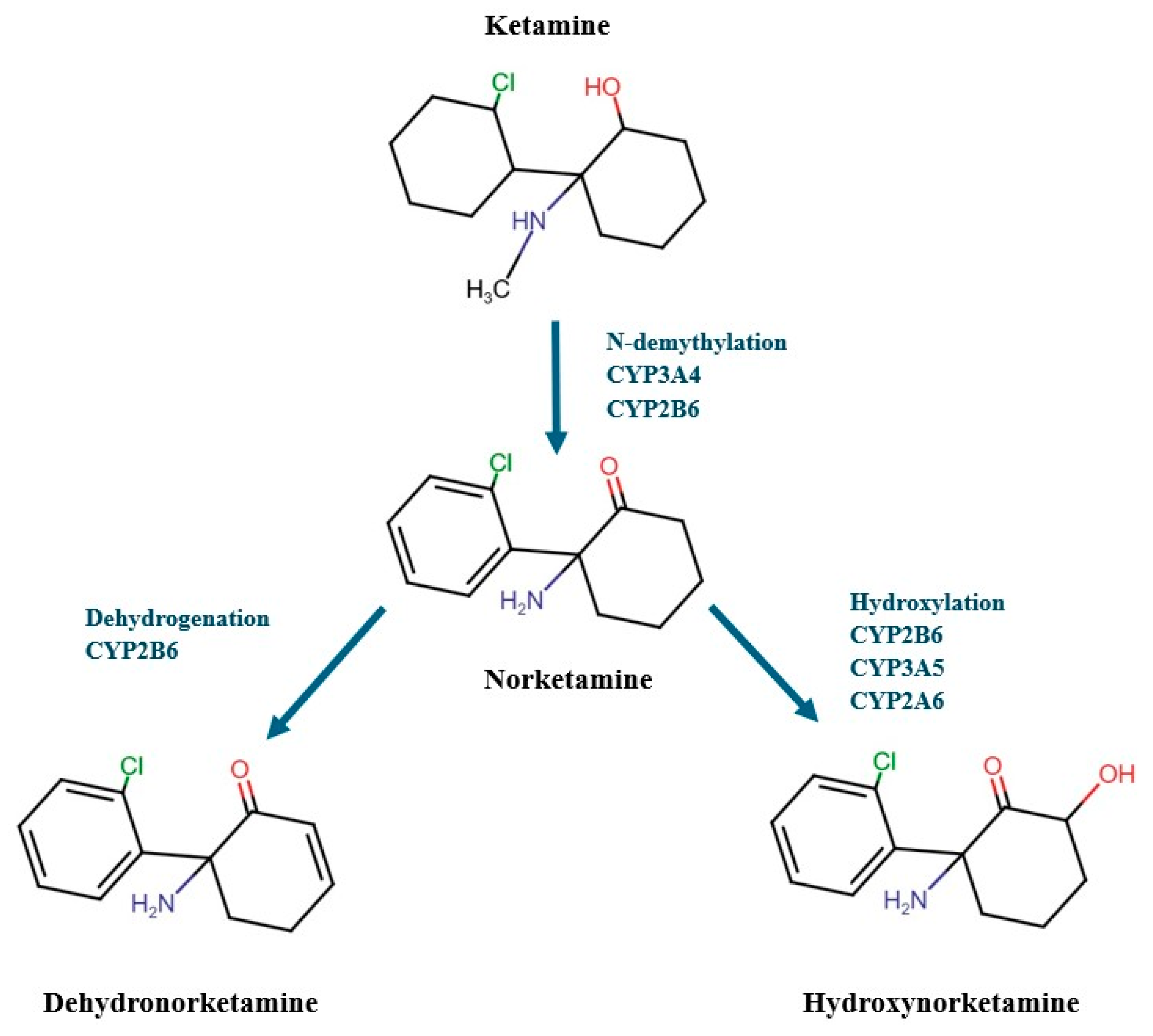
3.4. Distribution, Metabolism and Elimination
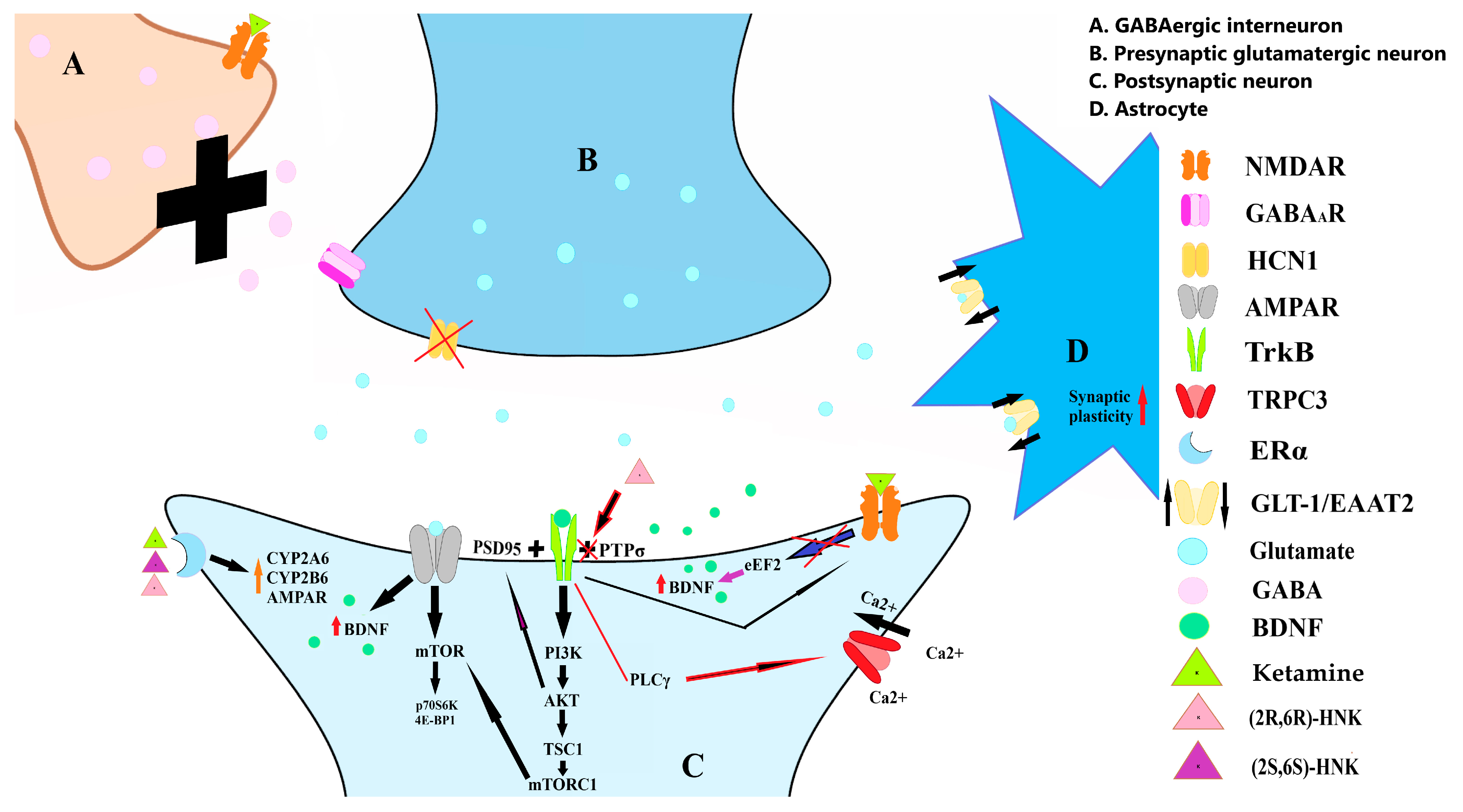
4. NMDARs Mechanism
Sex Differences in NMDARs Expression and Their Influence on Ketamine’s Action
5. Neuroplasticity
5.1. BDNF Role in Depression
5.2. Ketamine’s Impact on BDNF and Its Pathways
5.3. Glial Involvement
5.4. Structural Changes in Depression
5.5. Genetic Considerations
6. Opioid Receptor System Involvement in Ketamine Antidepressant Effect
7. Triple Network Dysfunction in Depression and Ketamine Influence
7.1. Structural and Functional Organization of Brain Networks in Depression
7.1.1. Default Mode Network
7.1.2. Central Executive Network
7.1.3. Salience Network
7.2. Ketamine’s Modulatory Effects on Triple Network Function
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACC | Anterior cingulate cortex. |
| AI | Anterior insula |
| AMPA | Alpha-amino-3-hydroxy-5-methyl-4-isooxazole-propionic acid |
| AMPARs | Alpha-amino-3-hydroxy-5-methyl-4-isooxazole-propionic acid receptors |
| Bcl2/Bax | B-cell lymphoma 2/Bcl-2-associated X protein |
| BDNF | Brain-derived neurotrophic factor |
| CA1.SR | CA1 stratum radiatum |
| Ca2+ | Calcium |
| cAMP | Cyclic adenosine monophosphate |
| CEN | Central Executive Network |
| CNS | Central nervous system |
| CREB | Phosphorylated cAMP response element binding protein |
| CUS | Chronic unpredictable stress |
| CYP | Cytochrome P450 |
| dACC | Dorsal anterior cingulate cortex |
| DA | Dopamine |
| DHNK | Dehydronorketamine |
| dlPFC | Dorsolateral prefrontal cortex |
| DMN | Default Mode Network |
| dmPFC | Dorsomedial prefrontal cortex |
| DOR | Delta |
| DTI | Diffusion tensor imaging |
| EAAT1 | Excitatory amino acid transporter 1 |
| EAAT3 | Excitatory amino acid transporter 3 |
| eEF2 | Eukaryotic elongation factor 2 |
| ERα | Estrogen receptor alpha |
| FC | Functional connectivity |
| FIC | Fronto-insular cortex |
| GABA | Gamma-aminobutyric acid |
| GFAP | Glial fibrillary acidic protein |
| GLT-1 | Glutamate transporter 1 |
| HCN1 | Hyperpolarization-activated cyclic nucleotide-gated channel 1 |
| HDRS | Hamilton Depression Rating Scale |
| HNK | Hydroxynorketamine |
| Ih | Hyperpolarization-activated current |
| IM | Intramuscular |
| IN | Intranasal |
| IV | Intravenous |
| K+ | Potassium |
| KOR | Kappa |
| LTD | Long-term depression |
| LTP | Long-term potentiation |
| MAOIs | Monoamine oxidase inhibitors |
| MDD | Major depressive disorder |
| Mg2⁺ | Magnesium |
| MMAs | Multimodal antidepressants |
| MOR | Mu |
| mPFC | Medial prefrontal cortex |
| MRI | Magnetic resonance imaging |
| mRNA | Messenger ribonucleic acid |
| mTORC1 | mTOR Complex 1 |
| mTOR | Mechanistic target of rapamycin |
| Na+ | Sodium |
| NASSAs | Specific serotonergic antidepressants |
| NDRIs | NE reuptake inhibitors |
| NE | Norepinephrine |
| NET | Norepinephrine transporter |
| mEPSCs | Miniature excitatory postsynaptic currents |
| NMDA | N-methyl-D-aspartate |
| NMDARs | N-methyl-D-aspartate acid glutamate receptors |
| NRIs | Selective norepinephrine reuptake inhibitors |
| OFC | Orbitofrontal cortex |
| p70S6K | p70S6 kinase |
| PCC | Posterior cingulate cortex |
| pERK | Phosphorylated extracellular signal-regulated kinase |
| PET | Positron emission tomography |
| PFC | Prefrontal cortex |
| pgACC | Pregenual anterior cingulate cortex |
| PI3K | Phosphatidylinositol 3-kinase |
| PLCγ | Phospholipase Cγ |
| PNs | Pyramidal neurons |
| PrL | Prelimbic |
| proBDNF | Brain-derived neurotrophic factor precursor |
| PSD95 | Postsynaptic density 95 |
| PTPσ | Protein tyrosine phosphatase sigma |
| p-TrkB | Phosphorylated tropomyosin-related kinase B |
| RasGrf1 | Ras protein-specific guanine nucleotide-releasing factor 1 |
| R-ketamine | Arketamine |
| RS-fMRI | Resting-state functional magnetic resonance imaging |
| SARIs | Serotonin-2 antagonists and reuptake inhibitors |
| SC | Subcutaneous |
| SERT | Serotonin transporter |
| sgACC | Subgenual anterior cingulate cortex |
| S-ketamine | Esketamine |
| SNRIs | Norepinephrine reuptake inhibitors |
| SN | Salience Network |
| SSRIs | Selective serotonin reuptake inhibitors |
| TCAs | Tricyclic antidepressants |
| TeCAs | Tetracyclic antidepressants |
| TPJ | Bilateral temporal parietal junction |
| TRD | Treatment-resistant depression |
| TrkB | Tropomyosin receptor kinase B |
| TRPC3 | Transient receptor potential canonical subfamily 3 |
| TSC2 | Tuberous Sclerosis Complex 2 |
| UGTs | Uridine-5′-diphospho-glucuronosyl-transferases |
| Val66Met | Substitution from valine to methionine at codon 66 |
| vlPFC | Ventrolateral prefrontal cortex |
| vmPFC | Ventromedial prefrontal cortex |
| WHO | World Health Organization |
| Zn2⁺ | Zinc |
| Δ-MD | Changes i[111]n DTI mean diffusivity |
| μOR | μ-opioid receptor |
| 4E-BP1 | Eukaryotic initiation factor 4E binding protein 1 |
| 5-HT | Serotonin |
References
- Institute of Health Metrics and Evaluation. Global Health Data Exchange (GHDx). Available online: https://vizhub.healthdata.org/gbd-results/ (accessed on 5 October 2024).
- Malhi, G.S.; Mann, J.J. Depression. Lancet 2018, 392, 2299–2312. [Google Scholar] [CrossRef]
- Moitra, M.; Santomauro, D.; Degenhardt, L.; Collins, P.Y.; Whiteford, H.; Vos, T.; Ferrari, A. Estimating the risk of suicide associated with mental disorders: A systematic review and meta-regression analysis. J. Psychiatr. Res. 2021, 137, 242–249. [Google Scholar] [CrossRef]
- Chesney, E.; Goodwin, G.M.; Fazel, S. Risks of all-cause and suicide mortality in mental disorders: A meta-review. World Psychiatry 2014, 13, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.J.; Norman, R.E.; Freedman, G.; Baxter, A.J.; Pirkis, J.E.; Harris, M.G.; Page, A.; Carnahan, E.; Degenhardt, L.; Vos, T.; et al. The burden attributable to mental and substance use disorders as risk factors for suicide: Findings from the Global Burden of Disease Study 2010. PLoS ONE 2014, 9, e91936. [Google Scholar] [CrossRef] [PubMed]
- Vigo, D.; Thornicroft, G.; Atun, R. Estimating the true global burden of mental illness. Lancet Psychiatry 2016, 3, 171–178. [Google Scholar] [CrossRef]
- Bromet, E.; Andrade, L.H.; Hwang, I.; Sampson, N.A.; Alonso, J.; de Girolamo, G.; de Graaf, R.; Demyttenaere, K.; Hu, C.; Iwata, N.; et al. Cross-national epidemiology of DSM-IV major depressive episode. BMC Med. 2011, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Johnston, K.M.; Powell, L.C.; Anderson, I.M.; Szabo, S.; Cline, S. The burden of treatment-resistant depression: A systematic review of the economic and quality of life literature. J. Affect. Disord. 2019, 242, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Hock, R.S.; Or, F.; Kolappa, K.; Burkey, M.D.; Surkan, P.J.; Eaton, W.W. A new resolution for global mental health. Lancet 2012, 379, 1367–1368. [Google Scholar] [CrossRef] [PubMed]
- Warden, D.; Rush, A.J.; Trivedi, M.H.; Fava, M.; Wisniewski, S.R. The STAR*D Project results: A comprehensive review of findings. Curr. Psychiatry Rep. 2007, 9, 449–459. [Google Scholar] [CrossRef]
- McIntyre, R.S.; Alsuwaidan, M.; Baune, B.T.; Berk, M.; Demyttenaere, K.; Goldberg, J.F.; Gorwood, P.; Ho, R.; Kasper, S.; Kennedy, S.H.; et al. Treatment-resistant depression: Definition, prevalence, detection, management, and investigational interventions. World Psychiatry 2023, 22, 394–412. [Google Scholar] [CrossRef] [PubMed]
- Sim, K.; Lau, W.K.; Sim, J.; Sum, M.Y.; Baldessarini, R.J. Prevention of Relapse and Recurrence in Adults with Major Depressive Disorder: Systematic Review and Meta-Analyses of Controlled Trials. Int. J. Neuropsychopharmacol. 2015, 19. [Google Scholar] [CrossRef] [PubMed]
- Schildkraut, J.J. The catecholamine hypothesis of affective disorders: A review of supporting evidence. Am. J. Psychiatry 1965, 122, 509–522. [Google Scholar] [CrossRef]
- Delgado, P.L. Depression: The case for a monoamine deficiency. J. Clin. Psychiatry 2000, 61 (Suppl. S6), 7–11. [Google Scholar] [PubMed]
- Hirschfeld, R.M. History and evolution of the monoamine hypothesis of depression. J. Clin. Psychiatry 2000, 61 (Suppl. S6), 4–6. [Google Scholar] [PubMed]
- Kupfer, D.J. The pharmacological management of depression. Dialogues Clin. Neurosci. 2005, 7, 191–205. [Google Scholar] [CrossRef]
- Hammad, T.A.; Laughren, T.; Racoosin, J. Suicidality in pediatric patients treated with antidepressant drugs. Arch. Gen. Psychiatry 2006, 63, 332–339. [Google Scholar] [CrossRef]
- Rucci, P.; Frank, E.; Scocco, P.; Calugi, S.; Miniati, M.; Fagiolini, A.; Cassano, G.B. Treatment-emergent suicidal ideation during 4 months of acute management of unipolar major depression with SSRI pharmacotherapy or interpersonal psychotherapy in a randomized clinical trial. Depress. Anxiety 2011, 28, 303–309. [Google Scholar] [CrossRef]
- McIntyre, R.S.; Jain, R. Glutamatergic Modulators for Major Depression from Theory to Clinical Use. CNS Drugs 2024, 38, 869–890. [Google Scholar] [CrossRef] [PubMed]
- Jarończyk, M.; Walory, J. Novel Molecular Targets of Antidepressants. Molecules 2022, 27, 533. [Google Scholar] [CrossRef]
- Kroeze, Y.; Zhou, H.; Homberg, J.R. The genetics of selective serotonin reuptake inhibitors. Pharmacol. Ther. 2012, 136, 375–400. [Google Scholar] [CrossRef] [PubMed]
- Edinoff, A.N.; Akuly, H.A.; Hanna, T.A.; Ochoa, C.O.; Patti, S.J.; Ghaffar, Y.A.; Kaye, A.D.; Viswanath, O.; Urits, I.; Boyer, A.G.; et al. Selective Serotonin Reuptake Inhibitors and Adverse Effects: A Narrative Review. Neurol. Int. 2021, 13, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Shulman, K.I.; Herrmann, N.; Walker, S.E. Current place of monoamine oxidase inhibitors in the treatment of depression. CNS Drugs 2013, 27, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Hillhouse, T.M.; Porter, J.H. A brief history of the development of antidepressant drugs: From monoamines to glutamate. Exp. Clin. Psychopharmacol. 2015, 23, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Cusack, B.; Nelson, A.; Richelson, E. Binding of antidepressants to human brain receptors: Focus on newer generation compounds. Psychopharmacology 1994, 114, 559–565. [Google Scholar] [CrossRef]
- Owens, M.J.; Morgan, W.N.; Plott, S.J.; Nemeroff, C.B. Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. J. Pharmacol. Exp. Ther. 1997, 283, 1305–1322. [Google Scholar]
- Tian, H.; Hu, Z.; Xu, J.; Wang, C. The molecular pathophysiology of depression and the new therapeutics. MedComm (2020) 2022, 3, e156. [Google Scholar] [CrossRef] [PubMed]
- Bymaster, F.P.; Dreshfield-Ahmad, L.J.; Threlkeld, P.G.; Shaw, J.L.; Thompson, L.; Nelson, D.L.; Hemrick-Luecke, S.K.; Wong, D.T. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology 2001, 25, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Millan, M.J.; Gobert, A.; Lejeune, F.; Newman-Tancredi, A.; Rivet, J.M.; Auclair, A.; Peglion, J.L. S33005, a novel ligand at both serotonin and norepinephrine transporters: I. Receptor binding, electrophysiological, and neurochemical profile in comparison with venlafaxine, reboxetine, citalopram, and clomipramine. J. Pharmacol. Exp. Ther. 2001, 298, 565–580. [Google Scholar] [PubMed]
- Feighner, J.P. Mechanism of action of antidepressant medications. J. Clin. Psychiatry 1999, 60 (Suppl. S4), 4–11; discussion 12–13. [Google Scholar]
- Croom, K.F.; Perry, C.M.; Plosker, G.L. Mirtazapine: A review of its use in major depression and other psychiatric disorders. CNS Drugs 2009, 23, 427–452. [Google Scholar] [CrossRef]
- Henssler, J.; Alexander, D.; Schwarzer, G.; Bschor, T.; Baethge, C. Combining Antidepressants vs Antidepressant Monotherapy for Treatment of Patients with Acute Depression: A Systematic Review and Meta-analysis. JAMA Psychiatry 2022, 79, 300–312. [Google Scholar] [CrossRef] [PubMed]
- McCormack, P.L. Vilazodone: A review in major depressive disorder in adults. Drugs 2015, 75, 1915–1923. [Google Scholar] [CrossRef] [PubMed]
- Gonda, X.; Sharma, S.R.; Tarazi, F.I. Vortioxetine: A novel antidepressant for the treatment of major depressive disorder. Expert. Opin. Drug Discov. 2019, 14, 81–89. [Google Scholar] [CrossRef]
- Becker, G.; Bolbos, R.; Costes, N.; Redouté, J.; Newman-Tancredi, A.; Zimmer, L. Selective serotonin 5-HT1A receptor biased agonists elicitdistinct brain activation patterns: A pharmacoMRI study. Sci. Rep. 2016, 6, 26633. [Google Scholar] [CrossRef] [PubMed]
- Bratsos, S.; Saleh, S.N. Clinical Efficacy of Ketamine for Treatment-resistant Depression. Cureus 2019, 11, e5189. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Cheng, L.; Ma, J.; Yuan, J.; Pi, C.; Xiong, L.; Chen, J.; Liu, H.; Tang, J.; Zhong, Y.; et al. Molecular mechanisms of rapid-acting antidepressants: New perspectives for developing antidepressants. Pharmacol. Res. 2023, 194, 106837. [Google Scholar] [CrossRef]
- Cavaleri, D.; Riboldi, I.; Crocamo, C.; Paglia, G.; Carrà, G.; Bartoli, F. Evidence from preclinical and clinical metabolomics studies on the antidepressant effects of ketamine and esketamine. Neurosci. Lett. 2024, 831, 137791. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Dong, Y.; Hu, H. The N-methyl-d-aspartate receptor hypothesis of ketamine’s antidepressant action: Evidence and controversies. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2024, 379, 20230225. [Google Scholar] [CrossRef]
- Scotton, E.; Casa, P.L.; de Abreu, F.P.; de Avila, E.S.S.; Wilges, R.L.B.; Rossetto, M.V.; Géa, L.P.; Rosa, A.R.; Colombo, R. Differentially regulated targets in the fast-acting antidepressant effect of (R)-ketamine: A systems biology approach. Pharmacol. Biochem. Behav. 2023, 223, 173523. [Google Scholar] [CrossRef]
- Yavi, M.; Lee, H.; Henter, I.D.; Park, L.T.; Zarate, C.A., Jr. Ketamine treatment for depression: A review. Discov. Ment. Health 2022, 2, 9. [Google Scholar] [CrossRef]
- Ren, L. The mechanistic basis for the rapid antidepressant-like effects of ketamine: From neural circuits to molecular pathways. Prog. Neuropsychopharmacol. Biol. Psychiatry 2024, 129, 110910. [Google Scholar] [CrossRef] [PubMed]
- Andrade, C. Ketamine for Depression, 2: Diagnostic and Contextual Indications. J. Clin. Psychiatry 2017, 78, e555–e558. [Google Scholar] [CrossRef]
- Zolghadriha, A.; Anjomshoaa, A.; Jamshidi, M.R.; Taherkhani, F. Rapid and sustained antidepressant effects of intravenous ketamine in treatment-resistant major depressive disorder and suicidal ideation: A randomized clinical trial. BMC Psychiatry 2024, 24, 341. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, R.S.; Rodrigues, N.B.; Lee, Y.; Lipsitz, O.; Subramaniapillai, M.; Gill, H.; Nasri, F.; Majeed, A.; Lui, L.M.W.; Senyk, O.; et al. The effectiveness of repeated intravenous ketamine on depressive symptoms, suicidal ideation and functional disability in adults with major depressive disorder and bipolar disorder: Results from the Canadian Rapid Treatment Center of Excellence. J. Affect. Disord. 2020, 274, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Lewis, V.; Rodrigue, B.; Arsenault, E.; Zhang, M.; Taghavi-Abkuh, F.F.; Silva, W.C.C.; Myers, M.; Matta-Camacho, E.; Aguilar-Valles, A. Translational control by ketamine and its implications for comorbid cognitive deficits in depressive disorders. J. Neurochem. 2023, 166, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Suzuki, K.; Kavalali, E.T.; Monteggia, L.M. Ketamine: Mechanisms and Relevance to Treatment of Depression. Annu. Rev. Med. 2024, 75, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Peltoniemi, M.A.; Hagelberg, N.M.; Olkkola, K.T.; Saari, T.I. Ketamine: A Review of Clinical Pharmacokinetics and Pharmacodynamics in Anesthesia and Pain Therapy. Clin. Pharmacokinet. 2016, 55, 1059–1077. [Google Scholar] [CrossRef] [PubMed]
- Gałecki, P.; Bliźniewska-Kowalska, K.M.; Cubała, W.J.; Depukat, A.; Mosiołek, A.; Rybakowski, J.; Samochowiec, J.; Sobolewski, B.; Szulc, A.; Dudek, D. Polski standard leczenia racemiczną ketaminą pacjentów z zaburzeniami depresyjnymi opracowany przez zespół roboczy powołany przez Konsultanta Krajowegow dziedzinie psychiatrii. Psychiatr. Pol. 2024, 58, 377–401. [Google Scholar] [CrossRef]
- McIntyre, R.S.; Rosenblat, J.D.; Nemeroff, C.B.; Sanacora, G.; Murrough, J.W.; Berk, M.; Brietzke, E.; Dodd, S.; Gorwood, P.; Ho, R.; et al. Synthesizing the Evidence for Ketamine and Esketamine in Treatment-Resistant Depression: An International Expert Opinion on the Available Evidence and Implementation. Am. J. Psychiatry 2021, 178, 383–399. [Google Scholar] [CrossRef]
- Zanos, P.; Moaddel, R.; Morris, P.J.; Riggs, L.M.; Highland, J.N.; Georgiou, P.; Pereira, E.F.R.; Albuquerque, E.X.; Thomas, C.J.; Zarate, C.A., Jr.; et al. Ketamine and Ketamine Metabolite Pharmacology: Insights into Therapeutic Mechanisms. Pharmacol. Rev. 2018, 70, 621–660. [Google Scholar] [CrossRef]
- Cavenaghi, V.B.; da Costa, L.P.; Lacerda, A.L.T.; Hirata, E.S.; Miguel, E.C.; Fraguas, R. Subcutaneous Ketamine in Depression: A Systematic Review. Front. Psychiatry 2021, 12, 513068. [Google Scholar] [CrossRef] [PubMed]
- Harihar, C.; Dasari, P.; Srinivas, J.S. Intramuscular ketamine in acute depression: A report on two cases. Indian. J. Psychiatry 2013, 55, 186–188. [Google Scholar] [CrossRef] [PubMed]
- Rasool, U.; Jan, N.; Zahoor, M.; Roub, F.; Rather, Y.; Hussain, A.; Najeeb, R. Effect of ketamine infusion in treatment resistant depression and in depressive patients with active suicidal ideations: A study from North India. Int. J. Res. Med. Sci. 2023, 11, 535–543. [Google Scholar] [CrossRef]
- Loo, C.K.; Gálvez, V.; O’Keefe, E.; Mitchell, P.B.; Hadzi-Pavlovic, D.; Leyden, J.; Harper, S.; Somogyi, A.A.; Lai, R.; Weickert, C.S.; et al. Placebo-controlled pilot trial testing dose titration and intravenous, intramuscular and subcutaneous routes for ketamine in depression. Acta Psychiatr. Scand. 2016, 134, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Daly, E.J.; Singh, J.B.; Fedgchin, M.; Cooper, K.; Lim, P.; Shelton, R.C.; Thase, M.E.; Winokur, A.; Van Nueten, L.; Manji, H.; et al. Efficacy and Safety of Intranasal Esketamine Adjunctive to Oral Antidepressant Therapy in Treatment-Resistant Depression: A Randomized Clinical Trial. JAMA Psychiatry 2018, 75, 139–148. [Google Scholar] [CrossRef]
- Lapidus, K.A.; Levitch, C.F.; Perez, A.M.; Brallier, J.W.; Parides, M.K.; Soleimani, L.; Feder, A.; Iosifescu, D.V.; Charney, D.S.; Murrough, J.W. A randomized controlled trial of intranasal ketamine in major depressive disorder. Biol. Psychiatry 2014, 76, 970–976. [Google Scholar] [CrossRef]
- Wang, S.M.; Kim, N.Y.; Na, H.R.; Lim, H.K.; Woo, Y.S.; Pae, C.U.; Bahk, W.M. Rapid Onset of Intranasal Esketamine in Patients with Treatment Resistant Depression and Major Depression with Suicide Ideation: A Meta-Analysis. Clin. Psychopharmacol. Neurosci. 2021, 19, 341–354. [Google Scholar] [CrossRef]
- Glue, P.; Neehoff, S.; Beaglehole, B.; Shadli, S.; McNaughton, N.; Hughes-Medlicott, N.J. Ketamine for treatment-resistant major depressive disorder: Double-blind active-controlled crossover study. J. Psychopharmacol. 2024, 38, 162–167. [Google Scholar] [CrossRef]
- Del Sant, L.C.; Sarin, L.M.; Lucchese, A.C.; Magalhães, E.J.M.; Tuena, M.A.; Nakahira, C.; Del Porto, J.A.; De Lacerda, A.L.T.; Mari, J.J. Impact of Repeated Doses of Subcutaneous Esketamine on Acute Dissociative Symptoms in Treatment-Resistant Depression. Pharmaceuticals 2022, 16, 31. [Google Scholar] [CrossRef] [PubMed]
- Glue, P.; Medlicott, N.J.; Surman, P.; Lam, F.; Hung, N.; Hung, C.T. Ascending-Dose Study of Controlled-Release Ketamine Tablets in Healthy Volunteers: Pharmacokinetics, Pharmacodynamics, Safety, and Tolerability. J. Clin. Pharmacol. 2020, 60, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Irwin, S.A.; Iglewicz, A.; Nelesen, R.A.; Lo, J.Y.; Carr, C.H.; Romero, S.D.; Lloyd, L.S. Daily oral ketamine for the treatment of depression and anxiety in patients receiving hospice care: A 28-day open-label proof-of-concept trial. J. Palliat. Med. 2013, 16, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Colla, M.; Offenhammer, B.; Scheerer, H.; Kronenberg, G.; Vetter, S.; Mutschler, J.; Mikoteit, T.; Bankwitz, A.; Adank, A.; Schaekel, L.; et al. Oral prolonged-release ketamine in treatment-resistant depression—A double-blind randomized placebo-controlled multicentre trial of KET01, a novel ketamine formulation—Clinical and safety results. J. Psychiatr. Res. 2024, 173, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Mansoor, S.; Khan, K.; Faridi, M. Tolerance and Acceptability of Intravenous Ketamine Therapy for Treatment Resistant Depression: A Mixed Methods Assessment. J. Pak. Psychiatr. Soc. 2023, 20, 13–17. [Google Scholar] [CrossRef]
- Kwaśna, J.; Cubała, W.J.; Kwaśny, A.; Wilkowska, A. The quest for optimal ketamine dosing formula in treatment-resistant major depressive disorder. Pharmacol. Rep. 2024, 76, 1318–1324. [Google Scholar] [CrossRef] [PubMed]
- Shiroma, P.R.; Thuras, P.; Wels, J.; Albott, C.S.; Erbes, C.; Tye, S.; Lim, K.O. A randomized, double-blind, active placebo-controlled study of efficacy, safety, and durability of repeated vs single subanesthetic ketamine for treatment-resistant depression. Transl. Psychiatry 2020, 10, 206. [Google Scholar] [CrossRef] [PubMed]
- Schep, L.J.; Slaughter, R.J.; Watts, M.; Mackenzie, E.; Gee, P. The clinical toxicology of ketamine. Clin. Toxicol. 2023, 61, 415–428. [Google Scholar] [CrossRef]
- Mion, G.; Villevieille, T. Ketamine pharmacology: An update (pharmacodynamics and molecular aspects, recent findings). CNS Neurosci. Ther. 2013, 19, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Lipsitz, O.; McIntyre, R.S.; Rodrigues, N.B.; Lee, Y.; Gill, H.; Subramaniapillai, M.; Kratiuk, K.; Nasri, F.; Mansur, R.B.; Rosenblat, J.D. Does body mass index predict response to intravenous ketamine treatment in adults with major depressive and bipolar disorder? Results from the Canadian Rapid Treatment Center of Excellence. CNS Spectr. 2022, 27, 322–330. [Google Scholar] [CrossRef]
- Barik, A.K.; Chauhan, R.; Reddy, A.; Kaloria, N. Concerns on the Use of Ketamine in the Treatment of Depression. Indian. J. Psychol. Med. 2024, 46, 94–95. [Google Scholar] [CrossRef] [PubMed]
- Sandbaumhüter, F.A.; Thormann, W. Enantioselective capillary electrophoresis provides insight into the phase II metabolism of ketamine and its metabolites in vivo and in vitro. Electrophoresis 2018, 39, 1478–1481. [Google Scholar] [CrossRef]
- Davydova, N.Y.; Hutner, D.A.; Gaither, K.A.; Singh, D.K.; Prasad, B.; Davydov, D.R. High-Throughput Assay of Cytochrome P450-Dependent Drug Demethylation Reactions and Its Use to Re-Evaluate the Pathways of Ketamine Metabolism. Biology 2023, 12, 1055. [Google Scholar] [CrossRef]
- Chue, P.; Andreiev, A.; Chue, J.; Tate, M. A Review of the Metabolism and Relevance to Form and Formulation of Ketamine. Eur. Psychiatry 2023, 66, S842. [Google Scholar] [CrossRef]
- Dinis-Oliveira, R.J. Metabolism and metabolomics of ketamine: A toxicological approach. Forensic Sci. Res. 2017, 2, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.F.; Neiner, A.; Kharasch, E.D. Stereoselective Ketamine Metabolism by Genetic Variants of Cytochrome P450 CYP2B6 and Cytochrome P450 Oxidoreductase. Anesthesiology 2018, 129, 756–768. [Google Scholar] [CrossRef]
- Kharasch, E.D.; Labroo, R. Metabolism of ketamine stereoisomers by human liver microsomes. Anesthesiology 1992, 77, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Ihmsen, H.; Geisslinger, G.; Schüttler, J. Stereoselective pharmacokinetics of ketamine: R(−)-ketamine inhibits the elimination of S(+)-ketamine. Clin. Pharmacol. Ther. 2001, 70, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Hess, E.M.; Riggs, L.M.; Michaelides, M.; Gould, T.D. Mechanisms of ketamine and its metabolites as antidepressants. Biochem. Pharmacol. 2022, 197, 114892. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Venkata, S.L.; Moaddel, R.; Luckenbaugh, D.A.; Brutsche, N.E.; Ibrahim, L.; Zarate, C.A., Jr.; Mager, D.E.; Wainer, I.W. Simultaneous population pharmacokinetic modelling of ketamine and three major metabolites in patients with treatment-resistant bipolar depression. Br. J. Clin. Pharmacol. 2012, 74, 304–314. [Google Scholar] [CrossRef]
- Zarate, C.A., Jr.; Brutsche, N.; Laje, G.; Luckenbaugh, D.A.; Venkata, S.L.; Ramamoorthy, A.; Moaddel, R.; Wainer, I.W. Relationship of ketamine’s plasma metabolites with response, diagnosis, and side effects in major depression. Biol. Psychiatry 2012, 72, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Moaddel, R.; Venkata, S.L.; Tanga, M.J.; Bupp, J.E.; Green, C.E.; Iyer, L.; Furimsky, A.; Goldberg, M.E.; Torjman, M.C.; Wainer, I.W. A parallel chiral-achiral liquid chromatographic method for the determination of the stereoisomers of ketamine and ketamine metabolites in the plasma and urine of patients with complex regional pain syndrome. Talanta 2010, 82, 1892–1904. [Google Scholar] [CrossRef] [PubMed]
- Vyklicky, V.; Korinek, M.; Smejkalova, T.; Balik, A.; Krausova, B.; Kaniakova, M.; Lichnerova, K.; Cerny, J.; Krusek, J.; Dittert, I.; et al. Structure, function, and pharmacology of NMDA receptor channels. Physiol. Res. 2014, 63, S191–S203. [Google Scholar] [CrossRef]
- Cull-Candy, S.; Brickley, S.; Farrant, M. NMDA receptor subunits: Diversity, development and disease. Curr. Opin. Neurobiol. 2001, 11, 327–335. [Google Scholar] [CrossRef]
- Niciu, M.J.; Kelmendi, B.; Sanacora, G. Overview of glutamatergic neurotransmission in the nervous system. Pharmacol. Biochem. Behav. 2012, 100, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, L.; Diaz Granados, N.; Jolkovsky, L.; Brutsche, N.; Luckenbaugh, D.A.; Herring, W.J.; Potter, W.Z.; Zarate, C.A., Jr. A Randomized, placebo-controlled, crossover pilot trial of the oral selective NR2B antagonist MK-0657 in patients with treatment-resistant major depressive disorder. J. Clin. Psychopharmacol. 2012, 32, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Talebian, A.; Robinson-Brookes, K.; Meakin, S.O. TrkB Regulates N-Methyl-D-Aspartate Receptor Signaling by Uncoupling and Recruiting the Brain-Specific Guanine Nucleotide Exchange Factor, RasGrf1. J. Mol. Neurosci. 2019, 67, 97–110. [Google Scholar] [CrossRef]
- Murrough, J.W.; Abdallah, C.G.; Mathew, S.J. Targeting glutamate signalling in depression: Progress and prospects. Nat. Rev. Drug Discov. 2017, 16, 472–486. [Google Scholar] [CrossRef] [PubMed]
- Newport, D.J.; Carpenter, L.L.; McDonald, W.M.; Potash, J.B.; Tohen, M.; Nemeroff, C.B. Ketamine and Other NMDA Antagonists: Early Clinical Trials and Possible Mechanisms in Depression. Am. J. Psychiatry 2015, 172, 950–966. [Google Scholar] [CrossRef]
- Sherwood, M.W.; Oliet, S.H.R.; Panatier, A. NMDARs, Coincidence Detectors of Astrocytic and Neuronal Activities. Int. J. Mol. Sci. 2021, 22, 7258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ye, F.; Zhang, T.; Lv, S.; Zhou, L.; Du, D.; Lin, H.; Guo, F.; Luo, C.; Zhu, S. Structural basis of ketamine action on human NMDA receptors. Nature 2021, 596, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Choi, S.; Lee, E.; Koh, W.; Lee, C.J. Tonic NMDA Receptor Currents in the Brain: Regulation and Cognitive Functions. Biol. Psychiatry 2024, 96, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Autry, A.E.; Adachi, M.; Nosyreva, E.; Na, E.S.; Los, M.F.; Cheng, P.F.; Kavalali, E.T.; Monteggia, L.M. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011, 475, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Gideons, E.S.; Kavalali, E.T.; Monteggia, L.M. Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc. Natl. Acad. Sci. USA 2014, 111, 8649–8654. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cui, Y.; Sang, K.; Dong, Y.; Ni, Z.; Ma, S.; Hu, H. Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature 2018, 554, 317–322. [Google Scholar] [CrossRef]
- Zanos, P.; Thompson, S.M.; Duman, R.S.; Zarate, C.A., Jr.; Gould, T.D. Convergent Mechanisms Underlying Rapid Antidepressant Action. CNS Drugs 2018, 32, 197–227. [Google Scholar] [CrossRef]
- Li, N.; Liu, R.-J.; Dwyer, J.M.; Banasr, M.; Lee, B.; Son, H.; Li, X.-Y.; Aghajanian, G.; Duman, R.S. Glutamate N-methyl-D-aspartate Receptor Antagonists Rapidly Reverse Behavioral and Synaptic Deficits Caused by Chronic Stress Exposure. Biol. Psychiatry 2011, 69, 754–761. [Google Scholar] [CrossRef]
- Miller, O.H.; Moran, J.T.; Hall, B.J. Two cellular hypotheses explaining the initiation of ketamine’s antidepressant actions: Direct inhibition and disinhibition. Neuropharmacology 2016, 100, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Miller, O.H.; Yang, L.; Wang, C.-C.; Hargroder, E.A.; Zhang, Y.; Delpire, E.; Hall, B.J. GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. eLife 2014, 3, e03581. [Google Scholar] [CrossRef] [PubMed]
- Kohtala, S. Ketamine—50 years in use: From anesthesia to rapid antidepressant effects and neurobiological mechanisms. Pharmacol. Rep. 2021, 73, 323–345. [Google Scholar] [CrossRef] [PubMed]
- Biel, M.; Wahl-Schott, C.; Michalakis, S.; Zong, X. Hyperpolarization-activated cation channels: From genes to function. Physiol. Rev. 2009, 89, 847–885. [Google Scholar] [CrossRef]
- Nolan, M.F.; Malleret, G.; Dudman, J.T.; Buhl, D.L.; Santoro, B.; Gibbs, E.; Vronskaya, S.; Buzsáki, G.; Siegelbaum, S.A.; Kandel, E.R.; et al. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell 2004, 119, 719–732. [Google Scholar] [CrossRef]
- Zhang, K.; Xu, T.; Yuan, Z.; Wei, Z.; Yamaki, V.N.; Huang, M.; Huganir, R.L.; Cai, X. Essential roles of AMPA receptor GluA1 phosphorylation and presynaptic HCN channels in fast-acting antidepressant responses of ketamine. Sci. Signal 2016, 9, ra123. [Google Scholar] [CrossRef] [PubMed]
- Guilloux, J.P.; Nguyen, T.M.L.; Gardier, A.M. [Ketamine: A neuropsychotropic drug with an innovative mechanism of action]. Biol. Aujourdhui 2023, 217, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Yao, X.; Li, B.; Cui, R.; Zhu, C.; Wang, Y.; Yang, W. Uncovering the Underlying Mechanisms of Ketamine as a Novel Antidepressant. Front. Pharmacol. 2021, 12, 740996. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lee, B.; Liu, R.J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.Y.; Aghajanian, G.; Duman, R.S. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Tizabi, Y.; Bhatti, B.H.; Manaye, K.F.; Das, J.R.; Akinfiresoye, L. Antidepressant-like effects of low ketamine dose is associated with increased hippocampal AMPA/NMDA receptor density ratio in female Wistar-Kyoto rats. Neuroscience 2012, 213, 72–80. [Google Scholar] [CrossRef]
- Du, J.; Machado-Vieira, R.; Maeng, S.; Martinowich, K.; Manji, H.K.; Zarate, C.A., Jr. Enhancing AMPA to NMDA throughput as a convergent mechanism for antidepressant action. Drug Discov. Today Ther. Strateg. 2006, 3, 519–526. [Google Scholar] [CrossRef]
- Zanos, P.; Moaddel, R.; Morris, P.J.; Georgiou, P.; Fischell, J.; Elmer, G.I.; Alkondon, M.; Yuan, P.; Pribut, H.J.; Singh, N.S.; et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 2016, 533, 481–486. [Google Scholar] [CrossRef]
- Akinfiresoye, L.; Tizabi, Y. Antidepressant effects of AMPA and ketamine combination: Role of hippocampal BDNF, synapsin, and mTOR. Psychopharmacology 2013, 230, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.F.; Correia, C.; Ingle, J.N.; Kaddurah-Daouk, R.; Wang, L.; Kaufmann, S.H.; Weinshilboum, R.M. Ketamine and ketamine metabolites as novel estrogen receptor ligands: Induction of cytochrome P450 and AMPA glutamate receptor gene expression. Biochem. Pharmacol. 2018, 152, 279–292. [Google Scholar] [CrossRef] [PubMed]
- O’Gorman, R.L.; Michels, L.; Edden, R.A.; Murdoch, J.B.; Martin, E. In vivo detection of GABA and glutamate with MEGA-PRESS: Reproducibility and gender effects. J. Magn. Reson. Imaging 2011, 33, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Gruenbaum, B.F.; Mohar, B.; Kuts, R.; Gruenbaum, S.E.; Ohayon, S.; Boyko, M.; Klin, Y.; Sheiner, E.; Shaked, G.; et al. The effects of estrogen and progesterone on blood glutamate levels: Evidence from changes of blood glutamate levels during the menstrual cycle in women. Biol. Reprod. 2011, 84, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Zahr, N.M.; Mayer, D.; Rohlfing, T.; Chanraud, S.; Gu, M.; Sullivan, E.V.; Pfefferbaum, A. In vivo glutamate measured with magnetic resonance spectroscopy: Behavioral correlates in aging. Neurobiol. Aging 2013, 34, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, Y.; Hu, J.; Cheng, W.; Jiang, H.; Zhang, X.; Li, M.; Ren, J.; Li, X. Prenatal chronic mild stress induces depression-like behavior and sex-specific changes in regional glutamate receptor expression patterns in adult rats. Neuroscience 2015, 301, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Woolley, C.S.; McEwen, B.S. Estradiol mediates fluctuation in hippocampal synapse density during the estrous cycle in the adult rat. J. Neurosci. 1992, 12, 2549–2554. [Google Scholar] [CrossRef]
- Oberlander, J.G.; Woolley, C.S. 17β-Estradiol Acutely Potentiates Glutamatergic Synaptic Transmission in the Hippocampus through Distinct Mechanisms in Males and Females. J. Neurosci. 2016, 36, 2677–2690. [Google Scholar] [CrossRef] [PubMed]
- Torrisi, S.A.; Rizzo, S.; Laudani, S.; Ieraci, A.; Drago, F.; Leggio, G.M. Acute stress alters recognition memory and AMPA/NMDA receptor subunits in a sex-dependent manner. Neurobiol. Stress. 2023, 25, 100545. [Google Scholar] [CrossRef]
- Wickens, M.M.; Bangasser, D.A.; Briand, L.A. Sex Differences in Psychiatric Disease: A Focus on the Glutamate System. Front. Mol. Neurosci. 2018, 11, 197. [Google Scholar] [CrossRef] [PubMed]
- Giacometti, L.L.; Barker, J.M. Sex differences in the glutamate system: Implications for addiction. Neurosci. Biobehav. Rev. 2020, 113, 157–168. [Google Scholar] [CrossRef]
- Picard, N.; Takesian, A.E.; Fagiolini, M.; Hensch, T.K. NMDA 2A receptors in parvalbumin cells mediate sex-specific rapid ketamine response on cortical activity. Mol. Psychiatry 2019, 24, 828–838. [Google Scholar] [CrossRef]
- El-Bakri, N.K.; Islam, A.; Zhu, S.; Elhassan, A.; Mohammed, A.; Winblad, B.; Adem, A. Effects of estrogen and progesterone treatment on rat hippocampal NMDA receptors: Relationship to Morris water maze performance. J. Cell Mol. Med. 2004, 8, 537–544. [Google Scholar] [CrossRef]
- Jelks, K.B.; Wylie, R.; Floyd, C.L.; McAllister, A.K.; Wise, P. Estradiol targets synaptic proteins to induce glutamatergic synapse formation in cultured hippocampal neurons: Critical role of estrogen receptor-alpha. J. Neurosci. 2007, 27, 6903–6913. [Google Scholar] [CrossRef]
- Saland, S.K.; Kabbaj, M. Sex Differences in the Pharmacokinetics of Low-dose Ketamine in Plasma and Brain of Male and Female Rats. J. Pharmacol. Exp. Ther. 2018, 367, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, D. Comparing the adverse effects of ketamine and esketamine between genders using FAERS data. Front. Pharmacol. 2024, 15, 1329436. [Google Scholar] [CrossRef]
- Ponton, E.; Turecki, G.; Nagy, C. Sex Differences in the Behavioral, Molecular, and Structural Effects of Ketamine Treatment in Depression. Int. J. Neuropsychopharmacol. 2022, 25, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.S.; Cardoso, A.; Vale, N. BDNF Unveiled: Exploring Its Role in Major Depression Disorder Serotonergic Imbalance and Associated Stress Conditions. Pharmaceutics 2023, 15, 2081. [Google Scholar] [CrossRef]
- Tyler, W.J.; Pozzo-Miller, L. Miniature synaptic transmission and BDNF modulate dendritic spine growth and form in rat CA1 neurones. J. Physiol. 2003, 553, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Woelfer, M.; Li, M.; Colic, L.; Liebe, T.; Di, X.; Biswal, B.; Murrough, J.; Lessmann, V.; Brigadski, T.; Walter, M. Ketamine-induced changes in plasma brain-derived neurotrophic factor (BDNF) levels are associated with the resting-state functional connectivity of the prefrontal cortex. World J. Biol. Psychiatry 2020, 21, 696–710. [Google Scholar] [CrossRef]
- Luo, T.; Deng, Z.; Ren, Q.; Mu, F.; Zhang, Y.; Wang, H. Effects of esketamine on postoperative negative emotions and early cognitive disorders in patients undergoing non-cardiac thoracic surgery: A randomized controlled trial. J. Clin. Anesth. 2024, 95, 111447. [Google Scholar] [CrossRef]
- Caliman-Fontes, A.T.; Leal, G.C.; Correia-Melo, F.S.; Paixão, C.S.; Carvalho, M.S.; Jesus-Nunes, A.P.; Vieira, F.; Magnavita, G.; Bandeira, I.D.; Mello, R.P.; et al. Brain-derived neurotrophic factor serum levels following ketamine and esketamine intervention for treatment-resistant depression: Secondary analysis from a randomized trial. Trends Psychiatry Psychother. 2023, 45, e20210298. [Google Scholar] [CrossRef]
- Jiang, Q.; Qi, Y.; Zhou, M.; Dong, Y.; Zheng, W.; Zhu, L.; Li, Y.; Zhou, H.; Wang, L. Effect of esketamine on serum neurotransmitters in patients with postpartum depression: A randomized controlled trial. BMC Anesthesiol. 2024, 24, 293. [Google Scholar] [CrossRef] [PubMed]
- Haile, C.N.; Murrough, J.W.; Iosifescu, D.V.; Chang, L.C.; Al Jurdi, R.K.; Foulkes, A.; Iqbal, S.; Mahoney, J.J., 3rd; De La Garza, R., 2nd; Charney, D.S.; et al. Plasma brain derived neurotrophic factor (BDNF) and response to ketamine in treatment-resistant depression. Int. J. Neuropsychopharmacol. 2014, 17, 331–336. [Google Scholar] [CrossRef]
- Chen, M.H.; Kao, C.F.; Tsai, S.J.; Li, C.T.; Lin, W.C.; Hong, C.J.; Bai, Y.M.; Tu, P.C.; Su, T.P. Treatment response to low-dose ketamine infusion for treatment-resistant depression: A gene-based genome-wide association study. Genomics 2021, 113, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Rybakowski, J.K.; Permoda-Osip, A.; Skibinska, M.; Adamski, R.; Bartkowska-Sniatkowska, A. Single ketamine infusion in bipolar depression resistant to antidepressants: Are neurotrophins involved? Hum. Psychopharmacol. 2013, 28, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Duncan, W.C.; Sarasso, S.; Ferrarelli, F.; Selter, J.; Riedner, B.A.; Hejazi, N.S.; Yuan, P.; Brutsche, N.; Manji, H.K.; Tononi, G.; et al. Concomitant BDNF and sleep slow wave changes indicate ketamine-induced plasticity in major depressive disorder. Int. J. Neuropsychopharmacol. 2013, 16, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Meshkat, S.; Alnefeesi, Y.; Jawad, M.Y.; Di Vincenzo, J.D.; Rodrigues, N.B.; Ceban, F.; Mw Lui, L.; McIntyre, R.S.; Rosenblat, J.D. Brain-Derived Neurotrophic Factor (BDNF) as a biomarker of treatment response in patients with Treatment Resistant Depression (TRD): A systematic review & meta-analysis. Psychiatry Res. 2022, 317, 114857. [Google Scholar] [CrossRef]
- Li, S.; Luo, X.; Hua, D.; Wang, Y.; Zhan, G.; Huang, N.; Jiang, R.; Yang, L.; Zhu, B.; Yuan, X.; et al. Ketamine Alleviates Postoperative Depression-Like Symptoms in Susceptible Mice: The Role of BDNF-TrkB Signaling. Front. Pharmacol. 2019, 10, 1702. [Google Scholar] [CrossRef] [PubMed]
- Ju, L.; Yang, J.; Zhu, T.; Liu, P. BDNF-TrkB signaling-mediated upregulation of Narp is involved in the antidepressant-like effects of (2R,6R)-hydroxynorketamine in a chronic restraint stress mouse model. BMC Psychiatry 2022, 22, 182. [Google Scholar] [CrossRef]
- Casarotto, P.C.; Girych, M.; Fred, S.M.; Kovaleva, V.; Moliner, R.; Enkavi, G.; Biojone, C.; Cannarozzo, C.; Sahu, M.P.; Kaurinkoski, K.; et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 2021, 184, 1299–1313.e19. [Google Scholar] [CrossRef] [PubMed]
- Fred, S.M.; Moliner, R.; Antila, H.; Engelhardt, K.A.; Schlüter, O.M.; Casarotto, P.C.; Castrén, E. TRKB interaction with PSD95 is associated with latency of fluoxetine and 2R,6R-hydroxynorketamine. Eur. J. Neurosci. 2023, 57, 1215–1224. [Google Scholar] [CrossRef]
- Yoshii, A.; Constantine-Paton, M. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev. Neurobiol. 2010, 70, 304–322. [Google Scholar] [CrossRef]
- Liu, W.X.; Wang, J.; Xie, Z.M.; Xu, N.; Zhang, G.F.; Jia, M.; Zhou, Z.Q.; Hashimoto, K.; Yang, J.J. Regulation of glutamate transporter 1 via BDNF-TrkB signaling plays a role in the anti-apoptotic and antidepressant effects of ketamine in chronic unpredictable stress model of depression. Psychopharmacology 2016, 233, 405–415. [Google Scholar] [CrossRef]
- Duman, R.S.; Li, N.; Liu, R.J.; Duric, V.; Aghajanian, G. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology 2012, 62, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Cannarozzo, C.; Rubiolo, A.; Casarotto, P.; Castrén, E. Ketamine and its metabolite 2R,6R-hydroxynorketamine promote ocular dominance plasticity and release tropomyosin-related kinase B from inhibitory control without reducing perineuronal nets enwrapping parvalbumin interneurons. Eur. J. Neurosci. 2023, 57, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.X.; Yao, L.H.; Xu, B.B.; Qian, K.; Wang, H.L.; Liu, Z.C.; Wang, X.P.; Wang, G.H. Glutamate transporter 1-mediated antidepressant-like effect in a rat model of chronic unpredictable stress. J. Huazhong Univ. Sci. Technolog Med. Sci. 2014, 34, 838–844. [Google Scholar] [CrossRef]
- Fullana, M.N.; Paz, V.; Artigas, F.; Bortolozzi, A. Ketamine triggers rapid antidepressant effects by modulating synaptic plasticity in a new depressive-like mouse model based on astrocyte glutamate transporter GLT-1 knockdown in infralimbic cortex. Rev. Psiquiatr. Salud Ment. 2022, 15, 94–100. [Google Scholar] [CrossRef]
- Ardalan, M.; Rafati, A.H.; Nyengaard, J.R.; Wegener, G. Rapid antidepressant effect of ketamine correlates with astroglial plasticity in the hippocampus. Br. J. Pharmacol. 2017, 174, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R. Glial cells. Int. J. Biochem. Cell Biol. 2004, 36, 1861–1867. [Google Scholar] [CrossRef] [PubMed]
- Hol, E.M.; Pekny, M. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr. Opin. Cell Biol. 2015, 32, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Hughes, E.G.; Maguire, J.L.; McMinn, M.T.; Scholz, R.E.; Sutherland, M.L. Loss of glial fibrillary acidic protein results in decreased glutamate transport and inhibition of PKA-induced EAAT2 cell surface trafficking. Brain Res. Mol. Brain Res. 2004, 124, 114–123. [Google Scholar] [CrossRef]
- Rajkowska, G.; Stockmeier, C.A. Astrocyte pathology in major depressive disorder: Insights from human postmortem brain tissue. Curr. Drug Targets 2013, 14, 1225–1236. [Google Scholar] [CrossRef]
- Lin, N.H.; Yang, A.W.; Chang, C.H.; Perng, M.D. Elevated GFAP isoform expression promotes protein aggregation and compromises astrocyte function. FASEB J. 2021, 35, e21614. [Google Scholar] [CrossRef] [PubMed]
- Hassinger, T.D.; Atkinson, P.B.; Strecker, G.J.; Whalen, L.R.; Dudek, F.E.; Kossel, A.H.; Kater, S.B. Evidence for glutamate-mediated activation of hippocampal neurons by glial calcium waves. J. Neurobiol. 1995, 28, 159–170. [Google Scholar] [CrossRef]
- Bremner, J.D.; Narayan, M.; Anderson, E.R.; Staib, L.H.; Miller, H.L.; Charney, D.S. Hippocampal volume reduction in major depression. Am. J. Psychiatry 2000, 157, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Botteron, K.N.; Raichle, M.E.; Drevets, W.C.; Heath, A.C.; Todd, R.D. Volumetric reduction in left subgenual prefrontal cortex in early onset depression. Biol. Psychiatry 2002, 51, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Drevets, W.C.; Price, J.L.; Simpson, J.R., Jr.; Todd, R.D.; Reich, T.; Vannier, M.; Raichle, M.E. Subgenual prefrontal cortex abnormalities in mood disorders. Nature 1997, 386, 824–827. [Google Scholar] [CrossRef] [PubMed]
- Holmes, S.E.; Scheinost, D.; Finnema, S.J.; Naganawa, M.; Davis, M.T.; DellaGioia, N.; Nabulsi, N.; Matuskey, D.; Angarita, G.A.; Pietrzak, R.H.; et al. Lower synaptic density is associated with depression severity and network alterations. Nat. Commun. 2019, 10, 1529. [Google Scholar] [CrossRef]
- Kang, H.J.; Voleti, B.; Hajszan, T.; Rajkowska, G.; Stockmeier, C.A.; Licznerski, P.; Lepack, A.; Majik, M.S.; Jeong, L.S.; Banasr, M.; et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat. Med. 2012, 18, 1413–1417. [Google Scholar] [CrossRef]
- Deyama, S.; Duman, R.S. Neurotrophic mechanisms underlying the rapid and sustained antidepressant actions of ketamine. Pharmacol. Biochem. Behav. 2020, 188, 172837. [Google Scholar] [CrossRef] [PubMed]
- Rajkowska, G.; Miguel-Hidalgo, J.J.; Wei, J.; Dilley, G.; Pittman, S.D.; Meltzer, H.Y.; Overholser, J.C.; Roth, B.L.; Stockmeier, C.A. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol. Psychiatry 1999, 45, 1085–1098. [Google Scholar] [CrossRef]
- Boldrini, M.; Galfalvy, H.; Dwork, A.J.; Rosoklija, G.B.; Trencevska-Ivanovska, I.; Pavlovski, G.; Hen, R.; Arango, V.; Mann, J.J. Resilience Is Associated With Larger Dentate Gyrus, While Suicide Decedents With Major Depressive Disorder Have Fewer Granule Neurons. Biol. Psychiatry 2019, 85, 850–862. [Google Scholar] [CrossRef]
- Boldrini, M.; Santiago, A.N.; Hen, R.; Dwork, A.J.; Rosoklija, G.B.; Tamir, H.; Arango, V.; John Mann, J. Hippocampal granule neuron number and dentate gyrus volume in antidepressant-treated and untreated major depression. Neuropsychopharmacology 2013, 38, 1068–1077. [Google Scholar] [CrossRef]
- Höflich, A.; Kraus, C.; Pfeiffer, R.M.; Seiger, R.; Rujescu, D.; Zarate, C.A., Jr.; Kasper, S.; Winkler, D.; Lanzenberger, R. Translating the immediate effects of S-Ketamine using hippocampal subfield analysis in healthy subjects-results of a randomized controlled trial. Transl. Psychiatry 2021, 11, 200. [Google Scholar] [CrossRef]
- Treccani, G.; Ardalan, M.; Chen, F.; Musazzi, L.; Popoli, M.; Wegener, G.; Nyengaard, J.R.; Müller, H.K. S-Ketamine Reverses Hippocampal Dendritic Spine Deficits in Flinders Sensitive Line Rats Within 1 h of Administration. Mol. Neurobiol. 2019, 56, 7368–7379. [Google Scholar] [CrossRef]
- Ardalan, M.; Wegener, G.; Rafati, A.H.; Nyengaard, J.R. S-Ketamine Rapidly Reverses Synaptic and Vascular Deficits of Hippocampus in Genetic Animal Model of Depression. Int. J. Neuropsychopharmacol. 2017, 20, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Moda-Sava, R.N.; Murdock, M.H.; Parekh, P.K.; Fetcho, R.N.; Huang, B.S.; Huynh, T.N.; Witztum, J.; Shaver, D.C.; Rosenthal, D.L.; Alway, E.J.; et al. Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science 2019, 364, eaat8078. [Google Scholar] [CrossRef]
- Liu, R.J.; Lee, F.S.; Li, X.Y.; Bambico, F.; Duman, R.S.; Aghajanian, G.K. Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol. Psychiatry 2012, 71, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Chen, L.; Yang, J.; Han, D.; Fang, D.; Qiu, X.; Yang, X.; Qiao, Z.; Ma, J.; Wang, L.; et al. BDNF Val66Met polymorphism, life stress and depression: A meta-analysis of gene-environment interaction. J. Affect. Disord. 2018, 227, 226–235. [Google Scholar] [CrossRef]
- Hosang, G.M.; Shiles, C.; Tansey, K.E.; McGuffin, P.; Uher, R. Interaction between stress and the BDNF Val66Met polymorphism in depression: A systematic review and meta-analysis. BMC Med. 2014, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Adzic, M.; Lukic, I.; Mitic, M.; Glavonic, E.; Dragicevic, N.; Ivkovic, S. Contribution of the opioid system to depression and to the therapeutic effects of classical antidepressants and ketamine. Life Sci. 2023, 326, 121803. [Google Scholar] [CrossRef]
- Williams, N.R.; Heifets, B.D.; Blasey, C.; Sudheimer, K.; Pannu, J.; Pankow, H.; Hawkins, J.; Birnbaum, J.; Lyons, D.M.; Rodriguez, C.I.; et al. Attenuation of Antidepressant Effects of Ketamine by Opioid Receptor Antagonism. Am. J. Psychiatry 2018, 175, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.E.; Chandra, J.; Sheriff, S.; Malinow, R. Opioid system is necessary but not sufficient for antidepressive actions of ketamine in rodents. Proc. Natl. Acad. Sci. USA 2020, 117, 2656–2662. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Hashimoto, K. Lack of Opioid System in the Antidepressant Actions of Ketamine. Biol. Psychiatry 2019, 85, e25–e27. [Google Scholar] [CrossRef]
- Di Ianni, T.; Ewbank, S.N.; Levinstein, M.R.; Azadian, M.M.; Budinich, R.C.; Michaelides, M.; Airan, R.D. Sex dependence of opioid-mediated responses to subanesthetic ketamine in rats. Nat. Commun. 2024, 15, 893. [Google Scholar] [CrossRef]
- Gomes, I.; Gupta, A.; Margolis, E.B.; Fricker, L.D.; Devi, L.A. Ketamine and Major Ketamine Metabolites Function as Allosteric Modulators of Opioid Receptors. Mol. Pharmacol. 2024, 106, 240. [Google Scholar] [CrossRef] [PubMed]
- Seminowicz, D.A.; Mayberg, H.S.; McIntosh, A.R.; Goldapple, K.; Kennedy, S.; Segal, Z.; Rafi-Tari, S. Limbic-frontal circuitry in major depression: A path modeling metanalysis. Neuroimage 2004, 22, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, J.; Chen, X.; Liu, R.; Zhang, Z.; Feng, L.; Feng, Y.; Wang, G.; Zhou, Y. Effects of escitalopram therapy on effective connectivity among core brain networks in major depressive disorder. J. Affect. Disord. 2024, 350, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dai, X.; Wu, H.; Wang, L. Establishment of Effective Biomarkers for Depression Diagnosis With Fusion of Multiple Resting-State Connectivity Measures. Front. Neurosci. 2021, 15, 729958. [Google Scholar] [CrossRef]
- Zheng, H.; Xu, L.; Xie, F.; Guo, X.; Zhang, J.; Yao, L.; Wu, X. The Altered Triple Networks Interaction in Depression under Resting State Based on Graph Theory. Biomed. Res. Int. 2015, 2015, 386326. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qian, L.; Li, G.; Zhang, Z. Frequency specificity of aberrant triple networks in major depressive disorder: A resting-state effective connectivity study. Front. Neurosci. 2023, 17, 1200029. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tepfer, L.J.; Taren, A.A.; Smith, D.V. Functional parcellation of the default mode network: A large-scale meta-analysis. Sci. Rep. 2020, 10, 16096. [Google Scholar] [CrossRef]
- Jeong, G.; Paolini, M. Dynamic Adaptation of Default Mode Network in Resting state and Autobiographical Episodic Memory Retrieval State. bioRxiv 2023. [Google Scholar] [CrossRef]
- Fang, X.; Zhang, Y.; Zhou, Y.; Cheng, L.; Li, J.; Wang, Y.; Friston, K.J.; Jiang, T. Resting-State Coupling between Core Regions within the Central-Executive and Salience Networks Contributes to Working Memory Performance. Front. Behav. Neurosci. 2016, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.K.; Welton, T.; Lyon, M.; McCorkindale, A.N.; Sutherland, G.T.; Burnham, S.; Fripp, J.; Martins, R.; Grieve, S.M. Structural core of the executive control network: A high angular resolution diffusion MRI study. Hum. Brain Mapp. 2020, 41, 1226–1236. [Google Scholar] [CrossRef]
- Uddin, L.Q.; Nomi, J.S.; Hébert-Seropian, B.; Ghaziri, J.; Boucher, O. Structure and Function of the Human Insula. J. Clin. Neurophysiol. 2017, 34, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Uddin, L.Q. Salience processing and insular cortical function and dysfunction. Nat. Rev. Neurosci. 2015, 16, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Snyder, W.; Uddin, L.Q.; Nomi, J.S. Dynamic functional connectivity profile of the salience network across the life span. Hum. Brain Mapp. 2021, 42, 4740–4749. [Google Scholar] [CrossRef]
- Sevinc, G.; Gurvit, H.; Spreng, R.N. Salience network engagement with the detection of morally laden information. Soc. Cogn. Affect. Neurosci. 2017, 12, 1118–1127. [Google Scholar] [CrossRef]
- Ince, S.; Steward, T.; Harrison, B.J.; Jamieson, A.J.; Davey, C.G.; Agathos, J.A.; Moffat, B.A.; Glarin, R.K.; Felmingham, K.L. Subcortical contributions to salience network functioning during negative emotional processing. Neuroimage 2023, 270, 119964. [Google Scholar] [CrossRef]
- Seeley, W.W. The Salience Network: A Neural System for Perceiving and Responding to Homeostatic Demands. J. Neurosci. 2019, 39, 9878–9882. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhang, S.; Yin, S.; Ren, W.; He, R.; Li, J. The fronto-insular cortex causally mediates the default-mode and central-executive networks to contribute to individual cognitive performance in healthy elderly. Hum. Brain Mapp. 2018, 39, 4302–4311. [Google Scholar] [CrossRef]
- Schmaal, L.; Hibar, D.P.; Sämann, P.G.; Hall, G.B.; Baune, B.T.; Jahanshad, N.; Cheung, J.W.; van Erp, T.G.M.; Bos, D.; Ikram, M.A.; et al. Cortical abnormalities in adults and adolescents with major depression based on brain scans from 20 cohorts worldwide in the ENIGMA Major Depressive Disorder Working Group. Mol. Psychiatry 2017, 22, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Auer, D.P.; Pütz, B.; Kraft, E.; Lipinski, B.; Schill, J.; Holsboer, F. Reduced glutamate in the anterior cingulate cortex in depression: An in vivo proton magnetic resonance spectroscopy study. Biol. Psychiatry 2000, 47, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Tripp, A.; Kota, R.S.; Lewis, D.A.; Sibille, E. Reduced somatostatin in subgenual anterior cingulate cortex in major depression. Neurobiol. Dis. 2011, 42, 116–124. [Google Scholar] [CrossRef]
- Tripp, A.; Oh, H.; Guilloux, J.P.; Martinowich, K.; Lewis, D.A.; Sibille, E. Brain-derived neurotrophic factor signaling and subgenual anterior cingulate cortex dysfunction in major depressive disorder. Am. J. Psychiatry 2012, 169, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Horn, D.I.; Yu, C.; Steiner, J.; Buchmann, J.; Kaufmann, J.; Osoba, A.; Eckert, U.; Zierhut, K.C.; Schiltz, K.; He, H.; et al. Glutamatergic and resting-state functional connectivity correlates of severity in major depression—The role of pregenual anterior cingulate cortex and anterior insula. Front. Syst. Neurosci. 2010, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.; Henning, A.; Grimm, S.; Schulte, R.F.; Beck, J.; Dydak, U.; Schnepf, B.; Boeker, H.; Boesiger, P.; Northoff, G. The relationship between aberrant neuronal activation in the pregenual anterior cingulate, altered glutamatergic metabolism, and anhedonia in major depression. Arch. Gen. Psychiatry 2009, 66, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wu, J.; Pei, C.; Ma, M.; Dong, Y.; Gao, M.; Zhang, H. Altered functional connectivity in emotional subregions of the anterior cingulate cortex in young and middle-aged patients with major depressive disorder: A resting-state fMRI study. Biol. Psychol. 2022, 175, 108426. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Lin, W.C.; Tu, P.C.; Li, C.T.; Bai, Y.M.; Tsai, S.J.; Su, T.P. Antidepressant and antisuicidal effects of ketamine on the functional connectivity of prefrontal cortex-related circuits in treatment-resistant depression: A double-blind, placebo-controlled, randomized, longitudinal resting fMRI study. J. Affect. Disord. 2019, 259, 15–20. [Google Scholar] [CrossRef]
- Mkrtchian, A.; Evans, J.W.; Kraus, C.; Yuan, P.; Kadriu, B.; Nugent, A.C.; Roiser, J.P.; Zarate, C.A., Jr. Ketamine modulates fronto-striatal circuitry in depressed and healthy individuals. Mol. Psychiatry 2021, 26, 3292–3301. [Google Scholar] [CrossRef]
- Tseng, H.J.; Lu, C.F.; Jeng, J.S.; Cheng, C.M.; Chu, J.W.; Chen, M.H.; Bai, Y.M.; Tsai, S.J.; Su, T.P.; Li, C.T. Frontal asymmetry as a core feature of major depression: A functional near-infrared spectroscopy study. J. Psychiatry Neurosci. 2022, 47, E186–E193. [Google Scholar] [CrossRef]
- Wise, T.; Radua, J.; Via, E.; Cardoner, N.; Abe, O.; Adams, T.M.; Amico, F.; Cheng, Y.; Cole, J.H.; de Azevedo Marques Périco, C.; et al. Common and distinct patterns of grey-matter volume alteration in major depression and bipolar disorder: Evidence from voxel-based meta-analysis. Mol. Psychiatry 2017, 22, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Li, W.C.; Chen, L.F.; Su, T.P.; Li, C.T.; Lin, W.C.; Wu, H.J.; Tsai, S.J.; Bai, Y.M.; Tu, P.C.; Chen, M.H. Right dorsolateral prefrontal cortex volumetric reduction is associated with antidepressant effect of low-dose ketamine infusion: A randomized, double-blind, midazolam-controlled PET-MRI clinical trial. J. Affect. Disord. 2023, 335, 105–110. [Google Scholar] [CrossRef]
- Li, C.-T.; Lin, C.-P.; Chou, K.-H.; Chen, I.Y.; Hsieh, J.-C.; Wu, C.-L.; Lin, W.-C.; Su, T.-P. Structural and cognitive deficits in remitting and non-remitting recurrent depression: A voxel-based morphometric study. NeuroImage 2010, 50, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.W.; Szczepanik, J.; Brutsché, N.; Park, L.T.; Nugent, A.C.; Zarate, C.A., Jr. Default Mode Connectivity in Major Depressive Disorder Measured Up to 10 Days After Ketamine Administration. Biol. Psychiatry 2018, 84, 582–590. [Google Scholar] [CrossRef]
- Reed, J.L.; Nugent, A.C.; Furey, M.L.; Szczepanik, J.E.; Evans, J.W.; Zarate, C.A., Jr. Effects of Ketamine on Brain Activity During Emotional Processing: Differential Findings in Depressed Versus Healthy Control Participants. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2019, 4, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Woelfer, M.; Colic, L.; Safron, A.; Chang, C.; Heinze, H.J.; Speck, O.; Mayberg, H.S.; Biswal, B.B.; Salvadore, G.; et al. Default mode network connectivity change corresponds to ketamine’s delayed glutamatergic effects. Eur. Arch. Psychiatry Clin. Neurosci. 2020, 270, 207–216. [Google Scholar] [CrossRef]
- Chen, M.H.; Chang, W.C.; Lin, W.C.; Tu, P.C.; Li, C.T.; Bai, Y.M.; Tsai, S.J.; Huang, W.S.; Su, T.P. Functional Dysconnectivity of Frontal Cortex to Striatum Predicts Ketamine Infusion Response in Treatment-Resistant Depression. Int. J. Neuropsychopharmacol. 2020, 23, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Mueller, F.; Musso, F.; London, M.; de Boer, P.; Zacharias, N.; Winterer, G. Pharmacological fMRI: Effects of subanesthetic ketamine on resting-state functional connectivity in the default mode network, salience network, dorsal attention network and executive control network. Neuroimage Clin. 2018, 19, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Li, C.T.; Chen, M.H.; Lin, W.C.; Hong, C.J.; Yang, B.H.; Liu, R.S.; Tu, P.C.; Su, T.P. The effects of low-dose ketamine on the prefrontal cortex and amygdala in treatment-resistant depression: A randomized controlled study. Hum. Brain Mapp. 2016, 37, 1080–1090. [Google Scholar] [CrossRef]
- Burrows, M.; Kotoula, V.; Dipasquale, O.; Stringaris, A.; Mehta, M.A. Ketamine-induced changes in resting state connectivity, 2 h after the drug administration in patients with remitted depression. J. Psychopharmacol. 2023, 37, 784–794. [Google Scholar] [CrossRef]
- Kopelman, J.; Keller, T.A.; Panny, B.; Griffo, A.; Degutis, M.; Spotts, C.; Cruz, N.; Bell, E.; Do-Nguyen, K.; Wallace, M.L.; et al. Rapid neuroplasticity changes and response to intravenous ketamine: A randomized controlled trial in treatment-resistant depression. Transl. Psychiatry 2023, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krystal, J.H. Synaptic plasticity and depression: New insights from stress and rapid-acting antidepressants. Nat. Med. 2016, 22, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Heninger, G.R.; Nestler, E.J. A molecular and cellular theory of depression. Arch. Gen. Psychiatry 1997, 54, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Monteggia, L.M. A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 2006, 59, 1116–1127. [Google Scholar] [CrossRef]
- MacQueen, G.M.; Yucel, K.; Taylor, V.H.; Macdonald, K.; Joffe, R. Posterior hippocampal volumes are associated with remission rates in patients with major depressive disorder. Biol. Psychiatry 2008, 64, 880–883. [Google Scholar] [CrossRef] [PubMed]
- Savitz, J.; Drevets, W.C. Bipolar and major depressive disorder: Neuroimaging the developmental-degenerative divide. Neurosci. Biobehav. Rev. 2009, 33, 699–771. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.J.; Aghajanian, G.K. Stress blunts serotonin- and hypocretin-evoked EPSCs in prefrontal cortex: Role of corticosterone-mediated apical dendritic atrophy. Proc. Natl. Acad. Sci. USA 2008, 105, 359–364. [Google Scholar] [CrossRef]
- Arnone, D. Functional MRI findings, pharmacological treatment in major depression and clinical response. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 91, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Price, R.B.; Duman, R. Neuroplasticity in cognitive and psychological mechanisms of depression: An integrative model. Mol. Psychiatry 2020, 25, 530–543. [Google Scholar] [CrossRef] [PubMed]
- Meiering, M.S.; Weigner, D.; Gärtner, M.; Carstens, L.; Keicher, C.; Hertrampf, R.; Beckmann, C.F.; Mennes, M.; Wunder, A.; Weigand, A.; et al. Functional activity and connectivity signatures of ketamine and lamotrigine during negative emotional processing: A double-blind randomized controlled fMRI study. Transl. Psychiatry 2024, 14, 436. [Google Scholar] [CrossRef]
- Alexander, L.; Hawkins, P.C.T.; Evans, J.W.; Mehta, M.A.; Zarate, C.A., Jr. Preliminary evidence that ketamine alters anterior cingulate resting-state functional connectivity in depressed individuals. Transl. Psychiatry 2023, 13, 371. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Demenescu, L.R.; Colic, L.; Metzger, C.D.; Heinze, H.J.; Steiner, J.; Speck, O.; Fejtova, A.; Salvadore, G.; Walter, M. Temporal Dynamics of Antidepressant Ketamine Effects on Glutamine Cycling Follow Regional Fingerprints of AMPA and NMDA Receptor Densities. Neuropsychopharmacology 2017, 42, 1201–1209. [Google Scholar] [CrossRef]
- Gärtner, M.; Aust, S.; Bajbouj, M.; Fan, Y.; Wingenfeld, K.; Otte, C.; Heuser-Collier, I.; Böker, H.; Hättenschwiler, J.; Seifritz, E.; et al. Functional connectivity between prefrontal cortex and subgenual cingulate predicts antidepressant effects of ketamine. Eur. Neuropsychopharmacol. 2019, 29, 501–508. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Groups of Antidepressants | Mechanism of Action | Example of a Drug | Reference |
|---|---|---|---|
| SSRI | Affinity for 5-HT2 and 5-HT1A receptors, inhibit serotonin transporter SERT | Fluoxetine Sertraline | [21,22] |
| MAOI | Monoamine oxidase enzyme inhibition | Selegiline Moclobemide | [23,24] |
| TCA | Blocking of presynaptic norepinephrine reuptake transporters and presynaptic serotonin reuptake transporters, antagonism of postsynaptic A1- and A2-adrenergic receptors, postsynaptic muscarinic receptors, postsynaptic histamine H1 receptors | Amitriptyline Desipramine | [24,25,26,27] |
| SNRI | Blocking of norepinephrine reuptake transporters and serotonin reuptake transporters, minimal or no effect on adrenergic (A1, A2 and Β), histamine (H1), muscarinic, dopaminergic or postsynaptic 5-HT receptors | Duloxetine Venlafaxine | [28,29] |
| NRI | Blocking of norepinephrine transporter NET | Reboxetine Nisoxetine | [20,29] |
| NDRI | Binding affinity to DA and NE transporters | Bupropion | [20] |
| SARI | Affinity for A1-adrenergic receptor | Trazodone Nefazodone | [30] |
| NASSA | Antagonism of presynaptic A2 adrenergic autoreceptors and 5-HT2, 5-HT3 receptors | Mirtazapine | [27,31] |
| MMA | Affinity for 5-HT receptors (5-HT1A, 5-HT1B, 5-HT3A, 5-HT7) | Vortioxetine Vilazodone | [33,34] |
| TeCA | Antagonism of presynaptic A2 autoreceptors | Mianserin | [32] |
| Other drugs | Agonism to the 5-HT1A receptor | Buspirone Tandospirone | [20,32,35] |
| Antagonism of presynaptic A2 autoreceptors | Trazodone (SARI) | [32] | |
| Dissociative anesthetics | Non-competitive NMDA receptor antagonist, activation of postsynaptic AMPA receptors | Ketamine | [36,37] |
| Mechanism | Main Findings | Studies Used | Substance Used |
|---|---|---|---|
| NMDA Receptor Antagonism | Ketamine functions as a non-competitive antagonist of NMDA receptors, resulting in reduced GABA release, enhanced glutamate release, and the promotion of synaptic plasticity | Zanos et al. [108] | (2R,6R)-HNK |
| Li et al. [105] Yang et al. [94] Li et al. [96] Miller et al. [98] Autry et al. [92] | Ketamine | ||
| AMPA Receptors | The activation of AMPA receptors by glutamate enhances synaptic signaling and initiates intracellular cascades, including the production of BDNF and the activation of the mTOR pathway. Ketamine increases the ratio of AMPA receptors to NMDA receptors. | Li et al. [105] Casarotto et al. [139] Tizabi [106] Ho [110] | Ketamine |
| Zanos et al. [108] Casarotto et al. [139] Ho [110] | Ketamine (2R,6R)-HNK | ||
| Ho [110] | (2S,6S)-HNK | ||
| mTOR Signaling | Ketamine activates mTOR signaling, which promotes synaptic protein synthesis and the formation of dendritic spines | Li et al. [105] Moda-Sava et al. [166] | Ketamine |
| BDNF-TrkB Pathway | Ketamine increases the expression of BDNF, which binds to TrkB receptors, thereby enhancing synaptic connectivity and structural plasticity | Caliman-Fontes et al. [130] Rybakowski et al. [134] Li et al. [137] Woelfer et al. [128] Casarotto et al. [139] Haile et al. [132] Chen et al. [133] Duncan et al. [135] Cannarozzo et al. [144] | Ketamine |
| Casarotto et al. [139] Jiang et al. [131] Caliman-Fontes et al. [130] Luo et al. [129] | Esketamine | ||
| Fred et al. [140] Casarotto et al. [139] Ju et al. [138] Cannarozzo et al. [144] | (2R,6R)-HNK | ||
| Glial Involvement | Ketamine restores GLT-1 levels in the infralimbic cortex | Fullana et al. [146] Liu et al. [142] | Ketamine |
| Opioid Receptor System Involvement | Ketamine interacts permissively with opioid receptors, facilitating its antidepressant effects without functioning as a direct opioid agonist | Klein et al. [172] Nolan et al. [101] Zhang et al. [173] Di Ianni et al. [174] | Ketamine |
| Authors | Study Design | Intervention | Main Findings | Limitations |
|---|---|---|---|---|
| Gärtner et al. [224] | Observational study | 0.5 mg/kg racemic or 0.25 mg/kg S-ketamine IV over 45 min | Changes in sgACC and right lateral PFC connectivity correlated with improved anhedonia symptoms. |
|
| Li et al. [223] | Double-blind, randomized, placebo-controlled study | 0.5 mg/kg racemic ketamine IV over 40 min | Ketamine administration 24 h after infusion specifically increased the glutamine/glutamate ratio in the pgACC but not in the aMCC. |
|
| Alexander et al. [222] | Randomized, double-blind, placebo-controlled crossover trial | 0.5 mg/kg ketamine IV over 40 min | Changes in subgenual ACC connectivity correlated with improvements in anhedonia symptoms. |
|
| Meiering et al. [221] | Double-blind, single-dose, randomized, placebo-controlled study |
| Ketamine increased amygdala-PFC connectivity and reduced activity in the hippocampus, and mPFC. |
|
| Kopelman et al. [212] | Randomized, double-blind, placebo-controlled trial | 0.5 mg/kg ketamine IV over 40 min | Changes in a marker of neuroplasticity (DTI-MD) were associated with improvements in depression scores, particularly in the left BA10 and left amygdala regions for the ketamine group. |
|
| Burrows et al. [211] | Double-blind, randomized, crossover, placebo-controlled study | 0.5 mg/kg ketamine IV over 45 min | Ketamine produced significant changes in brain connectivity, including decreased connectivity between the sgACC and amygdala, as well as altered connectivity in the ECN, approximately 2 h after administration in volunteers with remitted depression. |
|
| Li et al. [210] | Randomized, double-blind, placebo-controlled, parallel-group clinical trial | 0.5 mg/kg or 0.2 mg/kg of ketamine IV | The increase in PFC activity correlated with rapid antidepressant effects at 40- and 240-min post-treatment. |
|
| Mueller et al. [209] | Randomized, double-blind, placebo-controlled crossover trial | S-ketamine bolus (0.1 mg/kg) + continuous infusion (0.015625 mg/kg/min for up to 1 h, with a 10% dosage reduction every 10 min.) | Decreased SN connectivity linked to negative symptoms induced by ketamine. Ketamine increased FC in the ECN. |
|
| Chen et al. [208] | Randomized, double-blind, placebo-controlled trial | 0.5 or 0.2 mg/kg ketamine IV over 40 min |
|
|
| Li et al. [207] | Double-blind, randomized, placebo-controlled study | Single dose of ketamine 0.5 mg/kg over 40 min | Ketamine administration led to a decrease in FC between the dorsal posterior cingulate cortex and the dmPFC at 24 h post-infusion, which was correlated with an increase in the glutamine/glutamate ratio in the perigenual anterior cingulate cortex at the same time point. |
|
| Mkrtchian et al. [200] | Double-blind, placebo-controlled, crossover trial | 0.5 mg/kg ketamine IV over 40 min |
|
|
| Reed et al. [206] | Randomized, double-blind, placebo-controlled crossover trial | 0.5 mg/kg ketamine IV over 40 min |
|
|
| Evans et al. [205] | Double-blind, placebo-controlled, crossover study | 0.5 mg/kg of ketamine hydrochloride IV over 40 min |
|
|
| Chen et al. [199] | Double-blind, randomized, placebo-controlled, longitudinal study | 0.2 mg/kg or 0.5 mg/kg of ketamine IV over 40 min |
|
|
| Li et al. [203] | Double-blind, randomized, placebo-controlled trial | 0.5 mg/kg single infusion of ketamine or 0.045 mg/kg midazolam | A smaller decrease in the right dlPFC volume was associated with a more significant reduction in depressive symptoms in the ketamine group. |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antos, Z.; Żukow, X.; Bursztynowicz, L.; Jakubów, P. Beyond NMDA Receptors: A Narrative Review of Ketamine’s Rapid and Multifaceted Mechanisms in Depression Treatment. Int. J. Mol. Sci. 2024, 25, 13658. https://doi.org/10.3390/ijms252413658
Antos Z, Żukow X, Bursztynowicz L, Jakubów P. Beyond NMDA Receptors: A Narrative Review of Ketamine’s Rapid and Multifaceted Mechanisms in Depression Treatment. International Journal of Molecular Sciences. 2024; 25(24):13658. https://doi.org/10.3390/ijms252413658
Chicago/Turabian StyleAntos, Zuzanna, Xawery Żukow, Laura Bursztynowicz, and Piotr Jakubów. 2024. "Beyond NMDA Receptors: A Narrative Review of Ketamine’s Rapid and Multifaceted Mechanisms in Depression Treatment" International Journal of Molecular Sciences 25, no. 24: 13658. https://doi.org/10.3390/ijms252413658
APA StyleAntos, Z., Żukow, X., Bursztynowicz, L., & Jakubów, P. (2024). Beyond NMDA Receptors: A Narrative Review of Ketamine’s Rapid and Multifaceted Mechanisms in Depression Treatment. International Journal of Molecular Sciences, 25(24), 13658. https://doi.org/10.3390/ijms252413658