
Complement Cascade Proteins Correlate with Fibrosis and Inflammation in Early-Stage Type 1 Diabetic Kidney Disease in the Ins2Akita Mouse Model
 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Complement Cascade, Inflammation, and Fibrosis Correlate in DKD in Animal Models and Human Datasets
2.2. Positive Correlation of Expression of Complement Proteins with Fibrosis- and Inflammation-Related Proteins in Ins2Akita Mice
2.3. Validation of Complement Upregulation and Its Association to Fibrosis and Inflammation in Human Patients with DKD
3. Discussion
4. Materials and Methods
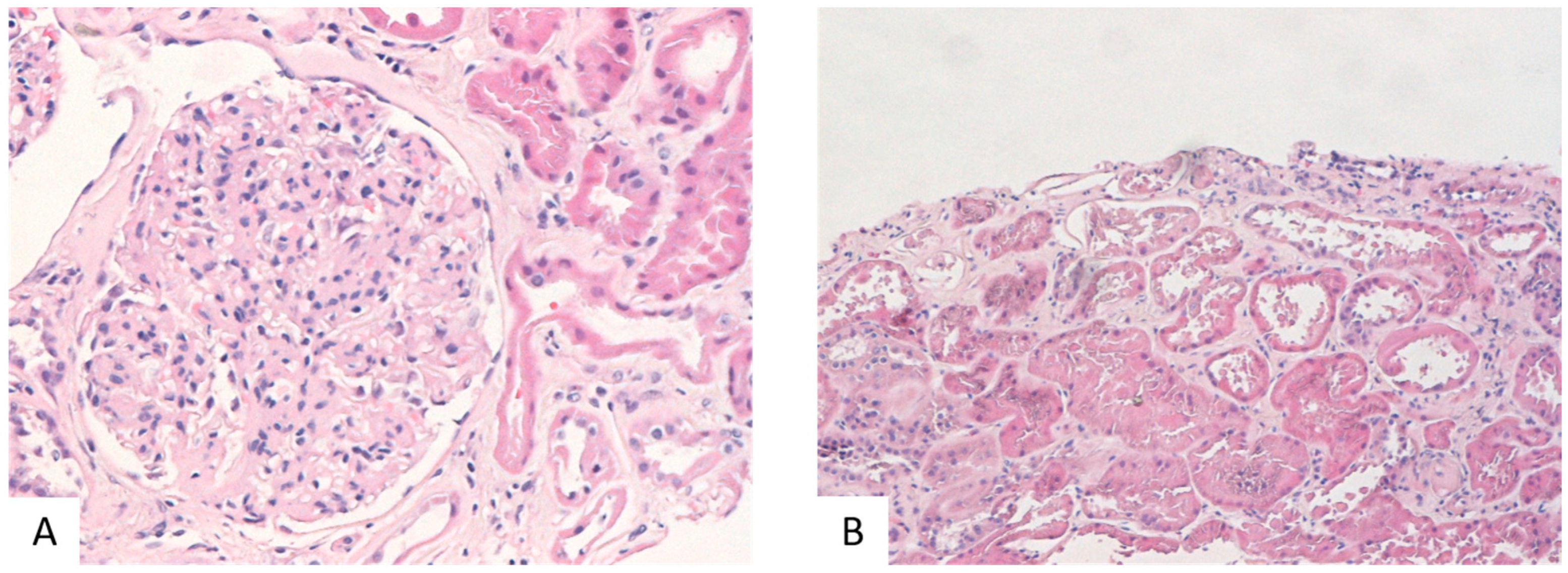
4.1. Samples, Glomeruli Preparation, and Biochemical Analysis
4.2. Sample Preparation and LC-MS/MS Analysis and MS Data Processing
4.3. Statistical Analysis
4.4. Cross-Correlation to Transcriptomics Data
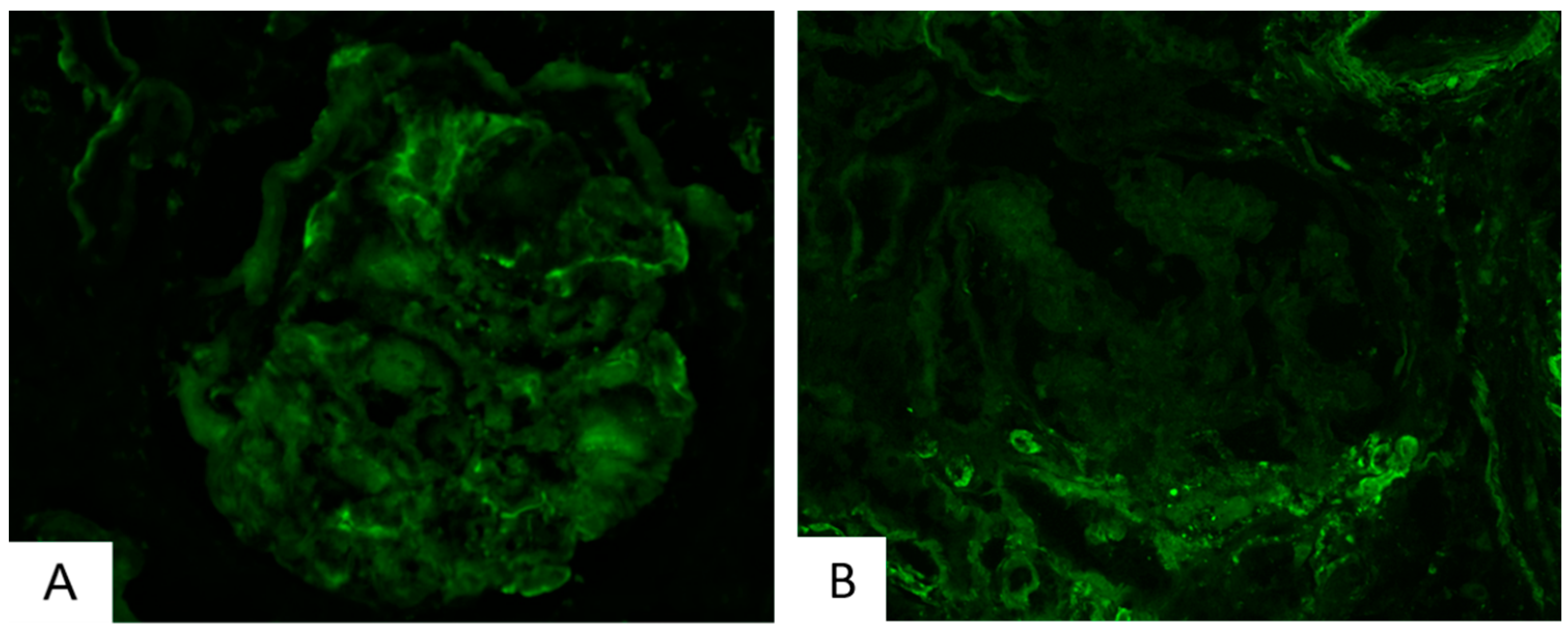
4.5. Clinical Material and Immunofluorescence
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Diabetes Control and Complications Trial Research Group; Nathan, D.M.; Genuth, S.; Lachin, J.; Cleary, P.; Crofford, O.; Davis, M.; Rand, L.; Siebert, C. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef] [PubMed]
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998, 352, 837–853. [Google Scholar] [CrossRef]
- Gaede, P.; Lund-Andersen, H.; Parving, H.H.; Pedersen, O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N. Engl. J. Med. 2008, 358, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Badal, S.S.; Danesh, F.R. New insights into molecular mechanisms of diabetic kidney disease. Am. J. Kidney Dis. 2014, 63 (Suppl. S2), S63–S83. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.W.; Yiu, W.H. Innate immunity in diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Tesch, G.H. Diabetic nephropathy—Is this an immune disorder? Clin. Sci. 2017, 131, 2183–2199. [Google Scholar] [CrossRef]
- Klein, J.; Caubet, C.; Camus, M.; Makridakis, M.; Denis, C.; Gilet, M.; Feuillet, G.; Rascalou, S.; Neau, E.; Garrigues, L.; et al. Connectivity mapping of glomerular proteins identifies dimethylaminoparthenolide as a new inhibitor of diabetic kidney disease. Sci. Rep. 2020, 10, 14898. [Google Scholar] [CrossRef]
- Hsieh, J.T.; Chang, F.P.; Yang, A.H.; Tarng, D.C.; Yang, C.Y. Timing of kidney biopsy in type 2 diabetic patients: A stepwise approach. BMC Nephrol. 2020, 21, 131. [Google Scholar] [CrossRef]
- Chen, J.; Liu, Q.; He, J.; Li, Y. Immune responses in diabetic nephropathy: Pathogenic mechanisms and therapeutic target. Front. Immunol. 2022, 13, 958790. [Google Scholar] [CrossRef]
- Guiteras, R.; Sola, A.; Flaquer, M.; Manonelles, A.; Hotter, G.; Cruzado, J.M. Exploring macrophage cell therapy on Diabetic Kidney Disease. J. Cell Mol. Med. 2019, 23, 841–851. [Google Scholar] [CrossRef]
- Chow, F.; Ozols, E.; Nikolic-Paterson, D.J.; Atkins, R.C.; Tesch, G.H. Macrophages in mouse type 2 diabetic nephropathy: Correlation with diabetic state and progressive renal injury. Kidney Int. 2004, 65, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Flyvbjerg, A. The role of the complement system in diabetic nephropathy. Nat. Rev. Nephrol. 2017, 13, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.K.; Tesch, G.H. Inflammation in diabetic nephropathy. Mediat. Inflamm. 2012, 2012, 146154. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, P.R.; Kramer, A.; Logtenberg, S.J.; Hoitsma, A.J.; Kleefstra, N.; Jager, K.J.; Bilo, H.J. Incidence of renal replacement therapy for diabetic nephropathy in the Netherlands: Dutch diabetes estimates (DUDE)-3. BMJ Open 2015, 5, e005624. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.F.; Chen, M. Complement Activation in Progression of Chronic Kidney Disease. Adv. Exp. Med. Biol. 2019, 1165, 423–441. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zheng, F. Immune Cells and Inflammation in Diabetic Nephropathy. J. Diabetes Res. 2016, 2016, 1841690. [Google Scholar] [CrossRef] [PubMed]
- Kolset, S.O.; Reinholt, F.P.; Jenssen, T. Diabetic nephropathy and extracellular matrix. J. Histochem. Cytochem. 2012, 60, 976–986. [Google Scholar] [CrossRef]
- Mason, R.M.; Wahab, N.A. Extracellular matrix metabolism in diabetic nephropathy. J. Am. Soc. Nephrol. 2003, 14, 1358–1373. [Google Scholar] [CrossRef]
- Jäckle-Meyer, I.; Szukics, B.; Neubauer, K.; Metze, V.; Petzoldt, R.; Stolte, H. Extracellular matrix proteins as early markers in diabetic nephropathy. Eur. J. Clin. Chem. Clin. Biochem. 1995, 33, 211–219. [Google Scholar] [CrossRef]
- Ziyadeh, F.N. The extracellular matrix in diabetic nephropathy. Am. J. Kidney Dis. 1993, 22, 736–744. [Google Scholar] [CrossRef]
- Navarro-González, J.F.; Mora-Fernández, C.; Muros de Fuentes, M.; García-Pérez, J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat. Rev. Nephrol. 2011, 7, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Budge, K.; Dellepiane, S.; Yu, S.M.; Cravedi, P. Complement, a Therapeutic Target in Diabetic Kidney Disease. Front. Med. 2021, 7, 599236. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.I.; Park, A.S.; Mohtat, D.; Thomas, D.B.; Pullman, J.M.; Susztak, K. Transcriptome analysis of human diabetic kidney disease. Diabetes 2011, 60, 2354–2369. [Google Scholar] [CrossRef] [PubMed]
- Xavier, S.; Sahu, R.K.; Landes, S.G.; Yu, J.; Taylor, R.P.; Ayyadevara, S.; Megyesi, J.; Stallcup, W.B.; Duffield, J.S.; Reis, E.S.; et al. Pericytes and immune cells contribute to complement activation in tubulointerstitial fibrosis. Am. J. Physiol. Renal Physiol. 2017, 312, F516–F532. [Google Scholar] [CrossRef] [PubMed]
- Giralt-López, A.; Molina-Van den Bosch, M.; Vergara, A.; García-Carro, C.; Seron, D.; Jacobs-Cachá, C.; Soler, M.J. Revisiting Experimental Models of Diabetic Nephropathy. Int. J. Mol. Sci. 2020, 21, 3587. [Google Scholar] [CrossRef]
- Gurley, S.B.; Mach, C.L.; Stegbauer, J.; Yang, J.; Snow, K.P.; Hu, A.; Meyer, T.W.; Coffman, T.M. Influence of genetic background on albuminuria and kidney injury in Ins2(+/C96Y) (Akita) mice. Am. J. Physiol. Renal Physiol. 2010, 298, F788–F795. [Google Scholar] [CrossRef] [PubMed]
- Alpers, C.E.; Hudkins, K.L. Mouse models of diabetic nephropathy. Curr. Opin. Nephrol. Hypertens. 2011, 20, 278–284. [Google Scholar] [CrossRef]
- Lin, D.W.; Chang, C.C.; Hsu, Y.C.; Lin, C.L. New Insights into the Treatment of Glomerular Diseases: When Mechanisms Become Vivid. Int. J. Mol. Sci. 2022, 23, 3525. [Google Scholar] [CrossRef]
- Welte, T.; Arnold, F.; Kappes, J.; Seidl, M.; Häffner, K.; Bergmann, C.; Walz, G.; Neumann-Haefelin, E. Treating C3 glomerulopathy with eculizumab. BMC Nephrol. 2018, 19, 7. [Google Scholar] [CrossRef]
- Pescovitz, M.D.; Greenbaum, C.J.; Krause-Steinrauf, H.; Becker, D.J.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Marks, J.B.; McGee, P.F.; Moran, A.M.; et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N. Engl. J. Med. 2009, 361, 2143–2152. [Google Scholar] [CrossRef]
- Hu, C.Y.; Rodriguez-Pinto, D.; Du, W.; Ahuja, A.; Henegariu, O.; Wong, F.S.; Shlomchik, M.J.; Wen, L. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J. Clin. Investig. 2007, 117, 3857–3867. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.N.; Fesseha, B.; Anzaldi, L.; Tsao, A.; Galiatsatos, P.; Sidhaye, A. Antibody-Mediated Extreme Insulin Resistance: A Report of Three Cases. Am. J. Med. 2018, 131, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Tserga, A.; Pouloudi, D.; Saulnier-Blache, J.S.; Stroggilos, R.; Theochari, I.; Gakiopoulou, H.; Mischak, H.; Zoidakis, J.; Schanstra, J.P.; Vlahou, A.; et al. Proteomic Analysis of Mouse Kidney Tissue Associates Peroxisomal Dysfunction with Early Diabetic Kidney Disease. Biomedicines 2022, 10, 216. [Google Scholar] [CrossRef] [PubMed]
- Schmid, H.; Boucherot, A.; Yasuda, Y.; Henger, A.; Brunner, B.; Eichinger, F.; Nitsche, A.; Kiss, E.; Bleich, M.; Gröne, H.J.; et al. Modular activation of nuclear factor-kappaB transcriptional programs in human diabetic nephropathy. Diabetes 2006, 55, 2993–3003. [Google Scholar] [CrossRef] [PubMed]
- Hodgin, J.B.; Nair, V.; Zhang, H.; Randolph, A.; Harris, R.C.; Nelson, R.G.; Weil, E.J.; Cavalcoli, J.D.; Patel, J.M.; Brosius, F.C., 3rd; et al. Identification of cross-species shared transcriptional networks of diabetic nephropathy in human and mouse glomeruli. Diabetes 2013, 62, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.; Greene, C.S.; Eichinger, F.; Nair, V.; Hodgin, J.B.; Bitzer, M.; Lee, Y.S.; Zhu, Q.; Kehata, M.; Li, M.; et al. Defining cell-type specificity at the transcriptional level in human disease. Genome Res. 2013, 23, 1862–1873. [Google Scholar] [CrossRef]
- Ju, W.; Nair, V.; Smith, S.; Zhu, L.; Shedden, K.; Song, P.X.K.; Mariani, L.H.; Eichinger, F.H.; Berthier, C.C.; Randolph, A.; et al. Tissue transcriptome-driven identification of epidermal growth factor as a chronic kidney disease biomarker. Sci. Transl. Med. 2015, 7, 316ra193. [Google Scholar] [CrossRef]
- Fujita, T.; Hemmi, S.; Kajiwara, M.; Yabuki, M.; Fuke, Y.; Satomura, A.; Soma, M. Complement-mediated chronic inflammation is associated with diabetic microvascular complication. Diabetes Metab. Res. Rev. 2013, 29, 220–226. [Google Scholar] [CrossRef]
- Rasmussen, K.L.; Nordestgaard, B.G.; Nielsen, S.F. Complement C3 and Risk of Diabetic Microvascular Disease: A Cohort Study of 95202 Individuals from the General Population. Clin. Chem. 2018, 64, 1113–1124. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Zhang, R.; Li, H.; Han, Q.; Wu, Y.; Wang, S.; Guo, R.; Wang, T.; Li, L.; et al. Serum levels of immunoglobulin G and complement 3 differentiate non-diabetic renal disease from diabetic nephropathy in patients with type 2 diabetes mellitus. Acta Diabetol. 2019, 56, 873–881. [Google Scholar] [CrossRef]
- Satoskar, A.A.; Shapiro, J.P.; Bott, C.N.; Song, H.; Nadasdy, G.M.; Brodsky, S.V.; Hebert, L.A.; Birmingham, D.J.; Nadasdy, T.; Freitas, M.A.; et al. Characterization of glomerular diseases using proteomic analysis of laser capture microdissected glomeruli. Mod. Pathol. 2012, 25, 709–721. [Google Scholar] [CrossRef]
- Nakatani, S.; Wei, M.; Ishimura, E.; Kakehashi, A.; Mori, K.; Nishizawa, Y.; Inaba, M.; Wanibuchi, H. Proteome analysis of laser microdissected glomeruli from formalin-fixed paraffin-embedded kidneys of autopsies of diabetic patients: Nephronectin is associated with the development of diabetic glomerulosclerosis. Nephrol. Dial. Transplant. 2012, 27, 1889–1897. [Google Scholar] [CrossRef]
- Mauer, S.M.; Steffes, M.W.; Sutherland, D.E.; Najarian, S.; Michael, A.F.; Brown, D.M. Studies of the rate of regression of the glomerular lesions in diabetic rats treated with pancreatic islet transplantation. Diabetes 1975, 24, 280–285. [Google Scholar] [CrossRef]
- Xiao, X.; Ma, B.; Dong, B.; Zhao, P.; Tai, N.; Chen, L.; Wong, F.S.; Wen, L. Cellular and humoral immune responses in the early stages of diabetic nephropathy in NOD mice. J. Autoimmun. 2009, 32, 85–93. [Google Scholar] [CrossRef]
- Yang, L.; Brozovic, S.; Xu, J.; Long, Y.; Kralik, P.M.; Waigel, S.; Zacharias, W.; Zheng, S.; Epstein, P.N. Inflammatory gene expression in OVE26 diabetic kidney during the development of nephropathy. Nephron Exp. Nephrol. 2011, 119, e8–e20. [Google Scholar] [CrossRef]
- Lim, A.K.; Ma, F.Y.; Nikolic-Paterson, D.J.; Kitching, A.R.; Thomas, M.C.; Tesch, G.H. Lymphocytes promote albuminuria, but not renal dysfunction or histological damage in a mouse model of diabetic renal injury. Diabetologia 2010, 53, 1772–1782. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, J.; Wu, X.; Chen, X.; Bai, X.; Zhuang, Y.; Fang, J.; Lin, X. High Expression of Complement Components in the Kidneys of Type 2 Diabetic Rats With Diabetic Nephropathy. Front. Endocrinol. 2019, 10, 459. [Google Scholar] [CrossRef]
- Tang, X.; Li, H.; Li, L.; Zhang, J.; Xu, H.; Li, L.; Liu, F. The Clinical Impact of Glomerular Immunoglobulin M Deposition in Patients with Type 2 Diabetic Nephropathy. Am. J. Med. Sci. 2018, 356, 365–373. [Google Scholar] [CrossRef]
- Wang, W.; Jia, Y.-J.; Yang, Y.-L.; Xue, M.; Zheng, Z.-J.; Wang, L.; Xue, Y.-M. LncRNA GAS5 exacerbates renal tubular epithelial fibrosis by acting as a competing endogenous RNA of miR-96-5p. Biomed. Pharmacother. 2020, 121, 109411. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Wang, W.; Feng, Z.; Liu, R.; Cheng, X.; Shen, W.; Dong, Z.; Cai, G.; Chen, X.; Hong, Q.; et al. Identification of key genes and pathways in diabetic nephropathy by bioinformatics analysis. J. Diabetes Investig. 2019, 10, 972–984. [Google Scholar] [CrossRef] [PubMed]
- Gerrits, T.; Zandbergen, M.; Wolterbeek, R.; Bruijn, J.A.; Baelde, H.J.; Scharpfenecker, M. Endoglin Promotes Myofibroblast Differentiation and Extracellular Matrix Production in Diabetic Nephropathy. Int. J. Mol. Sci. 2020, 21, 7713. [Google Scholar] [CrossRef]
- Bus, P.; Gerrits, T.; Heemskerk, S.A.C.; Zandbergen, M.; Wolterbeek, R.; Bruijn, J.A.; Baelde, H.J.; Scharpfenecker, M. Endoglin Mediates Vascular Endothelial Growth Factor-A-Induced Endothelial Cell Activation by Regulating Akt Signaling. Am. J. Pathol. 2018, 188, 2924–2935. [Google Scholar] [CrossRef]
- Liu, W.; Chen, X.; Wang, Y.; Chen, Y.; Chen, S.; Gong, W.; Chen, T.; Sun, L.; Zheng, C.; Yin, B.; et al. Micheliolide ameliorates diabetic kidney disease by inhibiting Mtdh-mediated renal inflammation in type 2 diabetic db/db mice. Pharmacol. Res. 2019, 150, 104506. [Google Scholar] [CrossRef]
- Wei, H.; Li, J.; Li, Y.; Song, J. MicroRNA-451 inhibits inflammation and proliferation of glomerular mesangial cells through down-regulating PSMD11 and NF-κB p65. Biosci. Rep. 2019, 39, BSR20191455. [Google Scholar] [CrossRef]
- Fan, Y.; Fei, Y.; Zheng, L.; Wang, J.; Xiao, W.; Wen, J.; Xu, Y.; Wang, Y.; He, L.; Guan, J.; et al. Expression of endothelial cell injury marker CD146 correlates with disease severity and predicts the renal outcomes in patients with diabetic nephropathy. Cell Physiol. Biochem. 2018, 48, 63–74. [Google Scholar] [CrossRef]
- Li, X.; Wen, J.; Dong, Y.; Zhang, Q.; Guan, J.; Liu, F.; Zhou, T.; Li, Z.; Fan, Y.; Wang, N. Wnt5a promotes renal tubular inflammation in diabetic nephropathy by binding to CD146 through noncanonical Wnt signaling. Cell Death Dis. 2021, 12, 92. [Google Scholar] [CrossRef]
- Wada, J.; Makino, H. Innate immunity in diabetes and diabetic nephropathy. Nat. Rev. Nephrol. 2016, 12, 13–26. [Google Scholar] [CrossRef]
- Fu, J.; Akat, K.M.; Sun, Z.; Zhang, W.; Schlondorff, D.; Liu, Z.; Tuschl, T.; Lee, K.; He, J.C. Single-Cell RNA Profiling of Glomerular Cells Shows Dynamic Changes in Experimental Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2019, 30, 533–545. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, M.; Zhang, Y. Identification of fibronectin 1 (FN1) and complement component 3 (C3) as immune infiltration-related biomarkers for diabetic nephropathy using integrated bioinformatic analysis. Bioengineered 2021, 12, 5386–5401. [Google Scholar] [CrossRef]
- Falk, R.J.; Dalmasso, A.P.; Kim, Y.; Tsai, C.H.; Scheinman, J.I.; Gewurz, H.; Michael, A.F. Neoantigen of the polymerized ninth component of complement. Characterization of a monoclonal antibody and immunohistochemical localization in renal disease. J. Clin. Investig. 1983, 72, 560–573. [Google Scholar] [CrossRef]
- Ghosh, P.; Sahoo, R.; Vaidya, A.; Chorev, M.; Halperin, J.A. Role of complement and complement regulatory proteins in the complications of diabetes. Endocr. Rev. 2015, 36, 272–288. [Google Scholar] [CrossRef]
- Ricardo, S.D.; van Goor, H.; Eddy, A.A. Macrophage diversity in renal injury and repair. J. Clin. Investig. 2008, 118, 3522–3530. [Google Scholar] [CrossRef]
- Bus, P.; Chua, J.S.; Klessens, C.Q.F.; Zandbergen, M.; Wolterbeek, R.; van Kooten, C.; Trouw, L.A.; Bruijn, J.A.; Baelde, H.J. Complement Activation in Patients with Diabetic Nephropathy. Kidney Int. Rep. 2017, 3, 302–313. [Google Scholar] [CrossRef]
- Qin, X.; Goldfine, A.; Krumrei, N.; Grubissich, L.; Acosta, J.; Chorev, M.; Hays, A.P.; Halperin, J.A. Glycation inactivation of the complement regulatory protein CD59: A possible role in the pathogenesis of the vascular complications of human diabetes. Diabetes 2004, 53, 2653–2661. [Google Scholar] [CrossRef]
- Kelly, K.J.; Liu, Y.; Zhang, J.; Dominguez, J.H. Renal C3 complement component: Feed forward to diabetic kidney disease. Am. J. Nephrol. 2015, 41, 48–56. [Google Scholar] [CrossRef]
- Jiao, Y.; Jiang, S.; Wang, Y.; Yu, T.; Zou, G.; Zhuo, L.; Li, W. Activation of complement C1q and C3 in glomeruli might accelerate the progression of diabetic nephropathy: Evidence from transcriptomic data and renal histopathology. J. Diabetes Investig. 2022, 13, 839–849. [Google Scholar] [CrossRef]
- Sun, Z.J.; Li, X.Q.; Chang, D.Y.; Wang, S.X.; Liu, G.; Chen, M.; Zhao, M.H. Complement deposition on renal histopathology of patients with diabetic nephropathy. Diabetes Metab. 2019, 45, 363–368. [Google Scholar] [CrossRef]
- Pelletier, K.; Bonnefoy, A.; Chapdelaine, H.; Pichette, V.; Lejars, M.; Madore, F.; Brachemi, S.; Troyanov, S. Clinical Value of Complement Activation Biomarkers in Overt Diabetic Nephropathy. Kidney Int. Rep. 2019, 4, 797–805. [Google Scholar] [CrossRef]
- Morigi, M.; Perico, L.; Corna, D.; Locatelli, M.; Cassis, P.; Carminati, C.E.; Bolognini, S.; Zoja, C.; Remuzzi, G.; Benigni, A.; et al. C3a receptor blockade protects podocytes from injury in diabetic nephropathy. JCI Insight 2020, 5, e131849. [Google Scholar] [CrossRef]
- Duan, S.; Sun, L.; Nie, G.; Chen, J.; Zhang, C.; Zhu, H.; Huang, Z.; Qian, J.; Zhao, X.; Xing, C.; et al. Association of Glomerular Complement C4c Deposition with the Progression of Diabetic Kidney Disease in Patients with Type 2 Diabetes. Front. Immunol. 2020, 11, 2073. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.S.; Kim, J.H.; Jeong, H.; Yu, J.; Yeom, J.; Song, S.H.; Kim, S.S.; Kim, I.J.; Kim, K. Differential Urinary Proteome Analysis for Predicting Prognosis in Type 2 Diabetes Patients with and without Renal Dysfunction. Int. J. Mol. Sci. 2020, 21, 4236. [Google Scholar] [CrossRef]
- Guillén-Gómez, E.; Bardají-de-Quixano, B.; Ferrer, S.; Brotons, C.; Knepper, M.A.; Carrascal, M.; Abian, J.; Mas, J.M.; Calero, F.; Ballarín, J.A.; et al. Urinary Proteome Analysis Identified Neprilysin and VCAM as Proteins Involved in Diabetic Nephropathy. J. Diabetes Res. 2018, 2018, 6165303. [Google Scholar] [CrossRef]
- Qi, W.; Keenan, H.A.; Li, Q.; Ishikado, A.; Kannt, A.; Sadowski, T.; Yorek, M.A.; Wu, I.H.; Lockhart, S.; Coppey, L.J.; et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat. Med. 2017, 23, 753–762. [Google Scholar] [CrossRef]
- Magagnotti, C.; Zerbini, G.; Fermo, I.; Carletti, R.M.; Bonfanti, R.; Vallone, F.; Andolfo, A. Identification of nephropathy predictors in urine from children with a recent diagnosis of type 1 diabetes. J. Proteomics. 2019, 193, 205–216. [Google Scholar] [CrossRef]
- Li, X.Q.; Chang, D.Y.; Chen, M.; Zhao, M.H. Complement activation in patients with diabetic nephropathy. Diabetes Metab. 2019, 45, 248–253. [Google Scholar] [CrossRef]
- Kelsh, R.; You, R.; Horzempa, C.; Zheng, M.; McKeown-Longo, P.J. Regulation of the innate immune response by fibronectin: Synergism between the III-1 and EDA domains. PLoS ONE 2014, 9, e102974. [Google Scholar] [CrossRef]
- Lee, S.; Ishitsuka, A.; Kuroki, T.; Lin, Y.H.; Shibuya, A.; Hongu, T.; Funakoshi, Y.; Kanaho, Y.; Nagata, K.; Kawaguchi, A. Arf6 exacerbates allergic asthma through cell-to-cell transmission of ASC inflammasomes. JCI Insight 2021, 6, e139190. [Google Scholar] [CrossRef]
- Gamara, J.; Davis, L.; Leong, A.Z.; Pagé, N.; Rollet-Labelle, E.; Zhao, C.; Hongu, T.; Funakoshi, Y.; Kanaho, Y.; Aoudji, F.; et al. Arf6 regulates energy metabolism in neutrophils. Free Radic. Biol. Med. 2021, 172, 550–561. [Google Scholar] [CrossRef]
- Li, R.; Shen, Q.; Wu, N.; He, M.; Liu, N.; Huang, J.; Lu, B.; Yao, Q.; Yang, Y.; Hu, R. MiR-145 improves macrophage-mediated inflammation through targeting Arf6. Endocrine 2018, 60, 73–82. [Google Scholar] [CrossRef]
- Wu, Z.; MacNeil, A.J.; Berman, J.N.; Lin, T.J. Syntaxin binding protein 1 is not required for allergic inflammation via IgE-mediated mast cell activation. PLoS ONE 2013, 8, e58560. [Google Scholar] [CrossRef]
- Zhang, P.; Yu, C.; Yu, J.; Li, Z.; Lan, H.Y.; Zhou, Q. Arid2-IR promotes NF-κB-mediated renal inflammation by targeting NLRC5 transcription. Cell Mol. Life Sci. 2021, 78, 2387–2404. [Google Scholar] [CrossRef]
- Han, S.; Xu, S.; Zhou, J.; Qiao, A.; Boriboun, C.; Ma, W.; Li, H.; Biyashev, D.; Yang, L.; Zhang, E.; et al. Sam68 impedes the recovery of arterial injury by augmenting inflammatory response. J. Mol. Cell Cardiol. 2019, 137, 82–92. [Google Scholar] [CrossRef]
- Nozato, Y.; Takami, Y.; Yamamoto, K.; Nagasawa, M.; Nozato, S.; Imaizumi, Y.; Takeshita, H.; Wang, C.; Ito, Y.; Takeda, S.; et al. Novel properties of myoferlin in glucose metabolism via pathways involving modulation of adipose functions. FASEB J. 2020, 34, 2792–2811. [Google Scholar] [CrossRef]
- Sivaraj, D.; Padmanabhan, J.; Chen, K.; Henn, D.; Noishiki, C.; Trotsyuk, A.A.; Kussie, H.C.; Leeolou, M.C.; Magbual, N.J.; Andrikopoulos, S.; et al. IQGAP1-mediated mechanical signaling promotes the foreign body response to biomedical implants. FASEB J. 2022, 36, e22007. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, G.; Sun, G. Actin-binding protein, IQGAP1, regulates LPS-induced RPMVECs hyperpermeability and ICAM-1 upregulation via Rap1/Src signalling pathway. Cell Signal. 2021, 85, 110067. [Google Scholar] [CrossRef]
- Yang, J.; Xu, C.; Wu, M.; Wu, Y.; Jia, X.; Zhou, C.; Zhang, X.; Ge, S.; Li, Z.; Zhang, L. MicroRNA-124 inhibits hepatic stellate cells inflammatory cytokines secretion by targeting IQGAP1 through NF-κB pathway. Int. Immunopharmacol. 2021, 95, 107520. [Google Scholar] [CrossRef]
- Yang, Z.; Zhou, X.; Liu, Y.; Gong, C.; Wei, X.; Zhang, T.; Ma, D.; Gao, Q. Activation of integrin β1 mediates the increased malignant potential of ovarian cancer cells exerted by inflammatory cytokines. Anticancer Agents Med. Chem. 2014, 14, 955–962. [Google Scholar] [CrossRef]
- Kong, J.S.; Park, J.H.; Yoo, S.A.; Kim, K.M.; Bae, Y.J.; Park, Y.J.; Cho, C.S.; Hwang, D.; Kim, W.U. Dynamic transcriptome analysis unveils key proresolving factors of chronic inflammatory arthritis. J. Clin. Investig. 2020, 130, 3974–3986. [Google Scholar] [CrossRef]
- Plosa, E.J.; Benjamin, J.T.; Sucre, J.M.; Gulleman, P.M.; Gleaves, L.A.; Han, W.; Kook, S.; Polosukhin, V.V.; Haake, S.M.; Guttentag, S.H.; et al. β1 Integrin regulates adult lung alveolar epithelial cell inflammation. JCI Insight 2020, 5, e129259. [Google Scholar] [CrossRef]
- Guo, Q.; Furuta, K.; Lucien, F.; Gutierrez Sanchez, L.H.; Hirsova, P.; Krishnan, A.; Kabashima, A.; Pavelko, K.D.; Madden, B.; Alhuwaish, H.; et al. Integrin β1-enriched extracellular vesicles mediate monocyte adhesion and promote liver inflammation in murine NASH. J. Hepatol. 2019, 71, 1193–1205. [Google Scholar] [CrossRef]
- de Fougerolles, A.R.; Sprague, A.G.; Nickerson-Nutter, C.L.; Chi-Rosso, G.; Rennert, P.D.; Gardner, H.; Gotwals, P.J.; Lobb, R.R.; Koteliansky, V.E. Regulation of inflammation by collagen-binding integrins alpha1beta1 and alpha2beta1 in models of hypersensitivity and arthritis. J. Clin. Investig. 2000, 105, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.C.; Isenman, D.E. Regulation of humoral immunity by complement. Immunity 2012, 37, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Sagi, Y.; Landrigan, A.; Levy, R.; Levy, S. Complementary costimulation of human T-cell subpopulations by cluster of differentiation 28 (CD28) and CD81. Proc. Natl. Acad. Sci. USA 2012, 109, 1613–1618. [Google Scholar] [CrossRef]
- Lu, H.J.; Tzeng, T.F.; Liou, S.S.; Da Lin, S.; Wu, M.C.; Liu, I.M. Ruscogenin ameliorates diabetic nephropathy by its anti-inflammatory and anti-fibrotic effects in streptozotocin-induced diabetic rat. BMC Complement. Altern. Med. 2014, 14, 110. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Qin, W.; Buya, M.; Dong, X.; Zheng, W.; Lu, W.; Chen, J.; Guo, Q.; Wu, Y. VNN1, a potential biomarker for pancreatic cancer-associated new-onset diabetes, aggravates paraneoplastic islet dysfunction by increasing oxidative stress. Cancer Lett. 2016, 373, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Berruyer, C.; Pouyet, L.; Millet, V.; Martin, F.M.; LeGoffic, A.; Canonici, A.; Garcia, S.; Bagnis, C.; Naquet, P.; Galland, F. Vanin-1 licenses inflammatory mediator production by gut epithelial cells and controls colitis by antagonizing peroxisome proliferator-activated receptor gamma activity. J. Exp. Med. 2006, 203, 2817–2827. [Google Scholar] [CrossRef]
- Boddul, S.V.; Meng, J.; Dolly, J.O.; Wang, J. SNAP-23 and VAMP-3 contribute to the release of IL-6 and TNFα from a human synovial sarcoma cell line. FEBS J. 2014, 281, 750–765. [Google Scholar] [CrossRef]
- Pei, W.; Tanaka, K.; Huang, S.C.; Xu, L.; Liu, B.; Sinclair, J.; Idol, J.; Varshney, G.K.; Huang, H.; Lin, S.; et al. Extracellular HSP60 triggers tissue regeneration and wound healing by regulating inflammation and cell proliferation. npj Regen. Med. 2016, 1, 16013. [Google Scholar] [CrossRef]
- Scheu, S.; Ruland, C.; Lahl, K.; Zimmer, A.; Werner, H.; Förster, I.; Sparwasser, T.; Alferink, J. Tspan2 is involved in anti-infectious immune responses and CNS autoimmunity. J. Immunol. 2020, 204 (Suppl. S1), 67.15. [Google Scholar] [CrossRef]
- Xiang, H.; Guo, F.; Tao, X.; Zhou, Q.; Xia, S.; Deng, D.; Li, L.; Shang, D. Pancreatic ductal deletion of S100A9 alleviates acute pancreatitis by targeting VNN1-mediated ROS release to inhibit NLRP3 activation. Theranostics 2021, 11, 4467–4482. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| COMPLEMENT | |||||
| Protein | Protein Name | MW_p_INS2vsWT2 | Ratio_INS2vsWT2 | MW_p_INS4vsWT4 (ns = not significant) | Ratio_INS4vsWT4 |
| C3 | Complement C3 | 0.001 | 5.073 | 0.058 (ns) | 1.885 |
| C4B | Complement C4-B | 0.009 | 5.568 | 0.003 | 4.425 |
| IGHM | Ig mu chain C region | 0.023 | 4.941 | 0.001 | 13.972 |
| FIBROSIS | |||||
| Protein | Protein Name | MW_p_INS2vsWT2 | Ratio_INS2vsWT2 (ns = not significant) | MW_p_INS4vsWT4 | Ratio_INS4vsWT4 |
| FN1 | Fibronectin | 0.0292 | 3.047 | 0.0005 | 103.182 |
| ENG | Endoglin | 0.028 | 6.629 | 0.525 (ns) | 1.276 |
| DYSF | Dysferlin | 0.032 | 2.156 | 0.004 | 4.548 |
| CORO-1C | Coronin-1C | 0.04 | 1.846 | 0.382 (ns) | 1.444 |
| FLNA | Filamin-A | 0.694 (ns) | 1.172 | 0.0003 | 2.612 |
| MYOF | Myoferlin | 0.463 (ns) | 1.176 | 0.0006 | 2.685 |
| INFLAMMATION | |||||
| Protein | Protein Name | MW_p_INS2vsWT2 | Ratio_INS2vsWT2 | MW_p_INS4vsWT4 (ns = not significant) | Ratio_INS4vsWT4 |
| PSMD11 | 26S proteasome non-ATPase regulatory subunit 11 | 0.008 | 3.654 | 0.792 (ns) | 0.863 |
| IFITM3 | Interferon-induced transmembrane protein 3 | 0.038 | 6.487 | 0.160 (ns) | 2.231 |
| TGFb1i1 | Transforming growth factor beta-1-induced transcript 1 | 0.029 | 3.777 | 0.072 (ns) | 1.967 |
| CLIC4 | Chloride intracellular channel protein 4 | 0.040 | 1.702 | 0.645 (ns) | 1.405 |
| ECSIT | Evolutionarily conserved signaling intermediate in Toll pathway | 0.042 | 3.356 | 0.562 (ns) | 0.811 |
| MTDH | Protein LYRIC | 0.043 | 25.173 | 0.653 (ns) | 0.743 |
| YTHDF1 | YTH domain-containing family protein 1 | 0.044 | 3.788 | 0.381 (ns) | 0 |
| MCAM | Cell surface glycoprotein MUC18 | 0.596 (ns) | 1.611 | 0.005 | 7.291 |
| TSPAN2 | Tetraspanin-2 | 0.08 (ns) | 4.429 | 0.004 | 3.465 |
| Protein | Transcriptomics Expression (Nephroseq; in DKD vs. Controls) [Ref.] | Protein Expression [Ref.] |
|---|---|---|
| Complement | ||
| C3 | increased (human DKD tubuli and glomeruli [23,36,37] and human tubuli ERCB) increased (mouse db/db glomeruli [35]) | increased (human T2D plasma [38], human DKD plasma [39], human T2D DKD serum [40], human T2D DKD glomeruli [41,42]) increased (rat/mouse T1D glomeruli [43,44,45,46], mouse T2D glomeruli [11], rat T2D DKD tubuli [47]) |
| IGHM | increased (human DKD tubuli and glomeruli [23], human T2D tubuli [36,37], and human tubuli ERCB) | increased (human T2D DKD glomeruli [48,49]) increased (mouse T1D DKD [44], mouse T1D DKD glomeruli [46], mouse T2D DKD glomeruli [11]) |
| Fibrosis | ||
| FN1 | increased (human DKD tubuli and glomeruli [23,34,36] and human tubuli ERCB) increased (mouse db/db glomeruli [35]) | increased (human T2D DKD glomeruli [42]) increased (mouse T2D kidney [49], mouse T2D kidney cortex [50]) |
| ENG | increased (human tubuli ERCB) decreased (human DKD glomeruli [23]) | increased (human T2D DKD kidney [51]) increased (mouse T1D DKD glomeruli [52]) |
| DYSF | increased (mouse db/db glomeruli [35]) | |
| CORO-1C | increased (human DKD glomeruli [23], human DKD glomeruli [36]) | |
| FLNA | increased (human tubuli ERCB) increased (mouse db/db glomeruli [35]) | increased (human DKD glomeruli [42]) |
| MYOF | increased (human DKD tubuli [23,34,37]—tubuli and human tubuli ERCB) decreased (human DKD glomeruli [23]) increased (mouse db/db glomeruli [35]) | |
| Inflammation | ||
| ECSIT | increased (mouse db/db glomeruli [35]) | |
| MTDH | increased (human DKD tubuli [23]) decreased (human DKD glomeruli [23], human DKD tubuli [34]) | increased (mouse db/db [53]) |
| PSMD11 | increased (glomerular mesangial cells (GMCs) under high-glucose condition [54]) | |
| MCAM | increased (human DKD tubuli [23], human DKD glomeruli [36], and human tubuli ERCB) increased (mouse db/db glomeruli [35]) | increased (human T2D DKD kidney [55] and human DKD [56]) increased (mouse db/db [56]) |
| TSPAN2 | decreased (human DKD glomeruli [23], human DKD glomeruli [36], and human tubuli ERCB) increased (mouse db/db glomeruli [35]) | |
| IFITM3 | increased (human DKD tubuli [23] and human tubuli ERCB) | |
| TGFB1i1 | increased (human DKD tubuli [23], human tubuli ERCB, mouse db/db glomeruli [37]) decreased (human DKD glomeruli [23]) | |
| CLIC4 | increased (human DKD tubuli [34]) decreased (human DKD tubuli [23]) | |
| Complement Cascade | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ins2Akita, 2 Months | Ins2Akita, 4 Months | Ins2Akita, 2 and 4 Months | |||||||
| Fibrosis-Related Proteins | C3 | C4B | IGHM | C3 | C4B | IGHM | C3 | C4B | IGHM |
| ABAT | ** | ns | *** | ns | ns | ns | * | ns | * |
| FN1 | ns | ns | ns | *** | * | ns | ns | ns | ** |
| ARF6 | ns | ns | ns | ns | * | ns | ns | ns | ns |
| STXBP1 | ns | ns | ns | ns | *** | ns | ns | ns | * |
| FLNA | ns | ns | ns | ns | * | ns | * | *** | *** |
| CYFIP1 | ns | ns | ns | ns | * | ns | ns | ns | ns |
| MYOF | * | * | ** | ns | ** | ns | * | ** | *** |
| IQGAP1 | ns | ns | ns | ns | * | ns | ns | ns | ns |
| ITGB1 | ns | ns | ns | ns | * | ns | ns | * | ** |
| ITGA1 | * | ns | ** | ns | ns | ns | ** | * | ** |
| Complement Cascade | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ins2Akita, 2 Months | Ins2Akita, 4 Months | Ins2Akita, 2 and 4 Months | |||||||
| Inflammation-Related Proteins | C3 | C4B | IGHM | C3 | C4B | IGHM | C3 | C4B | IGHM |
| CD81 | ns | ns | ns | ns | ns | ns | * | * | ** |
| ICAM1 | ns | ns | ns | ns | * | ns | ns | * | ** |
| VNN1 | * | * | ns | ns | ns | ns | * | ns | ns |
| SNAP23 | ns | ns | * | ns | ** | ns | * | * | ** |
| HSPD1 | * | ns | ** | ns | ns | ns | ns | ns | ns |
| TSPAN2 | * | * | ns | ns | * | ns | ns | ns | ns |
| Complement Cascade | ||
|---|---|---|
| Fibrosis-Related Proteins | Nephroseq Glomeruli | Nephroseq Tubuli |
| ABAT | rs = −0.791, p = 0.001 (C3, 37) | |
| FN1 | rs = 0.802, p = 0.001 (C3, 36), rs = 0.723, p = 0.003 (IGHM, 36) | rs = 0.733, p = 0.016 (IGHM, 23), rs = 0.571, p = 0.041 (C3, 34) |
| ARF6 | rs = −0.631, p = 0.016 (IGHM, 36) | rs = 0.63736, p = 0.01912 (C3, 37) |
| STXBP1 | rs = −0.569, p = 0.034 (IGHM, 36) | rs = 0.615, p = 0.025 (C3, 34) |
| rs = 0.667, p = 0.049 (C3, 23) | ||
| FLNA | rs = 0.571, p = 0.041 (C3, 37) | |
| CYFIP1 | rs = 0.78, p = 0.002 (C3, 37), rs = 0.745, p = 0.013 (C3, 23) | |
| MYOF | rs = 0.747, p = 0.003 (C3, 37) | |
| IQGAP1 | rs = 0.758, p = 0.003 (C3, 37), rs = 0.758, p = 0.011 (C3, 18) | |
| ITGB1 | rs = 0.906, p = 2 × 10−5 (C3, 34) | |
| Complement Cascade | ||
|---|---|---|
| Inflammation-Related Proteins | Nephroseq Glomeruli | Nephroseq Tubuli |
| CD81 | rs = −0.556, p = 0.039 (C3, 36) | rs = 0.852, p = 0.00022 (C3, 37),rs = 0.555, p = 0.049 (IGHM, 37), rs = 0.842, p = 0.0003 (C3, 34) |
| ICAM1 | rs = 0.56, p = 0.046 (C3, 37), rs = 0.692, p = 0.009 (C3, 34) | |
| VNN1 | rs = −0.632, p = 0.02 (C3, 37) | |
| TSPAN2 | rs = −0.543, p = 0.044 (C3, 36) | |
| SNAP23 | rs = 0.733, p = 0.016 (C3, 23) | |
| Glomerular C3 Deposition | Glomerular C1q Deposition | Glomerular C4 Deposition | Glomerular IgM Deposition | |
|---|---|---|---|---|
| DKD class | ||||
| I | 0/1 | 0/1 | 0/1 | 1/1 |
| II | 3/17 | 0/17 | 0/17 | 3/17 |
| III | 14/19 | 3/19 | 3/19 | 9/19 |
| IV | 5/6 | 2/6 | 0/6 | 5/6 |
| Fibrosis | rs = 0.343, p = 0.024 | rs = 0.379, p = 0.012 | rs = 0.21264, p = 0.171 | rs = 0.17478, p = 0.2623 |
| eGFR | rs = −0.05, p = 0.7597 | rs = −0.348, p = 0.03 | rs = −0.154, p = 0.349 | rs = −0.118, p = 0.472 |
| IFTA | rs = 0.337, p = 0.027 | rs = 0.245, p = 0.113 | rs = −0.028, p = 0.858 | rs = 0.082, p = 0.598 |
| GS | rs = 0.422, p = 0.0048 | rs = 0.163, p = 0.294 | rs = −0.018, p = 0.907 | rs = 0.174, p = 0.264 |
| Tubular atrophy | rs = 0.399, p = 0.008 | rs = 0.282, p = 0.067 | rs = 0.089, p = 0.567 | rs = 0.095, p = 0.543 |
| Study/Set/Reference | Group |
|---|---|
| Schmid Diabetes/Tubulointerstitium/[34] | DKD vs. Pretransplant kidney donors |
| Hodgin Diabetes Mouse/Glomerulus/[35] | DKD mouse model vs. Non-DKD |
| Ju CKD/Glomerulus/[36] | DKD human vs. Healthy |
| Ju CKD/Tubulointerstitium/[37] | DKD human vs. Healthy |
| Woroniecka Diabetes/Tubulointerstitium/[23] | DKD human vs. Healthy |
| Woroniecka Diabetes/Glomerulus/[23] | DKD human vs. Healthy |
| ERCB Nephrotic Syndrome/Tubulointerstitium (unpublished study; there are data available only in Nephroseq) | DKD human vs. Healthy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tserga, A.; Saulnier-Blache, J.S.; Palamaris, K.; Pouloudi, D.; Gakiopoulou, H.; Zoidakis, J.; Schanstra, J.P.; Vlahou, A.; Makridakis, M. Complement Cascade Proteins Correlate with Fibrosis and Inflammation in Early-Stage Type 1 Diabetic Kidney Disease in the Ins2Akita Mouse Model. Int. J. Mol. Sci. 2024, 25, 1387. https://doi.org/10.3390/ijms25031387
Tserga A, Saulnier-Blache JS, Palamaris K, Pouloudi D, Gakiopoulou H, Zoidakis J, Schanstra JP, Vlahou A, Makridakis M. Complement Cascade Proteins Correlate with Fibrosis and Inflammation in Early-Stage Type 1 Diabetic Kidney Disease in the Ins2Akita Mouse Model. International Journal of Molecular Sciences. 2024; 25(3):1387. https://doi.org/10.3390/ijms25031387
Chicago/Turabian StyleTserga, Aggeliki, Jean Sébastien Saulnier-Blache, Kostantinos Palamaris, Despoina Pouloudi, Harikleia Gakiopoulou, Jerome Zoidakis, Joost Peter Schanstra, Antonia Vlahou, and Manousos Makridakis. 2024. "Complement Cascade Proteins Correlate with Fibrosis and Inflammation in Early-Stage Type 1 Diabetic Kidney Disease in the Ins2Akita Mouse Model" International Journal of Molecular Sciences 25, no. 3: 1387. https://doi.org/10.3390/ijms25031387