

A Narrative Review: The Role of NETs in Acute Respiratory Distress Syndrome/Acute Lung Injury

Abstract
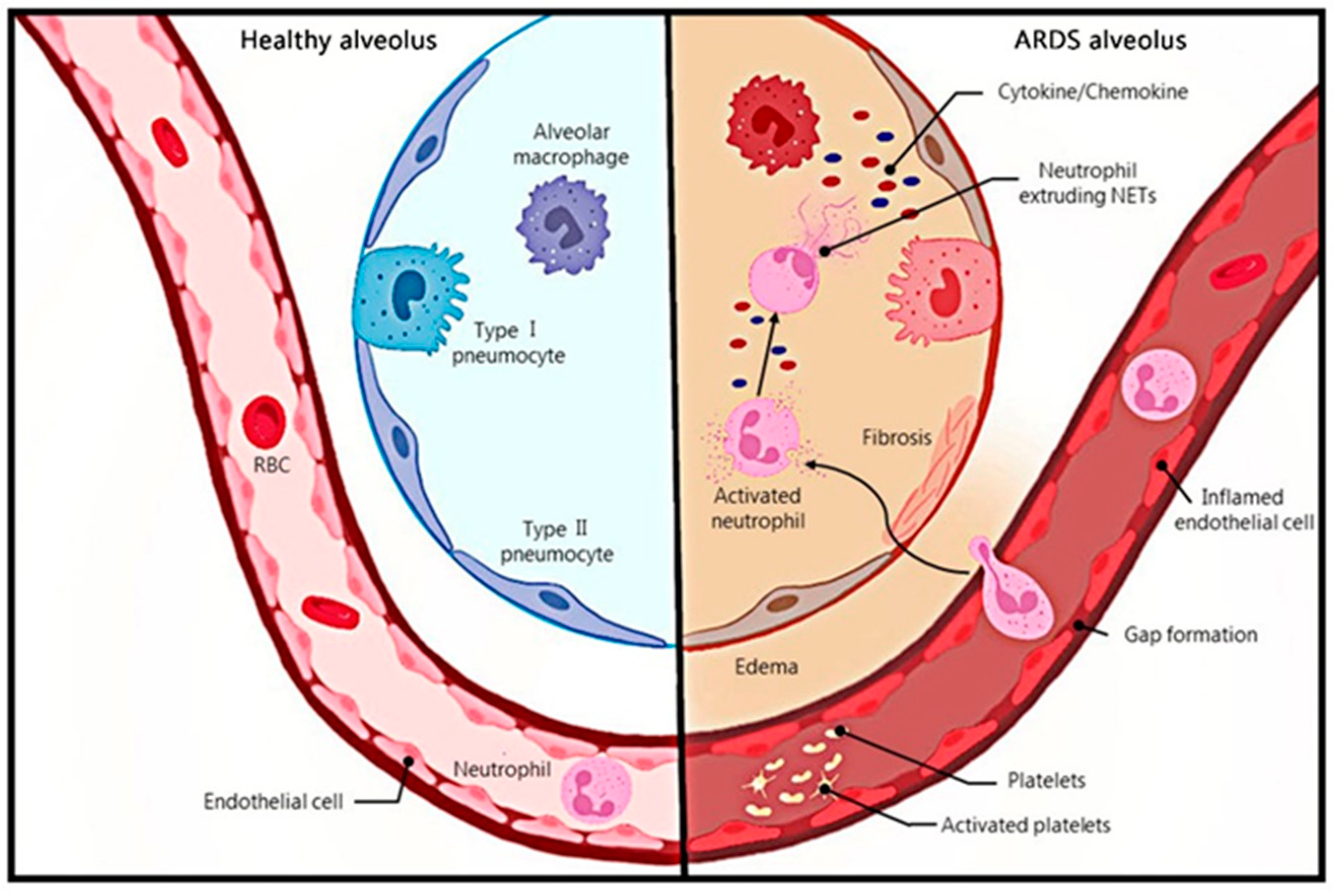
1. Introduction
2. Evidence on the Influence of NETs on ARDS
3. The Mechanism of NETs Influencing ARDS
3.1. Histones
3.2. NE
3.3. DNA
3.4. MPO
4. The Mechanism of NETs Production in ARDS and the Substances That Affect the Production

{kind=link}
{kind=link}
| Correlates | Effect | References | |
|---|---|---|---|
| PAD4 | Mediates histone citrullination, be involved in NF-κB activation and IL-1β release, PAD4 inhibitors reduce the formation of NETs, and PAD4 enzymes promote the production of NETs. | [35,36,79,80,82] | |
| ROS | Participates in classic NETosis, and triggers the separation, release, and migration of NE and MPO to the neutrophil nucleus. | [9,77,84] | |
| Platelets | Activate neutrophils via TLR4 to release NETs. | [29,31] | |
| Signaling pathways | Raf-MEK-ERK pathway | Activates NADPH oxidase and upregulates anti-apoptotic proteins, promoting the formation of NETs. | [30] |
| Lipoxin pathway | Regulates the calcium flux of neutrophils, participates in citrullination and ROS production, and regulates neutrophil apoptosis and promotes NET formation. | [20] | |
| NF-κB/CXCR1 pathway | Activation promotes the formation of NETs. | [85] | |
| Cytokines and others | IL-8 | Triggers the cleavage of gasdermin D by serine proteases, leading to pore formation and NET formation, the content was correlated with the concentration of NETs in BALF. | [82,91] |
| Gasdermin D (GSDMD) | Forms pores in the granular and plasma membranes, allowing NE to enter the cytoplasm and NETs to be released outside the neutrophils. | [90] | |
| IL17A | Induces NET production through STAT3 activation with other cofactors. | [92] | |
| Integrins and G-protein coupled receptors | Necessary for NET formation during the VILI process. | [31] | |
| CCL5/CXCL4 heterodimers | Blocking inhibits the formation of NETs. | [31] | |
| Complement | Inhibition of activation reduces the release of NETs. | [93] | |
| miR144, miR155 | Promotes NET formation. | [81,85] | |
| PD-L1 | Inhibits autophagy of neutrophils and maintain the release of NETs through the PI3K/Akt/mTOR pathway. | [94] | |
| Interferon inducer poly I:C | Induces the formation of NETs via activation of P38 MAPK and decreased expression of the tight-linking protein claudin-5. | [95] | |
5. Relationship between the Inflammatory Storm and NETs
6. Correlation between Immune Balance and Regulation in ARDS and NETs
7. Production and Role of NETs in COVID-19 ARDS
8. Clinical Research
9. Treatment
| Interventions | Effect | References | |
|---|---|---|---|
| Clinical trial | Dnase Ⅰ | Removes DNA components from NETs, degrades NETs, and reduces NET deposition. | [106] |
| Untravenous vitamin C | Inhibits NETosis, unspecified. 1 | [129] | |
| Observational Study | HMGB1 inhibitors, metformin | Improve efferocytosis and NET clearance. | [16] |
| Laboratory research | DNase I | Removes DNA components from NETs, degrades NETs, and reduces NET deposition. | [20,26,28,31,95,132,133] |
| PAD4 inhibitors | Reduce histone citrullination and thus block NETosis. | [79,135] | |
| NE inhibitors | Inhibit the formation of NETs and reduce cytotoxicity. | [26,31,62] | |
| HMGB1 inhibitors | Block the signaling pathway and inhibit the formation and release of NETs. | [36] | |
| anti-PD-L1 antibodies | [94] | ||
| inhibitors of p38 MAPK kinase | [95] | ||
| Osteopontin (OPN) | Phosphorylation binds to histones with high affinity, thereby reducing cytotoxicity and reducing the formation of NETs. | [142] | |
| Protectin D1 (PD1) | Inhibit NETosis, unspecified. | [139] | |
| Deferasirox | [140] | ||
| Mesenchymal stem cells (MSCs) | [141] | ||
| Colchicine | [143] | ||
| Disulfiram | [144] | ||
| Methoxyeugenol | [145] | ||
| Anti-CLEC5A | [146] | ||
| Selinexor | [147] | ||
| Stimulation of the ear vagus nerve | [148] | ||
10. Future Directions
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meyer, N.J.; Gattinoni, L.; Calfee, C.S. Acute respiratory distress syndrome. Lancet 2021, 398, 622–637. [Google Scholar] [CrossRef]
- ARDS Definition of Task Force; Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute Respiratory Distress Syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar] [CrossRef]
- Gorman, E.A.; O’kane, C.M.; McAuley, D.F. Acute respiratory distress syndrome in adults: Diagnosis, outcomes, long-term sequelae, and management. Lancet 2022, 400, 1157–1170. [Google Scholar] [CrossRef] [PubMed]
- Bos, L.D.J.; Ware, L.B. Acute respiratory distress syndrome: Causes, pathophysiology, and phenotypes. Lancet 2022, 400, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; Van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800, Erratum in JAMA 2016, 316, 350. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, K.P.; A Milberg, J.; Martin, T.R.; Maunder, R.J.; A Cockrill, B.; Hudson, L.D. Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1994, 150, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The Multifaceted Functions of Neutrophils. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 181–218. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Jo, A.; Kim, D.W. Neutrophil Extracellular Traps in Airway Diseases: Pathological Roles and Therapeutic Implications. Int. J. Mol. Sci. 2023, 24, 5034. [Google Scholar] [CrossRef]
- Liu, L.; Mao, Y.; Xu, B.; Zhang, X.; Fang, C.; Ma, Y.; Men, K.; Qi, X.; Yi, T.; Wei, Y.; et al. Induction of neutrophil extracellular traps during tissue injury: Involvement of STING and Toll-like receptor 9 pathways. Cell Prolif. 2019, 52, e12579, Erratum in Cell Prolif. 2020, 53, e12775. [Google Scholar] [CrossRef]
- Ronchetti, L.; Boubaker, N.S.; Barba, M.; Vici, P.; Gurtner, A.; Piaggio, G. Neutrophil extracellular traps in cancer: Not only catching microbes. J. Exp. Clin. Cancer Res. 2021, 40, 231. [Google Scholar] [CrossRef]
- Twaddell, S.H.; Baines, K.J.; Grainge, C.; Gibson, P.G. The Emerging Role of Neutrophil Extracellular Traps in Respiratory Disease. Chest 2019, 156, 774–782. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2017, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-C.; Tsai, Y.-F.; Pan, Y.-L.; Hwang, T.-L. Understanding the role of neutrophils in acute respiratory distress syndrome. Biomed. J. 2020, 44, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Scozzi, D.; Liao, F.; Krupnick, A.S.; Kreisel, D.; Gelman, A.E. The role of neutrophil extracellular traps in acute lung injury. Front. Immunol. 2022, 13, 953195. [Google Scholar] [CrossRef] [PubMed]
- Grégoire, M.; Uhel, F.; Lesouhaitier, M.; Gacouin, A.; Guirriec, M.; Mourcin, F.; Dumontet, E.; Chalin, A.; Samson, M.; Berthelot, L.-L.; et al. Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. Eur. Respir. J. 2018, 52, 1702590. [Google Scholar] [CrossRef] [PubMed]
- Ojima, M.; Yamamoto, N.; Hirose, T.; Hamaguchi, S.; Tasaki, O.; Kojima, T.; Tomono, K.; Ogura, H.; Shimazu, T. Serial change of neutrophil extracellular traps in tracheal aspirate of patients with acute respiratory distress syndrome: Report of three cases. J. Intensiv. Care 2020, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Li, H.; Mao, Z.; Peng, L.; Liu, B.; Lin, F.; Li, Y.; Dai, M.; Cui, Y.; Zhao, Y.; et al. Delayed neutrophil apoptosis may enhance NET formation in ARDS. Respir. Res. 2022, 23, 155. [Google Scholar] [CrossRef]
- Grunwell, J.R.; Stephenson, S.T.; Mohammad, A.F.; Jones, K.; Mason, C.; Opolka, C.; Fitzpatrick, A.M. Differential type I interferon response and primary airway neutrophil extracellular trap release in children with acute respiratory distress syndrome. Sci. Rep. 2020, 10, 19049. [Google Scholar] [CrossRef]
- Lefrançais, E.; Mallavia, B.; Zhuo, H.; Calfee, C.S.; Looney, M.R. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. J. Clin. Investig. 2018, 3, e98178. [Google Scholar] [CrossRef]
- Ode, Y.; Aziz, M.; Jin, H.; Arif, A.; Nicastro, J.G.; Wang, P. Cold-inducible RNA-binding Protein Induces Neutrophil Extracellular Traps in the Lungs during Sepsis. Sci. Rep. 2019, 9, 6252. [Google Scholar] [CrossRef]
- Song, C.; Li, H.; Li, Y.; Dai, M.; Zhang, L.; Liu, S.; Tan, H.; Deng, P.; Liu, J.; Mao, Z.; et al. NETs promote ALI/ARDS inflammation by regulating alveolar macrophage polarization. Exp. Cell Res. 2019, 382, 111486. [Google Scholar] [CrossRef]
- Li, H.; Li, Y.; Song, C.; Hu, Y.; Dai, M.; Liu, B.; Pan, P. Neutrophil Extracellular Traps Augmented Alveolar Macrophage Pyroptosis via AIM2 Inflammasome Activation in LPS-Induced ALI/ARDS. J. Inflamm. Res. 2021, 14, 4839–4858. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, J.; Zhou, Y.; Qu, M.; Wang, Y.; Guo, K.; Shen, R.; Sun, Z.; Cata, J.P.; Yang, S.; et al. Neutrophil extracellular traps mediate m6A modification and regulates sepsis-associated acute lung injury by activating ferroptosis in alveolar epithelial cells. Int. J. Biol. Sci. 2022, 18, 3337–3357. [Google Scholar] [CrossRef] [PubMed]
- Sercundes, M.K.; Ortolan, L.S.; Debone, D.; Soeiro-Pereira, P.V.; Gomes, E.; Aitken, E.H.; Neto, A.C.; Russo, M.; Lima, M.R.D.I.; Alvarez, J.M.; et al. Targeting Neutrophils to Prevent Malaria-Associated Acute Lung Injury/Acute Respiratory Distress Syndrome in Mice. PLoS Pathog. 2016, 12, e1006054, Erratum in PLoS Pathog. 2017, 13, e1006730. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Ding, W.; Hao, Y.; Cen, L.; Li, J.; Shi, X.; Wang, T.; Chen, D.; Zhu, H. Neutrophil extracellular traps mediate severe lung injury induced by influenza A virus H1N1 in mice coinfected with Staphylococcus aureus. Microb. Pathog. 2022, 166, 105558. [Google Scholar] [CrossRef]
- Toy, P.; Lowell, C. TRALI—Definition, mechanisms, incidence and clinical relevance. Best Pract. Res. Clin. Anaesthesiol. 2007, 21, 183–193. [Google Scholar] [CrossRef]
- Thomas, G.M.; Carbo, C.; Curtis, B.R.; Martinod, K.; Mazo, I.B.; Schatzberg, D.; Cifuni, S.M.; Fuchs, T.A.; von Andrian, U.H.; Hartwig, J.H.; et al. Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood 2012, 119, 6335–6343. [Google Scholar] [CrossRef] [PubMed]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef]
- Hakkim, A.; Fuchs, T.A.; Martinez, N.E.; Hess, S.; Prinz, H.; Zychlinsky, A.; Waldmann, H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2011, 7, 75–77. [Google Scholar] [CrossRef]
- Rossaint, J.; Herter, J.M.; Van Aken, H.; Napirei, M.; Döring, Y.; Weber, C.; Soehnlein, O.; Zarbock, A. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap–mediated sterile inflammation. Blood J. Am. Soc. Hematol. 2014, 123, 2573–2584. [Google Scholar] [CrossRef]
- Yildiz, C.; Palaniyar, N.; Otulakowski, G.; Khan, M.A.; Post, M.; Kuebler, W.M.; Tanswell, K.; Belcastro, R.; Masood, A.; Engelberts, D.; et al. Mechanical Ventilation Induces Neutrophil Extracellular Trap Formation. Anesthesiology 2015, 122, 864–875. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Pan, P.; Su, X.; Liu, S.; Zhang, L.; Wu, D.; Li, H.; Dai, M.; Li, Y.; Hu, C.; et al. Neutrophil Extracellular Traps Are Pathogenic in Ventilator-Induced Lung Injury and Partially Dependent on TLR4. BioMed Res. Int. 2017, 2017, 8272504. [Google Scholar] [CrossRef]
- Hayase, N.; Doi, K.; Hiruma, T.; Matsuura, R.; Hamasaki, Y.; Noiri, E.; Nangaku, M.; Morimura, N. Recombinant thrombomodulin prevents acute lung injury induced by renal ischemia-reperfusion injury. Sci. Rep. 2020, 10, 289. [Google Scholar] [CrossRef]
- Surolia, R.; Li, F.J.; Wang, Z.; Kashyap, M.; Srivastava, R.K.; Traylor, A.M.; Singh, P.; Dsouza, K.G.; Kim, H.; Pittet, J.-F.; et al. NETosis in the pathogenesis of acute lung injury following cutaneous chemical burns. J. Clin. Investig. 2021, 6, e147564. [Google Scholar] [CrossRef]
- Zhan, Y.; Ling, Y.; Deng, Q.; Qiu, Y.; Shen, J.; Lai, H.; Chen, Z.; Huang, C.; Liang, L.; Li, X.; et al. HMGB1-Mediated Neutrophil Extracellular Trap Formation Exacerbates Intestinal Ischemia/Reperfusion-Induced Acute Lung Injury. J. Immunol. 2022, 208, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil Extracellular Traps Directly Induce Epithelial and Endothelial Cell Death: A Predominant Role of Histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil Extracellular Traps Contain Calprotectin, a Cytosolic Protein Complex Involved in Host Defense against Candida albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef]
- Hoeksema, M.; van Eijk, M.; Haagsman, H.P.; Hartshorn, K.L. Histones as mediators of host defense, inflammation and thrombosis. Futur. Microbiol. 2016, 11, 441–453. [Google Scholar] [CrossRef]
- Sørensen, O.E.; Borregaard, N. Neutrophil extracellular traps—The dark side of neutrophils. J. Clin. Investig. 2016, 126, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, A.S.; Slade, D.J.; Thompson, P.R.; Mowen, K.A. Activation of PAD4 in NET formation. Front. Immunol. 2012, 3, 360. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Neeli, I.; Khan, S.N.; Radic, M. Histone Deimination As a Response to Inflammatory Stimuli in Neutrophils. J. Immunol. 2008, 180, 1895–1902. [Google Scholar] [CrossRef]
- Tatsiy, O.; McDonald, P.P. Physiological Stimuli Induce PAD4-Dependent, ROS-Independent NETosis, With Early and Late Events Controlled by Discrete Signaling Pathways. Front. Immunol. 2018, 9, 2036. [Google Scholar] [CrossRef]
- Biron, B.M.; Chung, C.-S.; O’Brien, X.M.; Chen, Y.; Reichner, J.S.; Ayala, A. Cl-Amidine Prevents Histone 3 Citrullination and Neutrophil Extracellular Trap Formation, and Improves Survival in a Murine Sepsis Model. J. Innate Immun. 2016, 9, 22–32. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Jenne, C.N.; Surewaard, B.G.J.; Thanabalasuriar, A.; Lee, W.-Y.; Sanz, M.-J.; Mowen, K.; Opdenakker, G.; Kubes, P. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat. Commun. 2015, 6, 6673. [Google Scholar] [CrossRef]
- Guiducci, E.; Lemberg, C.; Küng, N.; Schraner, E.; Theocharides, A.P.A.; LeibundGut-Landmann, S. Candida albicans-Induced NETosis Is Independent of Peptidylarginine Deiminase 4. Front. Immunol. 2018, 9, 1573. [Google Scholar] [CrossRef] [PubMed]
- Claushuis, T.A.M.; van der Donk, L.E.H.; Luitse, A.L.; van Veen, H.A.; van der Wel, N.N.; van Vught, L.A.; Roelofs, J.J.T.H.; de Boer, O.J.; Lankelma, J.M.; Boon, L.; et al. Role of Peptidylarginine Deiminase 4 in Neutrophil Extracellular Trap Formation and Host Defense during Klebsiella pneumoniae–Induced Pneumonia-Derived Sepsis. J. Immunol. 2018, 201, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Abrams, S.T.; Zhang, N.; Manson, J.; Liu, T.; Dart, C.; Baluwa, F.; Wang, S.S.; Brohi, K.; Kipar, A.; Yu, W.; et al. Circulating Histones Are Mediators of Trauma-associated Lung Injury. Am. J. Respir. Crit. Care Med. 2013, 187, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Bhandari, A.A.; Wagner, D.D. Histones induce rapid and profound thrombocytopenia in mice. Blood 2011, 118, 3708–3714. [Google Scholar] [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef]
- Huang, H.; Chen, H.-W.; Evankovich, J.; Yan, W.; Rosborough, B.R.; Nace, G.W.; Ding, Q.; Loughran, P.; Beer-Stolz, D.; Billiar, T.R.; et al. Histones Activate the NLRP3 Inflammasome in Kupffer Cells during Sterile Inflammatory Liver Injury. J. Immunol. 2013, 191, 2665–2679. [Google Scholar] [CrossRef]
- O’donoghue, A.J.; Jin, Y.; Knudsen, G.M.; Perera, N.C.; Jenne, D.E.; Murphy, J.E.; Craik, C.S.; Hermiston, T.W. Global Substrate Profiling of Proteases in Human Neutrophil Extracellular Traps Reveals Consensus Motif Predominantly Contributed by Elastase. PLoS ONE 2013, 8, e75141. [Google Scholar] [CrossRef]
- Suzuki, K.; Okada, H.; Takemura, G.; Takada, C.; Kuroda, A.; Yano, H.; Zaikokuji, R.; Morishita, K.; Tomita, H.; Oda, K.; et al. Neutrophil Elastase Damages the Pulmonary Endothelial Glycocalyx in Lipopolysaccharide-Induced Experimental Endotoxemia. Am. J. Pathol. 2019, 189, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Perl, M.; Lomas-Neira, J.; Chung, C.-S.; Ayala, A. Epithelial Cell Apoptosis and Neutrophil Recruitment in Acute Lung Injury—A Unifying Hypothesis? What We Have Learned from Small Interfering RNAs. Mol. Med. 2008, 14, 465–475. [Google Scholar] [CrossRef]
- Frantzeskaki, F.; Armaganidis, A.; E Orfanos, S. Immunothrombosis in Acute Respiratory Distress Syndrome: Cross Talks between Inflammation and Coagulation. Respiration 2017, 93, 212–225. [Google Scholar] [CrossRef]
- Hermant, B.; Bibert, S.; Concord, E.; Dublet, B.; Weidenhaupt, M.; Vernet, T.; Gulino-Debrac, D. Identification of Proteases Involved in the Proteolysis of Vascular Endothelium Cadherin during Neutrophil Transmigration. J. Biol. Chem. 2003, 278, 14002–14012. [Google Scholar] [CrossRef] [PubMed]
- Okeke, E.B.; Louttit, C.; Fry, C.; Najafabadi, A.H.; Han, K.; Nemzek, J.; Moon, J.J. Inhibition of neutrophil elastase prevents neutrophil extracellular trap formation and rescues mice from endotoxic shock. Biomaterials 2020, 238, 119836. [Google Scholar] [CrossRef] [PubMed]
- Martinod, K.; Witsch, T.; Farley, K.; Gallant, M.; Remold-O’Donnell, E.; Wagner, D.D. Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J. Thromb. Haemost. 2016, 14, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed]
- Gould, T.J.; Vu, T.T.; Swystun, L.L.; Dwivedi, D.J.; Mai, S.H.; Weitz, J.I.; Liaw, P.C. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arter. Thromb. Vasc. Biol. 2014, 34, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wu, Z.; Long, Q.; Huang, J.; Hong, T.; Liu, W.; Lin, J. Insights Into Immunothrombosis: The Interplay among Neutrophil Extracellular Trap, von Willebrand Factor, and ADAMTS13. Front. Immunol. 2020, 11, 610696. [Google Scholar] [CrossRef]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef]
- McIlroy, D.J.; Jarnicki, A.G.; Au, G.G.; Lott, N.; Smith, D.W.; Hansbro, P.M.; Balogh, Z.J. Mitochondrial DNA neutrophil extracellular traps are formed after trauma and subsequent surgery. J. Crit. Care 2014, 29, 1133.e1–1133.e5. [Google Scholar] [CrossRef]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Itagaki, K.; Hauser, C.J. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock 2010, 34, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Mallavia, B.; Liu, F.; Lefrançais, E.; Cleary, S.J.; Kwaan, N.; Tian, J.J.; Magnen, M.; Sayah, D.M.; Soong, A.; Chen, J.; et al. Mitochondrial DNA Stimulates TLR9-Dependent Neutrophil Extracellular Trap Formation in Primary Graft Dysfunction. Am. J. Respir. Cell Mol. Biol. 2020, 62, 364–372. [Google Scholar] [CrossRef]
- Itagaki, K.; Kaczmarek, E.; Lee, Y.T.; Tang, I.T.; Isal, B.; Adibnia, Y.; Sandler, N.; Grimm, M.J.; Segal, B.H.; Otterbein, L.E.; et al. Mitochondrial DNA Released by Trauma Induces Neutrophil Extracellular Traps. PLoS ONE 2015, 10, e0120549. [Google Scholar] [CrossRef]
- Nishinaka, Y.; Arai, T.; Adachi, S.; Takaori-Kondo, A.; Yamashita, K. Singlet oxygen is essential for neutrophil extracellular trap formation. Biochem. Biophys. Res. Commun. 2011, 413, 75–79. [Google Scholar] [CrossRef]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A.; Metzler, K.D.; et al. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chen, H.; Yang, Z.-T.; Mao, E.-Q.; Chen, Y.; Chen, E.-Z. Free fatty acids-induced neutrophil extracellular traps lead to dendritic cells activation and T cell differentiation in acute lung injury. Aging 2021, 13, 26148–26160. [Google Scholar] [CrossRef]
- Szturmowicz, M.; Demkow, U. Neutrophil Extracellular Traps (NETs) in Severe SARS-CoV-2 Lung Disease. Int. J. Mol. Sci. 2021, 22, 8854. [Google Scholar] [CrossRef] [PubMed]
- Hamam, H.J.; Palaniyar, N. Post-Translational Modifications in NETosis and NETs-Mediated Diseases. Biomolecules 2019, 9, 369. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Pan, B.; Alam, H.B.; Deng, Q.; Wang, Y.; Chen, E.; Liu, B.; Tian, Y.; Williams, A.M.; Duan, X.; et al. Inhibition of peptidylarginine deiminase alleviates LPS-induced pulmonary dysfunction and improves survival in a mouse model of lethal endotoxemia. Eur. J. Pharmacol. 2018, 833, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Burgener, S.S.; Schroder, K. Neutrophil Extracellular Traps in Host Defense. Cold Spring Harb. Perspect. Biol. 2020, 12, a037028. [Google Scholar] [CrossRef] [PubMed]
- Hawez, A.; Taha, D.; Algaber, A.; Madhi, R.; Rahman, M.; Thorlacius, H. MiR-155 regulates neutrophil extracellular trap formation and lung injury in abdominal sepsis. J. Leukoc. Biol. 2022, 111, 391–400. [Google Scholar] [CrossRef]
- Damascena, H.L.; Silveira, W.A.A.; Castro, M.S.; Fontes, W. Neutrophil Activated by the Famous and Potent PMA (Phorbol Myristate Acetate). Cells 2022, 11, 2889. [Google Scholar] [CrossRef]
- Poli, V.; Ma, V.P.-Y.; Di Gioia, M.; Broggi, A.; Benamar, M.; Chen, Q.; Mazitschek, R.; Haggarty, S.J.; Chatila, T.A.; Karp, J.M.; et al. Zinc-dependent histone deacetylases drive neutrophil extracellular trap formation and potentiate local and systemic inflammation. iScience 2021, 24, 103256. [Google Scholar] [CrossRef]
- Liu, S.; Yue, Y.; Pan, P.; Zhang, L.; Su, X.; Li, H.; Li, H.; Li, Y.; Dai, M.; Li, Q.; et al. IRF-1 Intervention in the Classical ROS-Dependent Release of NETs during LPS-Induced Acute Lung Injury in Mice. Inflammation 2018, 42, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Wu, Y.; Liu, W.; Wu, C.; Hu, P.; Zou, J.; Kuang, L. MiR-144-induced KLF2 inhibition and NF-kappaB/CXCR1 activation promote neutrophil extracellular trap–induced transfusion-related acute lung injury. J. Cell. Mol. Med. 2021, 25, 6511–6523. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-M.; Lu, H.-C.; Tung, Y.-T.; Chen, W. Antiplatelet Therapy for Acute Respiratory Distress Syndrome. Biomedicines 2020, 8, 230. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Wagner, D.D. Neutrophil Extracellular Trap (NET) Impact on Deep Vein Thrombosis. Arter. Thromb. Vasc. Biol. 2012, 32, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Burkard, P.; Schonhart, C.; Vögtle, T.; Köhler, D.; Tang, L.; Johnson, D.; Hemmen, K.; Heinze, K.G.; Zarbock, A.; Hermanns, H.M.; et al. A key role for platelet GPVI in neutrophil recruitment, migration and NETosis in the early stages of acute lung injury. Blood 2023, 142, 1463–1477. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef]
- Bendib, I.; de Chaisemartin, L.; Granger, V.; Schlemmer, F.; Maitre, B.; Hüe, S.; Surenaud, M.; Beldi-Ferchiou, A.; Carteaux, G.; Razazi, K.; et al. Neutrophil Extracellular Traps Are Elevated in Patients with Pneumonia-related Acute Respiratory Distress Syndrome. Anesthesiology 2019, 130, 581–591. [Google Scholar] [CrossRef]
- Li, Y.; Shen, Y.; Lin, D.; Zhang, H.; Wang, T.; Liu, H.; Wang, Y. Neutrophils and IL17A mediate flagellar hook protein FlgE-induced mouse acute lung inflammation. Cell. Microbiol. 2019, 21, e12975. [Google Scholar] [CrossRef]
- Cleary, S.J.; Kwaan, N.; Tian, J.J.; Calabrese, D.R.; Mallavia, B.; Magnen, M.; Greenland, J.R.; Urisman, A.; Singer, J.P.; Hays, S.R.; et al. Complement activation on endothelium initiates antibody-mediated acute lung injury. J. Clin. Investig. 2020, 130, 5909–5923. [Google Scholar] [CrossRef]
- Zhu, C.-L.; Xie, J.; Zhao, Z.-Z.; Li, P.; Liu, Q.; Guo, Y.; Meng, Y.; Wan, X.-J.; Bian, J.-J.; Deng, X.-M.; et al. PD-L1 maintains neutrophil extracellular traps release by inhibiting neutrophil autophagy in endotoxin-induced lung injury. Front. Immunol. 2022, 13, 949217, Erratum in Front. Immunol. 2022, 13, 1038083. [Google Scholar] [CrossRef]
- Gan, T.; Yang, Y.; Hu, F.; Chen, X.; Zhou, J.; Li, Y.; Xu, Y.; Wang, H.; Chen, Y.; Zhang, M. TLR3 Regulated Poly I:C-Induced Neutrophil Extracellular Traps and Acute Lung Injury Partly through p38 MAP Kinase. Front. Microbiol. 2018, 9, 3174. [Google Scholar] [CrossRef]
- Yang, L.; Xie, X.; Tu, Z.; Fu, J.; Xu, D.; Zhou, Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct. Target. Ther. 2021, 6, 255, Erratum in Signal Transduct. Target. Ther. 2021, 6, 326. [Google Scholar] [CrossRef]
- Montazersaheb, S.; Khatibi, S.M.H.; Hejazi, M.S.; Tarhriz, V.; Farjami, A.; Sorbeni, F.G.; Farahzadi, R.; Ghasemnejad, T. COVID-19 infection: An overview on cytokine storm and related interventions. Virol. J. 2022, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef] [PubMed]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Santana, E.; Ninan, J.R.; Snyder, C.M.; Okeke, E.B. Neutrophil Extracellular Traps, Sepsis and COVID-19—A Tripod Stand. Front. Immunol. 2022, 13, 902206. [Google Scholar] [CrossRef] [PubMed]
- Blanch-Ruiz, M.A.; Ortega-Luna, R.; Gómez-García, G.; Martínez-Cuesta, M.Á.; Álvarez, Á. Role of Neutrophil Extracellular Traps in COVID-19 Progression: An Insight for Effective Treatment. Biomedicines 2021, 10, 31. [Google Scholar] [CrossRef]
- Obermayer, A.; Jakob, L.-M.; Haslbauer, J.D.; Matter, M.S.; Tzankov, A.; Stoiber, W. Neutrophil Extracellular Traps in Fatal COVID-19-Associated Lung Injury. Dis. Markers 2021, 2021, 5566826. [Google Scholar] [CrossRef]
- Rudd, J.M.; Pulavendran, S.; Ashar, H.K.; Ritchey, J.W.; Snider, T.A.; Malayer, J.R.; Marie, M.; Chow, V.T.K.; Narasaraju, T. Neutrophils Induce a Novel Chemokine Receptors Repertoire during Influenza Pneumonia. Front. Cell. Infect. Microbiol. 2019, 9, 108. [Google Scholar] [CrossRef]
- Tomar, B.; Anders, H.-J.; Desai, J.; Mulay, S.R. Neutrophils and Neutrophil Extracellular Traps Drive Necroinflammation in COVID-19. Cells 2020, 9, 1383. [Google Scholar] [CrossRef] [PubMed]
- Silva, B.M.; Gomes, G.F.; Veras, F.P.; Cambier, S.; Silva, G.V.; Quadros, A.U.; Caetité, D.B.; Nascimento, D.C.; Silva, C.M.; Silva, J.C.C.; et al. C5aR1 signaling triggers lung immunopathology in COVID-19 through neutrophil extracellular traps. J. Clin. Investig. 2023, 133, e163105. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Park, H.H.; Park, W.; Kim, H.; Jang, J.G.; Hong, K.S.; Lee, J.-Y.; Seo, H.S.; Na, D.H.; Kim, T.-H.; et al. Long-acting nanoparticulate DNase-1 for effective suppression of SARS-CoV-2-mediated neutrophil activities and cytokine storm. Biomaterials 2020, 267, 120389. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Xi, L.; Liu, Y.; Mak, J.C.W.; Mao, S.; Wang, Z.; Zheng, Y. An Inhalable Hybrid Biomimetic Nanoplatform for Sequential Drug Release and Remodeling Lung Immune Homeostasis in Acute Lung Injury Treatment. ACS Nano 2023, 17, 11626–11644. [Google Scholar] [CrossRef] [PubMed]
- McKenna, E.; Wubben, R.; Isaza-Correa, J.M.; Melo, A.M.; Mhaonaigh, A.U.; Conlon, N.; O’donnell, J.S.; Cheallaigh, C.N.; Hurley, T.; Stevenson, N.J.; et al. Neutrophils in COVID-19: Not Innocent Bystanders. Front. Immunol. 2022, 13, 864387. [Google Scholar] [CrossRef]
- Panda, R.; Castanheira, F.V.; Schlechte, J.M.; Surewaard, B.G.; Shim, H.B.; Zucoloto, A.Z.; Slavikova, Z.; Yipp, B.G.; Kubes, P.; McDonald, B. A functionally distinct neutrophil landscape in severe COVID-19 reveals opportunities for adjunctive therapies. J. Clin. Investig. 2021, 7, e152291. [Google Scholar] [CrossRef] [PubMed]
- Florence, J.M.; Krupa, A.; Booshehri, L.M.; Davis, S.A.; Matthay, M.A.; Kurdowska, A.K. Inhibiting Bruton’s tyrosine kinase rescues mice from lethal influenza-induced acute lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2018, 315, L52–L58. [Google Scholar] [CrossRef]
- Pedersen, S.F.; Ho, Y.-C. SARS-CoV-2: A storm is raging. J. Clin. Investig. 2020, 130, 2202–2205. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.N.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef]
- Ouwendijk, W.J.D.; Raadsen, M.P.; A van Kampen, J.J.; Verdijk, R.M.; von der Thusen, J.H.; Guo, L.; Hoek, R.A.S.; van den Akker, J.P.C.; Endeman, H.; Langerak, T.; et al. High Levels of Neutrophil Extracellular Traps Persist in the Lower Respiratory Tract of Critically Ill Patients with Coronavirus Disease 2019. J. Infect. Dis. 2021, 223, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Arcanjo, A.; Logullo, J.; Menezes, C.C.B.; de Souza Carvalho Giangiarulo, T.C.; dos Reis, M.C.; de Castro, G.M.M.; Fontes, Y.d.S.; Todeschini, A.R.; Freire-De-Lima, L.; Decoté-Ricardo, D.; et al. The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID-19). Sci. Rep. 2020, 10, 19630. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.; Havervall, S.; Rosell, A.; Aguilera, K.; Parv, K.; von Meijenfeldt, F.A.; Lisman, T.; Mackman, N.; Thålin, C.; Phillipson, M. Circulating Markers of Neutrophil Extracellular Traps Are of Prognostic Value in Patients with COVID-19. Arter. Thromb. Vasc. Biol. 2021, 41, 988–994, Erratum in Arter. Thromb. Vasc. Biol. 2021, 41, e384. [Google Scholar] [CrossRef] [PubMed]
- Prével, R.; Dupont, A.; Labrouche-Colomer, S.; Garcia, G.; Dewitte, A.; Rauch, A.; Goutay, J.; Caplan, M.; Jozefowicz, E.; Lanoix, J.-P.; et al. Plasma Markers of Neutrophil Extracellular Trap Are Linked to Survival but Not to Pulmonary Embolism in COVID-19-Related ARDS Patients. Front. Immunol. 2022, 13, 851497. [Google Scholar] [CrossRef] [PubMed]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Gandet, F.F.; et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020, 46, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Bortolasci, C.C.; Puri, B.K.; Olive, L.; Marx, W.; O’Neil, A.; Athan, E.; Carvalho, A.; Maes, M.; Walder, K.; et al. Preventing the development of severe COVID-19 by modifying immunothrombosis. Life Sci. 2020, 264, 118617. [Google Scholar] [CrossRef]
- Cesta, M.C.; Zippoli, M.; Marsiglia, C.; Gavioli, E.M.; Cremonesi, G.; Khan, A.; Mantelli, F.; Allegretti, M.; Balk, R. Neutrophil activation and neutrophil extracellular traps (NETs) in COVID-19 ARDS and immunothrombosis. Eur. J. Immunol. 2022, 53, e2250010. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Yaqinuddin, A.; Kashir, J. Novel therapeutic targets for SARS-CoV-2-induced acute lung injury: Targeting a potential IL-1β/neutrophil extracellular traps feedback loop. Med. Hypotheses 2020, 143, 109906. [Google Scholar] [CrossRef]
- Kaiser, R.; Leunig, A.; Pekayvaz, K.; Popp, O.; Joppich, M.; Polewka, V.; Escaig, R.; Anjum, A.; Hoffknecht, M.-L.; Gold, C.; et al. Self-sustaining IL-8 loops drive a prothrombotic neutrophil phenotype in severe COVID-19. J. Clin. Investig. 2021, 6, e150862. [Google Scholar] [CrossRef]
- Keshari, R.S.; Jyoti, A.; Dubey, M.; Kothari, N.; Kohli, M.; Bogra, J.; Barthwal, M.K.; Dikshit, M. Cytokines Induced Neutrophil Extracellular Traps Formation: Implication for the Inflammatory Disease Condition. PLoS ONE 2012, 7, e48111. [Google Scholar] [CrossRef]
- Sung, P.-S.; Yang, S.-P.; Peng, Y.-C.; Sun, C.-P.; Tao, M.-H.; Hsieh, S.-L. CLEC5A and TLR2 are critical in SARS-CoV-2-induced NET formation and lung inflammation. J. Biomed. Sci. 2022, 29, 52. [Google Scholar] [CrossRef]
- Weber, A.G.; Chau, A.S.; Egeblad, M.; Barnes, B.J.; Janowitz, T. Nebulized in-line endotracheal dornase alfa and albuterol administered to mechanically ventilated COVID-19 patients: A case series. Mol. Med. 2020, 26, 91. [Google Scholar] [CrossRef]
- Holliday, Z.M.; Earhart, A.P.; Alnijoumi, M.M.; Krvavac, A.; Allen, L.-A.H.; Schrum, A.G. Non-Randomized Trial of Dornase Alfa for Acute Respiratory Distress Syndrome Secondary to COVID-19. Front. Immunol. 2021, 12, 714833. [Google Scholar] [CrossRef]
- Clemente-Moragón, A.; Martínez-Milla, J.; Oliver, E.; Santos, A.; Flandes, J.; Fernández, I.; Rodríguez-González, L.; del Castillo, C.S.; Ioan, A.-M.; López-Álvarez, M.; et al. Metoprolol in Critically Ill Patients With COVID-19. J. Am. Coll. Cardiol. 2021, 78, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Kashiouris, M.G.; L’heureux, M.; Fisher, B.J.; Leichtle, S.W.; Truwit, J.D.; Nanchal, R.; Hite, R.D.; Morris, P.E.; Martin, G.S.; et al. Biological Effects of Intravenous Vitamin C on Neutrophil Extracellular Traps and the Endothelial Glycocalyx in Patients with Sepsis-Induced ARDS. Nutrients 2022, 14, 4415. [Google Scholar] [CrossRef] [PubMed]
- Panka, B.A.; de Grooth, H.-J.; Spoelstra-de Man, A.M.E.; Looney, M.R.; Tuinman, P.-R. Prevention or Treatment of Ards with Aspirin: A Review of Preclinical Models and Meta-Analysis of Clinical Studies. Shock 2017, 47, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef]
- Liu, S.; Su, X.; Pan, P.; Zhang, L.; Hu, Y.; Tan, H.; Wu, D.; Liu, B.; Li, H.; Li, H.; et al. Neutrophil extracellular traps are indirectly triggered by lipopolysaccharide and contribute to acute lung injury. Sci. Rep. 2016, 6, 37252. [Google Scholar] [CrossRef]
- Park, H.H.; Park, W.; Lee, Y.Y.; Kim, H.; Seo, H.S.; Choi, D.W.; Kwon, H.; Na, D.H.; Kim, T.; Bin Choy, Y.; et al. Bioinspired DNase-I-Coated Melanin-Like Nanospheres for Modulation of Infection-Associated NETosis Dysregulation. Adv. Sci. 2020, 7, 2001940, Erratum in Adv. Sci. 2021, 19, e2103748. [Google Scholar] [CrossRef]
- Lasky, J.A.; Fuloria, J.; Morrison, M.E.; Lanier, R.; Naderer, O.; Brundage, T.; Melemed, A. Design and Rationale of a Randomized, Double-Blind, Placebo-Controlled, Phase 2/3 Study Evaluating Dociparstat in Acute Lung Injury Associated with Severe COVID-19. Adv. Ther. 2020, 38, 782–791. [Google Scholar] [CrossRef]
- Du, M.; Yang, L.; Gu, J.; Wu, J.; Ma, Y.; Wang, T. Inhibition of Peptidyl Arginine Deiminase-4 Prevents Renal Ischemia-Reperfusion-Induced Remote Lung Injury. Mediat. Inflamm. 2020, 2020, 1724206. [Google Scholar] [CrossRef]
- Wu, Z.; Deng, Q.; Pan, B.; Alam, H.B.; Tian, Y.; Bhatti, U.F.; Liu, B.; Mondal, S.; Thompson, P.R.; Li, Y. Inhibition of PAD2 Improves Survival in a Mouse Model of Lethal LPS-Induced Endotoxic Shock. Inflammation 2020, 43, 1436–1445. [Google Scholar] [CrossRef]
- Tian, Y.; Qu, S.; Alam, H.B.; Williams, A.M.; Wu, Z.; Deng, Q.; Pan, B.; Zhou, J.; Liu, B.; Duan, X.; et al. Peptidylarginine deiminase 2 has potential as both a biomarker and therapeutic target of sepsis. J. Clin. Investig. 2020, 5, e138873. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Hippensteel, J.; Leavitt, A.; Maloney, J.P.; Beckham, D.; Garcia, C.; Li, Q.; Freed, B.M.; Ordway, D.; Sandhaus, R.A.; et al. Hypothesis: Alpha-1-antitrypsin is a promising treatment option for COVID-19. Med. Hypotheses 2020, 146, 110394. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhang, L.; Zhao, X.; Li, Z.; Lu, H.; Bu, C.; Wang, R.; Wang, X.; Cai, T.; Wu, D. Protectin D1 protects against lipopolysaccharide-induced acute lung injury through inhibition of neutrophil infiltration and the formation of neutrophil extracellular traps in lung tissue. Exp. Ther. Med. 2021, 22, 1074. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Matsuhiroya, S.; Obuchi, A.; Takahashi, T.; Imoto, S.; Kawano, S.; Saigo, K. Deferasirox, an iron-chelating agent, alleviates acute lung inflammation by inhibiting neutrophil activation and extracellular trap formation. J. Int. Med. Res. 2020, 48, 300060520951015. [Google Scholar] [CrossRef]
- Pedrazza, L.; Cunha, A.A.; Luft, C.; Nunes, N.K.; Schimitz, F.; Gassen, R.B.; Breda, R.V.; Donadio, M.V.F.; de Souza Wyse, A.T.; Pitrez, P.M.C.; et al. Mesenchymal stem cells improves survival in LPS-induced acute lung injury acting through inhibition of NETs formation. J. Cell. Physiol. 2017, 232, 3552–3564. [Google Scholar] [CrossRef] [PubMed]
- Kasetty, G.; Papareddy, P.; Bhongir, R.K.V.; Ali, M.N.; Mori, M.; Wygrecka, M.; Erjefält, J.S.; Hultgårdh-Nilsson, A.; Palmberg, L.; Herwald, H.; et al. Osteopontin protects against lung injury caused by extracellular histones. Mucosal Immunol. 2019, 12, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, J.; Sirois, M.G.; Rhéaume, E.; Nguyen, Q.T.; Clavet-Lanthier, M.-É.; Brand, G.; Mihalache-Avram, T.; Théberge-Julien, G.; Charpentier, D.; Rhainds, D.; et al. Colchicine reduces lung injury in experimental acute respiratory distress syndrome. PLoS ONE 2020, 15, e0242318. [Google Scholar] [CrossRef] [PubMed]
- Adrover, J.M.; Carrau, L.; Daßler-Plenker, J.; Bram, Y.; Chandar, V.; Houghton, S.; Redmond, D.; Merrill, J.R.; Shevik, M.; Tenoever, B.R.; et al. Disulfiram inhibits neutrophil extracellular trap formation and protects rodents from acute lung injury and SARS-CoV-2 infection. J. Clin. Investig. 2022, 7, e157342. [Google Scholar] [CrossRef]
- Antunes, G.L.; Matzenbacher, L.S.; Costa, B.P.; Basso, B.d.S.; Levorse, V.G.S.; Antunes, K.H.; Costa-Ferro, Z.S.M.; de Oliveira, J.R. Methoxyeugenol Protects Against Lung Inflammation and Suppresses Neutrophil Extracellular Trap Formation in an LPS-Induced Acute Lung Injury Model. Inflammation 2022, 45, 1534–1547. [Google Scholar] [CrossRef]
- Sung, P.-S.; Peng, Y.-C.; Yang, S.-P.; Chiu, C.-H.; Hsieh, S.-L. CLEC5A is critical in Pseudomonas aeruginosa-induced NET formation and acute lung injury. J. Clin. Investig. 2022, 7, e156613. [Google Scholar] [CrossRef]
- Baron, S.; Rashal, T.; Vaisman, D.; Elhasid, R.; Shukrun, R. Selinexor, a selective inhibitor of nuclear export, inhibits human neutrophil extracellular trap formation in vitro. Front. Pharmacol. 2022, 13, 1030991. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lu, J.; Wu, Z. Auricular Vagus Nerve Stimulation Attenuates Lipopolysaccharide-Induced Acute Lung Injury by Inhibiting Neutrophil Infiltration and Neutrophil Extracellular Traps Formation. Shock 2021, 57, 427–434. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Jin, J.; Lv, T.; Song, Y. A Narrative Review: The Role of NETs in Acute Respiratory Distress Syndrome/Acute Lung Injury. Int. J. Mol. Sci. 2024, 25, 1464. https://doi.org/10.3390/ijms25031464
Zhou X, Jin J, Lv T, Song Y. A Narrative Review: The Role of NETs in Acute Respiratory Distress Syndrome/Acute Lung Injury. International Journal of Molecular Sciences. 2024; 25(3):1464. https://doi.org/10.3390/ijms25031464
Chicago/Turabian StyleZhou, Xinyu, Jiajia Jin, Tangfeng Lv, and Yong Song. 2024. "A Narrative Review: The Role of NETs in Acute Respiratory Distress Syndrome/Acute Lung Injury" International Journal of Molecular Sciences 25, no. 3: 1464. https://doi.org/10.3390/ijms25031464
APA StyleZhou, X., Jin, J., Lv, T., & Song, Y. (2024). A Narrative Review: The Role of NETs in Acute Respiratory Distress Syndrome/Acute Lung Injury. International Journal of Molecular Sciences, 25(3), 1464. https://doi.org/10.3390/ijms25031464