
CircRNAome of Childhood Acute Lymphoblastic Leukemia: Deciphering Subtype-Specific Expression Profiles and Involvement in TCF3::PBX1 ALL

 , , , , , , , , and
, , , , , , , , and
Abstract
:1. Introduction
2. Results
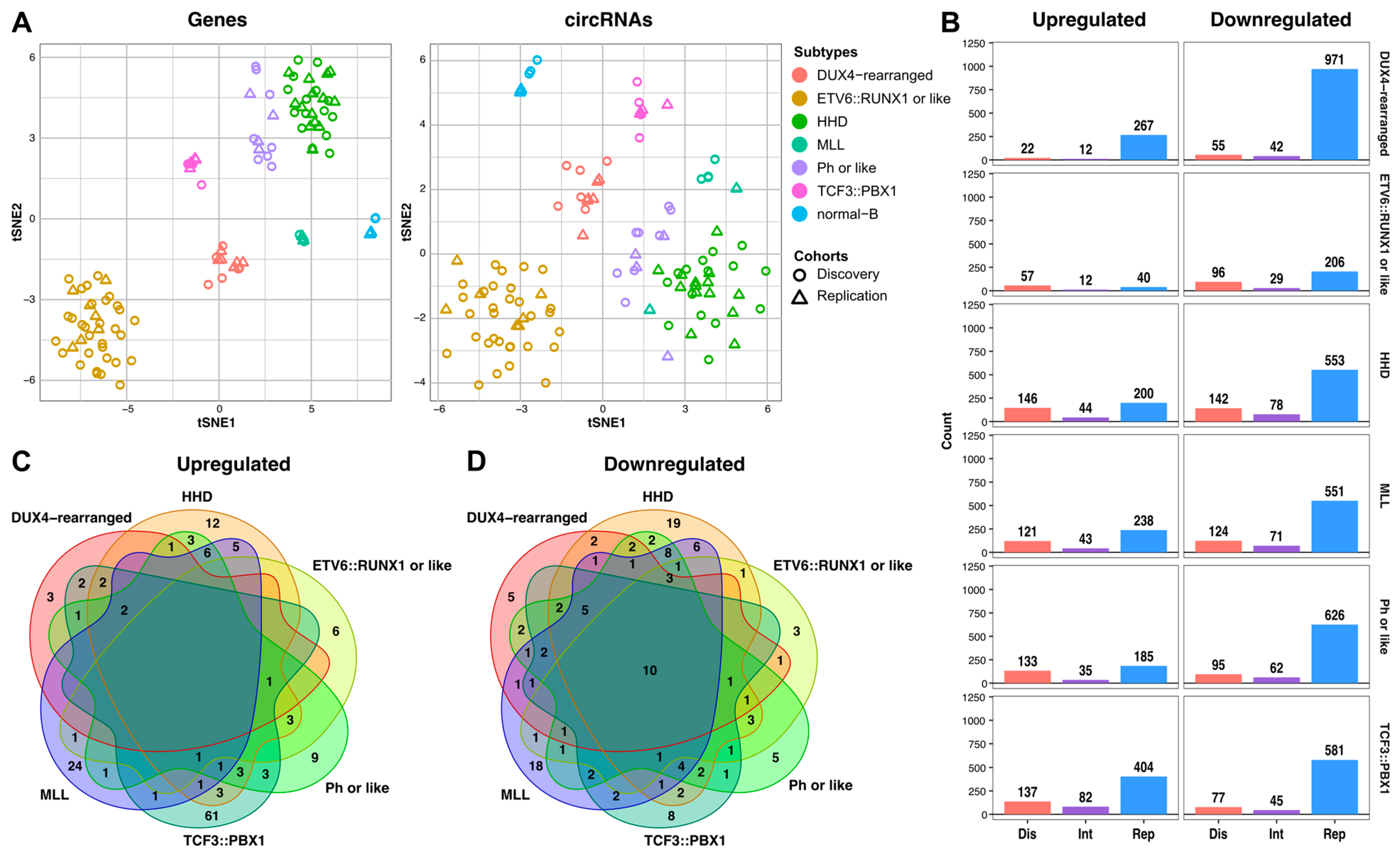
2.1. Characterization of the circRNAome in Childhood B-ALL
2.2. Subtype-Specific Profile of circRNAs in Childhood B-ALL
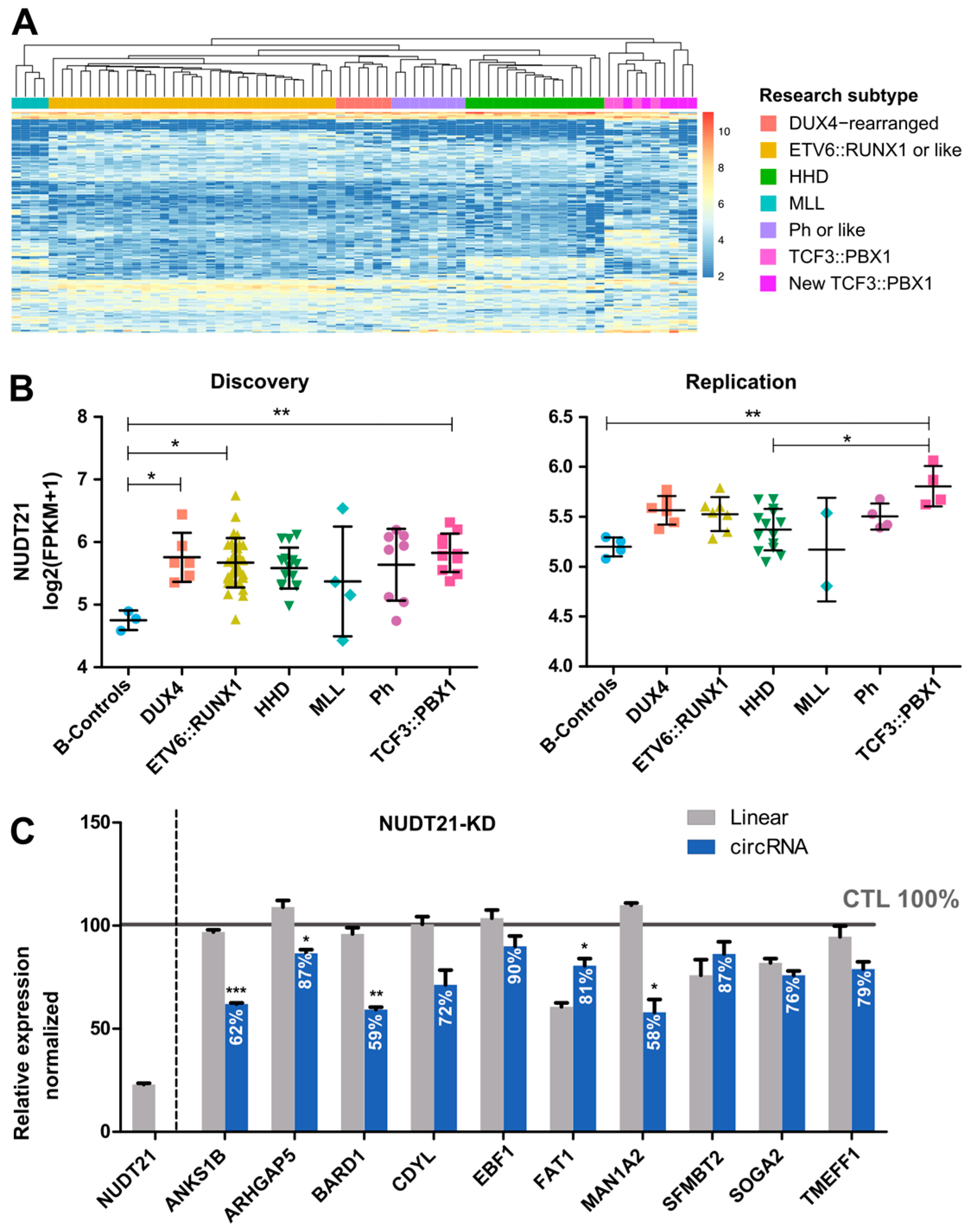
2.3. CircRNAome in TCF3::PBX1 B-ALL
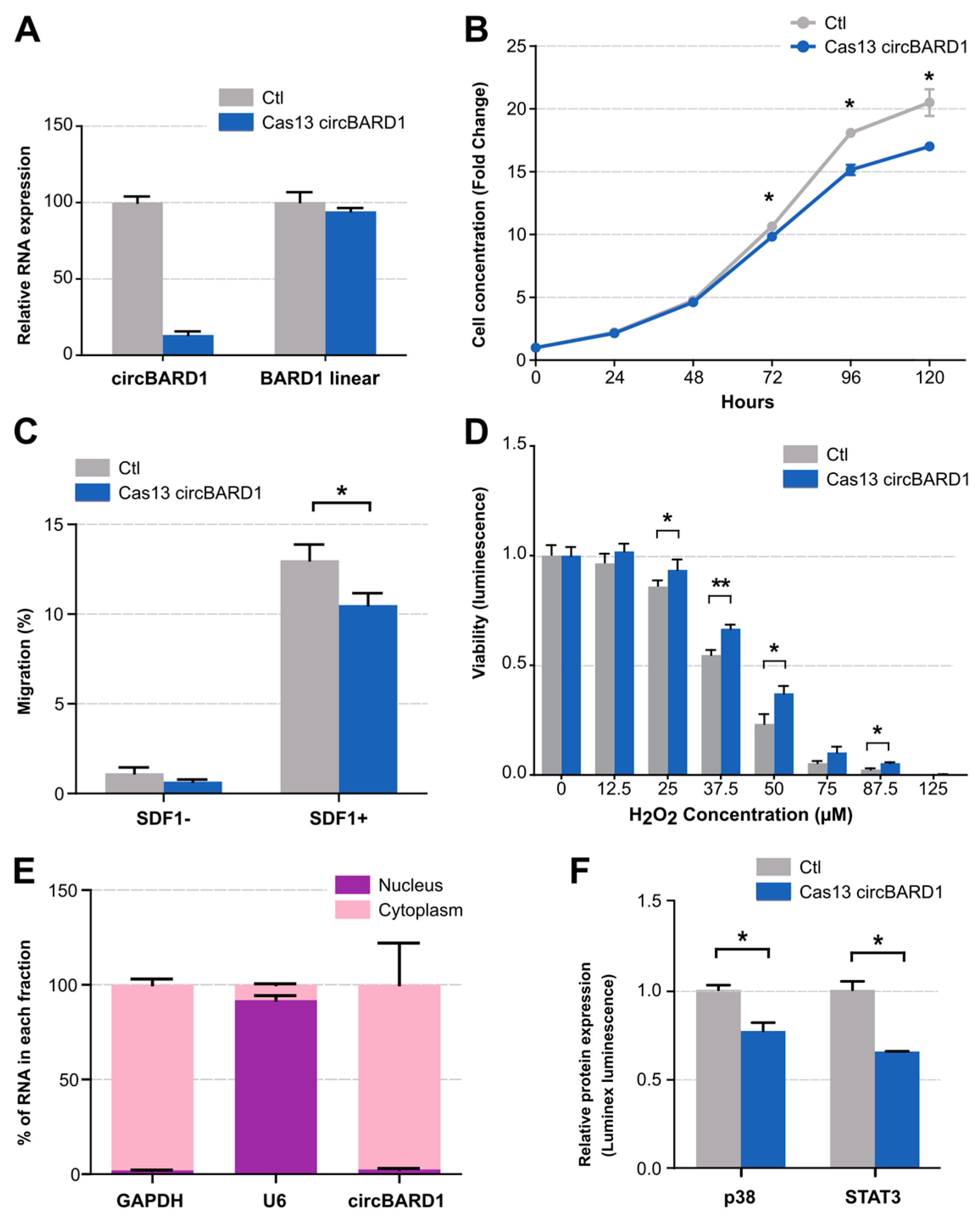
2.4. Functional Validation of circBARD1
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. Library Preparation
4.3. Bioinformatic Analyses
4.4. Quantification in Cell Lines
4.5. Cell Culture and Loss-of-Function Studies
4.6. In Vitro Assessment of circRNAs Silencing on Cancer Phenotypes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Woo, J.S.; Alberti, M.O.; Tirado, C.A. Childhood B-acute lymphoblastic leukemia: A genetic update. Exp. Hematol. Oncol. 2014, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, T.; Relling, M.V.; Yang, J.J. Inherited genetic variation in childhood acute lymphoblastic leukemia. Blood 2015, 125, 3988–3995. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G. Molecular genetics of B-precursor acute lymphoblastic leukemia. J. Clin. Investig. 2012, 122, 3407–3415. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Camino, A.; Garcia-Obregon, S.; Lopez-Lopez, E.; Astigarraga, I.; Garcia-Orad, A. miRNA deregulation in childhood acute lymphoblastic leukemia: A systematic review. Epigenomics 2020, 12, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, M.; Drouin, S.; Caron, M.; St-Onge, P.; Ouimet, M.; Gioia, R.; Lafond, M.H.; Vidal, R.; Richer, C.; Oualkacha, K.; et al. Specific expression of novel long non-coding RNAs in high-hyperdiploid childhood acute lymphoblastic leukemia. PLoS ONE 2017, 12, e0174124. [Google Scholar] [CrossRef]
- Casero, D.; Sandoval, S.; Seet, C.S.; Scholes, J.; Zhu, Y.; Ha, V.L.; Luong, A.; Parekh, C.; Crooks, G.M. Long non-coding RNA profiling of human lymphoid progenitor cells reveals transcriptional divergence of B cell and T cell lineages. Nat. Immunol. 2015, 16, 1282–1291. [Google Scholar] [CrossRef]
- Bonizzato, A.; Gaffo, E.; Te Kronnie, G.; Bortoluzzi, S. CircRNAs in hematopoiesis and hematological malignancies. Blood Cancer J. 2016, 6, e483. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Conn, S.J.; Pillman, K.A.; Toubia, J.; Conn, V.M.; Salmanidis, M.; Phillips, C.A.; Roslan, S.; Schreiber, A.W.; Gregory, P.A.; Goodall, G.J. The RNA binding protein quaking regulates formation of circRNAs. Cell 2015, 160, 1125–1134. [Google Scholar] [CrossRef]
- Yu, J.; Xie, D.; Huang, N.; Zhou, Q. Circular RNAs as Novel Diagnostic Biomarkers and Therapeutic Targets in Kidney Disease. Front. Med. 2021, 8, 714958. [Google Scholar] [CrossRef]
- Okholm, T.L.H.; Sathe, S.; Park, S.S.; Kamstrup, A.B.; Rasmussen, A.M.; Shankar, A.; Chua, Z.M.; Fristrup, N.; Nielsen, M.M.; Vang, S.; et al. Transcriptome-wide profiles of circular RNA and RNA-binding protein interactions reveal effects on circular RNA biogenesis and cancer pathway expression. Genome Med. 2020, 12, 112. [Google Scholar] [CrossRef]
- Aktas, T.; Avsar Ilik, I.; Maticzka, D.; Bhardwaj, V.; Pessoa Rodrigues, C.; Mittler, G.; Manke, T.; Backofen, R.; Akhtar, A. DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome. Nature 2017, 544, 115–119. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Nicolet, B.P.; Engels, S.; Aglialoro, F.; van den Akker, E.; von Lindern, M.; Wolkers, M.C. Circular RNA expression in human hematopoietic cells is widespread and cell-type specific. Nucleic Acids Res. 2018, 46, 8168–8180. [Google Scholar] [CrossRef]
- Dahl, M.; Husby, S.; Eskelund, C.W.; Besenbacher, S.; Fjelstrup, S.; Côme, C.; Ek, S.; Kolstad, A.; Räty, R.; Jerkeman, M.; et al. Expression patterns and prognostic potential of circular RNAs in mantle cell lymphoma: A study of younger patients from the MCL2 and MCL3 clinical trials. Leukemia 2022, 36, 177–188. [Google Scholar] [CrossRef]
- Hirsch, S.; Blätte, T.J.; Grasedieck, S.; Cocciardi, S.; Rouhi, A.; Jongen-Lavrencic, M.; Paschka, P.; Krönke, J.; Gaidzik, V.I.; Döhner, H.; et al. Circular RNAs of the nucleophosmin (NPM1) gene in acute myeloid leukemia. Haematologica 2017, 102, 2039–2047. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ma, Y.; Tan, Y.; Ma, X.; Zhao, M.; Chen, B.; Zhang, R.; Chen, Z.; Wang, K. Profiling and functional analysis of circular RNAs in acute promyelocytic leukemia and their dynamic regulation during all-trans retinoic acid treatment. Cell Death Dis. 2018, 9, 651. [Google Scholar] [CrossRef] [PubMed]
- Vo, J.N.; Cieslik, M.; Zhang, Y.; Shukla, S.; Xiao, L.; Wu, Y.M.; Dhanasekaran, S.M.; Engelke, C.G.; Cao, X.; Robinson, D.R.; et al. The Landscape of Circular RNA in Cancer. Cell 2019, 176, 869–881.e813. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Huang, V.; Xu, X.; Livingstone, J.; Soares, F.; Jeon, J.; Zeng, Y.; Hua, J.T.; Petricca, J.; Guo, H.; et al. Widespread and Functional RNA Circularization in Localized Prostate Cancer. Cell 2019, 176, 831–843.e822. [Google Scholar] [CrossRef] [PubMed]
- Buratin, A.; Paganin, M.; Gaffo, E.; Dal Molin, A.; Roels, J.; Germano, G.; Siddi, M.T.; Serafin, V.; De Decker, M.; Gachet, S.; et al. Large-scale circular RNA deregulation in T-ALL: Unlocking unique ectopic expression of molecular subtypes. Blood Adv. 2020, 4, 5902–5914. [Google Scholar] [CrossRef]
- Gaffo, E.; Boldrin, E.; Dal Molin, A.; Bresolin, S.; Bonizzato, A.; Trentin, L.; Frasson, C.; Debatin, K.M.; Meyer, L.H.; Te Kronnie, G.; et al. Circular RNA differential expression in blood cell populations and exploration of circRNA deregulation in pediatric acute lymphoblastic leukemia. Sci. Rep. 2019, 9, 14670. [Google Scholar] [CrossRef]
- Khabirova, E.; Jardine, L.; Coorens, T.H.H.; Webb, S.; Treger, T.D.; Engelbert, J.; Porter, T.; Prigmore, E.; Collord, G.; Piapi, A.; et al. Single-cell transcriptomics reveals a distinct developmental state of KMT2A-rearranged infant B-cell acute lymphoblastic leukemia. Nat. Med. 2022, 28, 743–751. [Google Scholar] [CrossRef]
- Glažar, P.; Papavasileiou, P.; Rajewsky, N. circBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef]
- Poubel, C.P.; Mansur, M.B.; Boroni, M.; Emerenciano, M. FLT3 overexpression in acute leukaemias: New insights into the search for molecular mechanisms. Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 80–88. [Google Scholar] [CrossRef]
- Huang, H.Y.; Lin, Y.C.; Cui, S.; Huang, Y.; Tang, Y.; Xu, J.; Bao, J.; Li, Y.; Wen, J.; Zuo, H.; et al. miRTarBase update 2022: An informative resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2022, 50, D222–D230. [Google Scholar] [CrossRef]
- Li, X.; Ding, J.; Wang, X.; Cheng, Z.; Zhu, Q. NUDT21 regulates circRNA cyclization and ceRNA crosstalk in hepatocellular carcinoma. Oncogene 2020, 39, 891–904. [Google Scholar] [CrossRef]
- Raimondi, V.; Ciccarese, F.; Ciminale, V. Oncogenic pathways and the electron transport chain: A dangeROS liaison. Br. J. Cancer 2020, 122, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lazaro, M. Dual role of hydrogen peroxide in cancer: Possible relevance to cancer chemoprevention and therapy. Cancer Lett. 2007, 252, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Doskey, C.M.; Buranasudja, V.; Wagner, B.A.; Wilkes, J.G.; Du, J.; Cullen, J.J.; Buettner, G.R. Tumor cells have decreased ability to metabolize H2O2: Implications for pharmacological ascorbate in cancer therapy. Redox Biol. 2016, 10, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.M.; Gilmartin, G.M. A mechanism for the regulation of pre-mRNA 3′ processing by human cleavage factor Im. Mol. Cell 2003, 12, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.T.; Lee, S.; Wang, E.; Lee, A.K.; Talbot, A.; Ma, C.; Tsopoulidis, N.; Brumbaugh, J.; Zhao, Y.; Roberts, K.G.; et al. NUDT21 limits CD19 levels through alternative mRNA polyadenylation in B cell acute lymphoblastic leukemia. Nat. Immunol. 2022, 23, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Nguyen, T.M.; Zhang, X.O.; Wang, L.; Phan, T.; Clohessy, J.G.; Pandolfi, P.P. Optimized RNA-targeting CRISPR/Cas13d technology outperforms shRNA in identifying functional circRNAs. Genome Biol. 2021, 22, 41. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Zeinalzadeh, E.; Valerievich Yumashev, A.; Rahman, H.S.; Marofi, F.; Shomali, N.; Kafil, H.S.; Solali, S.; Sajjadi-Dokht, M.; Vakili-Samiani, S.; Jarahian, M.; et al. The Role of Janus Kinase/STAT3 Pathway in Hematologic Malignancies With an Emphasis on Epigenetics. Front. Genet. 2021, 12, 703883. [Google Scholar] [CrossRef] [PubMed]
- Karvonen, H.; Perttila, R.; Niininen, W.; Hautanen, V.; Barker, H.; Murumagi, A.; Heckman, C.A.; Ungureanu, D. Wnt5a and ROR1 activate non-canonical Wnt signaling via RhoA in TCF3-PBX1 acute lymphoblastic leukemia and highlight new treatment strategies via Bcl-2 co-targeting. Oncogene 2019, 38, 3288–3300. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Hansen, T.B.; Venø, M.T.; Kjems, J. Circular RNAs in cancer: Opportunities and challenges in the field. Oncogene 2018, 37, 555–565. [Google Scholar] [CrossRef]
- Di Timoteo, G.; Dattilo, D.; Centron-Broco, A.; Colantoni, A.; Guarnacci, M.; Rossi, F.; Incarnato, D.; Oliviero, S.; Fatica, A.; Morlando, M.; et al. Modulation of circRNA Metabolism by m(6)A Modification. Cell Rep. 2020, 31, 107641. [Google Scholar] [CrossRef] [PubMed]
- Healy, J.; Bélanger, H.; Beaulieu, P.; Larivière, M.; Labuda, D.; Sinnett, D. Promoter SNPs in G1/S checkpoint regulators and their impact on the susceptibility to childhood leukemia. Blood 2007, 109, 683–692. [Google Scholar] [CrossRef]
- Tran, T.H.; Langlois, S.; Meloche, C.; Caron, M.; St-Onge, P.; Rouette, A.; Bataille, A.R.; Jimenez-Cortes, C.; Sontag, T.; Bittencourt, H.; et al. Whole-transcriptome analysis in acute lymphoblastic leukemia: A report from the DFCI ALL Consortium Protocol 16-001. Blood Adv. 2021, 6, 1329–1341. [Google Scholar] [CrossRef]
- Lilljebjorn, H.; Henningsson, R.; Hyrenius-Wittsten, A.; Olsson, L.; Orsmark-Pietras, C.; von Palffy, S.; Askmyr, M.; Rissler, M.; Schrappe, M.; Cario, G.; et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 11790. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Black, K.L.; Naqvi, A.S.; Asnani, M.; Hayer, K.E.; Yang, S.Y.; Gillespie, E.; Bagashev, A.; Pillai, V.; Tasian, S.K.; Gazzara, M.R.; et al. Aberrant splicing in B-cell acute lymphoblastic leukemia. Nucleic Acids Res. 2018, 46, 11357–11369. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.K.; Wang, M.R.; Liu, C.X.; Dong, R.; Carmichael, G.G.; Chen, L.L.; Yang, L. CIRCexplorer3: A CLEAR Pipeline for Direct Comparison of Circular and Linear RNA Expression. Genom. Proteom. Bioinform. 2019, 17, 511–521. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Benoit Bouvrette, L.P.; Bovaird, S.; Blanchette, M.; Lecuyer, E. oRNAment: A database of putative RNA binding protein target sites in the transcriptomes of model species. Nucleic Acids Res. 2020, 48, D166–D173. [Google Scholar] [CrossRef]
- Dudekula, D.B.; Panda, A.C.; Grammatikakis, I.; De, S.; Abdelmohsen, K.; Gorospe, M. CircInteractome: A web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol. 2016, 13, 34–42. [Google Scholar] [CrossRef]
- Liu, M.; Wang, Q.; Shen, J.; Yang, B.B.; Ding, X. Circbank: A comprehensive database for circRNA with standard nomenclature. RNA Biol. 2019, 16, 899–905. [Google Scholar] [CrossRef]
- Chen, X.; Han, P.; Zhou, T.; Guo, X.; Song, X.; Li, Y. circRNADb: A comprehensive database for human circular RNAs with protein-coding annotations. Sci. Rep. 2016, 6, 34985. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.C.; De, S.; Grammatikakis, I.; Munk, R.; Yang, X.; Piao, Y.; Dudekula, D.B.; Abdelmohsen, K.; Gorospe, M. High-purity circular RNA isolation method (RPAD) reveals vast collection of intronic circRNAs. Nucleic Acids Res. 2017, 45, e116. [Google Scholar] [CrossRef]
- Li, S.; Li, X.; Xue, W.; Zhang, L.; Yang, L.Z.; Cao, S.M.; Lei, Y.N.; Liu, C.X.; Guo, S.K.; Shan, L.; et al. Screening for functional circular RNAs using the CRISPR-Cas13 system. Nat. Methods 2021, 18, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Lotfy, P.; Brideau, N.J.; Oki, J.; Shokhirev, M.N.; Hsu, P.D. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 2018, 173, 665–676.e14. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Discovery | Replication | |
|---|---|---|
| No. of individuals | 74 | 37 |
| Mean age ± SE, y | 6.9 ± 4.4 | 6.3 ± 4.4 |
| Sex * | ||
| Males, n (%) | 34 (45.9) | 18 (48.6) |
| Females, n (%) | 38 (51.4) | 19 (51.4) |
| Genetic alterations | ||
| Hyperdiploid, n (%) | 15 (20.3) | 14 (34.1) |
| ETV6::RUNX1, n (%) | 31 (41.9) | 7 (17.1) |
| MLL, n (%) | 4 (5.4) | 2 (4.9) |
| BCR::ABL, n (%) | 8 (10.8) | 4 (9.8) |
| TCF3::PBX1, n (%) | 10 (13.5) | 4 (9.8) |
| DUX4-rearranged, n (%) | 6 (8.1) | 6 (14.6) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutierrez-Camino, A.; Caron, M.; Richer, C.; Fuchs, C.; Illarregi, U.; Poncelet, L.; St-Onge, P.; Bataille, A.R.; Tremblay-Dauphinais, P.; Lopez-Lopez, E.; et al. CircRNAome of Childhood Acute Lymphoblastic Leukemia: Deciphering Subtype-Specific Expression Profiles and Involvement in TCF3::PBX1 ALL. Int. J. Mol. Sci. 2024, 25, 1477. https://doi.org/10.3390/ijms25031477
Gutierrez-Camino A, Caron M, Richer C, Fuchs C, Illarregi U, Poncelet L, St-Onge P, Bataille AR, Tremblay-Dauphinais P, Lopez-Lopez E, et al. CircRNAome of Childhood Acute Lymphoblastic Leukemia: Deciphering Subtype-Specific Expression Profiles and Involvement in TCF3::PBX1 ALL. International Journal of Molecular Sciences. 2024; 25(3):1477. https://doi.org/10.3390/ijms25031477
Chicago/Turabian StyleGutierrez-Camino, Angela, Maxime Caron, Chantal Richer, Claire Fuchs, Unai Illarregi, Lucas Poncelet, Pascal St-Onge, Alain R. Bataille, Pascal Tremblay-Dauphinais, Elixabet Lopez-Lopez, and et al. 2024. "CircRNAome of Childhood Acute Lymphoblastic Leukemia: Deciphering Subtype-Specific Expression Profiles and Involvement in TCF3::PBX1 ALL" International Journal of Molecular Sciences 25, no. 3: 1477. https://doi.org/10.3390/ijms25031477
APA StyleGutierrez-Camino, A., Caron, M., Richer, C., Fuchs, C., Illarregi, U., Poncelet, L., St-Onge, P., Bataille, A. R., Tremblay-Dauphinais, P., Lopez-Lopez, E., Camos, M., Ramirez-Orellana, M., Astigarraga, I., Lécuyer, É., Bourque, G., Martin-Guerrero, I., & Sinnett, D. (2024). CircRNAome of Childhood Acute Lymphoblastic Leukemia: Deciphering Subtype-Specific Expression Profiles and Involvement in TCF3::PBX1 ALL. International Journal of Molecular Sciences, 25(3), 1477. https://doi.org/10.3390/ijms25031477