

Host Subcellular Organelles: Targets of Viral Manipulation



Abstract
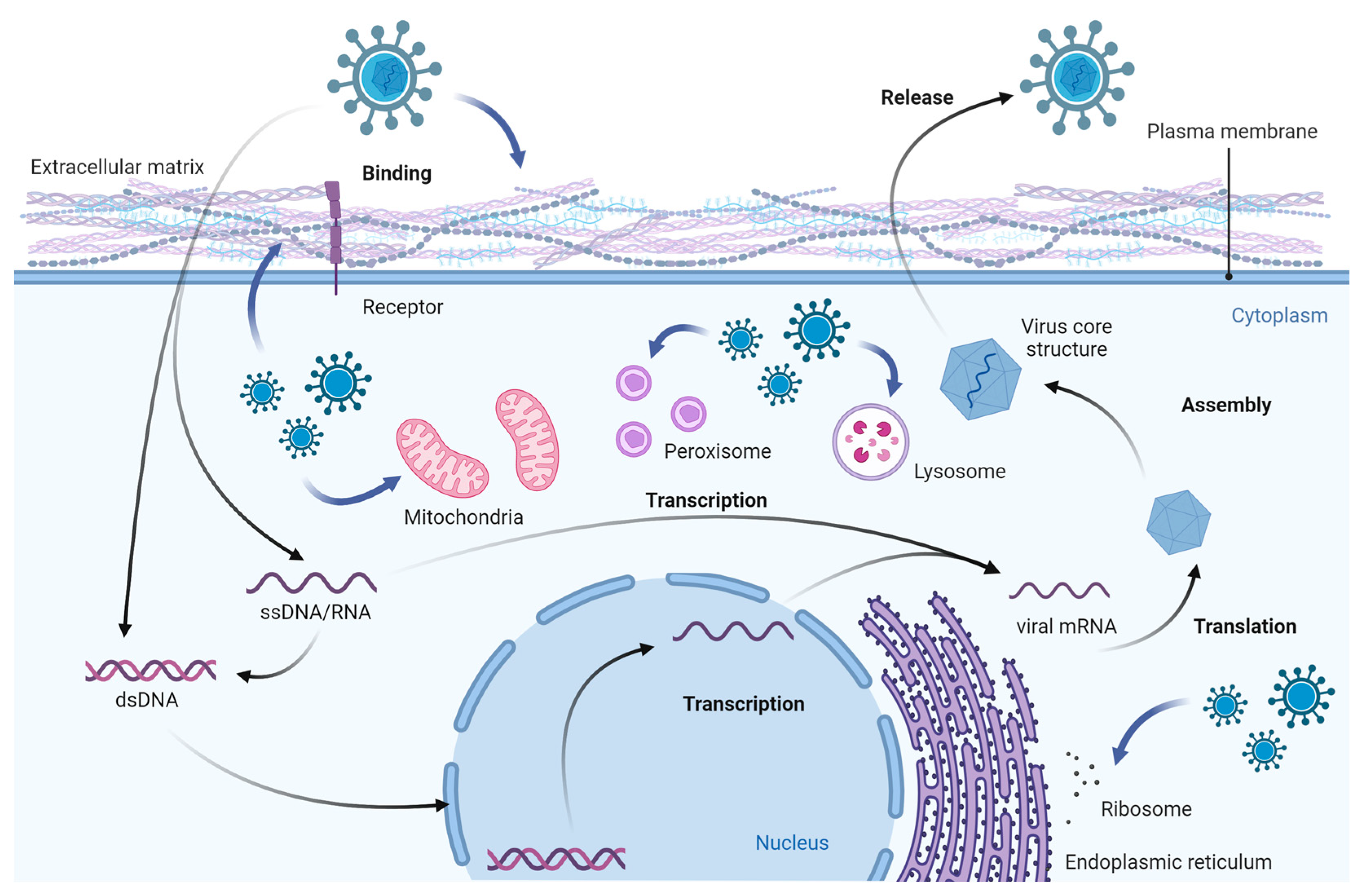
1. Introduction
2. Viral Infection and Its Effects on Cell Components
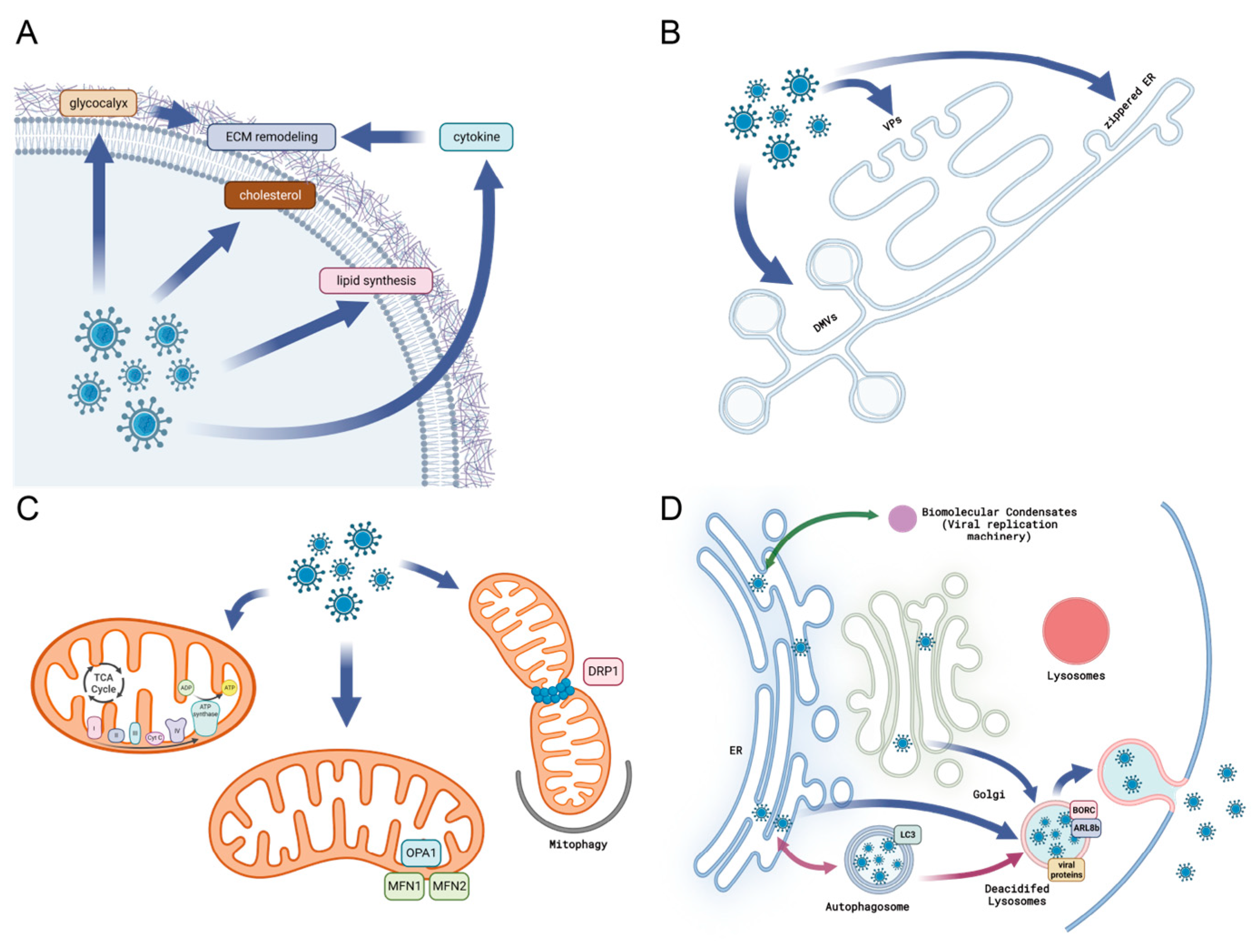
2.1. Viral Infection Remodels the Extracellular Matrix
2.2. Viral Manipulation and Interaction with Host Cell Membrane
2.3. Viral Infection Alters the Endocytic Pathways
2.4. Virus Remodels the Cytoskeleton
2.5. Virus and Nuclear Remodeling
2.6. Virus and Modification of Endoplasmic Reticulum
2.7. Alternation to Mitochondrial Morphology and Function during Viral Infection
2.8. Cellular Condensates and Viral Replication
2.9. Viral Manipulation of Autophagosomes and Viral Replication
2.10. Viral Exocytosis and Lysosome
2.11. Viral Manipulation of Extracellular Vesicles
3. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ginsberg, J.; Mohebbi, M.H.; Patel, R.S.; Brammer, L.; Smolinski, M.S.; Brilliant, L. Detecting influenza epidemics using search engine query data. Nature 2009, 457, 1012–1014. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Idoko, J.A.; Grinsztejn, B.; Phanuphak, N. The path to equitable HIV prevention. Commun. Med. 2022, 2, 161. [Google Scholar] [CrossRef] [PubMed]
- Majumder, K.; Morales, A.J. Utilization of Host Cell Chromosome Conformation by Viral Pathogens: Knowing When to Hold and When to Fold. Front. Immunol. 2021, 12, 633762. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Boutell, C.; Hale, B.G. Interplay between viruses and host sumoylation pathways. Nat. Rev. Microbiol. 2013, 11, 400–411. [Google Scholar] [CrossRef]
- McBride, A.A. Human papillomaviruses: Diversity, infection and host interactions. Nat. Rev. Microbiol. 2022, 20, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Marziano, N.K.; Hasegawa, D.K.; Phelan, P.; Turnbull, M.W. Functional interactions between polydnavirus and host cellular innexins. J. Virol. 2011, 85, 10222–10229. [Google Scholar] [CrossRef]
- Wang, J.; Fang, S.; Xiao, H.; Chen, B.; Tam, J.P.; Liu, D.X. Interaction of the coronavirus infectious bronchitis virus membrane protein with beta-actin and its implication in virion assembly and budding. PLoS ONE 2009, 4, e4908. [Google Scholar] [CrossRef]
- Heming, J.D.; Conway, J.F.; Homa, F.L. Herpesvirus Capsid Assembly and DNA Packaging. Adv. Anat. Embryol. Cell Biol. 2017, 223, 119–142. [Google Scholar] [CrossRef]
- Rowe, R.G.; Weiss, S.J. Navigating ECM Barriers at the Invasive Front: The Cancer Cell–Stroma Interface. Annu. Rev. Cell Dev. Biol. 2009, 25, 567–595. [Google Scholar] [CrossRef] [PubMed]
- Rozario, T.; DeSimone, D.W. The extracellular matrix in development and morphogenesis: A dynamic view. Dev. Biol. 2010, 341, 126–140. [Google Scholar] [CrossRef]
- Hällgren, R.; Samuelsson, T.; Laurent, T.C.; Modig, J. Accumulation of hyaluronan (hyaluronic acid) in the lung in adult respiratory distress syndrome. Am. Rev. Respir. Dis. 1989, 139, 682–687. [Google Scholar] [CrossRef]
- Hellman, U.; Karlsson, M.G.; Engström-Laurent, A.; Cajander, S.; Dorofte, L.; Ahlm, C.; Laurent, C.; Blomberg, A. Presence of hyaluronan in lung alveoli in severe COVID-19: An opening for new treatment options? J. Biol. Chem. 2020, 295, 15418–15422. [Google Scholar] [CrossRef]
- Syed, F.; Li, W.; Relich, R.F.; Russell, P.M.; Zhang, S.; Zimmerman, M.K.; Yu, Q. Excessive Matrix Metalloproteinase-1 and Hyperactivation of Endothelial Cells Occurred in COVID-19 Patients and Were Associated with the Severity of COVID-19. J. Infect. Dis. 2021, 224, 60–69. [Google Scholar] [CrossRef]
- Nasr El-Din, A.; Ata, K.A.E.; Abdel-Gawad, A.R.; Fahmy, N.F. Impact of High Serum Levels of MMP-7, MMP-9, TGF-β and PDGF Macrophage Activation Markers on Severity of COVID-19 in Obese-Diabetic Patients. Infect. Drug Resist. 2021, 14, 4015–4025. [Google Scholar] [CrossRef]
- Stroulios, G.; Brown, T.; Moreni, G.; Kondro, D.; Dei, A.; Eaves, A.; Louis, S.; Hou, J.; Chang, W.; Pajkrt, D.; et al. Apical-out airway organoids as a platform for studying viral infections and screening for antiviral drugs. Sci. Rep. 2022, 12, 7673. [Google Scholar] [CrossRef]
- Sugden, S.M.; Bego, M.G.; Pham, T.N.; Cohen, É.A. Remodeling of the Host Cell Plasma Membrane by HIV-1 Nef and Vpu: A Strategy to Ensure Viral Fitness and Persistence. Viruses 2016, 8, 67. [Google Scholar] [CrossRef]
- Spearman, P. Viral interactions with host cell Rab GTPases. Small GTPases 2018, 9, 192–201. [Google Scholar] [CrossRef]
- Young, J.M.; Zine El Abidine, A.; Gómez-Martinez, R.A.; Ozbun, M.A. The Known and Potential Intersections of Rab-GTPases in Human Papillomavirus Infections. Front. Cell Dev. Biol. 2019, 7, 139. [Google Scholar] [CrossRef] [PubMed]
- Mañes, S.; del Real, G.; Martínez, A.C. Pathogens: Raft hijackers. Nat. Rev. Immunol. 2003, 3, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Trbojević-Akmačić, I.; Petrović, T.; Lauc, G. SARS-CoV-2 S glycoprotein binding to multiple host receptors enables cell entry and infection. Glycoconj. J. 2021, 38, 611–623. [Google Scholar] [CrossRef]
- Millet, J.K.; Jaimes, J.A.; Whittaker, G.R. Molecular diversity of coronavirus host cell entry receptors. FEMS Microbiol. Rev. 2020, 45, fuaa057. [Google Scholar] [CrossRef]
- Cheng, Y.; Lou, J.-x.; Liu, C.-c.; Liu, Y.-y.; Chen, X.-n.; Liang, X.-d.; Zhang, J.; Yang, Q.; Go, Y.Y.; Zhou, B. Microfilaments and Microtubules Alternately Coordinate the Multistep Endosomal Trafficking of Classical Swine Fever Virus. J. Virol. 2021, 95, e02436-20. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Liu, W.-L.; Yang, D.; Shen, Z.-Q.; Qiu, Z.-G.; Jin, M.; Li, J.-W. Hepatitis C virus infection induces endoplasmic reticulum stress and apoptosis in human fetal liver stem cells. J. Pathol. 2019, 248, 155–163. [Google Scholar] [CrossRef]
- Peng, J.; Ran, Y.; Xie, H.; Deng, L.; Li, C.; Chen, L. Sarco/Endoplasmic Reticulum Ca2+-Transporting ATPase (SERCA) Modulates Autophagic, Inflammatory, and Mitochondrial Responses during Influenza A Virus Infection in Human Lung Cells. J. Virol. 2021, 95, e00217-21. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Men, Y.; Wang, D.; Xu, D.; Liu, S.; Xiao, S.; Fang, L. Porcine reproductive and respiratory syndrome virus infection induces endoplasmic reticulum stress, facilitates virus replication, and contributes to autophagy and apoptosis. Sci. Rep. 2020, 10, 13131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, H.; Pei, R.; Mao, B.; Zhao, Z.; Li, H.; Lin, Y.; Lu, K. The SARS-CoV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell Discov. 2021, 7, 31. [Google Scholar] [CrossRef]
- Yang, J. Viruses Binding to Host Receptors Interacts with Autophagy. Int. J. Mol. Sci. 2023, 24, 3423. [Google Scholar] [CrossRef]
- Beese, C.J.; Brynjólfsdóttir, S.H.; Frankel, L.B. Selective Autophagy of the Protein Homeostasis Machinery: Ribophagy, Proteaphagy and ER-Phagy. Front. Cell Dev. Biol. 2020, 7, 373. [Google Scholar] [CrossRef]
- Huang, Y.; Ji, J.; Zhao, Q.; Song, J. Editorial: Regulation of endoplasmic reticulum and mitochondria in cellular homeostasis. Front. Cell Dev. Biol. 2022, 10, 1004376. [Google Scholar] [CrossRef]
- Awadh, A.A. The Role of Cytosolic Lipid Droplets in Hepatitis C Virus Replication, Assembly, and Release. BioMed Res. Int. 2023, 2023, 5156601. [Google Scholar] [CrossRef]
- Altan-Bonnet, N.; Balla, T. Phosphatidylinositol 4-kinases: Hostages harnessed to build panviral replication platforms. Trends Biochem. Sci. 2012, 37, 293–302. [Google Scholar] [CrossRef]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in Membrane Traffic and Cell Physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef]
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. β-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 2020, 183, 1520–1535.e14. [Google Scholar] [CrossRef]
- Eastburn, D.J.; Mostov, K.E. Laying the foundation for epithelia: Insights into polarized basement membrane deposition. EMBO Rep. 2010, 11, 329–330. [Google Scholar] [CrossRef]
- Lee, S.J.; Atala, A. Scaffold technologies for controlling cell behavior in tissue engineering. Biomed. Mater. 2013, 8, 010201. [Google Scholar] [CrossRef]
- Van Hul, N.K.M.; Abarca-Quinones, J.; Sempoux, C.; Horsmans, Y.; Leclercq, I.A. Relation between liver progenitor cell expansion and extracellular matrix deposition in a CDE-induced murine model of chronic liver injury. Hepatology 2009, 49, 1625–1635. [Google Scholar] [CrossRef]
- Llacua, L.A.; Faas, M.M.; de Vos, P. Extracellular matrix molecules and their potential contribution to the function of transplanted pancreatic islets. Diabetologia 2018, 61, 1261–1272. [Google Scholar] [CrossRef]
- Chen, J.; He, W.-R.; Shen, L.; Dong, H.; Yu, J.; Wang, X.; Yu, S.; Li, Y.; Li, S.; Luo, Y.; et al. The Laminin Receptor Is a Cellular Attachment Receptor for Classical Swine Fever Virus. J. Virol. 2015, 89, 4894–4906. [Google Scholar] [CrossRef]
- Boshuizen, J.A.; Rossen, J.W.A.; Sitaram, C.K.; Kimenai, F.F.P.; Simons-Oosterhuis, Y.; Laffeber, C.; Büller, H.A.; Einerhand, A.W.C. Rotavirus Enterotoxin NSP4 Binds to the Extracellular Matrix Proteins Laminin-β3 and Fibronectin. J. Virol. 2004, 78, 10045–10053. [Google Scholar] [CrossRef]
- Liu, W.-J.; Li, Y.-C.; Kou, G.-H.; Lo, C.-F. Laminin Receptor in Shrimp Is a Cellular Attachment Receptor for White Spot Syndrome Virus. PLoS ONE 2016, 11, e0156375. [Google Scholar] [CrossRef]
- Manzoor, S.; Khalid, M.; Idrees, M. P2X4 receptors mediate induction of antioxidants, fibrogenic cytokines and ECM transcripts; in presence of replicating HCV in in vitro setting: An insight into role of P2X4 in fibrosis. PLoS ONE 2022, 17, e0259727. [Google Scholar] [CrossRef]
- Ahuja, S.; Lazar, I.M. Systems-Level Proteomics Evaluation of Microglia Response to Tumor-Supportive Anti-Inflammatory Cytokines. Front. Immunol. 2021, 12, 646043. [Google Scholar] [CrossRef]
- Walawalkar, S.; Almelkar, S. Re-Cellularised Kidney Scaffold for Chikungunya Virus Propagation: A Novel Approach. Tissue Eng. Regen. Med. 2022, 19, 769–779. [Google Scholar] [CrossRef]
- Gualberto Cavalcanti, N.; MeloVilar, K.; Branco Pinto Duarte, A.L.; Jesus Barreto de Melo Rêgo, M.; Cristiny Pereira, M.; da Rocha Pitta, I.; Diniz Lopes Marques, C.; Galdino da Rocha Pitta, M. IL-27 in patients with Chikungunya fever: A possible chronicity biomarker? Acta Trop. 2019, 196, 48–51. [Google Scholar] [CrossRef]
- Liu, X.; Poo, Y.-S.; Alves, J.C.; Almeida, R.P.; Mostafavi, H.; Tang, P.C.H.; Bucala, R.; Teixeira, M.M.; Taylor, A.; Zaid, A.; et al. Interleukin-17 Contributes to Chikungunya Virus-Induced Disease. mBio 2022, 13, e00289-22. [Google Scholar] [CrossRef]
- Miossec, P.; Kolls, J.K. Targeting IL-17 and TH17 cells in chronic inflammation. Nat. Rev. Drug Discov. 2012, 11, 763–776. [Google Scholar] [CrossRef]
- Wu, J.; Thabet, S.R.; Kirabo, A.; Trott, D.W.; Saleh, M.A.; Xiao, L.; Madhur, M.S.; Chen, W.; Harrison, D.G. Inflammation and Mechanical Stretch Promote Aortic Stiffening in Hypertension Through Activation of p38 Mitogen-Activated Protein Kinase. Circ. Res. 2014, 114, 616–625. [Google Scholar] [CrossRef]
- Okamoto, Y.; Hasegawa, M.; Matsushita, T.; Hamaguchi, Y.; Huu, D.L.; Iwakura, Y.; Fujimoto, M.; Takehara, K. Potential roles of interleukin-17A in the development of skin fibrosis in mice. Arthritis Rheum. 2012, 64, 3726–3735. [Google Scholar] [CrossRef]
- Hayn, M.; Hirschenberger, M.; Koepke, L.; Nchioua, R.; Straub, J.H.; Klute, S.; Hunszinger, V.; Zech, F.; Prelli Bozzo, C.; Aftab, W.; et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 2021, 35, 109126. [Google Scholar] [CrossRef]
- Gao, G.; Cao, L.; Du, X.; Xu, B.; Zhang, P.; Zhang, X.; Wang, R.; Quan, Z. Comparison of Minimally Invasive Surgery Transforaminal Lumbar Interbody Fusion and TLIF for Treatment of Lumbar Spine Stenosis. J. Heal. Eng. 2022, 2022, 9389239. [Google Scholar] [CrossRef]
- Liu, B.; Liu, T.; Liu, Y.; Feng, X.; Jiang, X.; Long, J.; Ye, S.; Chen, D.; Wang, J.; Yang, Z. TSG-6 promotes Cancer Cell aggressiveness in a CD44-Dependent Manner and Reprograms Normal Fibroblasts to create a Pro-metastatic Microenvironment in Colorectal Cancer. Int. J. Biol. Sci. 2022, 18, 1677–1694. [Google Scholar] [CrossRef]
- Hussain, K.M.; Lee, R.C.H.; Ng, M.M.-L.; Chu, J.J.H. Establishment of a Novel Primary Human Skeletal Myoblast Cellular Model for Chikungunya Virus Infection and Pathogenesis. Sci. Rep. 2016, 6, 21406. [Google Scholar] [CrossRef]
- Tourdot, S.; Mathie, S.; Hussell, T.; Edwards, L.; Wang, H.; Openshaw, P.J.M.; Schwarze, J.; Lloyd, C.M. Respiratory syncytial virus infection provokes airway remodelling in allergen-exposed mice in absence of prior allergen sensitization. Clin. Exp. Allergy 2008, 38, 1016–1024. [Google Scholar] [CrossRef]
- Monick, M.M.; Yarovinsky, T.O.; Powers, L.S.; Butler, N.S.; Carter, A.B.; Gudmundsson, G.; Hunninghake, G.W. Respiratory Syncytial Virus Up-regulates TLR4 and Sensitizes Airway Epithelial Cells to Endotoxin. J. Biol. Chem. 2003, 278, 53035–53044. [Google Scholar] [CrossRef]
- Reeves, S.R.; Barrow, K.A.; Rich, L.M.; White, M.P.; Shubin, N.J.; Chan, C.K.; Kang, I.; Ziegler, S.F.; Piliponsky, A.M.; Wight, T.N.; et al. Respiratory Syncytial Virus Infection of Human Lung Fibroblasts Induces a Hyaluronan-Enriched Extracellular Matrix That Binds Mast Cells and Enhances Expression of Mast Cell Proteases. Front. Immunol. 2020, 10, 3159. [Google Scholar] [CrossRef]
- Reeves, S.R.; Kang, I.; Chan, C.K.; Barrow, K.A.; Kolstad, T.K.; White, M.P.; Ziegler, S.F.; Wight, T.N.; Debley, J.S. Asthmatic bronchial epithelial cells promote the establishment of a Hyaluronan-enriched, leukocyte-adhesive extracellular matrix by lung fibroblasts. Respir. Res. 2018, 19, 146. [Google Scholar] [CrossRef]
- Wang, L.; Chu, C.-Y.; McCall, M.N.; Slaunwhite, C.; Holden-Wiltse, J.; Corbett, A.; Falsey, A.R.; Topham, D.J.; Caserta, M.T.; Mariani, T.J.; et al. Airway gene-expression classifiers for respiratory syncytial virus (RSV) disease severity in infants. BMC Med. Genom. 2021, 14, 57. [Google Scholar] [CrossRef]
- Song, X.; Wu, Y.; Wu, X.; Hu, G.; Zhang, T. Effects of Highly Pathogenic Porcine Reproductive and Respiratory Syndrome Virus Infection on the Surface Glycoprofiling of Porcine Pulmonary Microvascular Endothelial Cells. Viruses 2022, 14, 2569. [Google Scholar] [CrossRef]
- Rewisha, E.; Salman, T.; Alhaddad, O.; Abo Raia, G.; Naguib, M.; Rashad, S.; Abdelfattah, A.; Metwally, K.; Abdelsameea, E. Hyaluronic acid as a potential marker for assessment of fibrosis regression after direct acting antiviral drugs in chronic hepatitis C patients. Clin. Exp. Hepatol. 2021, 7, 320–327. [Google Scholar] [CrossRef]
- Rockey, D.C. Fibrosis reversal after hepatitis C virus elimination. Curr. Opin. Gastroenterol. 2019, 35, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Miyaaki, H.; Fukushima, M.; Sasaki, R.; Haraguchi, M.; Miuma, S.; Nakao, K. The impact of single-nucleotide polymorphisms on liver stiffness and controlled attenuation parameter in patients treated with direct-acting antiviral drugs for hepatitis C infection. Biomed. Rep. 2022, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Mohan, J.; Wollert, T. Membrane remodeling by SARS-CoV-2—Double-enveloped viral replication. Fac. Rev. 2021, 10, 17. [Google Scholar] [CrossRef]
- Yang, J.; Park, J.; Koehler, M.; Simpson, J.; Luque, D.; Rodríguez, J.M.; Alsteens, D. Rotavirus Binding to Cell Surface Receptors Directly Recruiting α2 Integrin. Adv. NanoBiomed Res. 2021, 1, 2100077. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, J.; Tillieux, S.; Guo, Z.; Natividade, R.d.S.; Koehler, M.; Petitjean, S.; Cui, Z.; Alsteens, D. Stepwise Enzymatic-Dependent Mechanism of Ebola Virus Binding to Cell Surface Receptors Monitored by AFM. Nano Lett. 2022, 22, 1641–1648. [Google Scholar] [CrossRef]
- Koehler, M.; Petitjean, S.J.L.; Yang, J.; Aravamudhan, P.; Somoulay, X.; Lo Giudice, C.; Poncin, M.A.; Dumitru, A.C.; Dermody, T.S.; Alsteens, D. Reovirus directly engages integrin to recruit clathrin for entry into host cells. Nat. Commun. 2021, 12, 2149. [Google Scholar] [CrossRef]
- Yang, J.; Petitjean, S.J.L.; Koehler, M.; Zhang, Q.; Dumitru, A.C.; Chen, W.; Derclaye, S.; Vincent, S.P.; Soumillion, P.; Alsteens, D. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Nat. Commun. 2020, 11, 4541. [Google Scholar] [CrossRef]
- Steffens, C.M.; Hope, T.J. Mobility of the Human Immunodeficiency Virus (HIV) Receptor CD4 and Coreceptor CCR5 in Living Cells: Implications for HIV Fusion and Entry Events. J. Virol. 2004, 78, 9573–9578. [Google Scholar] [CrossRef]
- Rogers, G.N.; Paulson, J.C. Receptor determinants of human and animal influenza virus isolates: Differences in receptor specificity of the H3 hemagglutinin based on species of origin. Virology 1983, 127, 361–373. [Google Scholar] [CrossRef]
- Connor, R.J.; Kawaoka, Y.; Webster, R.G.; Paulson, J.C. Receptor Specificity in Human, Avian, and Equine H2 and H3 Influenza Virus Isolates. Virology 1994, 205, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.; Blixt, O.; Glaser, L.; Taubenberger, J.K.; Palese, P.; Paulson, J.C.; Wilson, I.A. Glycan Microarray Analysis of the Hemagglutinins from Modern and Pandemic Influenza Viruses Reveals Different Receptor Specificities. J. Mol. Biol. 2006, 355, 1143–1155. [Google Scholar] [CrossRef]
- Palacios-Rápalo, S.N.; De Jesús-González, L.A.; Cordero-Rivera, C.D.; Farfan-Morales, C.N.; Osuna-Ramos, J.F.; Martínez-Mier, G.; Quistián-Galván, J.; Muñoz-Pérez, A.; Bernal-Dolores, V.; del Ángel, R.M.; et al. Cholesterol-Rich Lipid Rafts as Platforms for SARS-CoV-2 Entry. Front. Immunol. 2021, 12, 796855. [Google Scholar] [CrossRef]
- Li, Y.-J.; Chen, C.-Y.; Yang, J.-H.; Chiu, Y.-F. Modulating cholesterol-rich lipid rafts to disrupt influenza A virus infection. Front. Immunol. 2022, 13, 982264. [Google Scholar] [CrossRef]
- Peruzzu, D.; Amendola, A.; Venturi, G.; de Turris, V.; Marsili, G.; Fortuna, C.; Fecchi, K.; Gagliardi, M.C. Zika Virus Exploits Lipid Rafts to Infect Host Cells. Viruses 2022, 14, 2059. [Google Scholar] [CrossRef]
- Rex, D.A.B.; Keshava Prasad, T.S.; Kandasamy, R.K. Revisiting Regulated Cell Death Responses in Viral Infections. Int. J. Mol. Sci. 2022, 23, 7023. [Google Scholar] [CrossRef] [PubMed]
- Scarcella, M.; d’Angelo, D.; Ciampa, M.; Tafuri, S.; Avallone, L.; Pavone, L.M.; De Pasquale, V. The Key Role of Lysosomal Protease Cathepsins in Viral Infections. Int. J. Mol. Sci. 2022, 23, 9089. [Google Scholar] [CrossRef]
- Roe, B.; Kensicki, E.; Mohney, R.; Hall, W.W. Metabolomic Profile of Hepatitis C Virus-Infected Hepatocytes. PLoS ONE 2011, 6, e23641. [Google Scholar] [CrossRef] [PubMed]
- Moriishi, K.; Matsuura, Y. Exploitation of Lipid Components by Viral and Host Proteins for Hepatitis C Virus Infection. Front. Microbiol. 2012, 3, 54. [Google Scholar] [CrossRef] [PubMed]
- Kellermann, M.; Scharte, F.; Hensel, M. Manipulation of Host Cell Organelles by Intracellular Pathogens. Int. J. Mol. Sci. 2021, 22, 6484. [Google Scholar] [CrossRef]
- Neufeldt, C.J.; Joyce, M.A.; Van Buuren, N.; Levin, A.; Kirkegaard, K.; Gale, M., Jr.; Tyrrell, D.L.; Wozniak, R.W. The Hepatitis C Virus-Induced Membranous Web and Associated Nuclear Transport Machinery Limit Access of Pattern Recognition Receptors to Viral Replication Sites. PLoS Pathog. 2016, 12, e1005428. [Google Scholar] [CrossRef]
- Gouttenoire, J.; Penin, F.; Moradpour, D. Hepatitis C virus nonstructural protein 4B: A journey into unexplored territory. Rev. Med. Virol. 2010, 20, 117–129. [Google Scholar] [CrossRef]
- He, Y.; Staschke, K.A.; Tan, S.L. HCV NS5A: A Multifunctional Regulator of Cellular Pathways and Virus Replication. In Hepatitis C Viruses: Genomes and Molecular Biology; Tan, S.L., Ed.; Horizon Bioscience: Norfolk, UK, 2006. [Google Scholar]
- Huang, J.T.; Tseng, C.P.; Liao, M.H.; Lu, S.C.; Yeh, W.Z.; Sakamoto, N.; Chen, C.M.; Cheng, J.C. Hepatitis C virus replication is modulated by the interaction of nonstructural protein NS5B and fatty acid synthase. J. Virol. 2013, 87, 4994–5004. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.-M. Chapter Five—Hepatitis C Virus: Standard-of-Care Treatment. In Advances in Pharmacology; De Clercq, E., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 67, pp. 169–215. [Google Scholar]
- An, N.; Ge, Q.; Shao, H.; Li, Q.; Guo, F.; Liang, C.; Li, X.; Yi, D.; Yang, L.; Cen, S. Interferon-inducible SAMHD1 restricts viral replication through downregulation of lipid synthesis. Front. Immunol. 2022, 13, 1007718. [Google Scholar] [CrossRef] [PubMed]
- Aktepe, T.E.; Liebscher, S.; Prier, J.E.; Simmons, C.P.; Mackenzie, J.M. The Host Protein Reticulon 3.1A Is Utilized by Flaviviruses to Facilitate Membrane Remodelling. Cell Rep. 2017, 21, 1639–1654. [Google Scholar] [CrossRef]
- Peng, Y.; Yang, Y.; Li, Y.; Shi, T.; Luan, Y.; Yin, C. Exosome and virus infection. Front. Immunol. 2023, 14, 1154217. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, L.; Liang, W.; Liu, S.; Deng, W.; Liu, Y.; Liu, Y.; Song, M.; Guo, K.; Zhang, Y. Extracellular vesicles originating from autophagy mediate an antibody-resistant spread of classical swine fever virus in cell culture. Autophagy 2022, 18, 1433–1449. [Google Scholar] [CrossRef] [PubMed]
- Aydin, Y.; Koksal, A.R.; Reddy, V.; Lin, D.; Osman, H.; Heidari, Z.; Rhadhi, S.M.; Wimley, W.C.; Parsi, M.A.; Dash, S. Extracellular Vesicle Release Promotes Viral Replication during Persistent HCV Infection. Cells 2021, 10, 984. [Google Scholar] [CrossRef]
- Nikolic, D.S.; Lehmann, M.; Felts, R.; Garcia, E.; Blanchet, F.P.; Subramaniam, S.; Piguet, V. HIV-1 activates Cdc42 and induces membrane extensions in immature dendritic cells to facilitate cell-to-cell virus propagation. Blood 2011, 118, 4841–4852. [Google Scholar] [CrossRef]
- Businger, R.; Kivimäki, S.; Simeonov, S.; Vavouras Syrigos, G.; Pohlmann, J.; Bolz, M.; Müller, P.; Codrea, M.C.; Templin, C.; Messerle, M.; et al. Comprehensive Analysis of Human Cytomegalovirus- and HIV-Mediated Plasma Membrane Remodeling in Macrophages. mBio 2021, 12, e0177021. [Google Scholar] [CrossRef]
- Hsu, J.-L.; van den Boomen, D.J.H.; Tomasec, P.; Weekes, M.P.; Antrobus, R.; Stanton, R.J.; Ruckova, E.; Sugrue, D.; Wilkie, G.S.; Davison, A.J.; et al. Plasma Membrane Profiling Defines an Expanded Class of Cell Surface Proteins Selectively Targeted for Degradation by HCMV US2 in Cooperation with UL141. PLoS Pathog. 2015, 11, e1004811. [Google Scholar] [CrossRef]
- Griffiths, P.; Reeves, M. Pathogenesis of human cytomegalovirus in the immunocompromised host. Nat. Rev. Microbiol. 2021, 19, 759–773. [Google Scholar] [CrossRef]
- McCurdy, L.H.; Graham, B.S. Role of plasma membrane lipid microdomains in respiratory syncytial virus filament formation. J. Virol. 2003, 77, 1747–1756. [Google Scholar] [CrossRef]
- Linfield, D.T.; Gao, N.; Raduka, A.; Harford, T.J.; Piedimonte, G.; Rezaee, F. RSV attenuates epithelial cell restitution by inhibiting actin cytoskeleton-dependent cell migration. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2021, 321, L189–L203. [Google Scholar] [CrossRef]
- Vanover, D.; Smith, D.V.; Blanchard, E.L.; Alonas, E.; Kirschman, J.L.; Lifland, A.W.; Zurla, C.; Santangelo, P.J. RSV glycoprotein and genomic RNA dynamics reveal filament assembly prior to the plasma membrane. Nat. Commun. 2017, 8, 667. [Google Scholar] [CrossRef]
- Soh, T.K.; Davies, C.T.R.; Muenzner, J.; Hunter, L.M.; Barrow, H.G.; Connor, V.; Bouton, C.R.; Smith, C.; Emmott, E.; Antrobus, R.; et al. Temporal Proteomic Analysis of Herpes Simplex Virus 1 Infection Reveals Cell-Surface Remodeling via pUL56-Mediated GOPC Degradation. Cell Rep. 2020, 33, 108235. [Google Scholar] [CrossRef]
- Weed, D.J.; Nicola, A.V. Herpes simplex virus Membrane Fusion. Adv. Anat. Embryol. Cell Biol. 2017, 223, 29–47. [Google Scholar] [CrossRef]
- Naamati, A.; Williamson, J.C.; Greenwood, E.J.D.; Marelli, S.; Lehner, P.J.; Matheson, N.J. Functional proteomic atlas of HIV infection in primary human CD4+ T cells. eLife 2019, 8, e41431. [Google Scholar] [CrossRef]
- Kwon, Y.; Kaake, R.M.; Echeverria, I.; Suarez, M.; Karimian Shamsabadi, M.; Stoneham, C.; Ramirez, P.W.; Kress, J.; Singh, R.; Sali, A.; et al. Structural basis of CD4 downregulation by HIV-1 Nef. Nat. Struct. Mol. Biol. 2020, 27, 822–828. [Google Scholar] [CrossRef]
- Chang-Graham, A.L.; Perry, J.L.; Engevik, M.A.; Engevik, K.A.; Scribano, F.J.; Gebert, J.T.; Danhof, H.A.; Nelson, J.C.; Kellen, J.S.; Strtak, A.C.; et al. Rotavirus induces intercellular calcium waves through ADP signaling. Science 2020, 370, eabc3621. [Google Scholar] [CrossRef]
- Felli, C.; Vincentini, O.; Silano, M.; Masotti, A. HIV-1 Nef Signaling in Intestinal Mucosa Epithelium Suggests the Existence of an Active Inter-kingdom Crosstalk Mediated by Exosomes. Front. Microbiol. 2017, 8, 1022. [Google Scholar] [CrossRef]
- Hassan, Z.; Kumar, N.D.; Reggiori, F.; Khan, G. How Viruses Hijack and Modify the Secretory Transport Pathway. Cells 2021, 10, 2535. [Google Scholar] [CrossRef]
- Das, A.; Rivera-Serrano, E.E.; Yin, X.; Walker, C.M.; Feng, Z.; Lemon, S.M. Cell entry and release of quasi-enveloped human hepatitis viruses. Nat. Rev. Microbiol. 2023, 21, 573–589. [Google Scholar] [CrossRef]
- Motsa, B.B.; Stahelin, R.V. Lipid-protein interactions in virus assembly and budding from the host cell plasma membrane. Biochem. Soc. Trans. 2021, 49, 1633–1641. [Google Scholar] [CrossRef]
- Crawford, S.E.; Criglar, J.M.; Liu, Z.; Broughman, J.R.; Estes, M.K. COPII Vesicle Transport Is Required for Rotavirus NSP4 Interaction with the Autophagy Protein LC3 II and Trafficking to Viroplasms. J. Virol. 2019, 94, e01341-19. [Google Scholar] [CrossRef]
- Bagchi, P.; Torres, M.; Qi, L.; Tsai, B. Selective EMC subunits act as molecular tethers of intracellular organelles exploited during viral entry. Nat. Commun. 2020, 11, 1127. [Google Scholar] [CrossRef]
- Wei, X.; Li, R.; Qiao, S.; Chen, X.-x.; Xing, G.; Zhang, G. Porcine Reproductive and Respiratory Syndrome Virus Utilizes Viral Apoptotic Mimicry as an Alternative Pathway To Infect Host Cells. J. Virol. 2020, 94, e00709-20. [Google Scholar] [CrossRef]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef]
- Neil, S.J.D.; Eastman, S.W.; Jouvenet, N.; Bieniasz, P.D. HIV-1 Vpu Promotes Release and Prevents Endocytosis of Nascent Retrovirus Particles from the Plasma Membrane. PLoS Pathog. 2006, 2, e39. [Google Scholar] [CrossRef]
- López-Huertas, M.R.; Callejas, S.; Abia, D.; Mateos, E.; Dopazo, A.; Alcamí, J.; Coiras, M. Modifications in host cell cytoskeleton structure and function mediated by intracellular HIV-1 Tat protein are greatly dependent on the second coding exon. Nucleic Acids Res. 2010, 38, 3287–3307. [Google Scholar] [CrossRef]
- García-Dorival, I.; Cuesta-Geijo, M.Á.; Galindo, I.; del Puerto, A.; Barrado-Gil, L.; Urquiza, J.; Alonso, C. Elucidation of the Cellular Interactome of African Swine Fever Virus Fusion Proteins and Identification of Potential Therapeutic Targets. Viruses 2023, 15, 1098. [Google Scholar] [CrossRef]
- Bhagwat, A.R.; Le Sage, V.; Nturibi, E.; Kulej, K.; Jones, J.; Guo, M.; Tae Kim, E.; Garcia, B.A.; Weitzman, M.D.; Shroff, H.; et al. Quantitative live cell imaging reveals influenza virus manipulation of Rab11A transport through reduced dynein association. Nat. Commun. 2020, 11, 23. [Google Scholar] [CrossRef]
- Fukuda, M.; Longnecker, R. Epstein-Barr Virus (EBV) Latent Membrane Protein 2A Regulates B-Cell Receptor-Induced Apoptosis and EBV Reactivation through Tyrosine Phosphorylation. J. Virol. 2005, 79, 8655–8660. [Google Scholar] [CrossRef]
- Steffen, A.; Reusch, B.; Gruteser, N.; Mainz, D.; Roncarati, R.; Baumann, A.; Stradal, T.E.B.; Knebel-Mörsdorf, D. Baculovirus Actin Rearrangement-Inducing Factor 1 Can Remodel the Mammalian Actin Cytoskeleton. Microbiol. Spectr. 2023, 11, e05189-22. [Google Scholar] [CrossRef]
- Denes, C.E.; Miranda-Saksena, M.; Cunningham, A.L.; Diefenbach, R.J. Cytoskeletons in the Closet-Subversion in Alphaherpesvirus Infections. Viruses 2018, 10, 79. [Google Scholar] [CrossRef]
- Jolly, C.; Mitar, I.; Sattentau Quentin, J. Requirement for an Intact T-Cell Actin and Tubulin Cytoskeleton for Efficient Assembly and Spread of Human Immunodeficiency Virus Type 1. J. Virol. 2007, 81, 5547–5560. [Google Scholar] [CrossRef]
- De Conto, F.; Fazzi, A.; Razin, S.V.; Arcangeletti, M.C.; Medici, M.C.; Belletti, S.; Chezzi, C.; Calderaro, A. Mammalian Diaphanous-related formin-1 restricts early phases of influenza A/NWS/33 virus (H1N1) infection in LLC-MK2 cells by affecting cytoskeleton dynamics. Mol. Cell. Biochem. 2018, 437, 185–201. [Google Scholar] [CrossRef]
- Scherer, K.M.; Manton, J.D.; Soh, T.K.; Mascheroni, L.; Connor, V.; Crump, C.M.; Kaminski, C.F. A fluorescent reporter system enables spatiotemporal analysis of host cell modification during herpes simplex virus-1 replication. J. Biol. Chem. 2021, 296, 100236. [Google Scholar] [CrossRef]
- Condemine, W.; Eguether, T.; Couroussé, N.; Etchebest, C.; Gardet, A.; Trugnan, G.; Chwetzoff, S. The C Terminus of Rotavirus VP4 Protein Contains an Actin Binding Domain Which Requires Cooperation with the Coiled-Coil Domain for Actin Remodeling. J. Virol. 2018, 93, e01598-18. [Google Scholar] [CrossRef]
- Chevalier, S.A.; Turpin, J.; Cachat, A.; Afonso, P.V.; Gessain, A.; Brady, J.N.; Pise-Masison, C.A.; Mahieux, R. Gem-Induced Cytoskeleton Remodeling Increases Cellular Migration of HTLV-1-Infected Cells, Formation of Infected-to-Target T-Cell Conjugates and Viral Transmission. PLoS Pathog. 2014, 10, e1003917. [Google Scholar] [CrossRef]
- Xie, W.; Chen, M.; Zhai, Z.; Li, H.; Song, T.; Zhu, Y.; Dong, D.; Zhou, P.; Duan, L.; Zhang, Y.; et al. HIV-1 exposure promotes PKG1-mediated phosphorylation and degradation of stathmin to increase epithelial barrier permeability. J. Biol. Chem. 2021, 296, 100644. [Google Scholar] [CrossRef]
- Xie, W.; Li, D.; Dong, D.; Li, Y.; Zhang, Y.; Duan, L.; Liu, X.; Meng, W.; Liu, M.; Zhou, J. HIV-1 exposure triggers autophagic degradation of stathmin and hyperstabilization of microtubules to disrupt epithelial cell junctions. Signal Transduct. Target. Ther. 2020, 5, 79. [Google Scholar] [CrossRef]
- Haffar, O.K.; Popov, S.; Dubrovsky, L.; Agostini, I.; Tang, H.; Pushkarsky, T.; Nadler, S.G.; Bukrinsky, M. Two nuclear localization signals in the HIV-1 matrix protein regulate nuclear import of the HIV-1 pre-integration complex. J. Mol. Biol. 2000, 299, 359–368. [Google Scholar] [CrossRef]
- Wu, W.W.H.; Sun, Y.-H.B.; Panté, N. Nuclear import of influenza A viral ribonucleoprotein complexes is mediated by two nuclear localization sequences on viral nucleoprotein. Virol. J. 2007, 4, 49. [Google Scholar] [CrossRef]
- Sodeik, B.; Ebersold, M.W.; Helenius, A. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 1997, 136, 1007–1021. [Google Scholar] [CrossRef]
- Trotman, L.C.; Mosberger, N.; Fornerod, M.; Stidwill, R.P.; Greber, U.F. Import of adenovirus DNA involves the nuclear pore complex receptor CAN/Nup214 and histone H1. Nat. Cell Biol. 2001, 3, 1092–1100. [Google Scholar] [CrossRef]
- Schmitz, A.; Schwarz, A.; Foss, M.; Zhou, L.; Rabe, B.; Hoellenriegel, J.; Stoeber, M.; Panté, N.; Kann, M. Nucleoporin 153 arrests the nuclear import of hepatitis B virus capsids in the nuclear basket. PLoS Pathog. 2010, 6, e1000741. [Google Scholar] [CrossRef]
- Cohen, S.; Behzad, A.R.; Carroll, J.B.; Panté, N. Parvoviral nuclear import: Bypassing the host nuclear-transport machinery. J. Gen. Virol. 2006, 87, 3209–3213. [Google Scholar] [CrossRef]
- Callé, A.; Ugrinova, I.; Epstein, A.L.; Bouvet, P.; Diaz, J.J.; Greco, A. Nucleolin is required for an efficient herpes simplex virus type 1 infection. J. Virol. 2008, 82, 4762–4773. [Google Scholar] [CrossRef]
- Matthews, D.A. Adenovirus protein V induces redistribution of nucleolin and B23 from nucleolus to cytoplasm. J. Virol. 2001, 75, 1031–1038. [Google Scholar] [CrossRef]
- Matthews, D.; Emmott, E.; Hiscox, J. Viruses and the Nucleolus. In The Nucleolus; Olson, M.O.J., Ed.; Springer: New York, NY, USA, 2011; pp. 321–345. [Google Scholar]
- Arizala, J.A.C.; Takahashi, M.; Burnett, J.C.; Ouellet, D.L.; Li, H.; Rossi, J.J. Nucleolar Localization of HIV-1 Rev Is Required, Yet Insufficient for Production of Infectious Viral Particles. AIDS Res. Hum. Retrovir. 2018, 34, 961–981. [Google Scholar] [CrossRef]
- Fitzgerald, K.D.; Semler, B.L. Re-localization of Cellular Protein SRp20 during Poliovirus Infection: Bridging a Viral IRES to the Host Cell Translation Apparatus. PLoS Pathog. 2011, 7, e1002127. [Google Scholar] [CrossRef] [PubMed]
- Groppo, R.; Brown, B.A.; Palmenberg, A.C. Mutational analysis of the EMCV 2A protein identifies a nuclear localization signal and an eIF4E binding site. Virology 2011, 410, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Maul, G.G. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J. 1994, 13, 5062–5069. [Google Scholar] [CrossRef] [PubMed]
- Doucas, V.; Ishov, A.M.; Romo, A.; Juguilon, H.; Weitzman, M.D.; Evans, R.M.; Maul, G.G. Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes. Dev. 1996, 10, 196–207. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; de Thé, H. PML nuclear bodies. Cold Spring Harb. Perspect. Biol. 2010, 2, a000661. [Google Scholar] [CrossRef]
- Lettin, L.; Erbay, B.; Blair, G.E. Viruses and Cajal Bodies: A Critical Cellular Target in Virus Infection? Viruses 2023, 15, 2311. [Google Scholar] [CrossRef]
- Horníková, L.; Bruštíková, K.; Huérfano, S.; Forstová, J. Nuclear Cytoskeleton in Virus Infection. Int. J. Mol. Sci. 2022, 23, 578. [Google Scholar] [CrossRef]
- Lieberman, P.M. Chromatin regulation of virus infection. Trends Microbiol. 2006, 14, 132–140. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Fagone, P.; Jackowski, S. Membrane phospholipid synthesis and endoplasmic reticulum function. J. Lipid Res. 2009, 50, S311–S316. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.W.; Nicchitta, C.V. Diversity and selectivity in mRNA translation on the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2015, 16, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Penke, B.; Bogar, F.; Fulop, L. Protein Folding and Misfolding, Endoplasmic Reticulum Stress in Neurodegenerative Diseases: In Trace of Novel Drug Targets. Curr. Protein Pept. Sci. 2016, 17, 169–182. [Google Scholar] [CrossRef]
- Cortese, M.; Goellner, S.; Acosta, E.G.; Neufeldt, C.J.; Oleksiuk, O.; Lampe, M.; Haselmann, U.; Funaya, C.; Schieber, N.; Ronchi, P.; et al. Ultrastructural Characterization of Zika Virus Replication Factories. Cell Rep. 2017, 18, 2113–2123. [Google Scholar] [CrossRef] [PubMed]
- Roingeard, P.; Eymieux, S.; Burlaud-Gaillard, J.; Hourioux, C.; Patient, R.; Blanchard, E. The double-membrane vesicle (DMV): A virus-induced organelle dedicated to the replication of SARS-CoV-2 and other positive-sense single-stranded RNA viruses. Cell Mol. Life Sci. 2022, 79, 425. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, M.; Morita, E. Flavivirus Replication Organelle Biogenesis in the Endoplasmic Reticulum: Comparison with Other Single-Stranded Positive-Sense RNA Viruses. Int. J. Mol. Sci. 2019, 20, 2336. [Google Scholar] [CrossRef] [PubMed]
- Snijder, E.J.; Limpens, R.; de Wilde, A.H.; de Jong, A.W.M.; Zevenhoven-Dobbe, J.C.; Maier, H.J.; Faas, F.; Koster, A.J.; Barcena, M. A unifying structural and functional model of the coronavirus replication organelle: Tracking down RNA synthesis. PLoS Biol. 2020, 18, e3000715. [Google Scholar] [CrossRef] [PubMed]
- Wolff, G.; Melia, C.E.; Snijder, E.J.; Barcena, M. Double-Membrane Vesicles as Platforms for Viral Replication. Trends Microbiol. 2020, 28, 1022–1033. [Google Scholar] [CrossRef]
- Nagy, P.D.; Strating, J.R.; van Kuppeveld, F.J. Building Viral Replication Organelles: Close Encounters of the Membrane Types. PLoS Pathog. 2016, 12, e1005912. [Google Scholar] [CrossRef]
- Paul, D.; Bartenschlager, R. Architecture and biogenesis of plus-strand RNA virus replication factories. World J. Virol. 2013, 2, 32–48. [Google Scholar] [CrossRef]
- Pedersen, K.W.; van der Meer, Y.; Roos, N.; Snijder, E.J. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J. Virol. 1999, 73, 2016–2026. [Google Scholar] [CrossRef]
- Snijder, E.J.; van der Meer, Y.; Zevenhoven-Dobbe, J.; Onderwater, J.J.; van der Meulen, J.; Koerten, H.K.; Mommaas, A.M. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 2006, 80, 5927–5940. [Google Scholar] [CrossRef]
- Laurent, T.; Kumar, P.; Liese, S.; Zare, F.; Jonasson, M.; Carlson, A.; Carlson, L.A. Architecture of the chikungunya virus replication organelle. Elife 2022, 11, e83042. [Google Scholar] [CrossRef]
- Maier, H.J.; Hawes, P.C.; Keep, S.M.; Britton, P. Spherules and IBV. Bioengineered 2014, 5, 288–292. [Google Scholar] [CrossRef]
- Mihelc, E.M.; Baker, S.C.; Lanman, J.K. Coronavirus infection induces progressive restructuring of the endoplasmic reticulum involving the formation and degradation of double membrane vesicles. Virology 2021, 556, 9–22. [Google Scholar] [CrossRef]
- Huttunen, M.; Waris, M.; Kajander, R.; Hyypia, T.; Marjomaki, V. Coxsackievirus A9 infects cells via nonacidic multivesicular bodies. J. Virol. 2014, 88, 5138–5151. [Google Scholar] [CrossRef]
- Kinast, V.; Plociennikowska, A.; Anggakusuma; Bracht, T.; Todt, D.; Brown, R.J.P.; Boldanova, T.; Zhang, Y.; Bruggemann, Y.; Friesland, M.; et al. C19orf66 is an interferon-induced inhibitor of HCV replication that restricts formation of the viral replication organelle. J. Hepatol. 2020, 73, 549–558. [Google Scholar] [CrossRef]
- Bamunusinghe, D.; Chaturvedi, S.; Seo, J.K.; Rao, A.L. Mutations in the capsid protein of Brome mosaic virus affecting encapsidation eliminate vesicle induction in planta: Implications for virus cell-to-cell spread. J. Virol. 2013, 87, 8982–8992. [Google Scholar] [CrossRef]
- Cao, X.; Jin, X.; Zhang, X.; Li, Y.; Wang, C.; Wang, X.; Hong, J.; Wang, X.; Li, D.; Zhang, Y. Morphogenesis of Endoplasmic Reticulum Membrane-Invaginated Vesicles during Beet Black Scorch Virus Infection: Role of Auxiliary Replication Protein and New Implications of Three-Dimensional Architecture. J. Virol. 2015, 89, 6184–6195. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.L. Ultrastructural studies of Kunjin virus-infected Aedes albopictus cells. J. Gen. Virol. 1987, 68 Pt 2, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Kaufusi, P.H.; Kelley, J.F.; Yanagihara, R.; Nerurkar, V.R. Induction of endoplasmic reticulum-derived replication-competent membrane structures by West Nile virus non-structural protein 4B. PLoS ONE 2014, 9, e84040. [Google Scholar] [CrossRef] [PubMed]
- Dunoyer, P.; Ritzenthaler, C.; Hemmer, O.; Michler, P.; Fritsch, C. Intracellular localization of the peanut clump virus replication complex in tobacco BY-2 protoplasts containing green fluorescent protein-labeled endoplasmic reticulum or Golgi apparatus. J. Virol. 2002, 76, 865–874. [Google Scholar] [CrossRef]
- Wan, J.; Basu, K.; Mui, J.; Vali, H.; Zheng, H.; Laliberte, J.F. Ultrastructural Characterization of Turnip Mosaic Virus-Induced Cellular Rearrangements Reveals Membrane-Bound Viral Particles Accumulating in Vacuoles. J. Virol. 2015, 89, 12441–12456. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tukachinsky, H.; Romano, F.B.; Rapoport, T.A. Cooperation of the ER-shaping proteins atlastin, lunapark, and reticulons to generate a tubular membrane network. eLife 2016, 5, e18605. [Google Scholar] [CrossRef] [PubMed]
- Monel, B.; Rajah, M.M.; Hafirassou, M.L.; Sid Ahmed, S.; Burlaud-Gaillard, J.; Zhu, P.P.; Nevers, Q.; Buchrieser, J.; Porrot, F.; Meunier, C.; et al. Atlastin Endoplasmic Reticulum-Shaping Proteins Facilitate Zika Virus Replication. J. Virol. 2019, 93, e01047-19. [Google Scholar] [CrossRef] [PubMed]
- Long, W.Y.; Zhao, G.H.; Wu, Y. Endoplasmic Reticulum-Shaping Atlastin Proteins Facilitate KSHV Replication. Front. Cell Infect. Microbiol. 2021, 11, 790243. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.M.; Chen, Y.J.; Cho, W.J.; Tai, A.W.; Tsai, B. Reticulons promote formation of ER-derived double-membrane vesicles that facilitate SARS-CoV-2 replication. J. Cell Biol. 2023, 222, e202203060. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, P.; Liu, X.; Cho, W.J.; Tsai, B. Lunapark-dependent formation of a virus-induced ER exit site contains multi-tubular ER junctions that promote viral ER-to-cytosol escape. Cell Rep. 2021, 37, 110077. [Google Scholar] [CrossRef]
- Scorrano, L.; De Matteis, M.A.; Emr, S.; Giordano, F.; Hajnóczky, G.; Kornmann, B.; Lackner, L.L.; Levine, T.P.; Pellegrini, L.; Reinisch, K.; et al. Coming together to define membrane contact sites. Nat. Commun. 2019, 10, 1287. [Google Scholar] [CrossRef]
- Wu, H.; Carvalho, P.; Voeltz, G.K. Here, there, and everywhere: The importance of ER membrane contact sites. Science 2018, 361, eaan5835. [Google Scholar] [CrossRef]
- Kim, S.; Coukos, R.; Gao, F.; Krainc, D. Dysregulation of organelle membrane contact sites in neurological diseases. Neuron 2022, 110, 2386–2408. [Google Scholar] [CrossRef]
- Chen, Y.; Williams, V.; Filippova, M.; Filippov, V.; Duerksen-Hughes, P. Viral carcinogenesis: Factors inducing DNA damage and virus integration. Cancers 2014, 6, 2155–2186. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef]
- Alavi, M.V.; Bette, S.; Schimpf, S.; Schuettauf, F.; Schraermeyer, U.; Wehrl, H.F.; Ruttiger, L.; Beck, S.C.; Tonagel, F.; Pichler, B.J.; et al. A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain 2007, 130, 1029–1042. [Google Scholar] [CrossRef]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Rahn, J.J.; Stackley, K.D.; Chan, S.S. Opa1 is required for proper mitochondrial metabolism in early development. PLoS ONE 2013, 8, e59218. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.C.; Hu, J.P. FISSION1A and FISSION1B proteins mediate the fission of peroxisomes and mitochondria in Arabidopsis. Mol. Plant 2008, 1, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Khan, M.; Syed, G.H.; Kim, S.J.; Siddiqui, A. Mitochondrial dynamics and viral infections: A close nexus. Biochim. Biophys. Acta 2015, 1853, 2822–2833. [Google Scholar] [CrossRef]
- Eisner, V.; Picard, M.; Hajnoczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef]
- Yu, C.Y.; Liang, J.J.; Li, J.K.; Lee, Y.L.; Chang, B.L.; Su, C.I.; Huang, W.J.; Lai, M.M.; Lin, Y.L. Dengue Virus Impairs Mitochondrial Fusion by Cleaving Mitofusins. PLoS Pathog. 2015, 11, e1005350. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef]
- Vilmen, G.; Glon, D.; Siracusano, G.; Lussignol, M.; Shao, Z.; Hernandez, E.; Perdiz, D.; Quignon, F.; Mouna, L.; Pous, C.; et al. BHRF1, a BCL2 viral homolog, disturbs mitochondrial dynamics and stimulates mitophagy to dampen type I IFN induction. Autophagy 2021, 17, 1296–1315. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, Y.; Ren, C.; Yang, S.; Tian, S.; Chen, H.; Jin, M.; Zhou, H. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy 2021, 17, 496–511. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Syed, G.H.; Siddiqui, A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 2013, 9, e1003285. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Zhang, L.; Li, Z.; Zhong, Y.; Tang, Q.; Qin, Y.; Chen, M. The Matrix Protein of Human Parainfluenza Virus Type 3 Induces Mitophagy that Suppresses Interferon Responses. Cell Host Microbe 2017, 21, 538–547 e534. [Google Scholar] [CrossRef] [PubMed]
- Pant, A.; Dsouza, L.; Cao, S.; Peng, C.; Yang, Z. Viral growth factor- and STAT3 signaling-dependent elevation of the TCA cycle intermediate levels during vaccinia virus infection. PLoS Pathog. 2021, 17, e1009303. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Roh, J.Y.; Ryu, J.; Shin, H.J.; Hong, E.J. Activation of TCA cycle restrains virus-metabolic hijacking and viral replication in mouse hepatitis virus-infected cells. Cell Biosci. 2022, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Sekiya, T.; Nomura, N.; Daito, T.J.; Shingai, M.; Kida, H. Influenza virus infection affects insulin signaling, fatty acid-metabolizing enzyme expressions, and the tricarboxylic acid cycle in mice. Sci. Rep. 2020, 10, 10879. [Google Scholar] [CrossRef]
- Masip, J.; Rallon, N.; Yeregui, E.; Olona, M.; Resino, S.; Benito, J.M.; Vilades, C.; Garcia-Pardo, G.; Alcami, J.; Ruiz-Mateos, E.; et al. Elevated alpha-Ketoglutaric Acid Concentrations and a Lipid-Balanced Signature Are the Key Factors in Long-Term HIV Control. Front. Immunol. 2022, 13, 822272. [Google Scholar] [CrossRef]
- Sun, N.; Shen, C.; Zhang, L.; Wu, X.; Yu, Y.; Yang, X.; Yang, C.; Zhong, C.; Gao, Z.; Miao, W.; et al. Hepatic Kruppel-like factor 16 (KLF16) targets PPARalpha to improve steatohepatitis and insulin resistance. Gut 2021, 70, 2183–2195. [Google Scholar] [CrossRef]
- Yamane, D.; Hayashi, Y.; Matsumoto, M.; Nakanishi, H.; Imagawa, H.; Kohara, M.; Lemon, S.M.; Ichi, I. FADS2-dependent fatty acid desaturation dictates cellular sensitivity to ferroptosis and permissiveness for hepatitis C virus replication. Cell Chem. Biol. 2022, 29, 799–810.e4. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Sorouri, M.; Chang, T.; Hancks Dustin, C. Mitochondria and Viral Infection: Advances and Emerging Battlefronts. mBio 2022, 13, e02096-21. [Google Scholar] [CrossRef] [PubMed]
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.X.; Chen, Z.J. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011, 146, 448–461. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, G.; Xu, Z.G.; Tu, H.; Hu, F.; Dai, J.; Chang, Y.; Chen, Y.; Lu, Y.; Zeng, H.; et al. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 2019, 178, 176–189.e15. [Google Scholar] [CrossRef] [PubMed]
- Abrantes, J.L.; Alves, C.M.; Costa, J.; Almeida, F.C.; Sola-Penna, M.; Fontes, C.F.; Souza, T.M. Herpes simplex type 1 activates glycolysis through engagement of the enzyme 6-phosphofructo-1-kinase (PFK-1). Biochim. Biophys. Acta 2012, 1822, 1198–1206. [Google Scholar] [CrossRef]
- Rajan, R.S.; Illing, M.E.; Bence, N.F.; Kopito, R.R. Specificity in intracellular protein aggregation and inclusion body formation. Proc. Natl. Acad. Sci. USA 2001, 98, 13060–13065. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Kriwacki, R.W. Phase separation in biology; functional organization of a higher order. Cell Commun. Signal 2016, 14, 1. [Google Scholar] [CrossRef]
- Guo, Q.; Shi, X.; Wang, X. RNA and liquid-liquid phase separation. Noncoding RNA Res. 2021, 6, 92–99. [Google Scholar] [CrossRef]
- Sagan, S.M.; Weber, S.C. Let’s phase it: Viruses are master architects of biomolecular condensates. Trends Biochem. Sci. 2023, 48, 229–243. [Google Scholar] [CrossRef]
- Perdikari, T.M.; Murthy, A.C.; Ryan, V.H.; Watters, S.; Naik, M.T.; Fawzi, N.L. SARS-CoV-2 nucleocapsid protein phase-separates with RNA and with human hnRNPs. EMBO J. 2020, 39, e106478. [Google Scholar] [CrossRef]
- Monette, A.; Niu, M.; Nijhoff Asser, M.; Gorelick, R.J.; Mouland, A.J. Scaffolding viral protein NC nucleates phase separation of the HIV-1 biomolecular condensate. Cell Rep. 2022, 40, 111251. [Google Scholar] [CrossRef]
- Visentin, A.; Demitroff, N.; Salgueiro, M.; Borkosky, S.S.; Uversky, V.N.; Camporeale, G.; de Prat-Gay, G. Assembly of the Tripartite and RNA Condensates of the Respiratory Syncytial Virus Factory Proteins In Vitro: Role of the Transcription Antiterminator M(2-1). Viruses 2023, 15, 1329. [Google Scholar] [CrossRef]
- Gaete-Argel, A.; Márquez, C.L.; Barriga, G.P.; Soto-Rifo, R.; Valiente-Echeverría, F. Strategies for Success. Viral Infections and Membraneless Organelles. Front. Cell. Infect. Microbiol. 2019, 9, 336. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Mittasch, M.; Gomes, B.F.; Klein, I.A.; Murcko, M.A. Modulating biomolecular condensates: A novel approach to drug discovery. Nat. Rev. Drug Discov. 2022, 21, 841–862. [Google Scholar] [CrossRef]
- Dolnik, O.; Gerresheim, G.K.; Biedenkopf, N. New Perspectives on the Biogenesis of Viral Inclusion Bodies in Negative-Sense RNA Virus Infections. Cells 2021, 10, 1460. [Google Scholar] [CrossRef]
- Heinrich, B.S.; Cureton, D.K.; Rahmeh, A.A.; Whelan, S.P. Protein expression redirects vesicular stomatitis virus RNA synthesis to cytoplasmic inclusions. PLoS Pathog. 2010, 6, e1000958. [Google Scholar] [CrossRef]
- Hoenen, T.; Shabman, R.S.; Groseth, A.; Herwig, A.; Weber, M.; Schudt, G.; Dolnik, O.; Basler, C.F.; Becker, S.; Feldmann, H. Inclusion bodies are a site of ebolavirus replication. J. Virol. 2012, 86, 11779–11788. [Google Scholar] [CrossRef] [PubMed]
- Guseva, S.; Milles, S.; Jensen, M.R.; Salvi, N.; Kleman, J.P.; Maurin, D.; Ruigrok, R.W.H.; Blackledge, M. Measles virus nucleo- and phosphoproteins form liquid-like phase-separated compartments that promote nucleocapsid assembly. Sci. Adv. 2020, 6, eaaz7095. [Google Scholar] [CrossRef]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of Cellular Autophagosomal Machinery by RNA Viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef]
- Alirezaei, M.; Flynn, C.T.; Wood, M.R.; Harkins, S.; Whitton, J.L. Coxsackievirus can exploit LC3 in both autophagy-dependent and -independent manners in vivo. Autophagy 2015, 11, 1389–1407. [Google Scholar] [CrossRef]
- Abernathy, E.; Mateo, R.; Majzoub, K.; van Buuren, N.; Bird, S.W.; Carette, J.E.; Kirkegaard, K. Differential and convergent utilization of autophagy components by positive-strand RNA viruses. PLoS Biol. 2019, 17, e2006926. [Google Scholar] [CrossRef]
- Scutigliani, E.M.; Kikkert, M. Interaction of the innate immune system with positive-strand RNA virus replication organelles. Cytokine Growth Factor. Rev. 2017, 37, 17–27. [Google Scholar] [CrossRef]
- Prentice, E.; Jerome, W.G.; Yoshimori, T.; Mizushima, N.; Denison, M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004, 279, 10136–10141. [Google Scholar] [CrossRef]
- Blanchard, E.; Roingeard, P. Virus-induced double-membrane vesicles. Cell. Microbiol. 2015, 17, 45–50. [Google Scholar] [CrossRef]
- Suhy, D.A.; Giddings, T.H., Jr.; Kirkegaard, K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: An autophagy-like origin for virus-induced vesicles. J. Virol. 2000, 74, 8953–8965. [Google Scholar] [CrossRef]
- O’Donnell, V.; Pacheco, J.M.; LaRocco, M.; Burrage, T.; Jackson, W.; Rodriguez, L.L.; Borca, M.V.; Baxt, B. Foot-and-mouth disease virus utilizes an autophagic pathway during viral replication. Virology 2011, 410, 142–150. [Google Scholar] [CrossRef]
- Robinson, S.M.; Tsueng, G.; Sin, J.; Mangale, V.; Rahawi, S.; McIntyre, L.L.; Williams, W.; Kha, N.; Cruz, C.; Hancock, B.M.; et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014, 10, e1004045. [Google Scholar] [CrossRef]
- Wong, J.; Zhang, J.; Si, X.; Gao, G.; Mao, I.; McManus, B.M.; Luo, H. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 2008, 82, 9143–9153. [Google Scholar] [CrossRef]
- Berryman, S.; Brooks, E.; Burman, A.; Hawes, P.; Roberts, R.; Netherton, C.; Monaghan, P.; Whelband, M.; Cottam, E.; Elazar, Z.; et al. Foot-and-mouth disease virus induces autophagosomes during cell entry via a class III phosphatidylinositol 3-kinase-independent pathway. J. Virol. 2012, 86, 12940–12953. [Google Scholar] [CrossRef]
- Koci, J.; Novotova, M.; Slavikova, M.; Klempa, B.; Zahradnik, I. SARS-CoV-2 Exploits Non-Canonical Autophagic Processes to Replicate, Mature, and Egress the Infected Vero E6 Cells. Pathogens 2022, 11, 1535. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Kastner, S.; Krijnse-Locker, J.; Buhler, S.; Bartenschlager, R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 2007, 282, 8873–8882. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, A.B.; Martin-Acebes, M.A.; Saiz, J.C. Amino acid substitutions in the non-structural proteins 4A or 4B modulate the induction of autophagy in West Nile virus infected cells independently of the activation of the unfolded protein response. Front. Microbiol. 2014, 5, 797. [Google Scholar] [CrossRef]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed]
- Lennemann, N.J.; Coyne, C.B. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy 2017, 13, 322–332. [Google Scholar] [CrossRef]
- Neufeldt, C.J.; Cortese, M.; Scaturro, P.; Cerikan, B.; Wideman, J.G.; Tabata, K.; Moraes, T.; Oleksiuk, O.; Pichlmair, A.; Bartenschlager, R. ER-shaping atlastin proteins act as central hubs to promote flavivirus replication and virion assembly. Nat. Microbiol. 2019, 4, 2416–2429. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Zhang, G.; Yang, X.; Zhang, S.; Chen, L.; Yan, Q.; Xu, M.; Banerjee, A.K.; Chen, M. Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe 2014, 15, 564–577. [Google Scholar] [CrossRef]
- Wang, L.; Tian, Y.; Ou, J.H. HCV induces the expression of Rubicon and UVRAG to temporally regulate the maturation of autophagosomes and viral replication. PLoS Pathog. 2015, 11, e1004764. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, E.M.; Jarosinski, K.W.; Jackson, W.; Carpenter, J.E.; Grose, C. Exocytosis of Varicella-Zoster Virus Virions Involves a Convergence of Endosomal and Autophagy Pathways. J. Virol. 2016, 90, 8673–8685. [Google Scholar] [CrossRef]
- Shrivastava, S.; Devhare, P.; Sujijantarat, N.; Steele, R.; Kwon, Y.C.; Ray, R.; Ray, R.B. Knockdown of Autophagy Inhibits Infectious Hepatitis C Virus Release by the Exosomal Pathway. J. Virol. 2016, 90, 1387–1396. [Google Scholar] [CrossRef]
- Lemberg, M.K.; Strisovsky, K. Maintenance of organellar protein homeostasis by ER-associated degradation and related mechanisms. Mol. Cell 2021, 81, 2507–2519. [Google Scholar] [CrossRef]
- Ciechanover, A.; Schwartz, A.L. The ubiquitin-proteasome pathway: The complexity and myriad functions of proteins death. Proc. Natl. Acad. Sci. USA 1998, 95, 2727–2730. [Google Scholar] [CrossRef]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Fregno, I.; Molinari, M. Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, X. Lysosome biogenesis: Regulation and functions. J. Cell Biol. 2021, 220, e202102001. [Google Scholar] [CrossRef]
- Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. Int. J. Mol. Sci. 2020, 21, 2576. [Google Scholar] [CrossRef]
- Fernandez de Castro, I.; Tenorio, R.; Ortega-Gonzalez, P.; Knowlton, J.J.; Zamora, P.F.; Lee, C.H.; Fernandez, J.J.; Dermody, T.S.; Risco, C. A modified lysosomal organelle mediates nonlytic egress of reovirus. J. Cell Biol. 2020, 219, e201910131. [Google Scholar] [CrossRef]
- Labadie, T.; Roy, P. A non-enveloped arbovirus released in lysosome-derived extracellular vesicles induces super-infection exclusion. PLoS Pathog. 2020, 16, e1009015. [Google Scholar] [CrossRef]
- Mindell, J.A. Lysosomal acidification mechanisms. Annu. Rev. Physiol. 2012, 74, 69–86. [Google Scholar] [CrossRef]
- Chen, D.; Zheng, Q.; Sun, L.; Ji, M.; Li, Y.; Deng, H.; Zhang, H. ORF3a of SARS-CoV-2 promotes lysosomal exocytosis-mediated viral egress. Dev. Cell 2021, 56, 3250–3263 e3255. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Kowal, J.; Tkach, M.; Théry, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef]
- Veettil, M.V.; Kumar, B.; Ansari, M.A.; Dutta, D.; Iqbal, J.; Gjyshi, O.; Bottero, V.; Chandran, B. ESCRT-0 Component Hrs Promotes Macropinocytosis of Kaposi’s Sarcoma-Associated Herpesvirus in Human Dermal Microvascular Endothelial Cells. J. Virol. 2016, 90, 3860–3872. [Google Scholar] [CrossRef]
- Kharkwal, H.; Smith, C.G.; Wilson, D.W. Herpes Simplex Virus Capsid Localization to ESCRT-VPS4 Complexes in the Presence and Absence of the Large Tegument Protein UL36p. J. Virol. 2016, 90, 7257–7267. [Google Scholar] [CrossRef]
- Johnson, D.C.; Baines, J.D. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 2011, 9, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Inoue, J.; Ninomiya, M.; Umetsu, T.; Nakamura, T.; Kogure, T.; Kakazu, E.; Iwata, T.; Takai, S.; Sano, A.; Fukuda, M.; et al. Small Interfering RNA Screening for the Small GTPase Rab Proteins Identifies Rab5B as a Major Regulator of Hepatitis B Virus Production. J. Virol. 2019, 93, e00621-19. [Google Scholar] [CrossRef] [PubMed]
- Zeyen, L.; Prange, R. Host Cell Rab GTPases in Hepatitis B Virus Infection. Front. Cell Dev. Biol. 2018, 6, 154. [Google Scholar] [CrossRef]
- Sanz-Ros, J.; Mas-Bargues, C.; Romero-García, N.; Huete-Acevedo, J.; Dromant, M.; Borrás, C. Extracellular Vesicles as Therapeutic Resources in the Clinical Environment. Int. J. Mol. Sci. 2023, 24, 2344. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, I.K.; Wood, M.J.A.; Fuhrmann, G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021, 16, 748–759. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Host Subcellular Organelles | Highlights | Viruses | References |
|---|---|---|---|
| ECM |
| HCV, CHIKV | [42,43,44,45,46,47,48,49,50,51,52,53] |
| RSV | [54,55,56,57,58] | |
| HP-PRRSV | [59] | |
| Cell Membrane |
| HCV | [78,79,80,81,82,83,84,85] |
| HIV, HCMV, etc. | [63,88,89,90,91,92,93,94,95,96,97,98] | |
| HIV | [99,100] | |
| Cytoskeleton |
| AcMNPV, HSV, etc. | [115,116,117,118,119,120,121,122,123] |
| Nucleus |
| HIV, HSV-1, etc. | [124,125,126,127,128,129] |
| HSV, HIV-1, etc. | [130,131,132,133,134,135,136,137,138] | |
| Endoplasmic Reticulum |
| BMV, BBSV, etc. | [149,160,161,162,163,164,165] |
| ZIKV, KSHV, etc. | [86,166,167,168,169] | |
| Mitochondria |
| HCV, HBV, etc. | [187,188,189] |
| HCV, HBV, etc. | [185,187,190,191,192,193,194,195] | |
| HIV, IV, etc. | [196,197,198,199,200,201] | |
| Cellular condensates |
| SARS-CoV-2, HIV-1, etc. | [210,211,212,213,214,215,216] |
| Autophagosome |
| SARS-CoV-2, MHV, etc. | [50,146,232,233,234,235,236,237,238] |
| HCV, HPIV3 | [238,239] | |
| Lysosome |
| SARS-CoV-1, SARS-CoV-2 | [249,250,251,252] |
| Extracellular Vesicles |
| HSV-1, HBV | [256,257,258,259] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, M.S.; Lee, D.-K.; Lee, C.-Y.; Park, S.-C.; Yang, J. Host Subcellular Organelles: Targets of Viral Manipulation. Int. J. Mol. Sci. 2024, 25, 1638. https://doi.org/10.3390/ijms25031638
Song MS, Lee D-K, Lee C-Y, Park S-C, Yang J. Host Subcellular Organelles: Targets of Viral Manipulation. International Journal of Molecular Sciences. 2024; 25(3):1638. https://doi.org/10.3390/ijms25031638
Chicago/Turabian StyleSong, Min Seok, Dong-Kun Lee, Chung-Young Lee, Sang-Cheol Park, and Jinsung Yang. 2024. "Host Subcellular Organelles: Targets of Viral Manipulation" International Journal of Molecular Sciences 25, no. 3: 1638. https://doi.org/10.3390/ijms25031638
APA StyleSong, M. S., Lee, D.-K., Lee, C.-Y., Park, S.-C., & Yang, J. (2024). Host Subcellular Organelles: Targets of Viral Manipulation. International Journal of Molecular Sciences, 25(3), 1638. https://doi.org/10.3390/ijms25031638