
Low-Density Neutrophils Contribute to Subclinical Inflammation in Patients with Type 2 Diabetes

Abstract
:1. Introduction
2. Results
2.1. Study Population
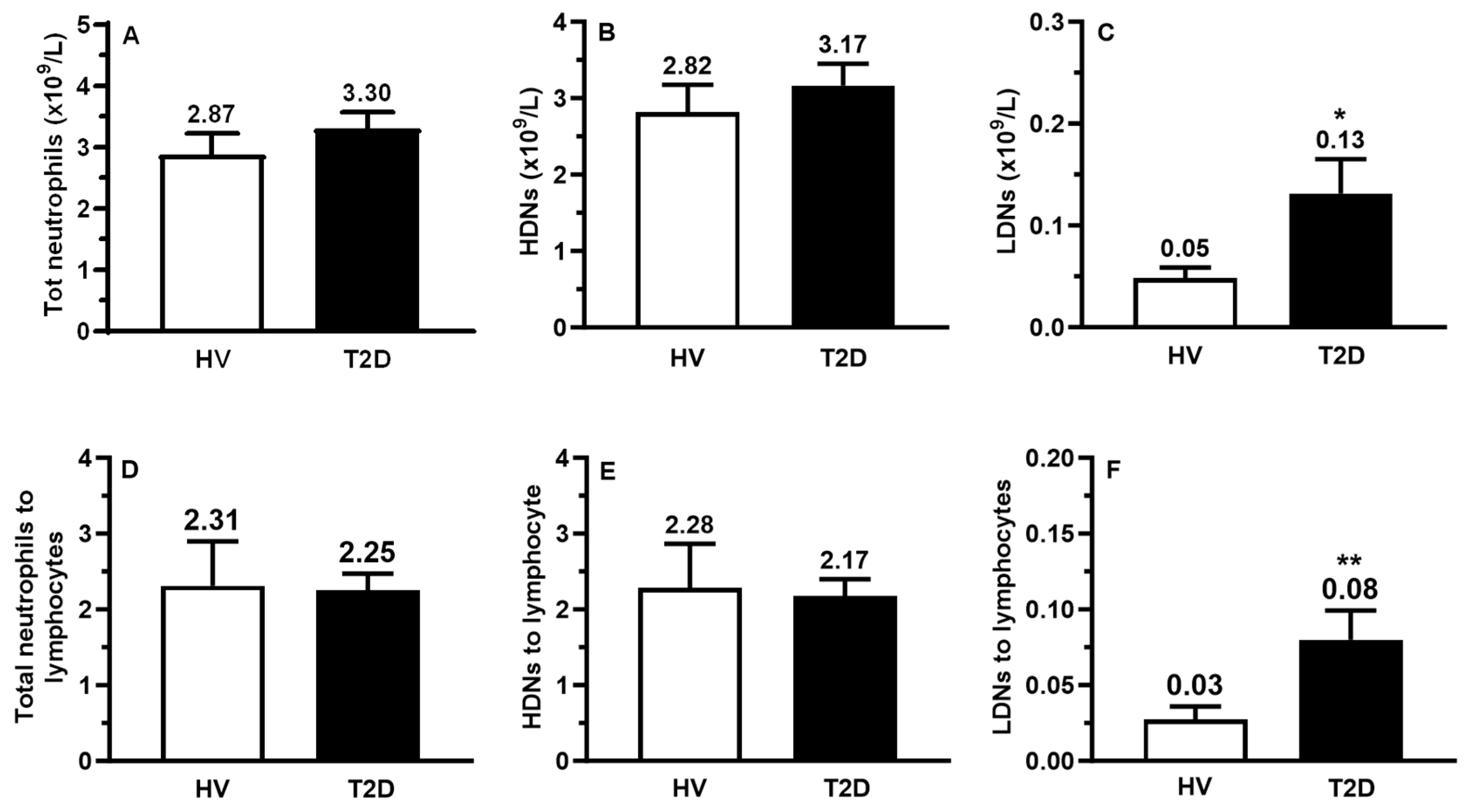
2.2. Circulating Counts of LDNs, HDNs, and Lymphocytes
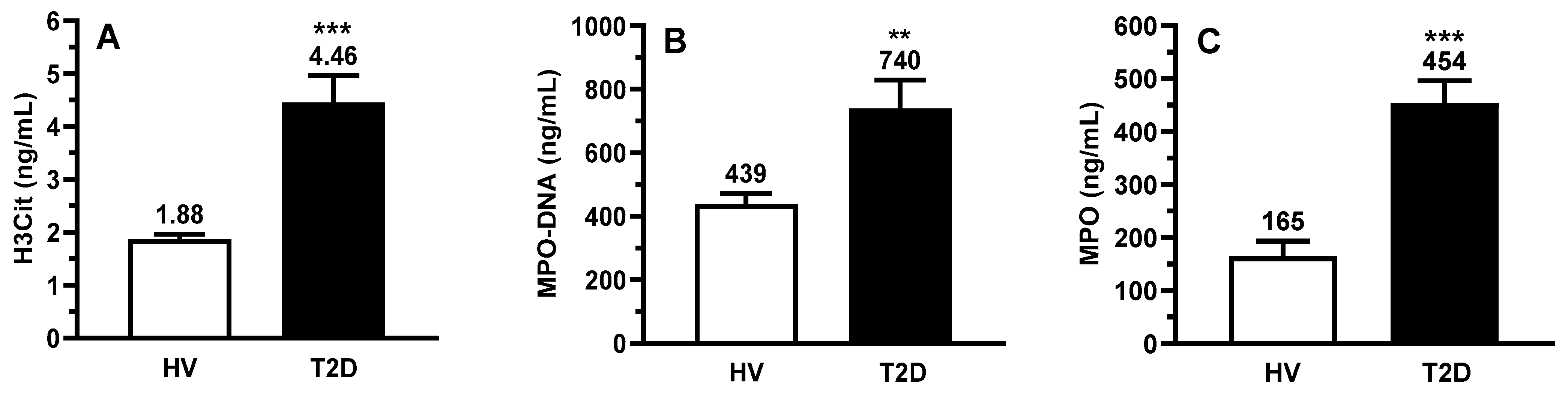
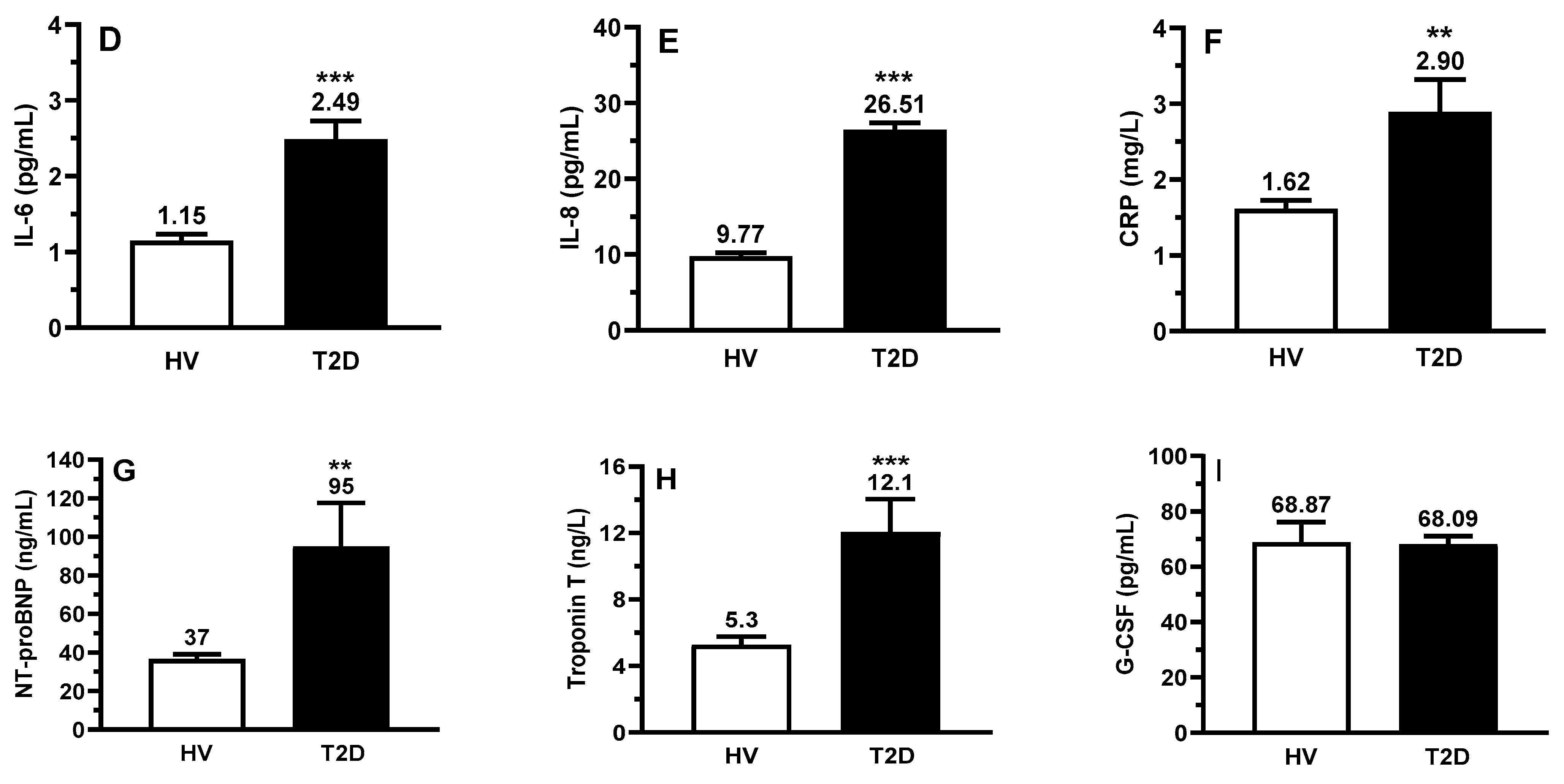
2.3. Circulating Biomarkers of Inflammation and NETs
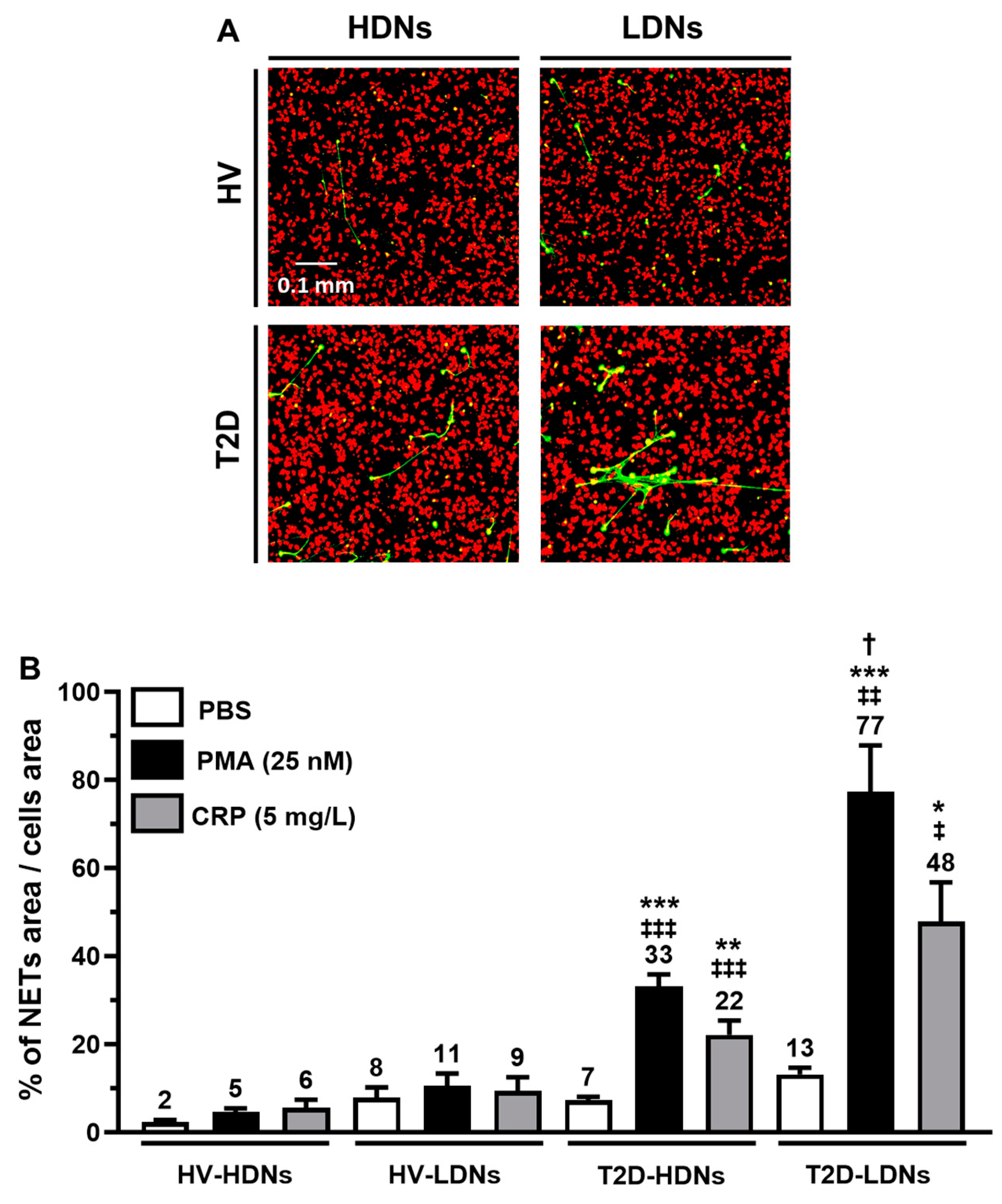
2.4. Basal NETosis in Isolated HDNs and LDNs
2.5. In Vitro NETosis from Stimulated HDNs and LDNs
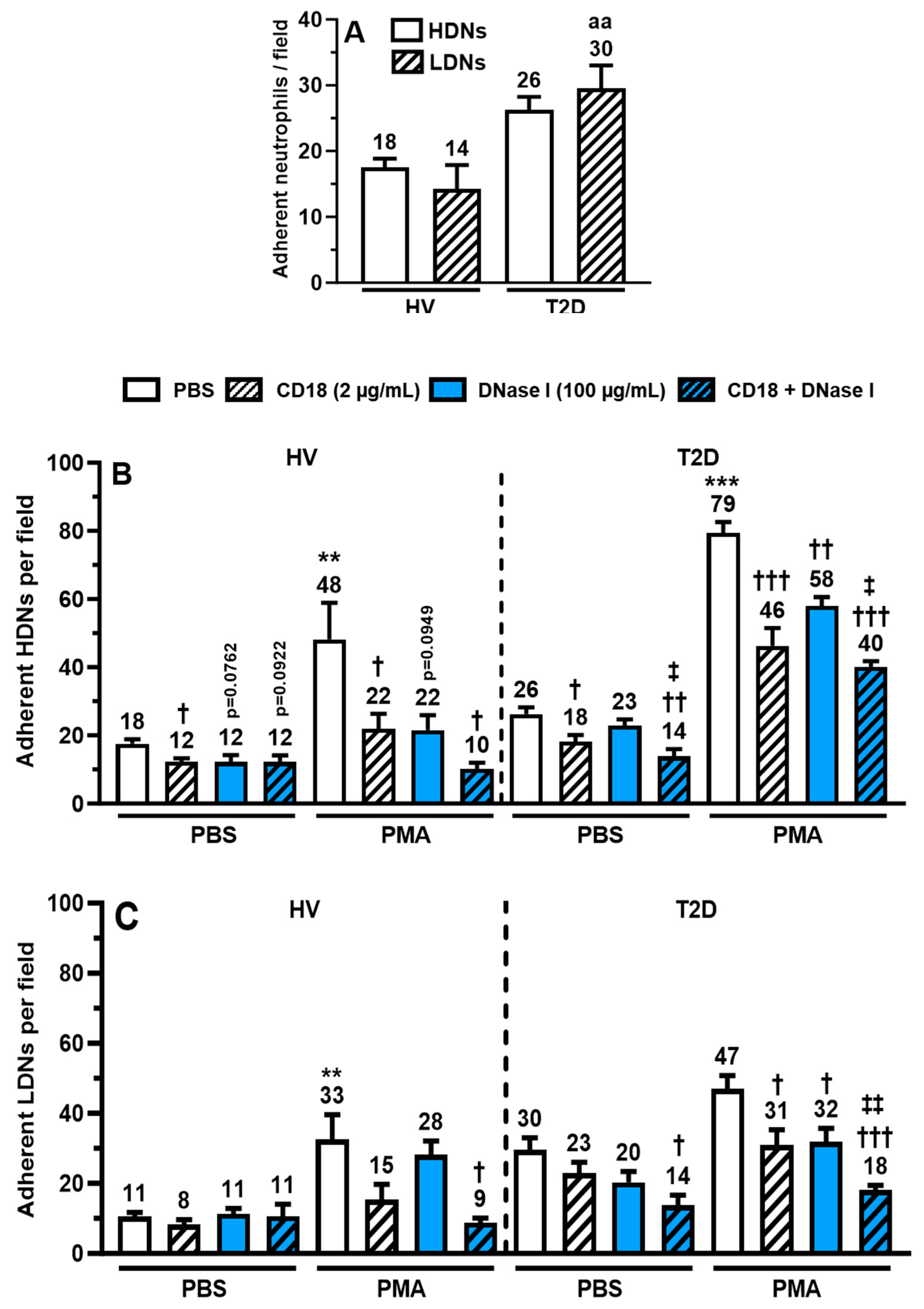
2.6. Binding of HDNs and LDNs onto Human Extracellular Matrix (hECM)
3. Discussion
4. Materials and Methods
4.1. Population
4.2. Selection Criteria of Healthy Volunteers and Patients
4.3. Serum and Neutrophil Collection
4.4. HDN and LDNs Isolation by Cell Sorting
4.5. Serum Biomarker Quantification
4.6. Flow Cytometry
4.7. NETosis Assay by Confocal Microscopy
4.8. LDN and HDN Adhesion on hECM
4.9. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chan, J.C.N.; Lim, L.L.; Wareham, N.J.; Shaw, J.E.; Orchard, T.J.; Zhang, P.; Lau, E.S.H.; Eliasson, B.; Kong, A.P.S.; Ezzati, M.; et al. The Lancet Commission on diabetes: Using data to transform diabetes care and patient lives. Lancet 2021, 396, 2019–2082. [Google Scholar] [CrossRef]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Ong, K.L.; Stafford, L.K.; McLaughlin, S.A.; Boyko, E.J.; Vollset, S.E.; Smith, A.E.; Dalton, B.E.; Duprey, J.; Cruz, J.A.; Hagins, H.; et al. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: A systematic analysis for the Global Burden of Disease Study 2021. Lancet 2023, 402, 203–234. [Google Scholar] [CrossRef]
- Squires, E.; Duber, H.; Campbell, M.; Cao, J.; Chapin, A.; Horst, C.; Li, Z.; Matyasz, T.; Reynolds, A.; Hirsch, I.B.; et al. Health Care Spending on Diabetes in the U.S. 1996–2013. Diabetes Care 2018, 41, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Dieleman, J.L.; Cao, J.; Chapin, A.; Chen, C.; Li, Z.; Liu, A.; Horst, C.; Kaldjian, A.; Matyasz, T.; Scott, K.W.; et al. US Health Care Spending by Payer and Health Condition, 1996–2016. JAMA 2020, 323, 863–884. [Google Scholar] [CrossRef] [PubMed]
- Afroz, A.; Alramadan, M.J.; Hossain, M.N.; Romero, L.; Alam, K.; Magliano, D.J.; Billah, B. Cost-of-illness of type 2 diabetes mellitus in low and lower-middle income countries: A systematic review. BMC Health Serv. Res. 2018, 18, 972. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, N.; Gao, P.; Seshasai, S.R.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; Stampfer, M.; et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar] [PubMed]
- Vos, T.; Lim, S.S.; Abbafati, C.; Abbas, K.M.; Abbasi, M.; Abbasifard, M.; Abbasi-Kangevari, M.; Abbastabar, H.; Abd-Allah, F.; Abdelalim, A.; et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef] [PubMed]
- Lempesis, I.G.; Georgakopoulou, V.E. Physiopathological mechanisms related to inflammation in obesity and type 2 diabetes mellitus. World J. Exp. Med. 2023, 13, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef]
- Dorcely, B.; Katz, K.; Jagannathan, R.; Chiang, S.S.; Oluwadare, B.; Goldberg, I.J.; Bergman, M. Novel biomarkers for prediabetes, diabetes, and associated complications. Diabetes Metab. Syndr. Obes. 2017, 10, 345–361. [Google Scholar] [CrossRef]
- Wang, X.; Bao, W.; Liu, J.; Ouyang, Y.Y.; Wang, D.; Rong, S.; Xiao, X.; Shan, Z.L.; Zhang, Y.; Yao, P.; et al. Inflammatory markers and risk of type 2 diabetes: A systematic review and meta-analysis. Diabetes Care 2013, 36, 166–175. [Google Scholar] [CrossRef]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 2001, 286, 327–334. [Google Scholar] [CrossRef]
- Adabimohazab, R.; Garfinkel, A.; Milam, E.C.; Frosch, O.; Mangone, A.; Convit, A. Does Inflammation Mediate the Association between Obesity and Insulin Resistance? Inflammation 2016, 39, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Vozarova, B.; Weyer, C.; Lindsay, R.S.; Pratley, R.E.; Bogardus, C.; Tataranni, P.A. High white blood cell count is associated with a worsening of insulin sensitivity and predicts the development of type 2 diabetes. Diabetes 2002, 51, 455–461. [Google Scholar] [CrossRef]
- Lorenzo, C.; Hanley, A.J.; Haffner, S.M. Differential white cell count and incident type 2 diabetes: The Insulin Resistance Atherosclerosis Study. Diabetologia 2014, 57, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Gkrania-Klotsas, E.; Ye, Z.; Cooper, A.J.; Sharp, S.J.; Luben, R.; Biggs, M.L.; Chen, L.K.; Gokulakrishnan, K.; Hanefeld, M.; Ingelsson, E.; et al. Differential white blood cell count and type 2 diabetes: Systematic review and meta-analysis of cross-sectional and prospective studies. PLoS ONE 2010, 5, e13405. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.I.; Duncan, B.B.; Sharrett, A.R.; Lindberg, G.; Savage, P.J.; Offenbacher, S.; Azambuja, M.I.; Tracy, R.P.; Heiss, G. Markers of inflammation and prediction of diabetes mellitus in adults (Atherosclerosis Risk in Communities study): A cohort study. Lancet 1999, 353, 1649–1652. [Google Scholar] [CrossRef] [PubMed]
- Marini, O.; Costa, S.; Bevilacqua, D.; Calzetti, F.; Tamassia, N.; Spina, C.; De Sabata, D.; Tinazzi, E.; Lunardi, C.; Scupoli, M.T.; et al. Mature CD10(+) and immature CD10(−) neutrophils present in G-CSF-treated donors display opposite effects on T cells. Blood 2017, 129, 1343–1356. [Google Scholar] [CrossRef]
- Sagiv, J.Y.; Michaeli, J.; Assi, S.; Mishalian, I.; Kisos, H.; Levy, L.; Damti, P.; Lumbroso, D.; Polyansky, L.; Sionov, R.V.; et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015, 10, 562–573. [Google Scholar] [CrossRef]
- Denny, M.F.; Yalavarthi, S.; Zhao, W.; Thacker, S.G.; Anderson, M.; Sandy, A.R.; McCune, W.J.; Kaplan, M.J. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J. Immunol. 2010, 184, 3284–3297. [Google Scholar] [CrossRef]
- Hong, C.W. Current Understanding in Neutrophil Differentiation and Heterogeneity. Immune Netw. 2017, 17, 298–306. [Google Scholar] [CrossRef]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Grassle, S.; Huck, V.; Pappelbaum, K.I.; Gorzelanny, C.; Aponte-Santamaria, C.; Baldauf, C.; Grater, F.; Schneppenheim, R.; Obser, T.; Schneider, S.W. von Willebrand factor directly interacts with DNA from neutrophil extracellular traps. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Jenne, C.N.; Surewaard, B.G.; Thanabalasuriar, A.; Lee, W.Y.; Sanz, M.J.; Mowen, K.; Opdenakker, G.; Kubes, P. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat. Commun. 2015, 6, 6673. [Google Scholar] [CrossRef]
- Lavoie, S.S.; Dumas, E.; Vulesevic, B.; Neagoe, P.E.; White, M.; Sirois, M.G. Synthesis of Human Neutrophil Extracellular Traps Contributes to Angiopoietin-Mediated In Vitro Proinflammatory and Proangiogenic Activities. J. Immunol. 2018, 200, 3801–3813. [Google Scholar] [CrossRef]
- Sorensen, O.E.; Borregaard, N. Neutrophil extracellular traps—The dark side of neutrophils. J. Clin. Investig. 2016, 126, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef]
- Rodríguez-Espinosa, O.; Rojas-Espinosa, O.; Moreno-Altamirano, M.M.; López-Villegas, E.O.; Sánchez-García, F.J. Metabolic requirements for neutrophil extracellular traps formation. Immunology 2015, 145, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Berezin, A. Neutrophil extracellular traps: The core player in vascular complications of diabetes mellitus. Diabetes Metab. Syndr. 2019, 13, 3017–3023. [Google Scholar] [CrossRef]
- Shafqat, A.; Abdul Rab, S.; Ammar, O.; Al Salameh, S.; Alkhudairi, A.; Kashir, J.; Alkattan, K.; Yaqinuddin, A. Emerging role of neutrophil extracellular traps in the complications of diabetes mellitus. Front. Med. 2022, 9, 995993. [Google Scholar] [CrossRef]
- Sanchez-Pino, M.D.; Richardson, W.S.; Zabaleta, J.; Puttalingaiah, R.T.; Chapple, A.G.; Liu, J.; Kim, Y.; Ponder, M.; DeArmitt, R.; Baiamonte, L.B.; et al. Increased inflammatory low-density neutrophils in severe obesity and effect of bariatric surgery: Results from case-control and prospective cohort studies. EBioMedicine 2022, 77, 103910. [Google Scholar] [CrossRef]
- Cerecedo, D.; Martínez-Vieyra, I.; López-Villegas, E.O.; Hernández-Cruz, A.; Loza-Huerta, A.D.C. Heterogeneity of neutrophils in arterial hypertension. Exp. Cell Res. 2021, 402, 112577. [Google Scholar] [CrossRef]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef]
- Joshi, M.B.; Lad, A.; Bharath Prasad, A.S.; Balakrishnan, A.; Ramachandra, L.; Satyamoorthy, K. High glucose modulates IL-6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS Lett. 2013, 587, 2241–2246. [Google Scholar] [CrossRef]
- Vulesevic, B.; Lavoie, S.S.; Neagoe, P.E.; Dumas, E.; Rakel, A.; White, M.; Sirois, M.G. CRP Induces NETosis in Heart Failure Patients with or without Diabetes. Immunohorizons 2019, 3, 378–388. [Google Scholar] [CrossRef]
- Summers, C.; Rankin, S.M.; Condliffe, A.M.; Singh, N.; Peters, A.M.; Chilvers, E.R. Neutrophil kinetics in health and disease. Trends Immunol. 2010, 31, 318–324. [Google Scholar] [CrossRef]
- Terashima, T.; English, D.; Hogg, J.C.; van Eeden, S.F. Release of polymorphonuclear leukocytes from the bone marrow by interleukin-8. Blood 1998, 92, 1062–1069. [Google Scholar] [CrossRef]
- Walker, F.; Zhang, H.H.; Matthews, V.; Weinstock, J.; Nice, E.C.; Ernst, M.; Rose-John, S.; Burgess, A.W. IL6/sIL6R complex contributes to emergency granulopoietic responses in G-CSF- and GM-CSF-deficient mice. Blood 2008, 111, 3978–3985. [Google Scholar] [CrossRef]
- Yiu, J.Y.T.; Hally, K.E.; Larsen, P.D.; Holley, A.S. Increased levels of low density neutrophils (LDNs) in myocardial infarction. Acta Cardiol. 2023, 78, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Arruda-Olson, A.M.; Reeder, G.S.; Bell, M.R.; Weston, S.A.; Roger, V.L. Neutrophilia predicts death and heart failure after myocardial infarction: A community-based study. Circ. Cardiovasc. Qual. Outcomes 2009, 2, 656–662. [Google Scholar] [CrossRef]
- Abdalla, H.; Ramy, Z.; Dana, V. Prognostic significance of Absolute Neutrophil Count in Patients with Heart Failure with Preserved Ejection Fraction. Asian Pac. J. Health Sci. 2016, 3, 104–108. [Google Scholar]
- Tasal, A.; Erturk, M.; Uyarel, H.; Karakurt, H.; Bacaksiz, A.; Vatankulu, M.A.; Turfan, M.; Sonmez, O.; Erdogan, E.; Ergelen, M. Utility of the neutrophil to lymphocyte ratio for predicting in-hospital mortality after levosimendan infusion in patients with acute decompensated heart failure. J. Cardiol. 2014, 63, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Tobin, M.C.; Thomas, L.L. Neutrophil-like low-density granulocytes are elevated in patients with moderate to severe persistent asthma. Ann. Allergy Asthma Immunol. 2014, 113, 635–640.e2. [Google Scholar] [CrossRef] [PubMed]
- Hacbarth, E.; Kajdacsy-Balla, A. Low density neutrophils in patients with systemic lupus erythematosus, rheumatoid arthritis, and acute rheumatic fever. Arthritis Rheum. 1986, 29, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Mauracher, L.-M.; Hell, L.; Moik, F.; Krall, M.; Englisch, C.; Roiß, J.; Grilz, E.; Hofbauer, T.M.; Brostjan, C.; Knapp, S.; et al. Neutrophils in lung cancer patients: Activation potential and neutrophil extracellular trap formation. Res. Pract. Thromb. Haemost. 2023, 7, 100126. [Google Scholar] [CrossRef] [PubMed]
- McKenna, E.; Mhaonaigh, A.U.; Wubben, R.; Dwivedi, A.; Hurley, T.; Kelly, L.A.; Stevenson, N.J.; Little, M.A.; Molloy, E.J. Neutrophils: Need for Standardized Nomenclature. Front. Immunol. 2021, 12, 602963. [Google Scholar] [CrossRef]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef]
- Novotny, J.; Oberdieck, P.; Titova, A.; Pelisek, J.; Chandraratne, S.; Nicol, P.; Hapfelmeier, A.; Joner, M.; Maegdefessel, L.; Poppert, H.; et al. Thrombus NET content is associated with clinical outcome in stroke and myocardial infarction. Neurology 2020, 94, e2346–e2360. [Google Scholar] [CrossRef]
- Bonaventura, A.; Vecchie, A.; Abbate, A.; Montecucco, F. Neutrophil Extracellular Traps and Cardiovascular Diseases: An Update. Cells 2020, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tao, W.; Shen, F.; Du, W.; Xu, Z.; Liu, Z. The Emerging Role of Neutrophil Extracellular Traps in Arterial, Venous and Cancer-Associated Thrombosis. Front. Cardiovasc. Med. 2021, 8, 786387. [Google Scholar] [CrossRef] [PubMed]
- Frid, A.; Hirsch, L.; Gaspar, R.; Hicks, D.; Kreugel, G.; Liersch, J.; Letondeur, C.; Sauvanet, J.P.; Tubiana-Rufi, N.; Strauss, K. New injection recommendations for patients with diabetes. Diabetes Metab. 2010, 36 (Suppl. 2), S3–S18. [Google Scholar] [CrossRef] [PubMed]
- Béjot, Y.; Giroud, M. Stroke in diabetic patients. Diabetes Metab. 2010, 36 (Suppl. 3), S84–S87. [Google Scholar] [CrossRef] [PubMed]
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Investig. 2013, 123, 3446–3458. [Google Scholar] [CrossRef] [PubMed]
- Štulc, T.; Svobodová, H.; Krupičková, Z.; Doležalová, R.; Marinov, I.; Češka, R. Rosiglitazone influences the expression of leukocyte adhesion molecules and CD14 receptor in type 2 diabetes mellitus patients. Physiol. Res. 2014, 63 (Suppl. 2), S293–S298. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HV (n = 22) | T2D (n = 18) | p Value | |
|---|---|---|---|
| Age (years) | 39.1 ± 2.9 | 63.4 ± 2.8 | <0.001 |
| Male | 11 (50.0%) | 14 (77.8%) | 0.1040 |
| Body Mass Index (BMI) | - | 30.3 ± 1.3 | |
| Smoker | - | 3 (16.7%) | |
| Insulin treatment | - | 5 (27.8%) | |
| Dyslipidemia | - | 11 (61.1%) | |
| Statin | - | 12 (66.7%) | |
| Hematology and Biochemistry | |||
| Leukocytes (×109/L) | 6.18 ± 0.45 | 6.47 ± 0.34 | 0.6141 |
| CBC neutrophils (×109/L) | 4.29 ± 0.29 | 4.28 ± 0.26 | 0.9833 |
| Lymphocytes (×109/L) | 1.63 ± 0.04 | 1.57 ± 0.10 | 0.6046 |
| CBC neutrophils/lymphocytes ratio | 2.66 ± 0.21 | 2.93 ± 0.24 | 0.4283 |
| HbA1c (%) | - | 7.22 ± 0.28 | |
| Glycemia (mmol/L) | - | 8.67 ± 0.60 | |
| Total cholesterol (mmol/L) | - | 3.65 ± 0.13 | |
| HDL cholesterol (mmol/L) | - | 1.27 ± 0.07 | |
| LDL cholesterol (mmol/L) | - | 1.68 ± 0.12 | |
| Triglycerides (mmol/L) | - | 1.62 ± 0.21 | |
| T2D Medication | |||
| Metformin | - | 12 (66.7%) | |
| Sulfonylurea | - | 4 (22.2%) | |
| DPP-4 inhibitor | - | 4 (22.2%) | |
| GLP-1 receptor agonist | - | 7 (38.9%) | |
| SGLT2 inhibitor | - | 9 (50.0%) | |
| Insulin | - | 5 (27.8%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dumont, B.L.; Neagoe, P.-E.; Charles, E.; Villeneuve, L.; Tardif, J.-C.; Räkel, A.; White, M.; Sirois, M.G. Low-Density Neutrophils Contribute to Subclinical Inflammation in Patients with Type 2 Diabetes. Int. J. Mol. Sci. 2024, 25, 1674. https://doi.org/10.3390/ijms25031674
Dumont BL, Neagoe P-E, Charles E, Villeneuve L, Tardif J-C, Räkel A, White M, Sirois MG. Low-Density Neutrophils Contribute to Subclinical Inflammation in Patients with Type 2 Diabetes. International Journal of Molecular Sciences. 2024; 25(3):1674. https://doi.org/10.3390/ijms25031674
Chicago/Turabian StyleDumont, Benjamin L., Paul-Eduard Neagoe, Elcha Charles, Louis Villeneuve, Jean-Claude Tardif, Agnès Räkel, Michel White, and Martin G. Sirois. 2024. "Low-Density Neutrophils Contribute to Subclinical Inflammation in Patients with Type 2 Diabetes" International Journal of Molecular Sciences 25, no. 3: 1674. https://doi.org/10.3390/ijms25031674