
In-Depth Genomic Analysis: The New Challenge in Congenital Heart Disease

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Current Knowledge
Genetics


2. Development of the Heart
2.1. A Look at the Lines of Progenitor Cells and Their Morphogenesis
2.2. Heart Morphogenesis under Genetic Control
3. Interference with Human Cardiac Development and the Occurrence of Specific Anatomic Congenital Cardiac Disorders
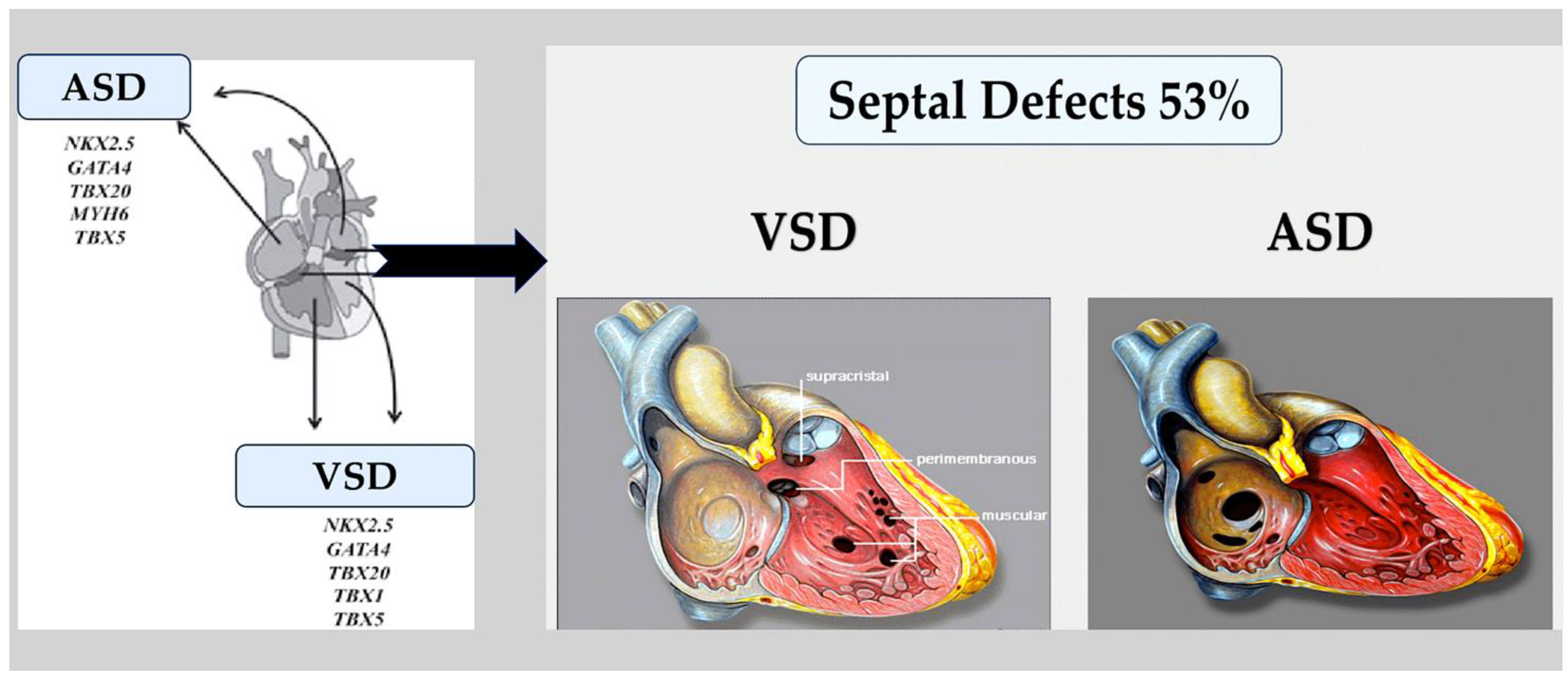
- Atrial and ventricular septal defects.
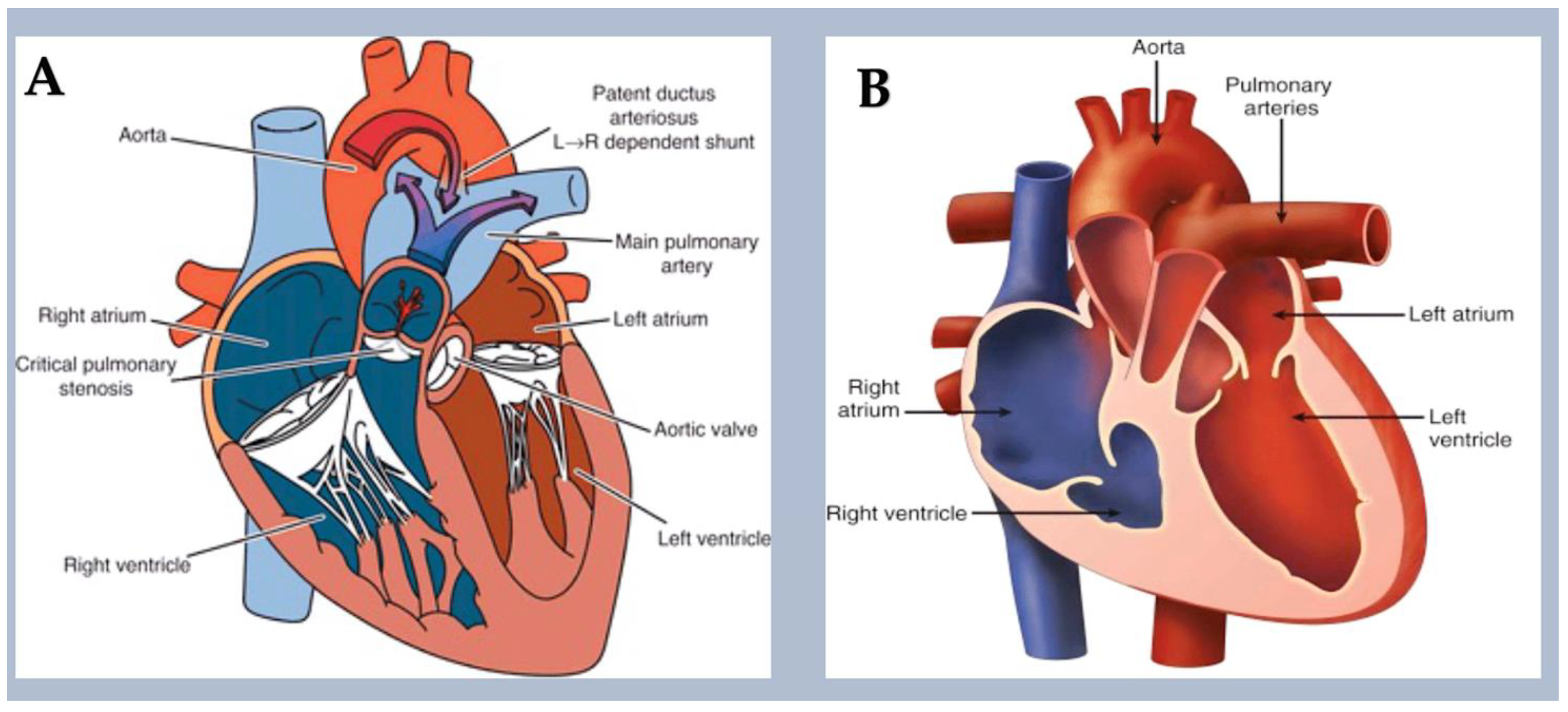
- Left-sided obstructive lesions.
- Right-sided obstructive lesions.
- Defects in the left–right pattern.
- Conotruncal defects.
4. New CHD Genes Discovered in Cohort Studies
- CHD definitive genes.
- Genes linked to CHD that have deleterious variants in humans.
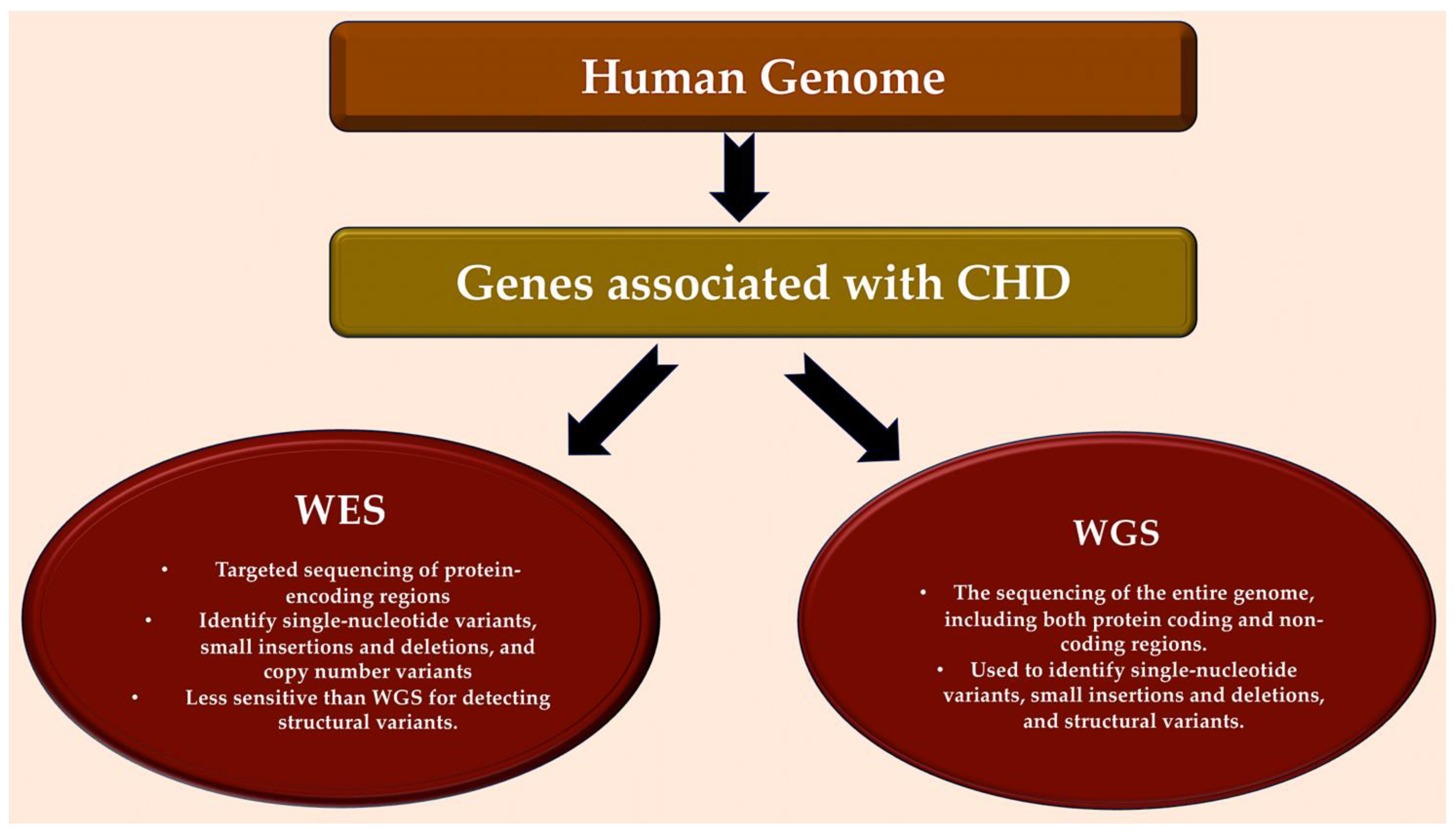
5. Assessing Whole-Genome Sequencing: A New Pathway of Investigation
5.1. Variation in Non-Coding Sequences through Whole-Genome Sequencing
5.2. Using Whole-Genome Sequencing to Identify Structural Variation
5.3. Future Uses of Genomics
6. Deriving Causality by Integrating Biology
6.1. Insights Gained from Single-Cell Transcriptomics
6.2. Findings from Pluripotent Stem Cell Models of Congenital Heart Disease
7. Moving Genomics into the Clinical Setting
7.1. CHD in Association with Extracardiac Abnormalities or Neurodevelopmental Disabilities
7.2. De Novo Copy Number Variations Have an Impact on Perioperative Outcomes
7.3. Cancer Risk in Patients with Congenital Heart Disease
7.4. Forthcoming Applications in the Clinic
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Ibrahim, S.; Gaborit, B.; Lenoir, M.; Collod-Beroud, G.; Stefanovic, S. Maternal Pre-Existing Diabetes: A Non-Inherited Risk Factor for Congenital Cardiopathies. Int. J. Mol. Sci. 2023, 24, 16258. [Google Scholar] [CrossRef] [PubMed]
- Dilli, D.; Akduman, H.; Zenciroğlu, A.; Çetinkaya, M.; Okur, N.; Turan, Ö.; Özlü, F.; Çalkavur, Ş.; Demirel, G.; Koksal, N.; et al. Neonatal Outcomes of Critical Congenital Heart Defects: A Multicenter Epidemiological Study of Turkish Neonatal Society: Neonatal Outcomes of CCHD. Pediatr. Cardiol. 2024, 45, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Hossin, M.Z.; de la Cruz, L.F.; McKay, K.A.; Oberlander, T.F.; Sandström, A.; Razaz, N. Association of pre-existing maternal cardiovascular diseases with neurodevelopmental disorders in offspring: A cohort study in Sweden and British Columbia, Canada. Int. J. Epidemiol. 2023, dyad184. [Google Scholar] [CrossRef]
- Bakker, M.K.; Bergman, J.E.H.; Krikov, S.; Amar, E.; Cocchi, G.; Cragan, J.; de Walle, H.E.K.; Gatt, M.; Groisman, B.; Liu, S.; et al. Prenatal diagnosis and prevalence of critical congenital heart defects: An international retrospective cohort study. BMJ Open 2019, 9, e028139. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.I.E.; Kaplan, S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900. [Google Scholar] [CrossRef]
- Reller, M.D.; Strickland, M.J.; Riehle-Colarusso, T.; Mahle, W.T.; Correa, A. Prevalence of congenital heart defects in metropolitan Atlanta. J. Pediatr. 2008, 153, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Spector, L.G.; Menk, J.S.; Knight, J.H.; McCracken, C.; Thomas, A.S.; Vinocur, J.M.; Oster, M.E.; St Louis, J.D.; Moller, J.H.; Kochilas, L. Trends in Long-Term Mortality after Congenital Heart Surgery. J. Am. Coll. Cardiol. 2018, 71, 2434–2446. [Google Scholar] [CrossRef]
- Egbe, A.; Lee, S.; Ho, D.; Uppu, S.; Srivastava, S. Prevalence of congenital anomalies in newborns with congenital heart disease diagnosis. Ann. Pediatr. Cardiol. 2014, 7, 86–91. [Google Scholar] [CrossRef]
- Wang, H.; Lin, X.; Lyu, G.; He, S.; Dong, B.; Yang, Y. Chromosomal abnormalities in fetuses with congenital heart disease: A meta-analysis. Arch. Gynecol. Obstet. 2023, 308, 797–811. [Google Scholar] [CrossRef]
- Diniz, B.L.; Deconte, D.; Gadelha, K.A.; Glaeser, A.B.; Guaraná, B.B.; de Moura, A.Á.; Rosa, R.F.M.; Zen, P.R.G. Congenital Heart Defects and 22q11.2 Deletion Syndrome: A 20-Year Update and New Insights to Aid Clinical Diagnosis. J. Pediatr. Genet. 2023, 12, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Thienpont, B.; Mertens, L.; de Ravel, T.; Eyskens, B.; Boshoff, D.; Maas, N.; Fryns, J.P.; Gewillig, M.; Vermeesch, J.R.; Devriendt, K. Submicroscopic chromosomal imbalances detected by array-CGH are a frequent cause of congenital heart defects in selected patients. Eur. Heart J. 2007, 28, 2778–2784. [Google Scholar] [CrossRef]
- Wilson, D.I.; Cross, I.E.; Goodship, J.A.; Brown, J.; Scambler, P.J.; Bain, H.H.; Taylor, J.F.; Walsh, K.; Bankier, A.; Burn, J.; et al. A prospective cytogenetic study of 36 cases of DiGeorge syndrome. Am. J. Hum. Genet. 1992, 51, 957–963. [Google Scholar]
- Sgardioli, I.C.; Vieira, T.P.; Simioni, M.; Monteiro, F.P. Gil-da-Silva-Lopes VL 22q11.2 Deletion Syndrome: Laboratory Diagnosis and TBX1 and FGF8 Mutation Screening. J. Pediatr. Genet. 2015, 4, 17–22. [Google Scholar] [CrossRef]
- Agergaard, P.; Olesen, C.; Østergaard, J.R.; Christiansen, M.; Sørensen, K.M. The prevalence of chromosome 22q11.2 deletions in 2478 children with cardiovascular malformations. A population-based study. Am. J. Med. Genet. A 2012, 158, 498–508. [Google Scholar] [CrossRef]
- Agergaard, P.; Hebert, A.; Sørensen, K.M.; Østergaard, J.R.; Olesen, C. Can clinical assessment detect 22q11.2 deletions in patients with cardiac malformations? A review. Eur. J. Med. Genet. 2011, 54, 3–8. [Google Scholar] [CrossRef]
- Peyvandi, S.; Lupo, P.J.; Garbarini, J.; Woyciechowski, S.; Edman, S.; Emanuel, B.S.; Mitchell, L.E.; Goldmuntz, E. 22q11.2 deletions in patients with conotruncal defects: Data from 1610 consecutive cases. Pediatr. Cardiol. 2013, 34, 1687–1694. [Google Scholar] [CrossRef]
- Dehghan, B.; Sabri, M.R.; Ahmadi, A.; Ghaderian, M.; Mahdavi, C.; Ramezani Nejad, D.; Sattari, M. Identifying the Factors Affecting the Incidence of Congenital Heart Disease Using Support Vector Machine and Particle Swarm Optimization. Adv. Biomed. Res. 2023, 12, 130. [Google Scholar] [CrossRef]
- Pierpont, M.E.; Basson, C.T.; Benson, D.W., Jr.; Gelb, B.D.; Giglia, T.M.; Goldmuntz, E.; McGee, G.; Sable, C.A.; Srivastava, D.; Webb, C.L.; et al. Genetic basis for congenital heart defects: Current knowledge: A scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: Endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 3015–3038. [Google Scholar] [CrossRef]
- Goldmuntz, E. 22q11.2 deletion syndrome and congenital heart disease. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Fahed, A.C.; Gelb, B.D.; Seidman, J.G.; Seidman, C.E. Genetics of congenital heart disease: The glass half empty. Circ. Res. 2013, 112, 707–720. [Google Scholar] [CrossRef] [PubMed]
- LaHaye, S.; Corsmeier, D.; Basu, M.; Bowman, J.L.; Fitzgerald-Butt, S.; Zender, G.; Bosse, K.; McBride, K.L.; White, P.; Garg, V. Utilization of Whole Exome Sequencing to Identify Causative Mutations in Familial Congenital Heart Disease. Circ. Cardiovasc. Genet. 2016, 9, 320–329. [Google Scholar] [CrossRef]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of rare inherited and de novo variants in 2871 congenital heart disease probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef]
- Homsy, J.; Zaidi, S.; Shen, Y.; Ware, J.S.; Samocha, K.E.; Karczewski, K.J.; DePalma, S.R.; McKean, D.; Wakimoto, H.; Gorham, J.; et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015, 350, 1262–1266. [Google Scholar] [CrossRef]
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef]
- Sifrim, A.; Hitz, M.P.; Wilsdon, A.; Breckpot, J.; Turki, S.H.; Thienpont, B.; McRae, J.; Fitzgerald, T.W.; Singh, T.; Swaminathan, G.J.; et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 2016, 48, 1060–1065. [Google Scholar] [CrossRef]
- Shan, W.; Yuanqing, X.; Jing, Z.; Xi, W.; Huifeng, G.; Yi, W. Risk factor analysis for adverse prognosis of the fetal ventricular septal defect (VSD). BMC Pregnancy Childbirth 2023, 23, 683. [Google Scholar] [CrossRef]
- International Society of Ultrasound in Obstetrics & Gynecology. Cardiac screening examination of the fetus: Guidelines for performing the ‘basic’ and ‘extended basic’ cardiac scan. Ultrasound Obstet. Gynecol. 2006, 27, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Meilhac, S.M.; Buckingham, M.E. The deployment of cell lineages that form the mammalian heart. Nat. Rev. Cardiol. 2018, 15, 705–724. [Google Scholar] [CrossRef] [PubMed]
- Kathiriya, I.S.; Nora, E.P.; Bruneau, B.G. Investigating the transcriptional control of cardiovascular development. Circ. Res. 2015, 116, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Maas, R.G.C.; van den Dolder, F.W.; Yuan, Q.; van der Velden, J.; Wu, S.M.; Sluijter, J.P.G.; Buikema, J.W. Harnessing developmental cues for cardiomyocyte production. Development 2023, 150, dev201483. [Google Scholar] [CrossRef] [PubMed]
- Günthel, M.; Barnett, P.; Christoffels, V.M. Development, proliferation, and growth of the mammalian heart. Mol. Ther. 2018, 26, 1599–1609. [Google Scholar] [CrossRef]
- Cui, M.; Wang, Z.; Bassel-Duby, R.; Olson, E.N. Genetic and epigenetic regulation of cardiomyocytes in development, regeneration and disease. Development 2018, 145, dev171983. [Google Scholar] [CrossRef]
- Chien, K.R.; Domian, I.J.; Parker, K.K. Cardiogenesis and the complex biology of regenerative cardiovascular medicine. Science 2008, 322, 1494–1497. [Google Scholar] [CrossRef]
- Jain, R.; Epstein, J.A. Competent for commitment: You’ve got to have heart! Genes Dev. 2018, 32, 4–13. [Google Scholar] [CrossRef]
- Zaffran, S.; Kelly, R.G.; Meilhac, S.M.; Buckingham, M.E.; Brown, N.A. Right ventricular myocardium derives from the anterior heart field. Circ. Res. 2004, 95, 261–268. [Google Scholar] [CrossRef]
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013, 498, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Hedermann, G.; Hedley, P.L.; Thagaard, I.N.; Krebs, L.; Ekelund, C.K.; Sørensen, T.I.A.; Christiansen, M. Maternal obesity and metabolic disorders associate with congenital heart defects in the offspring: A systematic review. PLoS ONE 2021, 16, e0252343. [Google Scholar] [CrossRef] [PubMed]
- Cavadino, A.; Sandberg, L.; Öhman, I.; Bergvall, T.; Star, K.; Dolk, H.; Loane, M.; Addor, M.C.; Barisic, I.; Cavero-Carbonell, C.; et al. Signal Detection in EUROmediCAT: Identification and Evaluation of Medication-Congenital Anomaly Associations and Use of VigiBase as a Complementary Source of Reference. Drug Saf. 2021, 44, 765–785. [Google Scholar] [CrossRef] [PubMed]
- Kalisch-Smith, J.I.; Ved, N.; Sparrow, D.B. Environmental Risk Factors for Congenital Heart Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a037234. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, K.J.; Correa, A.; Feinstein, J.A.; Botto, L.; Britt, A.E.; Daniels, S.R.; Elixson, M.; Warnes, C.A.; Webb, C.L. American Heart Association Council on Cardiovascular Disease in the Y: Noninherited risk factors and congenital cardiovascular defects: Current knowledge: A scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: Endorsed by the American Academy of Pediatrics. Circulation 2007, 115, 2995–3014. [Google Scholar]
- Hutson, M.R.; Kirby, M.L. Model systems for the study of heart development and disease. Semin. Cell Dev. Biol. 2007, 18, 101–110. [Google Scholar] [CrossRef]
- de la Pompa, J.L.; Timmerman, L.A.; Takimoto, H.; Yoshida, H.; Elia, A.J.; Samper, E.; Potter, J.; Wakeham, A.; Marengere, L.; Langille, B.L.; et al. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature 1998, 392, 182–186. [Google Scholar] [CrossRef]
- Lin, C.-J.; Lin, C.-Y.; Chen, C.-H.; Zhou, B.; Chang, C.-P. Partitioning the heart: Mechanisms of cardiac septation and valve development. Development 2012, 139, 3277–3299. [Google Scholar] [CrossRef]
- Wu, B.; Wu, B.; Benkaci, S.; Shi, L.; Lu, P.; Park, T.; Morrow, B.E.; Wang, Y.; Zhou, B. Crk and Crkl Are Required in the Endocardial Lineage for Heart Valve Development. J. Am. Heart Assoc. 2023, 12, e029683. [Google Scholar] [CrossRef] [PubMed]
- Christoffels, V.M.; Habets, P.E.; Franco, D.; Campione, M.; de Jong, F.; Lamers, W.H.; Bao, Z.Z.; Palmer, S.; Biben, C.; Harvey, R.P.; et al. Chamber formation and morphogenesis in the developing mammalian heart. Dev. Biol. 2000, 223, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Alsan, B.H.; Schultheiss, T.M. Regulation of avian cardiogenesis by Fgf8 signaling. Development 2002, 129, 1935–1943. [Google Scholar] [CrossRef] [PubMed]
- Astrof, S.; Arriagada, C.; Saijoh, Y.; Francou, A.; Kelly, R.G.; Moon, A. Aberrant differentiation of second heart field mesoderm prefigures cellular defects in the outflow tract in response to loss of FGF8. Dev. Biol. 2023, 499, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Ivanovitch, K.; Soro-Barrio, P.; Chakravarty, P.; Jones, R.A.; Bell, D.M.; Mousavy Gharavy, S.N.; Stamataki, D.; Delile, J.; Smith, J.C.; Briscoe, J. Ventricular, atrial, and outflow tract heart progenitors arise from spatially and molecularly distinct regions of the primitive streak. PLoS Biol. 2021, 19, e3001200. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N.; Ohta, H.; Nakayama, Y.; Konishi, M. Roles of FGF signals in heart development, health, and disease. Front. Cell Dev. Biol. 2016, 4, 110. [Google Scholar] [CrossRef] [PubMed]
- de Pater, E.; Ciampricotti, M.; Priller, F.; Veerkamp, J.; Strate, I.; Smith, K.; Lagendijk, A.K.; Schilling, T.F.; Herzog, W.; Abdelilah-Seyfried, S.; et al. Bmp signaling exerts opposite effects on cardiac differentiation. Circ. Res. 2012, 110, 578–587. [Google Scholar] [CrossRef]
- Targoff, K.L.; Colombo, S.; George, V.; Schell, T.; Kim, S.H.; Solnica-Krezel, L.; Yelon, D. Nkx genes are essential for maintenance of ventricular identity. Development 2013, 140, 4203–4213. [Google Scholar] [CrossRef]
- Nelson, D.O.; Lalit, P.A.; Biermann, M.; Markandeya, Y.S.; Capes, D.L.; Addesso, L.; Patel, G.; Han, T.; John, M.C.; Powers, P.A.; et al. Irx4 Marks a Multipotent, Ventricular-Specific Progenitor Cell. Stem Cells 2016, 34, 2875–2888. [Google Scholar] [CrossRef]
- Goldfracht, I.; Protze, S.; Shiti, A.; Setter, N.; Gruber, A.; Shaheen, N.; Nartiss, Y.; Keller, G.; Gepstein, L. Generating ring-shaped engineered heart tissues from ventricular and atrial human pluripotent stem cell-derived cardiomyocytes. Nat. Commun. 2020, 11, 75. [Google Scholar] [CrossRef]
- Giacomelli, E.; Bellin, M.; Orlova, V.V.; Mummery, C.L. Co-Differentiation of Human Pluripotent Stem Cells-Derived Cardiomyocytes and Endothelial Cells from Cardiac Mesoderm Provides a Three-Dimensional Model of Cardiac Microtissue. Curr. Protoc. Hum. Genet. 2017, 95, 21.9.1–21.9.22. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Wang, J.; Su, D.; Pan, H.; Huang, G.; Li, X.; Li, Z.; Shen, A.; Xie, X.; Wang, B.; et al. Two novel mutations of the IRX4 gene in patients with congenital heart disease. Hum. Genet. 2011, 130, 657–662. [Google Scholar] [CrossRef] [PubMed]
- de Soysa, T.Y.; Ranade, S.S.; Okawa, S.; Ravichandran, S.; Huang, Y.; Salunga, H.T.; Srivastava, D. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature 2019, 572, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Ishii, M.; Sun, J.; Sucov, H.M.; Maxson, R.E. Msx1 and Msx2 regulate survival of secondary heart field precursors and post-migratory proliferation of cardiac neural crest in the outflow tract. Dev. Biol. 2007, 308, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Wasson, L.K.; Willcox, J.A.; Morton, S.U.; Gorham, J.M.; DeLaughter, D.M. Pediatric Cardiac Genomics Consortium. GATA6 mutations in hiPSCs inform mechanisms for maldevelopment of the heart, pancreas, and diaphragm. eLife 2020, 9, e53278. [Google Scholar] [CrossRef] [PubMed]
- Uribe, V.; Badía-Careaga, C.; Casanova, J.C.; Domínguez, J.N.; de la Pompa, J.L.; Sanz-Ezquerro, J.J. Arid3b is essential for second heart field cell deployment and heart patterning. Development 2014, 141, 4168–4181. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, D.Z.; Hockemeyer, D.; McAnally, J.; Nordheim, A.; Olson, E.N. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 2004, 428, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Felker, A.; Prummel, K.D.; Merks, A.M.; Mickoleit, M.; Brombacher, E.C.; Huisken, J.; Panáková, D.; Mosimann, C. Continuous addition of progenitors forms the cardiac ventricle in zebrafish. Nat. Commun. 2018, 9, 2001. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Iranzo, H.; Galardi-Castilla, M.; Minguillón, C.; Sanz-Morejón, A.; González-Rosa, J.M.; Felker, A.; Ernst, A.; Guzmán-Martínez, G.; Mosimann, C.; Mercader, N. Tbx5a lineage tracing shows cardiomyocyte plasticity during zebrafish heart regeneration. Nat. Commun. 2018, 9, 428. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Choudhary, B.; Merki, E.; Chien, K.R.; Maxson, R.E.; Sucov, H.M. Normal fate and altered function of the cardiac neural crest cell lineage in retinoic acid receptor mutant embryos. Mech. Dev. 2002, 117, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Häneke, T.; Rohner, E.; Sohlmér, J.; Kameneva, P.; Artemov, A.; Adameyko, I.; Sahara, M. STRA6 is essential for induction of vascular smooth muscle lineages in human embryonic cardiac outflow tract development. Cardiovasc. Res. 2023, 119, 1202–1217. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.; Miyake, R.; Cheng, A.; Liu, T.; Iseki, S.; Kume, T. Conditional inactivation of Foxc1 and Foxc2 in neural crest cells leads to cardiac abnormalities. Genesis 2020, 58, e23364. [Google Scholar] [CrossRef] [PubMed]
- Kodo, K.; Shibata, S.; Miyagawa-Tomita, S.; Ong, S.G.; Takahashi, H.; Kume, T.; Okano, H.; Matsuoka, R.; Yamagishi, H. Regulation of Sema3c and the interaction between cardiac neural crest and second heart field during outflow tract development. Sci. Rep. 2017, 7, 6771. [Google Scholar] [CrossRef] [PubMed]
- Greulich, F.; Rudat, C.; Kispert, A. Mechanisms of T-box gene function in the developing heart. Cardiovasc. Res. 2011, 91, 212–222. [Google Scholar] [CrossRef] [PubMed]
- MacGrogan, D.; Nus, M.; de la Pompa, J.L. Notch signaling in cardiac development and disease. Curr. Top. Dev. Biol. 2010, 92, 333–365. [Google Scholar] [CrossRef]
- Stankunas, K.; Ma, G.K.; Kuhnert, F.J.; Kuo, C.J.; Chang, C.P. VEGF signaling has distinct spatiotemporal roles during heart valve development. Dev. Biol. 2010, 347, 325–336. [Google Scholar] [CrossRef]
- Alvandi, Z.; Bischoff, J. Endothelial-Mesenchymal Transition in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2357–2369. [Google Scholar] [CrossRef]
- Singh, N.; Trivedi, C.M.; Lu, M.; Mullican, S.E.; Lazar, M.A.; Epstein, J.A. Histone deacetylase 3 regulates smooth muscle differentiation in neural crest cells and development of the cardiac outflow tract. Circ. Res. 2011, 109, 1240–1249. [Google Scholar] [CrossRef]
- Kelly, R.G.; Papaioannou, V.E. Visualization of outflow tract development in the absence of Tbx1 using an FgF10 enhancer trap transgene. Dev. Dyn. 2007, 236, 821–828. [Google Scholar] [CrossRef]
- Peng, T.; Tian, Y.; Boogerd, C.J.; Lu, M.M.; Kadzik, R.S.; Stewart, K.M.; Evans, S.M.; Morrisey, E. Coordination of heart and lung co-development by a multipotent cardiopulmonary progenitor. Nature 2013, 500, 589–592. [Google Scholar] [CrossRef]
- Liu, X.; Chen, W.; Li, W.; Li, Y.; Priest, J.R.; Zhou, B.; Wang, J.; Zhou, Z. Single-Cell RNA-seq of the developing cardiac outflow tract reveals convergent development of the vascular smooth muscle cells. Cell Rep. 2019, 28, 1346–1361.e4. [Google Scholar] [CrossRef]
- Forrest, K.; Barricella, A.C.; Pohar, S.A.; Hinman, A.M.; Amack, J.D. Understanding laterality disorders and the left-right organizer: Insights from zebrafish. Front. Cell Dev. Biol. 2022, 10, 1035513. [Google Scholar] [CrossRef]
- Lopes Floro, K.; Artap, S.T.; Preis, J.I.; Fatkin, D.; Chapman, G.; Furtado, M.B.; Harvey, R.P.; Hamada, H.; Sparrow, D.B.; Dunwoodie, S. Loss of Cited2 causes congenital heart disease by perturbing left-right patterning of the body axis. Hum. Mol. Genet. 2011, 20, 1097–1110. [Google Scholar] [CrossRef] [PubMed]
- Bellchambers, H.M.; Ware, S.M. ZIC3 in Heterotaxy. Adv. Exp. Med. Biol. 2018, 1046, 301–327. [Google Scholar] [CrossRef] [PubMed]
- Levin, M. Left-right asymmetry in vertebrate embryogenesis. Bioessays 1997, 19, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Zwijsen, A.; Vogel, H.; Huylebroeck, D.; Matzuk, M.M. Smad5 is essential for left-right asymmetry in mice. Dev. Biol. 2000, 219, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Donofrio, M.; Frank, L.; He, D.; Jonas, R. Complete atrioventricular canal with guarded primum septal defect. Pediatr. Cardiol. 2011, 32, 503–505. [Google Scholar] [CrossRef] [PubMed]
- Paladini, D.; Tartaglione, A.; Agangi, A.; Teodoro, A.; Forleo, F.; Borghese, A.; Martinelli, P. The association between congenital heart disease and Down syndrome in prenatal life. Ultrasound. Obstet. Gynecol. 2000, 15, 104–108. [Google Scholar] [CrossRef]
- Santoro, M.; Coi, A.; Spadoni, I.; Bianchi, F.; Pierini, A. Sex differences for major congenital heart defects in Down Syndrome: A population based study. Eur. J. Med. Genet. 2018, 61, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Pelleri, M.C.; Locatelli, C.; Mattina, T.; Bonaglia, M.C.; Piazza, F.; Magini, P.; Antonaros, F.; Ramacieri, G.; Vione, B.; Vitale, L.; et al. Partial trisomy 21 with or without highly restricted Down syndrome critical region (HR-DSCR): Report of two new cases and reanalysis of the genotype-phenotype association. BMC Med. Genom. 2022, 15, 266. [Google Scholar] [CrossRef] [PubMed]
- Ang, Y.-S.; Rivas, R.N.; Ribeiro, A.J.S.; Srivas, R.; Rivera, J.; Stone, N.R.; Pratt, K.; Mohamed, T.M.A.; Fu, J.D.; Spencer, C.I.; et al. Disease model of GATA4 mutation reveals transcription factor cooperativity in human cardiogenesis. Cell 2016, 167, 1734–1749.e22. [Google Scholar] [CrossRef] [PubMed]
- Misra, C.; Sachan, N.; McNally, C.R.; Koenig, S.N.; Nichols, H.A.; Guggilam, A.; Lucchesi, P.A.; Pu, W.T.; Srivastava, D.; Garg, V. Congenital heart disease-causing Gata4 mutation displays functional deficits in vivo. PLoS Genet. 2012, 8, e1002690. [Google Scholar] [CrossRef]
- Garg, V.; Kathiriya, I.S.; Barnes, R.; Schluterman, M.K.; King, I.N.; Butler, C.A.; Rothrock, C.R.; Eapen, R.S.; Hirayama-Yamada, K.; Joo, K.; et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 2003, 424, 443–447. [Google Scholar] [CrossRef]
- Brown, C.O., 3rd; Chi, X.; Garcia-Gras, E.; Shirai, M.; Feng, X.H.; Schwartz, R.J. The cardiac determination factor, Nkx2-5, is activated by mutual cofactors GATA-4 and Smad1/4 via a novel upstream enhancer. J. Biol. Chem. 2004, 279, 10659–10669. [Google Scholar] [CrossRef]
- Vecoli, C.; Pulignani, S.; Foffa, I.; Andreassi, M.G. Congenital heart disease: The crossroads of genetics, epigenetics and environment. Curr. Genom. 2014, 15, 390–399. [Google Scholar] [CrossRef]
- James, K.; Kirklin, M.D. Kirklin/Barratt-Boyes Cardiac Surgery, 4th ed.; James, K., Kirklin, M.D., Eugene, H., Blackstone, M.D., Eds.; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- McBride, K.L.; Pignatelli, R.; Lewin, M.; Ho, T.; Fernbach, S.; Menesses, A.; Lam, W.; Leal, S.M.; Kaplan, N.; Schliekelman, P.; et al. Inheritance analysis of congenital left ventricular outflow tract obstruction malformations: Segregation, multiplex relative risk, and heritability. Am. J. Med. Genet. A 2005, 134, 180–186. [Google Scholar] [CrossRef]
- Nappi, F.; Giacinto, O.; Lusini, M.; Garo, M.; Caponio, C.; Nenna, A.; Nappi, P.; Rousseau, J.; Spadaccio, C.; Chello, M. Patients with Bicuspid Aortopathy and Aortic Dilatation. J. Clin. Med. 2022, 11, 6002. [Google Scholar] [CrossRef]
- Agasthi, P.; Pujari, S.H.; Tseng, A.; Graziano, J.N.; Marcotte, F.; Majdalany, D.; Mookadam, F.; Hagler, D.J.; Arsanjani, R. Management of adults with coarctation of aorta. World J. Cardiol. 2020, 12, 167–191. [Google Scholar] [CrossRef]
- Silberbach, M.; Roos-Hesselink, J.W.; Andersen, N.H.; Braverman, A.C.; Brown, N.; Collins, R.T.; De Backer, J.; Eagle, K.A.; Hiratzka, L.F.; Johnson, W.H., Jr.; et al. Cardiovascular health in Turner syndrome: A scientific statement from the American Heart Association. Circ. Genom. Precis. Med. 2018, 11, e000048. [Google Scholar] [CrossRef] [PubMed]
- Lara, D.A.; Ethen, M.K.; Canfield, M.A.; Nembhard, W.N.; Morris, S.A. A population-based analysis of mortality in patients with Turner syndrome and hypoplastic left heart syndrome using the Texas Birth Defects Registry. Congenit. Heart Dis. 2017, 12, 105–112. [Google Scholar] [CrossRef]
- Bouayed Abdelmoula, N.; Abdelmoula, B.; Smaoui, W.; Trabelsi, I.; Louati, R.; Aloulou, S.; Aloulou, W.; Abid, F.; Kammoun, S.; Trigui, K.; et al. Left-sided congenital heart lesions in mosaic Turner syndrome. Mol. Genet. Genom. 2018, 293, 495–501. [Google Scholar] [CrossRef]
- Ribé, L.; Shihadeh, F.D.; Afifi, R.O.; Estrera, A.L.; Prakash, S.K. Outcomes of cardiothoracic surgery in women with Turner syndrome. Ann. Cardiothorac. Surg. 2023, 12, 569–576. [Google Scholar] [CrossRef]
- Prakash, S.K.; Bondy, C.A.; Maslen, C.L.; Silberbach, M.; Lin, A.E.; Perrone, L.; Limongelli, G.; Michelena, H.I.; Bossone, E.; Citro, R.; et al. Autosomal and X chromosome structural variants are associated with congenital heart defects in Turner syndrome: The NHLBI GenTAC registry. Am. J. Med. Genet. A 2016, 170, 3157–3164. [Google Scholar] [CrossRef]
- Wenger, S.L.; Grossfeld, P.D.; Siu, B.L.; Coad, J.E.; Keller, F.G.; Hummel, M. Molecular characterization of an 11q interstitial deletion in a patient with the clinical features of Jacobsen syndrome. Am. J. Med. Genet. A 2006, 140, 704–708. [Google Scholar] [CrossRef]
- Crucean, A.; Alqahtani, A.; Barron, D.J.; Brawn, W.J.; Richardson, R.V.; O’Sullivan, J.; Anderson, R.H.; Henderson, D.J.; Chaudhry, B. Re-evaluation of hypoplastic left heart syndrome from a developmental and morphological perspective. Orphanet. J. Rare Dis. 2017, 12, 138. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Tian, L.; Martin, M.; Paige, S.L.; Galdos, F.X.; Li, J.; Klein, A.; Zhang, H.; Ma, N.; Wei, Y.; et al. Intrinsic endocardial defects contribute to hypoplastic left heart syndrome. Cell Stem Cell 2020, 27, 574–589.e8. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Habibollah, S.; Tilgner, K.; Collin, J.; Barta, T.; Al-Aama, J.Y.; Tesarov, L.; Hussain, R.; Trafford, A.W.; Kirkwood, G.; et al. An induced pluripotent stem cell model of hypoplastic left heart syndrome (HLHS) reveals multiple expression and functional differences in HLHS-derived cardiac myocytes. Stem Cells Transl. Med. 2014, 3, 416–423. [Google Scholar] [CrossRef]
- Shi, L.M.; Tao, J.W.; Qiu, X.B.; Wang, J.; Yuan, F.; Xu, L.; Liu, H.; Li, R.G.; Xu, Y.J.; Wang, Q.; et al. GATA5 loss-of-function mutations associated with congenital bicuspid aortic valve. Int. J. Mol. Med. 2014, 33, 1219–1226. [Google Scholar] [CrossRef]
- Bonachea, E.M.; Chang, S.-W.; Zender, G.; LaHaye, S.; Fitzgerald-Butt, S.; McBride, K.L.; Garg, V. Rare GATA5 sequence variants identified in individuals with bicuspid aortic valve. Pediatr. Res. 2014, 76, 211–216. [Google Scholar] [CrossRef]
- Huang, M.; Akerberg, A.A.; Zhang, X.; Yoon, H.; Joshi, S.; Hallinan, C.; Nguyen, C.; Pu, W.T.; Haigis, M.C.; Burns, C.G.; et al. Intrinsic myocardial defects underlie an Rbfox-deficient zebrafish model of hypoplastic left heart syndrome. Nat. Commun. 2022, 13, 5877. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.J.; Di, R.M.; Qiao, Q.; Li, X.M.; Huang, R.T.; Xue, S.; Liu, X.Y.; Wang, J.; Yang, Y.Q. GATA6 loss-of-function mutation contributes to congenital bicuspid aortic valve. Gene 2018, 663, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Theis, J.L.; Hu, J.J.; Sundsbak, R.S.; Evans, J.M.; Bamlet, W.R.; Qureshi, M.Y.; O’Leary, P.W.; Olson, T.M. Genetic Association between Hypoplastic Left Heart Syndrome and Cardiomyopathies. Circ. Genom. Precis. Med. 2021, 14, e003126. [Google Scholar] [CrossRef] [PubMed]
- Theis, J.L.; Zimmermann, M.T.; Evans, J.M.; Eckloff, B.W.; Wieben, E.D.; Qureshi, M.Y.; O’Leary, P.W.; Olson, T.M. Recessive MYH6 mutations in hypoplastic left heart with reduced ejection fraction. Circ. Cardiovasc. Genet. 2015, 8, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Berg, C.; Lachmann, R.; Kaiser, C.; Kozlowski, P.; Stressig, R.; Schneider, M.; Asfour, B.; Herberg, U.; Breuer, J.; Gembruch, U.; et al. Prenatal diagnosis of tricuspid atresia: Intrauterine course and outcome. Ultrasound Obstet. Gynecol. 2010, 35, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, A.; Conti, E.; D’Agostino, R.; Digilio, M.C.; Formigari, R.; Picchio, F.; Marino, B.; Pizzuti, A.; Dallapiccola, B. ZFPM2/FOG2 and HEY2 genes analysis in nonsyndromic tricuspid atresia. Am. J. Med. Genet. A 2005, 133, 68–70. [Google Scholar] [CrossRef]
- Pierpont, M.E.; Digilio, M.C. Cardiovascular disease in Noonan syndrome. Curr. Opin. Pediatr. 2018, 30, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Ki, C.S.; Lee, H.J. Mutation analysis of the genes involved in the Ras-mitogen-activated protein kinase (MAPK) pathway in Korean patients with Noonan syndrome. Clin. Genet. 2007, 72, 150–155. [Google Scholar] [CrossRef]
- Pandit, B.; Sarkozy, A.; Pennacchio, L.A.; Carta, C.; Oishi, K.; Martinelli, S.; Pogna, E.A.; Schackwitz, W.; Ustaszewska, A.; Landstrom, A.; et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat. Genet. 2007, 39, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Allanson, J.; Jadico, S.K.; Kavamura, M.I.; Noonan, J.; Opitz, J.M.; Young, T.; Neri, G. The cardiofaciocutaneous syndrome. J. Med. Genet. 2006, 43, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Tidyman, W.E.; Rauen, K.A. Noonan, Costello and cardio-facio-cutaneous syndromes: Dysregulation of the Ras-MAPK pathway. Expert Rev. Mol. Med. 2008, 10, e37. [Google Scholar] [CrossRef] [PubMed]
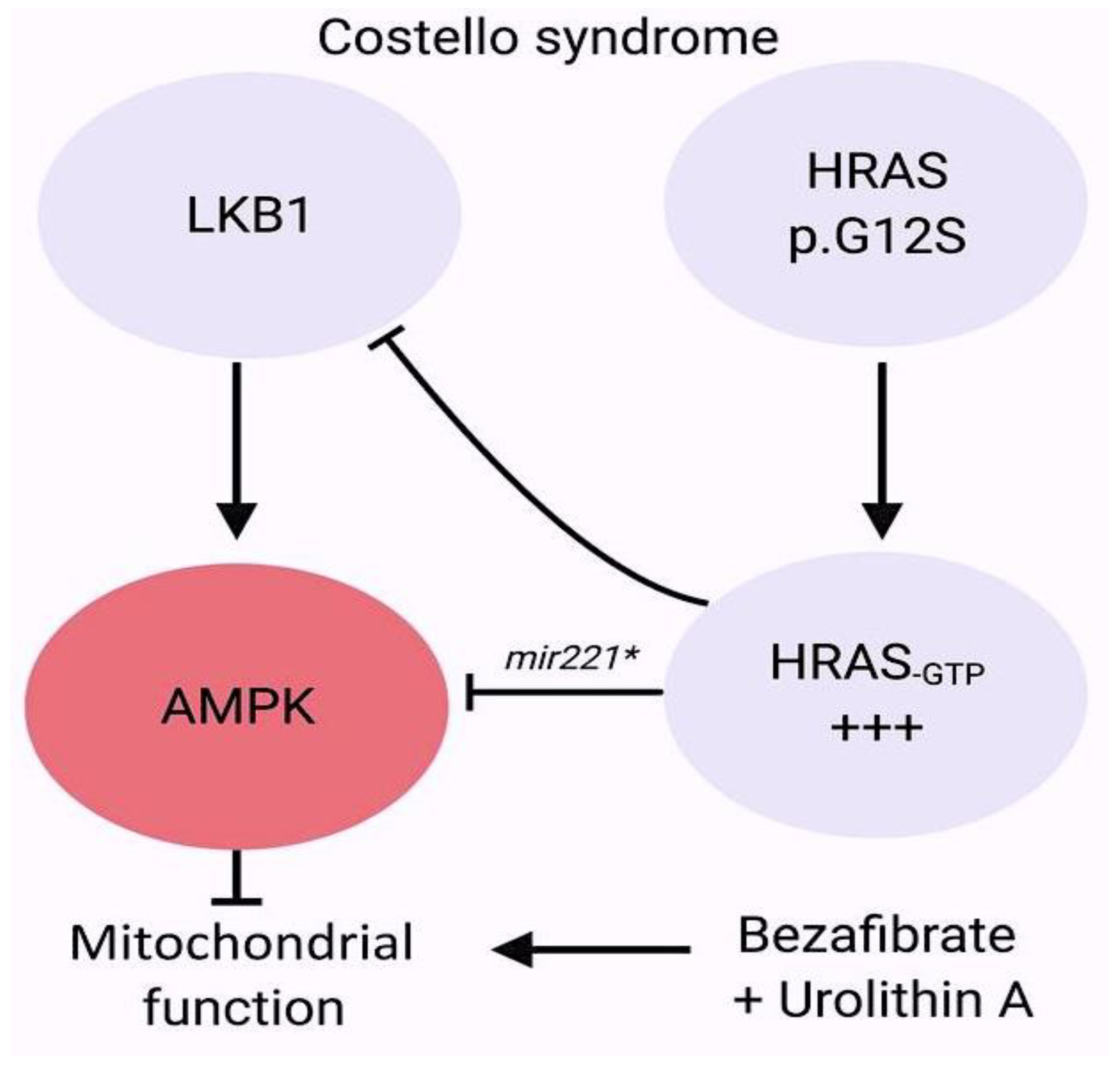
- Dard, L.; Hubert, C.; Esteves, P.; Blanchard, W.; Bou About, G.; Baldasseroni, L.; Dumon, E.; Angelini, C.; Delourme, M.; Guyonnet-Dupérat, V.; et al. HRAS germline mutations impair LKB1/AMPK signaling and mitochondrial homeostasis in Costello syndrome models. J. Clin. Investig. 2022, 132, e131053. [Google Scholar] [CrossRef] [PubMed]
- White, J.J.; Mazzeu, J.F.; Hoischen, A.; Bayram, Y.; Withers, M.; Gezdirici, A.; Kimonis, V.; Steehouwer, M.; Jhangiani, S.N.; Muzny, D.M.; et al. DVL3 Alleles Resulting in a −1 Frameshift of the Last Exon Mediate Autosomal-Dominant Robinow Syndrome. Am. J. Hum. Genet. 2016, 98, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Patil, S.J.; Srivastava, P.; Gaurishankar, K.; Phadke, S.R. Clinical and molecular characterization of four patients with Robinow syndrome from different families. Am. J. Med. Genet. A 2021, 185, 1105–1112. [Google Scholar] [CrossRef]
- Hu, R.; Qiu, Y.; Li, Y.; Li, J. A novel frameshift mutation of DVL1-induced Robinow syndrome: A case report and literature review. Mol. Genet. Genom. Med. 2022, 10, e1886. [Google Scholar] [CrossRef]
- Danyel, M.; Kortüm, F.; Dathe, K.; Kutsche, K.; Horn, D. Autosomal dominant Robinow syndrome associated with a novel DVL3 splice mutation. Am. J. Med. Genet. A 2018, 176, 992–996. [Google Scholar] [CrossRef]
- Mašek, J.; Andersson, E.R. The developmental biology of genetic Notch disorders. Development 2017, 144, 1743–1763. [Google Scholar] [CrossRef]
- Hofmann, J.J.; Briot, A.; Enciso, J.; Zovein, A.C.; Ren, S.; Zhang, Z.W.; Radtke, F.; Simons, M.; Wang, Y.; Iruela-Arispe, M.L. Endothelial deletion of murine Jag1 leads to valve calcification and congenital heart defects associated with Alagille syndrome. Development 2012, 139, 4449–4460. [Google Scholar] [CrossRef]
- McCright, B.; Lozier, J.; Gridley, T. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development 2002, 129, 1075–1082. [Google Scholar] [CrossRef]
- Liu, X.; Chen, W.; Li, W.; Priest, J.R.; Fu, Y.; Pang, K.; Ma, B.; Han, B.; Liu, X.; Hu, S.; et al. Exome-based case-control analysis highlights the pathogenic role of ciliary genes in transposition of the great arteries. Circ. Res. 2020, 126, 811–821. [Google Scholar] [CrossRef]
- Blue, G.M.; Mekel, M.; Das, D.; Troup, M.; Rath, E.; Ip, E.; Gudkov, M.; Perumal, G.; Harvey, R.P.; Sholler, G.F.; et al. Whole genome sequencing in transposition of the great arteries and associations with clinically relevant heart, brain and laterality genes. Am. Heart J. 2022, 244, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, A.H.; Hanchard, N.A.; Azamian, M.; D’Alessandro, L.C.A.; Coban-Akdemir, Z.; Lopez, K.N.; Hall, N.J.; Dickerson, H.; Nicosia, A.; Fernbach, S.; et al. Genetic architecture of laterality defects revealed by whole exome sequencing. Eur. J. Hum. Genet. 2019, 27, 563–573. [Google Scholar] [CrossRef]
- Yi, T.; Sun, H.; Fu, Y.; Hao, X.; Sun, L.; Zhang, Y.; Han, J.; Gu, X.; Liu, X.; Guo, Y.; et al. Genetic and Clinical Features of Heterotaxy in a Prenatal Cohort. Front. Genet. 2022, 13, 818241. [Google Scholar] [CrossRef]
- Papanayotou, C.; Collignon, J. Activin/Nodal signalling before implantation: Setting the stage for embryo patterning. Biol. Sci. 2014, 369, 20130539. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, B.; Casey, B.; Li, H.; Ho-Dawson, T.; Smith, L.; Fernbach, S.D.; Molinari, L.; Niesh, S.R.; Jefferies, J.L.; Craigen, W.J.; et al. Identification and functional characterization of NODAL rare variants in heterotaxy and isolated cardiovascular malformations. Hum. Mol. Genet. 2009, 18, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Bedard, J.E.; Haaning, A.M.; Ware, S.M. Identification of a novel ZIC3 isoform and mutation screening in patients with heterotaxy and congenital heart disease. PLoS ONE 2011, 6, e23755. [Google Scholar] [CrossRef]
- D’Alessandro, L.C.; Latney, B.C.; Paluru, P.C.; Goldmuntz, E. The phenotypic spectrum of ZIC3 mutations includes isolated d-transposition of the great arteries and double outlet right ventricle. Am. J. Med. Genet. A 2013, 161, 792–802. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Y.; Shi, L.; McDonald-McGinn, D.M.; Crowley, T.B.; McGinn, D.E.; Tran, O.T.; Miller, D.; Lin, J.R.; Zackai, E.; et al. Chromatin regulators in the TBX1 network confer risk for conotruncal heart defects in 22q11.2DS. NPJ Genom. Med. 2023, 8, 17. [Google Scholar] [CrossRef]
- Khositseth, A.; Tocharoentanaphol, C.; Khowsathit, P.; Ruangdaraganon, N. Chromosome 22q11 deletions in patients with conotruncal heart defects. Pediatr. Cardiol. 2005, 26, 570–573. [Google Scholar] [CrossRef]
- Ziolkowska, L.; Kawalec, W.; Turska-Kmiec, A.; Krajewska-Walasek, M.; Brzezinska-Rajszys, G.; Daszkowska, J.; Maruszewski, B.; Burczynski, P. Chromosome 22q11.2 microdeletion in children with conotruncal heart defects: Frequency, associated cardiovascular anomalies, and outcome following cardiac surgery. Eur. J. Pediatr. 2008, 167, 1135–1140. [Google Scholar] [CrossRef]
- Carotti, A.; Digilio, M.C.; Piacentini, G.; Saffirio, C.; Di Donato, R.M.; Marino, B. Cardiac defects and results of cardiac surgery in 22q11.2 deletion syndrome. Dev. Disabil. Res. Rev. 2008, 14, 35–42. [Google Scholar] [CrossRef]
- Corsten-Janssen, N.; du Marchie Sarvaas, G.J.; Kerstjens-Frederikse, W.S.; Hoefsloot, L.H.; van Beynum, I.M.; Kapusta, L.; van Ravenswaaij-Arts, C.M. CHD7 mutations are not a major cause of atrioventricular septal and conotruncal heart defects. Am. J. Med. Genet. A 2014, 164, 3003–3009. [Google Scholar] [CrossRef]
- Corsten-Janssen, N.; Scambler, P.J. Clinical and molecular effects of CHD7 in the heart. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 487–495. [Google Scholar] [CrossRef]
- Jongmans, M.C.; Admiraal, R.J.; van der Donk, K.P.; Vissers, L.E.; Baas, A.F.; Kapusta, L.; van Hagen, J.M.; Donnai, D.; de Ravel, T.J.; Veltman, J.A.; et al. CHARGE syndrome: The phenotypic spectrum of mutations in the CHD7 gene. J. Med. Genet. 2006, 43, 306–314. [Google Scholar] [CrossRef]
- Meisner, J.K.; Martin, D.M. Congenital heart defects in CHARGE: The molecular role of CHD7 and effects on cardiac phenotype and clinical outcomes. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 81–89. [Google Scholar] [CrossRef]
- Zentner, G.E.; Layman, W.S.; Martin, D.M.; Scacheri, P.C. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am. J. Med. Genet. A 2010, 152, 674–686. [Google Scholar] [CrossRef]
- Rozas, M.F.; Benavides, F.; León, L.; Repetto, G.M. Association between phenotype and deletion size in 22q11.2 microdeletion syndrome: Systematic review and meta-analysis. Orphanet. J. Rare Dis. 2019, 14, 195. [Google Scholar] [CrossRef]
- Zhao, Y.; Diacou, A.; Johnston, H.R.; Musfee, F.I.; McDonald-McGinn, D.M.; McGinn, D.; Crowley, T.B.; Repetto, G.M.; Swillen, A.; Breckpot, J.; et al. Complete sequence of the 22q11.2 allele in 1,053 subjects with 22q11.2 deletion syndrome reveals modifiers of conotruncal heart defects. Am. J. Hum. Genet. 2020, 106, 26–40. [Google Scholar] [CrossRef]
- He, G.W.; Maslen, C.L.; Chen, H.X.; Hou, H.T.; Bai, X.Y.; Wang, X.L.; Liu, X.C.; Lu, W.L.; Chen, X.X.; Chen, W.D.; et al. Identification of novel rare copy number variants associated with sporadic tetralogy of Fallot and clinical implications. Clin. Genet. 2022, 102, 391–403. [Google Scholar] [CrossRef]
- Mefford, H.C.; Sharp, A.J.; Baker, C.; Itsara, A.; Jiang, Z.; Buysse, K.; Huang, S.; Maloney, V.K.; Crolla, J.A.; Baralle, D.; et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 2008, 359, 1685–1699. [Google Scholar] [CrossRef]
- Ceylan, A.C.; Sahin, I.; Erdem, H.B.; Kayhan, G.; Simsek-Kiper, P.O.; Utine, G.E.; Percin, F.; Boduroglu, K.; Alikasifoglu, M.J. An eight-case 1q21 region series: Novel aberrations and clinical variability with new features. Intellect. Disabil. Res. 2019, 63, 548–557. [Google Scholar] [CrossRef]
- Reuter, M.S.; Chaturvedi, R.R.; Jobling, R.K.; Pellecchia, G.; Hamdan, O.; Sung, W.W.L.; Nalpathamkalam, T.; Attaluri, P.; Silversides, C.K.; Wald, R.M.; et al. Clinical Genetic Risk Variants Inform a Functional Protein Interaction Network for Tetralogy of Fallot. Circ. Genom. Precis. Med. 2021, 14, e003410. [Google Scholar] [CrossRef]
- Page, D.J.; Miossec, M.J.; Williams, S.G.; Monaghan, R.M.; Fotiou, E.; Cordell, H.J.; Sutcliffe, L.; Topf, A.; Bourgey, M.; Bourque, G.; et al. Whole exome sequencing reveals the major genetic contributors to nonsyndromic tetralogy of Fallot. Circ. Res. 2019, 124, 553–563. [Google Scholar] [CrossRef]
- Reuter, M.S.; Jobling, R.; Chaturvedi, R.R.; Manshaei, R.; Costain, G.; Heung, T.; Curtis, M.; Hosseini, S.M.; Liston, E.; Lowther, C.; et al. Haploinsufficiency of vascular endothelial growth factor related signaling genes is associated with tetralogy of Fallot. Genet. Med. 2019, 21, 1001–1007. [Google Scholar] [CrossRef]
- Škorić-Milosavljević, D.; Lahrouchi, N.; Bosada, F.M.; Dombrowsky, G.; Williams, S.G.; Lesurf, R.; Tjong, F.V.Y.; Walsh, R.; El Bouchikhi, I.; Breckpot, J.; et al. Rare variants in KDR, encoding VEGF Receptor 2, are associated with tetralogy of Fallot. Genet. Med. 2021, 23, 1952–1960. [Google Scholar] [CrossRef]
- Tan, Z.P.; Huang, C.; Xu, Z.B.; Yang, J.F.; Yang, Y.F. Novel ZFPM2/FOG2 variants in patients with double outlet right ventricle. Clin. Genet. 2012, 82, 466–471. [Google Scholar] [CrossRef]
- Huang, X.; Niu, W.; Zhang, Z.; Zhou, C.; Xu, Z.; Liu, J.; Su, Z.; Ding, W.; Zhang, H. Identification of novel significant variants of ZFPM2/FOG2 in non-syndromic Tetralogy of Fallot and double outlet right ventricle in a Chinese Han population. Mol. Biol. Rep. 2014, 41, 2671–2677. [Google Scholar] [CrossRef]
- Su, W.; Zhu, P.; Wang, R.; Wu, Q.; Wang, M.; Zhang, X.; Mei, L.; Tang, J.; Kumar, M.; Wang, X.; et al. Congenital heart diseases and their association with the variant distribution features on susceptibility genes. Clin. Genet. 2017, 91, 349–354. [Google Scholar] [CrossRef]
- Yang, Y.Q.; Gharibeh, L.; Li, R.G.; Xin, Y.F.; Wang, J.; Liu, Z.M.; Qiu, X.B.; Xu, Y.J.; Xu, L.; Qu, X.K.; et al. GATA4 loss-of-function mutations underlie familial tetralogy of fallot. Hum. Mutat. 2013, 34, 1662–1671. [Google Scholar] [CrossRef] [PubMed]
- Kodo, K.; Nishizawa, T.; Furutani, M.; Arai, S.; Yamamura, E.; Joo, K.; Takahashi, T.; Matsuoka, R.; Yamagishi, H. GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorinplexin signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 13933–13938. [Google Scholar] [CrossRef]
- Nataf, P.; Guettier, C.; Bourbon, A.; Nappi, F.; Lima, L.; Dorent, R.; Pavie, A.; Gandjbakhch, I. Influence of arterial allograft preparation techniques on chronic vascular rejection: A histological study. Transplant Proc. 1996, 28, 2890–2892. [Google Scholar]
- Hoang, T.T.; Goldmuntz, E.; Roberts, A.E.; Chung, W.K.; Kline, J.K.; Deanfield, J.E.; Giardini, A.; Aleman, A.; Gelb, B.D.; Mac Neal, M.; et al. The congenital heart disease genetic network study: Cohort description. PLoS ONE 2018, 13, e0191319. [Google Scholar] [CrossRef]
- Preuss, C.; Capredon, M.; Wünnemann, F.; Chetaille, P.; Prince, A.; Godard, B.; Leclerc, S.; Sobreira, N.; Ling, H.; Awadalla, P.; et al. MIBAVA Leducq consortium; Samuels ME, Andelfinger G Family based whole exome sequencing reveals the multifaceted role of Notch signaling in congenital heart disease. PLoS Genet. 2016, 12, e1006335. [Google Scholar] [CrossRef]
- Sevim Bayrak, C.; Zhang, P.; Tristani-Firouzi, M.; Gelb, B.D.; Itan, Y. De novo variants in exomes of congenital heart disease patients identify risk genes and pathways. Genome Med. 2020, 12, 9. [Google Scholar] [CrossRef]
- Morton, S.U.; Shimamura, A.; Newburger, P.E.; Opotowsky, A.R.; Quiat, D.; Pereira, A.C.; Jin, S.C.; Gurvitz, M.; Brueckner, M.; Chung, W.K.; et al. Association of damaging variants in genes with increased cancer risk among patients with congenital heart disease. JAMA Cardiol. 2021, 6, 457–462. [Google Scholar] [CrossRef]
- Park, J.E.; Park, J.S.; Jang, S.Y.; Park, S.H.; Kim, J.W.; Ki, C.S.; Kim, D.K. A novel SMAD6 variant in a patient with severely calcified bicuspid aortic valve and thoracic aortic aneurysm. Mol. Genet. Genom. Med. 2019, 7, e620. [Google Scholar] [CrossRef]
- Raya, A.; Kawakami, Y.; Rodriguez-Esteban, C.; Buscher, D.; Koth, C.M.; Itoh, T.; Morita, M.; Raya, R.M.; Dubova, I.; Bessa, J.G.; et al. Notch activity induces Nodal expression and mediates the establishment of left-right asymmetry in vertebrate embryos. Genes Dev. 2003, 17, 1213–1218. [Google Scholar] [CrossRef]
- Galvin, K.M.; Donovan, M.J.; Lynch, C.A.; Meyer, R.I.; Paul, R.J.; Lorenz, J.N.; Fairchild-Huntress, V.; Dixon, K.L.; Dunmore, J.H.; Gimbrone, M.A.; et al. A role for Smad6 in development and homeostasis of the cardiovascular system. Nat. Genet. 2000, 24, 171–174. [Google Scholar] [CrossRef]
- McKean, D.M.; Homsy, J.; Wakimoto, H.; Patel, N.; Gorham, J.; DePalma, S.R.; Ware, J.S.; Zaidi, S.; Ma, W.; Patel, N.; et al. Loss of RNA expression and allele-specific expression associated with congenital heart disease. Nat. Commun. 2016, 7, 12824. [Google Scholar] [CrossRef]
- Brault, V.; Nguyen, T.L.; Flores-Gutiérrez, J.; Iacono, G.; Birling, M.C.; Lalanne, V.; Meziane, H.; Manousopoulou, A.; Pavlovic, G.; Lindner, L.; et al. Dyrk1a gene dosage in glutamatergic neurons has key effects in cognitive deficits observed in mouse models of MRD7 and Down syndrome. PLoS Genet. 2021, 17, e1009777. [Google Scholar] [CrossRef] [PubMed]
- Feki, A.; Hibaoui, Y. DYRK1A Protein, A Promising Therapeutic Target to Improve Cognitive Deficits in Down Syndrome. Brain Sci. 2018, 8, 187. [Google Scholar] [CrossRef]
- Vandeweyer, G.; Helsmoortel, C.; Van Dijck, A.; Vulto-van Silfhout, A.T.; Coe, B.P.; Bernier, R.; Gerdts, J.; Rooms, L.; van den Ende, J.; Bakshi, M.; et al. The transcriptional regulator ADNP links the BAF (SWI/SNF) complexes with autism. Am. J. Med. Genet. C Semin. Med. Genet. 2014, 166, 315–326. [Google Scholar] [CrossRef]
- Ockeloen, C.W.; Willemsen, M.H.; de Munnik, S.; van Bon, B.W.; de Leeuw, N.; Verrips, A.; Kant, S.G.; Jones, E.A.; Brunner, H.G.; van Loon, R.L.; et al. Further delineation of the KBG syndrome phenotype caused by ANKRD11 aberrations. Eur. J. Hum. Genet. 2015, 23, 1176–1185. [Google Scholar] [CrossRef]
- Hamilton, M.J.; Caswell, R.C.; Canham, N.; Cole, T.; Firth, H.V.; Foulds, N.; Heimdal, K.; Hobson, E.; Houge, G.; Joss, S.; et al. Heterozygous mutations affecting the protein kinase domain of CDK13 cause a syndromic form of developmental delay and intellectual disability. J. Med. Genet. 2018, 55, 28–38. [Google Scholar] [CrossRef]
- van den Akker, W.M.R.; Brummelman, I.; Martis, L.M.; Timmermans, R.N.; Pfundt, R.; Kleefstra, T.; Willemsen, M.H.; Gerkes, E.H.; Herkert, J.C.; van Essen, A.J.; et al. De novo variants in CDK13 associated with syndromic ID/DD: Molecular and clinical delineation of 15 individuals and a further review. Clin. Genet. 2018, 93, 1000–1007. [Google Scholar] [CrossRef]
- Beal, B.; Hayes, I.; McGaughran, J.; Amor, D.J.; Miteff, C.; Jackson, V.; van Reyk, O.; Subramanian, G.; Hildebrand, M.S.; Morgan, A.T.; et al. Expansion of phenotype of DDX3X syndrome: Six new cases. Clin. Dysmorphol. 2019, 28, 169–174. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Belkadi, A.; Bolze, A.; Itan, Y.; Cobat, A.; Vincent, Q.B.; Antipenko, A.; Shang, L.; Boisson, B.; Casanova, J.L.; Abel, L. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 5473–5478. [Google Scholar] [CrossRef]
- Hsieh, A.; Morton, S.U.; Willcox, J.A.L.; Gorham, J.M.; Tai, A.C.; Qi, H.; DePalma, S.; McKean, D.; Griffin, E.; Manheimer, K.B.; et al. EM-mosaic detects mosaic point mutations that contribute to congenital heart disease. Genome Med. 2020, 12, 42. [Google Scholar] [CrossRef]
- Manheimer, K.B.; Richter, F.; Edelmann, L.J.; D’Souza, S.L.; Shi, L.; Shen, Y.; Homsy, J.; Boskovski, M.T.; Tai, A.C.; Gorham, J.; et al. Robust identification of mosaic variants in congenital heart disease. Hum. Genet. 2018, 137, 183–193. [Google Scholar] [CrossRef]
- Wei, W.; Keogh, M.J.; Aryaman, J.; Golder, Z.; Kullar, P.J.; Wilson, I.; Talbot, K.; Turner, M.R.; McKenzie, C.A.; Troakes, C.; et al. Frequency and signature of somatic variants in 1461 human brain exomes. Genet. Med. 2019, 21, 904–912. [Google Scholar] [CrossRef]
- Zech, M.; Jech, R.; Boesch, S.; Škorvánek, M.; Weber, S.; Wagner, M.; Zhao, C.; Jochim, A.; Necpál, J.; Dincer, Y.; et al. Monogenic variants in dystonia: An exome-wide sequencing study. Lancet Neurol. 2020, 19, 908–918. [Google Scholar] [CrossRef]
- Gardner, E.J.; Sifrim, A.; Lindsay, S.J.; Prigmore, E.; Rajan, D.; Danecek, P.; Gallone, G.; Eberhardt, R.Y.; Martin, H.C.; Wright, C.F.; et al. Detecting cryptic clinically relevant structural variation in exome-sequencing data increases diagnostic yield for developmental disorders. Am. J. Hum. Genet. 2021, 108, 2186–2194. [Google Scholar] [CrossRef]
- Wright, C.F.; Fitzgerald, T.W.; Jones, W.D.; Clayton, S.; McRae, J.F.; van Kogelenberg, M.; King, D.A.; Ambridge, K.; Barrett, D.M.; Bayzetinova, T.; et al. Genetic diagnosis of developmental disorders in the DDD study: A scalable analysis of genome-wide research data. Lancet 2015, 385, 1305–1314. [Google Scholar] [CrossRef]
- Padhi, E.M.; Hayeck, T.J.; Cheng, Z.; Chatterjee, S.; Mannion, B.J.; Byrska-Bishop, M.; Willems, M.; Pinson, L.; Redon, S.; Benech, C.; et al. Coding and noncoding variants in EBF3 are involved in HADDS and simplex autism. Hum. Genom. 2021, 15, 44. [Google Scholar] [CrossRef]
- Choy, M.K.; Javierre, B.M.; Williams, S.G.; Baross, S.L.; Liu, Y.; Wingett, S.W.; Akbarov, A.; Wallace, C.; Freire-Pritchett, P.; Rugg-Gunn, P.J.; et al. Promoter interactome of human embryonic stem cell-derived cardiomyocytes connects GWAS regions to cardiac gene networks. Nat. Commun. 2018, 9, 2526. [Google Scholar] [CrossRef]
- Hoelscher, S.C.; Stich, T.; Diehm, A.; Lahm, H.; Dreßen, M.; Zhang, Z.; Neb, I.; Aherrahrou, Z.; Erdmann, J.; Schunkert, H.; et al. miR-128a Acts as a Regulator in Cardiac Development by Modulating Differentiation of Cardiac Progenitor Cell Populations. Int. J. Mol. Sci. 2020, 21, 1158. [Google Scholar] [CrossRef]
- Kheradpour, P.; Ernst, J.; Melnikov, A.; Rogov, P.; Wang, L.; Zhang, X.; Alston, J.; Mikkelsen, T.S.; Kellis, M. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res. 2013, 23, 800–811. [Google Scholar] [CrossRef]
- Akerberg, B.N.; Gu, F.; VanDusen, N.J.; Zhang, X.; Dong, R.; Li, K.; Zhang, B.; Zhou, B.; Sethi, I.; Ma, Q.; et al. A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers. Nat. Commun. 2019, 10, 4907. [Google Scholar] [CrossRef]
- Vanoudenhove, J.; Yankee, T.N.; Wilderman Cotney, J. Epigenomic and transcriptomic dynamics during human heart organogenesis. Circ. Res. 2020, 127, e184–e209. [Google Scholar] [CrossRef]
- Hussein, I.R.; Bader, R.S.; Chaudhary, A.G.; Bassiouni, R.; Alquaiti, M.; Ashgan, F.; Schulten, H.J.; Al Qahtani, M.H. Dentification of De Novo and Rare Inherited Copy Number Variants in Children with Syndromic Congenital Heart Defects. Pediatr. Cardiol. 2018, 39, 924–940. [Google Scholar] [CrossRef]
- van Ouwerkerk, A.F.; Bosada, F.M.; van Duijvenboden, K.; Houweling, A.C.; Scholman, K.T.; Wakker, V.; Allaart, C.P.; Uhm, J.S.; Mathijssen, I.B.; Baartscheer, T.; et al. Patient-Specific TBX5-G125R Variant Induces Profound Transcriptional Deregulation and Atrial Dysfunction. Circulation 2022, 145, 606–619. [Google Scholar] [CrossRef]
- Richter, F.; Morton, S.U.; Kim, S.W.; Kitaygorodsky, A.; Wasson, L.K.; Chen, K.M.; Zhou, J.; Qi, H.; Patel, N.; DePalma, S.R.; et al. Genomic analyses implicate noncoding denovo variants in congenital heart disease. Nat. Genet. 2020, 52, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Morton, S.U.; Pereira, A.C.; Quiat, D.; Richter, F.; Kitaygorodsky, A.; Hagen, J.; Bernstein, D.; Brueckner, M.; Goldmuntz, E.; Kim, R.W.; et al. Genome-Wide De Novo Variants in Congenital Heart Disease Are Not Associated with Maternal Diabetes or Obesity. Circ. Genom. Precis. Med. 2022, 15, e003500. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhao, P.A.; Eichler, E.E. Rare variants and the oligogenic architecture of autism. Trends Genet. 2022, 38, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; MacArthur, D.G. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alföldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Hureaux, M.; Guterman, S.; Hervé, B.; Till, M.; Jaillard, S.; Redon, S.; Valduga, M.; Coutton, C.; Missirian, C.; Prieur, F.; et al. Chromosomal microarray analysis in fetuses with an isolated congenital heart defect: A retrospective, nationwide, multicenter study in France. Prenat. Diagn. 2019, 39, 464–470. [Google Scholar] [CrossRef]
- Hatim, O.; Pavlinov, I.; Xu, M.; Linask, K.; Beers, J.; Liu, C.; Baumgärtel, K.; Gilbert, M.; Spinner, N.; Chen, C.; et al. Generation of an Alagille syndrome (ALGS) patient-derived induced pluripotent stem cell line (TRNDi032-A) carrying a heterozygous mutation (p.Cys682Leufs*7) in the JAG1 gene. Stem Cell Res. 2023, 73, 103231. [Google Scholar] [CrossRef]
- Legoff, L.; D’Cruz, S.C.; Tevosian, S.; Primig, M.; Smagulova, F. Transgenerational inheritance of environmentally induced epigenetic alterations during mammalian development. Cells 2019, 8, 1559. [Google Scholar] [CrossRef]
- Barua, S.; Junaid, M.A. Lifestyle, pregnancy and epigenetic effects. Epigenomics 2015, 7, 85–102. [Google Scholar] [CrossRef]
- Yokouchi-Konishi, T.; Yoshimatsu, J.; Sawada, M.; Shionoiri, T.; Nakanishi, A.; Horiuchi, C.; Tsuritani, M.; Iwanaga, N.; Kamiya, C.A.; Neki, R.; et al. Recurrent congenital heart diseases among neonates born to mothers with congenital heart diseases. Pediatr. Cardiol. 2019, 40, 865–870. [Google Scholar] [CrossRef]
- Ellesøe, S.G.; Workman, C.T.; Bouvagnet, P.; Loffredo, C.A.; McBride, K.L.; Hinton, R.B.; van Engelen, K.; Gertsen, E.C.; Mulder, B.J.M.; Postma, A.V.; et al. Familial co-occurrence of congenital heart defects follows distinct patterns. Eur. Heart J. 2018, 39, 1015–1022. [Google Scholar] [CrossRef]
- Øyen, N.; Poulsen, G.; Boyd, H.A.; Wohlfahrt, J.; Jensen, P.K.; Melbye, M. Recurrence of congenital heart defects in families. Circulation 2009, 120, 295–301. [Google Scholar] [CrossRef]
- Argelaguet, R.; Clark, S.J.; Mohammed, H.; Stapel, L.C.; Krueger, C.; Kapourani, C.A.; Imaz-Rosshandler, I.; Lohoff, T.; Xiang, Y.; Hanna, C.W.; et al. Multi-omics profiling of mouse gastrulation at single-cell resolution. Nature 2019, 576, 487–491. [Google Scholar] [CrossRef]
- Tucker, N.R.; Chaffin, M.; Fleming, S.J.; Hall, A.W.; Parsons, V.A.; Bedi, K.C., Jr.; Akkad, A.D.; Herndon, C.N.; Arduini, A.; Papangeli, I.; et al. Transcriptional and Cellular Diversity of the Human Heart. Circulation 2020, 142, 466–482. [Google Scholar] [CrossRef]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef]
- Cui, Y.; Zheng, Y.; Liu, X.; Yan, L.; Fan, X.; Yong, J.; Hu, Y.; Dong, J.; Li, Q.; Wu, X.; et al. Single-cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. 2019, 26, 1934–1950.e5. [Google Scholar] [CrossRef]
- Lescroart, F.; Wang, X.; Lin, X.; Swedlund, B.; Gargouri, S.; Sànchez-Dànes, A.; Moignard, V.; Dubois, C.; Paulissen, C.; Kinston, S.; et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 2018, 359, 1177–1181. [Google Scholar] [CrossRef]
- DeLaughter, D.M.; Bick, A.G.; Wakimoto, H.; McKean, D.; Gorham, J.M.; Kathiriya, I.S.; Hinson, J.T.; Homsy, J.; Gray, J.; Pu, W.; et al. Single-cell resolution of temporal gene expression during heart development. Dev. Cell. 2016, 39, 480–490. [Google Scholar] [CrossRef]
- Pijuan-Sala, B.; Griffiths, J.A.; Guibentif, C.; Hiscock, T.W.; Jawaid, W.; Calero-Nieto, F.J.; Mulas, C.; Ibarra-Soria, X.; Tyser, R.C.V.; Ho, D.L.L.; et al. A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 2019, 566, 490–495. [Google Scholar] [CrossRef]
- Ulirsch, J.C.; Verboon, J.M.; Kazerounian, S.; Guo, M.H.; Yuan, D.; Ludwig, L.S.; Handsaker, R.E.; Abdulhay, N.J.; Fiorini, C.; Genovese, G.; et al. The genetic landscape of Diamond-Blackfan anemia. Am. J. Hum. Genet. 2018, 103, 930–947. [Google Scholar] [CrossRef]
- Bramel, E.E.; Creamer, T.J.; Saqib, M.; Camejo Nunez, W.A.; Bagirzadeh, R.; Roker, L.A.; Goff, L.A.; MacFarlane, E.G. Postnatal Smad3 Inactivation in Murine Smooth Muscle Cells Elicits a Temporally and Regionally Distinct Transcriptional Response. Front. Cardiovasc. Med. 2022, 9, 826495. [Google Scholar] [CrossRef]
- Bissoli, I.; D’Adamo, S.; Pignatti, C.; Agnetti, G.; Flamigni, F.; Cetrullo, S. Induced pluripotent stem cell-based models: Are we ready for that heart in a dish? Front. Cell Dev. Biol. 2023, 11, 1129263. [Google Scholar] [CrossRef]
- Baumann, K. Achieving pluripotency. Nat. Rev. Mol. Cell Biol. 2010, 11, 677. [Google Scholar] [CrossRef]
- Zhang, J.; Tao, R.; Campbell, K.F.; Carvalho, J.L.; Ruiz, E.C.; Kim, G.C.; Schmuck, E.G.; Raval, A.N.; da Rocha, A.M.; Herron, T.J.; et al. Functional cardiac fibroblasts derived from human pluripotent stem cells via second heart field progenitors. Nat. Commun. 2019, 10, 2238. [Google Scholar] [CrossRef]
- Liu, J.A.; Cheung, M. Neural crest stem cells and their potential therapeutic applications. Dev. Biol. 2016, 419, 199–216. [Google Scholar] [CrossRef] [PubMed]
- Neri, T.; Hiriart, E.; van Vliet, P.P.; Faure, E.; Norris, R.A.; Farhat, B.; Jagla, B.; Lefrancois, J.; Sugi, Y.; Moore-Morris, T.; et al. Human pre-valvular endocardial cells derived from pluripotent stem cells recapitulate cardiac pathophysiological valvulogenesis. Nat. Commun. 2019, 10, 1929. [Google Scholar] [CrossRef]
- Kathiriya, I.S.; Rao, K.S.; Iacono, G.; Devine, W.P.; Blair, A.P.; Hota, S.K.; Lai, M.H.; Garay, B.I.; Thomas, R.; Gong, H.Z.; et al. Modeling Human TBX5 Haploinsufficiency Predicts Regulatory Networks for Congenital Heart Disease. Dev. Cell. 2021, 56, 292–309.e9. [Google Scholar] [CrossRef] [PubMed]
- Rydzanicz, M.; Zwoliński, P.; Gasperowicz, P.; Pollak, A.; Kostrzewa, G.; Walczak, A.; Konarzewska, M.; Płoski, R. A recurrent de novo variant supports KCNC2 involvement in the pathogenesis of developmental and epileptic encephalopathy. Am. J. Med. Genet. A 2021, 185, 3384–3389. [Google Scholar] [CrossRef]
- Pierpont, M.E.; Brueckner, M.; Chung, W.K.; Garg, V.; Lacro, R.V.; McGuire, A.L.; Mital, S.; Priest, J.R.; Pu, W.T.; Roberts, A.; et al. Genetic basis for congenital heart disease: Revisited: A scientific statement from the American Heart Association. Circulation 2018, 138, e653–e711. [Google Scholar] [CrossRef] [PubMed]
- DiStefano, M.T.; Hemphill, S.E.; Oza, A.M.; Siegert, R.K.; Grant, A.R.; Hughes, M.Y.; Cushman, B.J.; Azaiez, H.; Booth, K.T.; Chapin, A.; et al. ClinGen expert clinical validity curation of 164 hearing loss gene-disease pairs. Genet. Med. 2019, 21, 2239–2247. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Boycott, K.M.; Azzariti, D.R.; Hamosh, A.; Rehm, H.L. Seven years since the launch of the Matchmaker Exchange: The evolution of genomic matchmaking. Hum. Mutat. 2022, 43, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.Y.; Shah, N.; Jackson, A.R.; Ghosh, R.; Pawliczek, P.; Paithankar, S.; Baker, A.; Riehle, K.; Chen, H.; Milosavljevic, S.; et al. ClinGen Pathogenicity Calculator: A configurable system for assessing pathogenicity of genetic variants. Genome Med. 2017, 9, 3. [Google Scholar] [CrossRef]
- Yu, Y.; Lei, W.; Yang, J.; Wei, Y.C.; Zhao, Z.L.; Zhao, Z.A.; Hu, S. Functional mutant GATA4 identification and potential application in preimplantation diagnosis of congenital heart. Gene 2018, 641, 349–354. [Google Scholar] [CrossRef]
- Boskovski, M.T.; Homsy, J.; Nathan, M.; Sleeper, L.A.; Morton, S.; Manheimer, K.B.; Tai, A.; Gorham, J.; Lewis, M.; Swartz, M.; et al. De novo damaging variants, clinical phenotypes and post-operative outcomes in congenital heart disease. Circ. Genom. Precis. Med. 2020, 13, e002836. [Google Scholar] [CrossRef]
- Zomer, A.C.; Ionescu-Ittu, R.; Vaartjes, I.; Pilote, L.; Mackie, A.S.; Therrien, J.; Langemeijer, M.M.; Grobbee, D.E.; Mulder, B.J.; Marelli, A.J. Sex differences in hospital mortality in adults with congenital heart disease: The impact of reproductive health. J. Am. Coll. Cardiol. 2013, 62, 58–67. [Google Scholar] [CrossRef]
- Gurvitz, M.; Ionescu-Ittu, R.; Guo, L.; Eisenberg, M.J.; Abrahamowicz, M.; Pilote, L.; Marelli, A.J. Prevalence of cancer in adults with congenital heart disease compared with the general population. Am. J. Cardiol. 2016, 118, 1742–1750. [Google Scholar] [CrossRef]
- Mandalenakis, Z.; Karazisi, C.; Skoglund, K.; Rosengren, A.; Lappas, G.; Eriksson, P.; Dellborg, M. Risk of cancer among children and young adults with congenital heart disease compared with healthy controls. JAMA Netw. Open 2019, 2, e196762. [Google Scholar] [CrossRef]
- Cohen, S.; Gurvitz, M.Z.; Beauséjour-Ladouceur, V.; Lawler, P.R.; Therrien, J.; Marelli, A.J. Cancer Risk in Congenital Heart Disease-What Is the Evidence? Can. J. Cardiol. 2019, 35, 1750–1761. [Google Scholar] [CrossRef]
- Lim, E.T.; Uddin, M.; De Rubeis, S.; Chan, Y.; Kamumbu, A.S.; Zhang, X.; D’Gama, A.M.; Kim, S.N.; Hill, R.S.; Goldberg, A.P.; et al. Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat. Neurosci. 2017, 20, 1217–1224. [Google Scholar] [CrossRef]
- Freed, D.; Pevsner, J. The Contribution of Mosaic Variants to Autism Spectrum Disorder. PLoS Genet. 2016, 12, e1006245. [Google Scholar] [CrossRef]
- Dou, Y.; Yang, X.; Li, Z.; Wang, S.; Zhang, Z.; Ye, A.Y.; Yan, L.; Yang, C.; Wu, Q.; Li, J.; et al. Postzygotic single-nucleotide mosaicisms contribute to the etiology of autism spectrum disorder and autistic traits and the origin of mutations. Hum. Mutat. 2017, 38, 1002–1013. [Google Scholar] [CrossRef]
- Yu, X.; Tao, Y.; Liu, X.; Yu, F.; Jiang, C.; Xiao, Y.; Zhang, H.; He, Y.; Ye, L.; Wang, Y.; et al. The implication of chromosomal abnormalities in the surgical outcomes of Chinese pediatric patients with congenital heart disease. Front. Cardiovasc. Med. 2023, 10, 1164577. [Google Scholar] [CrossRef] [PubMed]
- Putotto, C.; Pugnaloni, F.; Unolt, M.; Maiolo, S.; Trezzi, M.; Digilio, M.C.; Cirillo, A.; Limongelli, G.; Marino, B.; Calcagni, G.; et al. 22q11.2 Deletion Syndrome: Impact of Genetics in the Treatment of Conotruncal Heart Defects. Children 2022, 9, 772. [Google Scholar] [CrossRef]
- Mercer-Rosa, L.; Pinto, N.; Yang, W.; Tanel, R. Goldmuntz E 22q11.2 deletion syndrome is associated with perioperative outcome in tetralogy of Fallot. J. Thorac. Cardiovasc. Surg. 2013, 146, 868–873. [Google Scholar] [CrossRef]
- O’Byrne, M.L.; O’Byrne, M.L.; Yang, W.; Mercer-Rosa, L.; Parnell, A.S.; Oster, M.E.; Levenbrown, Y.; Tanel, R.E.; Goldmuntz, E. 22q11.2 deletion syndrome is associated with increased perioperative events and more complicated postoperative course in infants undergoing infant operative correction of truncus arteriosus communis or interrupted aortic arch. J. Thorac. Cardiovasc. Surg. 2014, 148, 1597–1605. [Google Scholar] [CrossRef]
- Kim, D.S.; Kim, J.H.; Burt, A.A.; Crosslin, D.R.; Burnham, N.; Kim, C.E.; McDonald-McGinn, D.M.; Zackai, E.H.; Nicolson, S.C.; Spray, T.L.; et al. Burden of potentially pathologic copy number variants is higher in children with isolated congenital heart disease and significantly impairs covariate-adjusted transplant-free survival. J. Thorac. Cardiovasc. Surg. 2016, 151, 1147–1151.e4. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.; Suzuki, H.; Huang, J.; Francis, C.; Wang, S.; Tarroni, G.; Guitton, F.; Aung, N.; Fung, K.; Petersen, S.E.; et al. A population-based phenome-wide association study of cardiac and aortic structure and function. Nat. Med. 2020, 26, 1654–1662. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Liu, A.; Gurvitz, M.; Guo, L.; Therrien, J.; Laprise, C.; Kaufman, J.S.; Abrahamowicz, M.; Marelli, A.J. Exposure to Low-Dose Ionizing Radiation from Cardiac Procedures and Malignancy Risk in Adults with Congenital Heart Disease. Circulation 2018, 137, 1334–1345. [Google Scholar] [CrossRef] [PubMed]
- Danieli, C.; Cohen, S.; Liu, A.; Pilote, L.; Guo, L.; Beauchamp, M.E.; Marelli, A.J.; Abrahamowicz, M. Flexible Modeling of the Association between Cumulative Exposure to Low-Dose Ionizing Radiation from Cardiac Procedures and Risk of Cancer in Adults with Congenital Heart Disease. Am. J. Epidemiol. 2019, 188, 1552–1562. [Google Scholar] [CrossRef]
- Gerull, B.; Klassen, S.; Brodhek, A. The Genetic Landscape of Cardiomyopathies; Springer Nature: Berlin/Heidelberg, Germany, 2019; pp. 45–49. Available online: https://link.springer.com/chapter/10.1007/978-3-030-27371-2_2 (accessed on 26 January 2024).







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nappi, F. In-Depth Genomic Analysis: The New Challenge in Congenital Heart Disease. Int. J. Mol. Sci. 2024, 25, 1734. https://doi.org/10.3390/ijms25031734
Nappi F. In-Depth Genomic Analysis: The New Challenge in Congenital Heart Disease. International Journal of Molecular Sciences. 2024; 25(3):1734. https://doi.org/10.3390/ijms25031734
Chicago/Turabian StyleNappi, Francesco. 2024. "In-Depth Genomic Analysis: The New Challenge in Congenital Heart Disease" International Journal of Molecular Sciences 25, no. 3: 1734. https://doi.org/10.3390/ijms25031734
APA StyleNappi, F. (2024). In-Depth Genomic Analysis: The New Challenge in Congenital Heart Disease. International Journal of Molecular Sciences, 25(3), 1734. https://doi.org/10.3390/ijms25031734