
Secondary Modification of S100B Influences Anti Amyloid-β Aggregation Activity and Alzheimer’s Disease Pathology

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
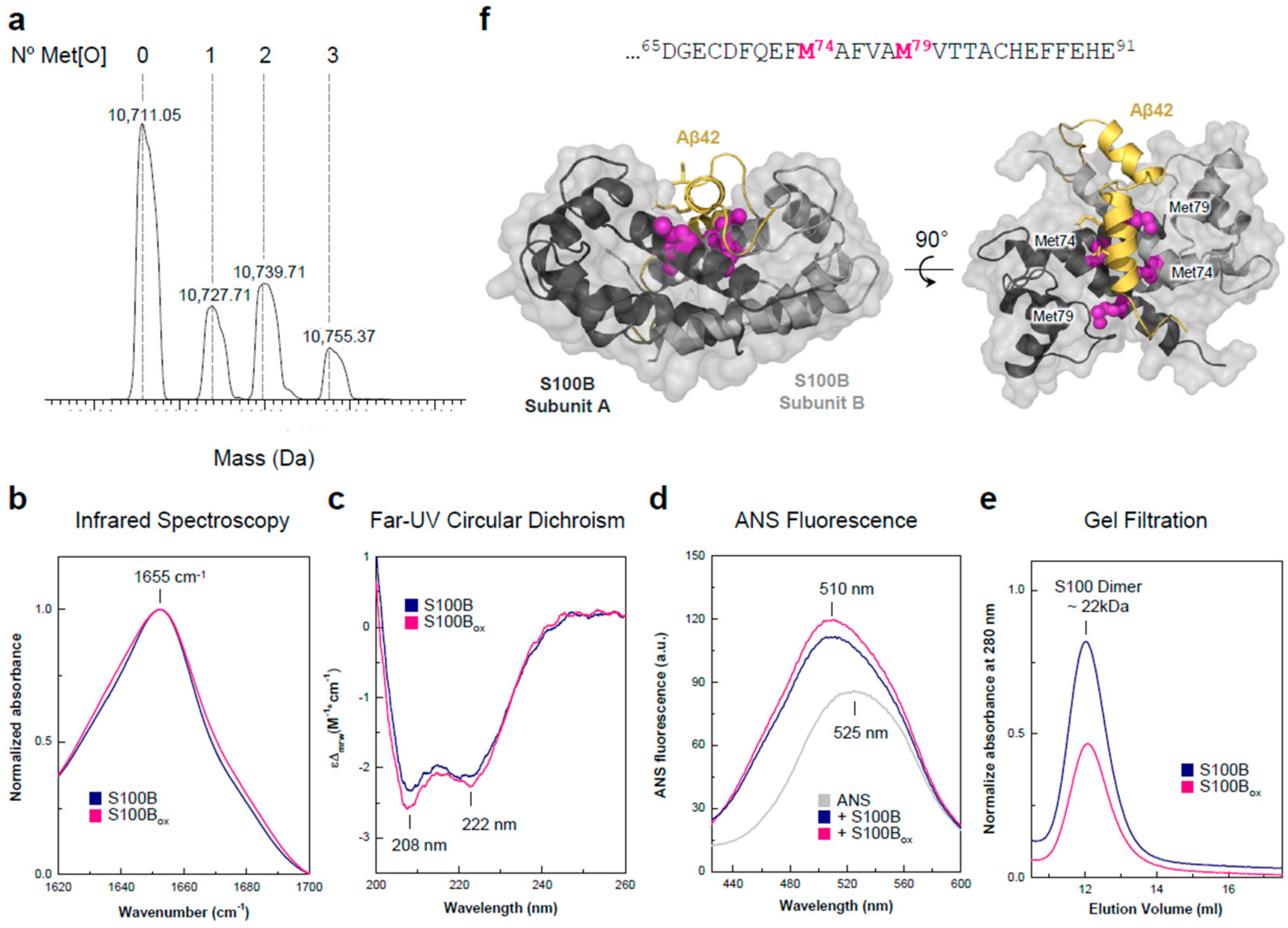
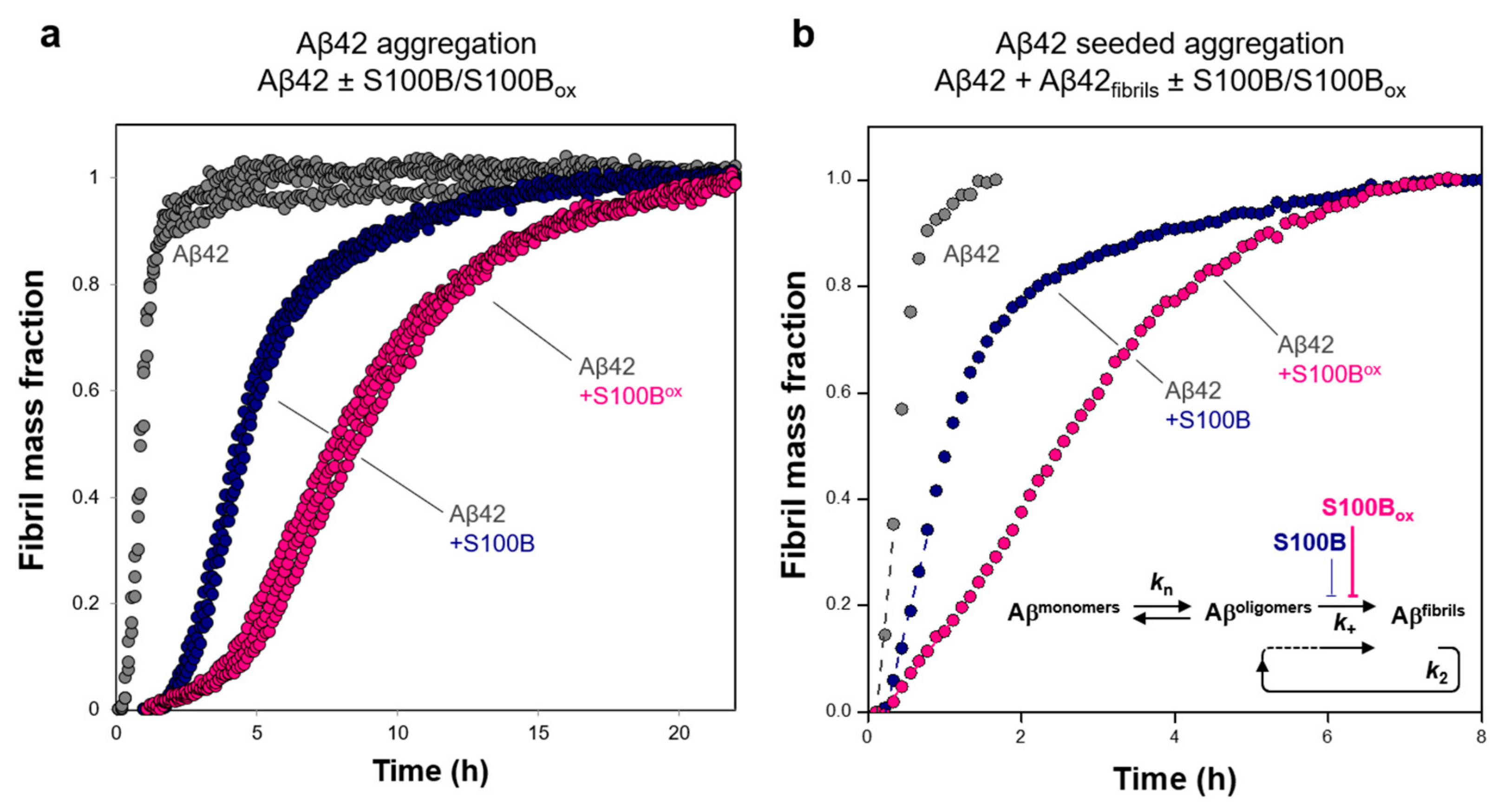
2.1. Effects of Oxidized and Non-Oxidized S100B on Aβ42 Aggregation and Toxicity
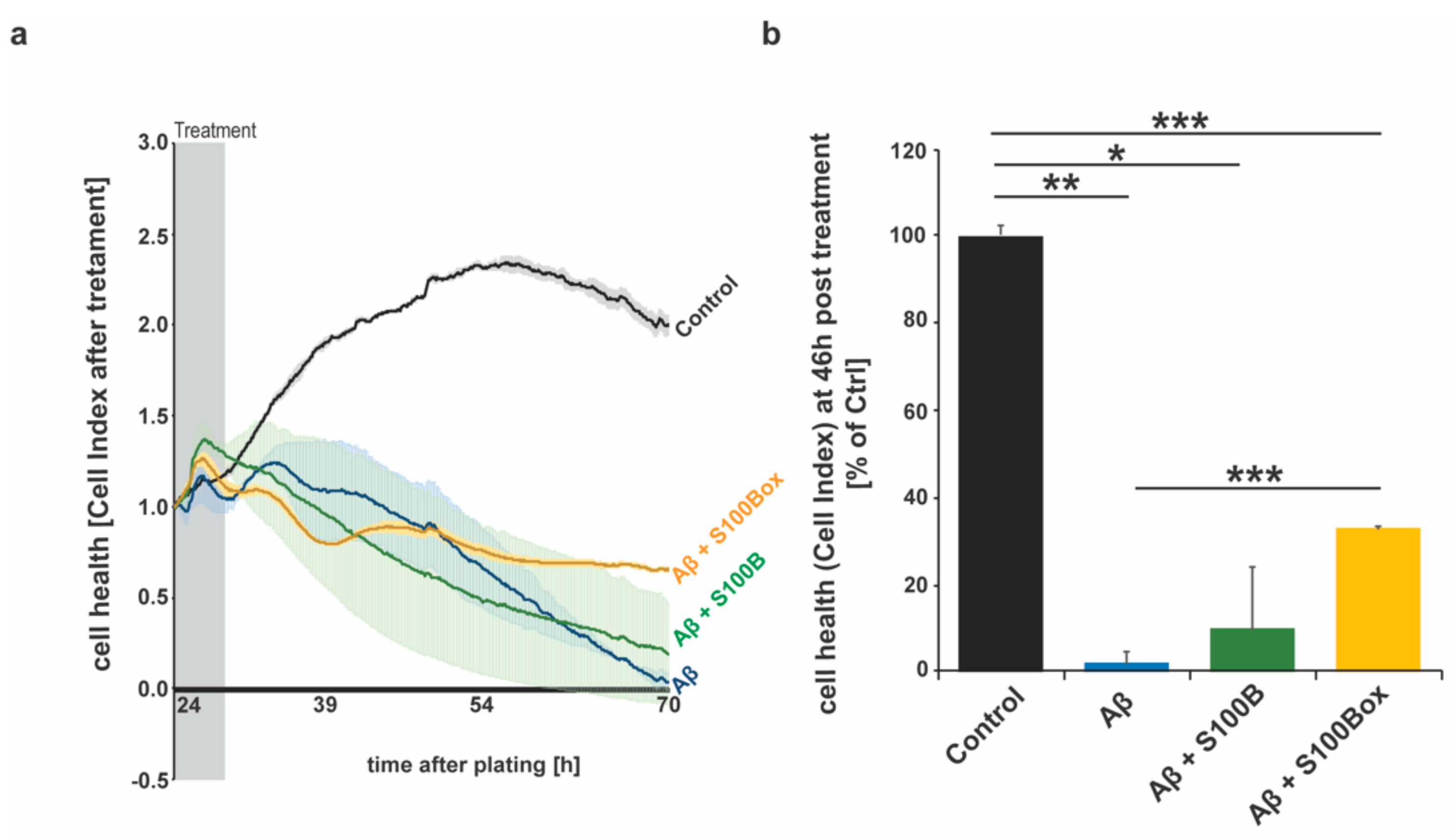
2.2. Effects of Oxidized and Non-Oxidized S100B on Cell Heath in AD
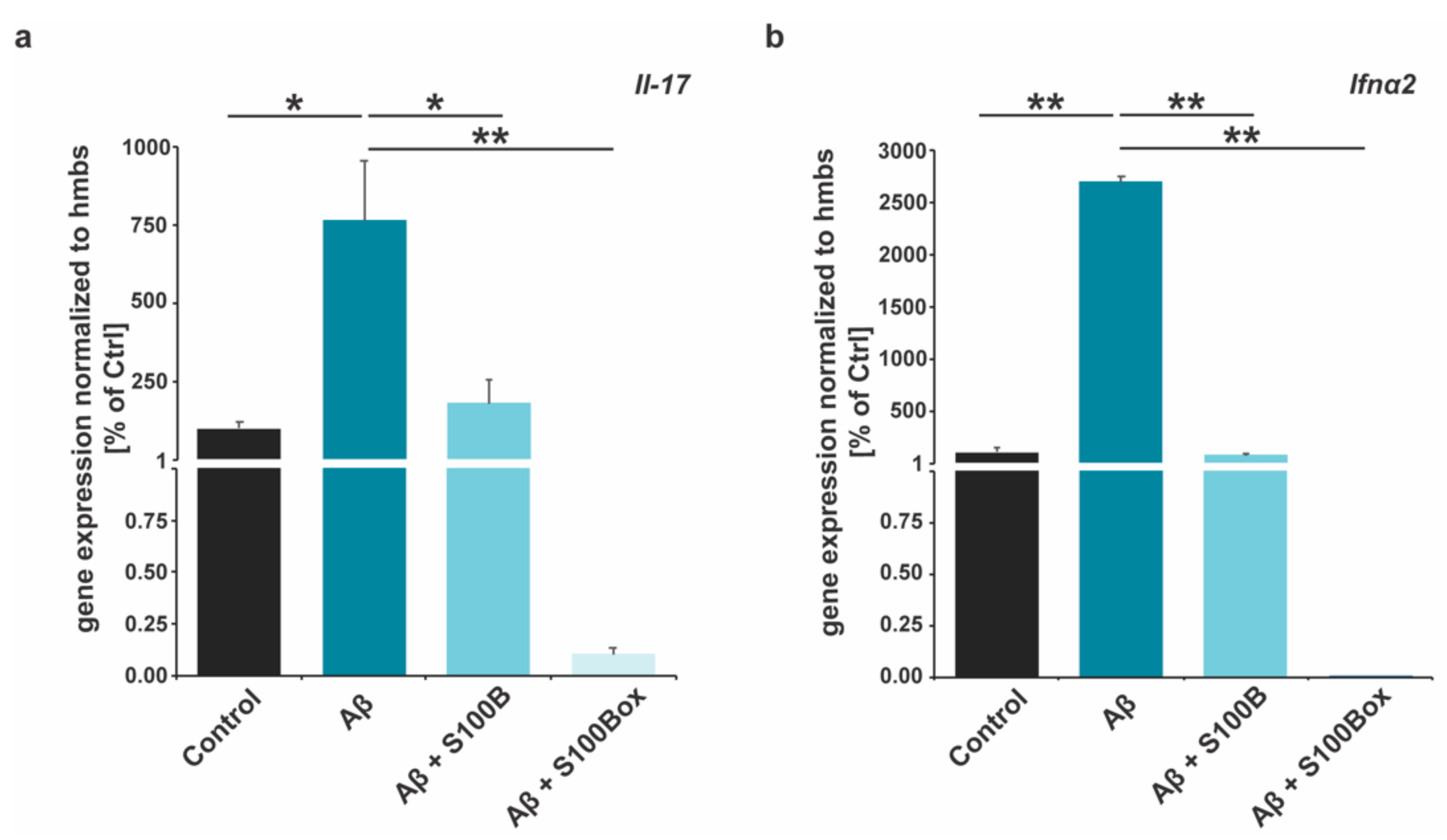
2.3. Cytokine Gene Expression Is Modified by S100B Oxidation
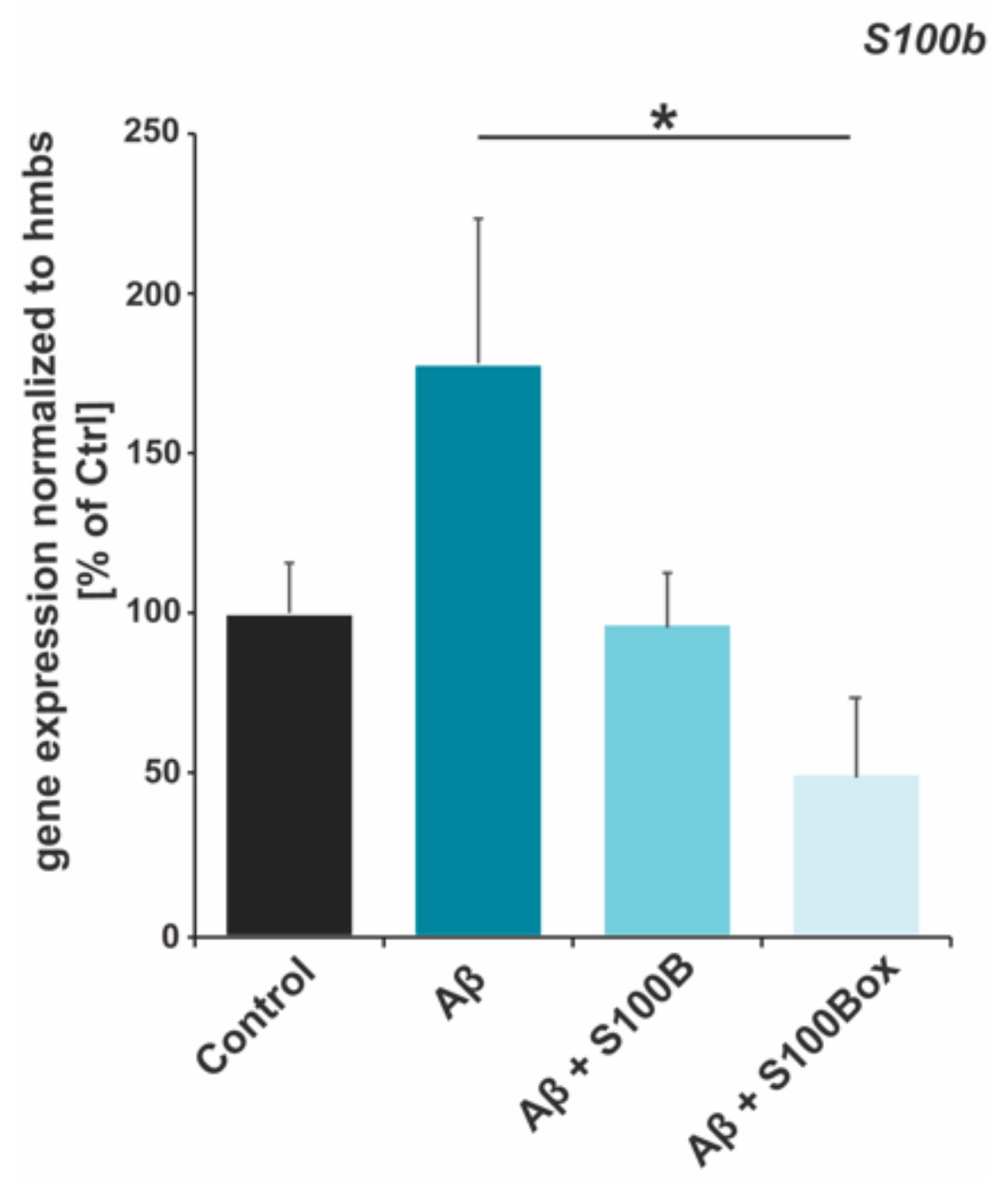
2.4. Autoregulation of S100B Gene Expression
3. Discussion
4. Materials and Methods
4.1. Recombinant Protein Generation
4.2. Mass Spectrometry
4.3. Aggregation Kinetics
4.4. ANS Fluorescence
4.5. Analytical Size-Exclusion Chromatography
4.6. Circular Dichroism (CD)
4.7. Fourier-Transformed Infrared Spectroscopy (FTIR)
4.8. Cell Culture
4.9. Quantitative Real-Time PCR (qRT-PCR)
4.10. Impedance-Based Cell Health Assay
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; Schelle, J.; Jucker, M. The Prion-Like Properties of Amyloid-β Assemblies: Implications for Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2016, 6, a024398. [Google Scholar] [CrossRef] [PubMed]
- Domert, J.; Rao, S.B.; Agholme, L.; Brorsson, A.C.; Marcusson, J.; Hallbeck, M.; Nath, S. Spreading of amyloid-β peptides via neuritic cell-to-cell transfer is dependent on insufficient cellular clearance. Neurobiol. Dis. 2014, 65, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Cuello, A.C. Early and Late CNS Inflammation in Alzheimer’s Disease: Two Extremes of a Continuum? Trends Pharmacol. Sci. 2017, 38, 956–966. [Google Scholar] [CrossRef]
- Su, F.; Bai, F.; Zhang, Z. Inflammatory Cytokines and Alzheimer’s Disease: A Review from the Perspective of Genetic Polymorphisms. Neurosci. Bull. 2016, 32, 469–480. [Google Scholar] [CrossRef]
- Cristóvão, J.S.; Gomes, C.M. S100 Proteins in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 463. [Google Scholar] [CrossRef]
- Singh, P.; Ali, S.A. Multifunctional Role of S100 Protein Family in the Immune System: An Update. Cells 2022, 11, 2274. [Google Scholar] [CrossRef]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [PubMed]
- Kira, G.H.; Paul, T.W.; Kristen, V.; Alexander, D.J.M.; Andrew, C.; Danna, Z.; Rena, L.; David, J.W. Inhibiting S100B in Malignant Melanoma. In Melanoma; Guy Huynh Thien, D., Ed.; IntechOpen: Rijeka, Croatia, 2013; Chapter 24. [Google Scholar]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Botelho, H.M.; Morozova-Roche, L.A.; Gomes, C.M. Natural and amyloid self-assembly of S100 proteins: Structural basis of functional diversity. FEBS J. 2010, 277, 4578–4590. [Google Scholar] [CrossRef] [PubMed]
- Donato, R. Functional roles of S100 proteins, calcium-binding proteins of the EF-hand type. Biochim. Biophys. Acta 1999, 1450, 191–231. [Google Scholar] [CrossRef] [PubMed]
- Ostendorp, T.; Leclerc, E.; Galichet, A.; Koch, M.; Demling, N.; Weigle, B.; Heizmann, C.W.; Kroneck, P.M.; Fritz, G. Structural and functional insights into RAGE activation by multimeric S100B. EMBO J. 2007, 26, 3868–3878. [Google Scholar] [CrossRef]
- Leclerc, E.; Sturchler, E.; Vetter, S.W. The S100B/RAGE Axis in Alzheimer’s Disease. Cardiovasc. Psychiatry Neurol. 2010, 2010, 539581. [Google Scholar] [CrossRef] [PubMed]
- Swindell, W.R.; Johnston, A.; Xing, X.; Little, A.; Robichaud, P.; Voorhees, J.J.; Fisher, G.; Gudjonsson, J.E. Robust shifts in S100a9 expression with aging: A novel mechanism for chronic inflammation. Sci. Rep. 2013, 3, 1215. [Google Scholar] [CrossRef]
- Daini, E.; Hagmeyer, S.; De Benedictis, C.A.; Cristóvão, J.S.; Bodria, M.; Ross, A.M.; Raab, A.; Boeckers, T.M.; Feldmann, J.; Gomes, C.M.; et al. S100B dysregulation during brain development affects synaptic SHANK protein networks via alteration of zinc homeostasis. Transl. Psychiatry 2021, 11, 562. [Google Scholar] [CrossRef]
- Botelho, H.M.; Leal, S.S.; Cardoso, I.; Yanamandra, K.; Morozova-Roche, L.A.; Fritz, G.; Gomes, C.M. S100A6 amyloid fibril formation is calcium-modulated and enhances superoxide dismutase-1 (SOD1) aggregation. J. Biol. Chem. 2012, 287, 42233–42242. [Google Scholar] [CrossRef]
- Cristóvão, J.S.; Morris, V.K.; Cardoso, I.; Leal, S.S.; Martínez, J.; Botelho, H.M.; Göbl, C.; David, R.; Kierdorf, K.; Alemi, M.; et al. The neuronal S100B protein is a calcium-tuned suppressor of amyloid-β aggregation. Sci. Adv. 2018, 4, eaaq1702. [Google Scholar] [CrossRef] [PubMed]
- Hagmeyer, S.; Cristóvão, J.S.; Mulvihill, J.J.E.; Boeckers, T.M.; Gomes, C.M.; Grabrucker, A.M. Zinc Binding to S100B Affords Regulation of Trace Metal Homeostasis and Excitotoxicity in the Brain. Front. Mol. Neurosci. 2018, 10, 456. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Naviaux, R.K. Metabolic features of the cell danger response. Mitochondrion 2014, 16, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Bajor, M.; Zaręba-Kozioł, M.; Zhukova, L.; Goryca, K.; Poznański, J.; Wysłouch-Cieszyńska, A. An Interplay of S-Nitrosylation and Metal Ion Binding for Astrocytic S100B Protein. PLoS ONE 2016, 11, e0154822. [Google Scholar] [CrossRef] [PubMed]
- Van Dieck, J.; Fernandez-Fernandez, M.R.; Veprintsev, D.B.; Fersht, A.R. Modulation of the Oligomerization State of p53 by Differential Binding of Proteins of the S100 Family to p53 Monomers and Tetramers. J. Biol. Chem. 2009, 284, 13804–13811. [Google Scholar] [CrossRef] [PubMed]
- Sroussi, H.Y.; Berline, J.; Palefsky, J.M. Oxidation of methionine 63 and 83 regulates the effect of S100A9 on the migration of neutrophils in vitro. J. Leukoc. Biol. 2007, 81, 818–824. [Google Scholar] [CrossRef]
- Lim, S.Y.; Raftery, M.J.; Goyette, J.; Hsu, K.; Geczy, C.L. Oxidative modifications of S100 proteins: Functional regulation by redox. J. Leukoc. Biol. 2009, 86, 577–587. [Google Scholar] [CrossRef]
- Wu, H.P. Correlation of serum uric acid level with neurotrophy, nerve injury and systemic oxidative stress response in patients with Parkinson’s disease. J. Hainan Med. Univ. 2018, 24, 25–28. [Google Scholar]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef]
- Bai, R.; Guo, J.; Ye, X.-Y.; Xie, Y.; Xie, T. Oxidative stress: The core pathogenesis and mechanism of Alzheimer’s disease. Ageing Res. Rev. 2022, 77, 101619. [Google Scholar] [CrossRef]
- Caruso, G.; Grasso, M.; Fidilio, A.; Torrisi, S.A.; Musso, N.; Geraci, F.; Tropea, M.R.; Privitera, A.; Tascedda, F.; Puzzo, D.; et al. Antioxidant Activity of Fluoxetine and Vortioxetine in a Non-Transgenic Animal Model of Alzheimer’s Disease. Front. Pharmacol. 2021, 12, 809541. [Google Scholar] [CrossRef] [PubMed]
- Raftery, M.J.; Yang, Z.; Valenzuela, S.M.; Geczy, C.L. Novel intra- and inter-molecular sulfinamide bonds in S100A8 produced by hypochlorite oxidation. J. Biol. Chem. 2001, 276, 33393–33401. [Google Scholar] [CrossRef] [PubMed]
- Ravi, J.; Hills, A.E.; Cerasoli, E.; Rakowska, P.D.; Ryadnov, M.G. FTIR markers of methionine oxidation for early detection of oxidized protein therapeutics. Eur. Biophys. J. EBJ 2011, 40, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Parmar, S.; Pawar, S.P.; Iyer, R.; Kalia, D. Aldehyde-mediated bioconjugation via in situ generated ylides. Chem. Commun. 2019, 55, 14926–14929. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2005, 1751, 119–139. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, T.H.; Wilder, P.T.; Liriano, M.A.; Varney, K.M.; Zhong, S.; Coop, A.; Pozharski, E.; MacKerell, A.D., Jr.; Toth, E.A.; Weber, D.J. Small Molecules Bound to Unique Sites in the Target Protein Binding Cleft of Calcium-Bound S100B As Characterized by Nuclear Magnetic Resonance and X-ray Crystallography. Biochemistry 2009, 48, 6202–6212. [Google Scholar] [CrossRef] [PubMed]
- Younan, N.D.; Viles, J.H. A Comparison of Three Fluorophores for the Detection of Amyloid Fibers and Prefibrillar Oligomeric Assemblies. ThT (Thioflavin T); ANS (1-Anilinonaphthalene-8-sulfonic Acid); and bisANS (4,4′-Dianilino-1,1′-binaphthyl-5,5′-disulfonic Acid). Biochemistry 2015, 54, 4297–4306. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.E.P.; Figueira, A.J.; Gomes, C.M.; Machuqueiro, M. Computational Analysis of the Interactions between the S100B Extracellular Chaperone and Its Amyloid β Peptide Client. Int. J. Mol. Sci. 2021, 22, 3629. [Google Scholar] [CrossRef]
- Gade Malmos, K.; Blancas-Mejia, L.M.; Weber, B.; Buchner, J.; Ramirez-Alvarado, M.; Naiki, H.; Otzen, D. ThT 101: A primer on the use of thioflavin T to investigate amyloid formation. Amyloid 2017, 24, 1–16. [Google Scholar] [CrossRef]
- Biancalana, M.; Koide, S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta 2010, 1804, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Knowles, T.P.; Linse, S. On the lag phase in amyloid fibril formation. Phys. Chem. Chem. Phys. 2015, 17, 7606–7618. [Google Scholar] [CrossRef]
- Cohen, S.I.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P. Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [PubMed]
- Chia, S.; Habchi, J.; Michaels, T.C.T.; Cohen, S.I.A.; Linse, S.; Dobson, C.M.; Knowles, T.P.J.; Vendruscolo, M. SAR by kinetics for drug discovery in protein misfolding diseases. Proc. Natl. Acad. Sci. USA 2018, 115, 10245–10250. [Google Scholar] [CrossRef] [PubMed]
- Meisl, G.; Yang, X.; Hellstrand, E.; Frohm, B.; Kirkegaard, J.B.; Cohen, S.I.; Dobson, C.M.; Linse, S.; Knowles, T.P. Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides. Proc. Natl. Acad. Sci. USA 2014, 111, 9384–9389. [Google Scholar] [CrossRef] [PubMed]
- Limbocker, R.; Chia, S.; Ruggeri, F.S.; Perni, M.; Cascella, R.; Heller, G.T.; Meisl, G.; Mannini, B.; Habchi, J.; Michaels, T.C.T.; et al. Trodusquemine enhances Abeta42 aggregation but suppresses its toxicity by displacing oligomers from cell membranes. Nat. Commun. 2019, 10, 225. [Google Scholar] [CrossRef] [PubMed]
- Stine, W.B.; Jungbauer, L.; Yu, C.; LaDu, M.J. Preparing synthetic Aβ in different aggregation states. Methods Mol. Biol. 2011, 670, 13–32. [Google Scholar]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef]
- Dubenko, O.E.; Chyniak, O.S.; Potapov, O.O. Levels of proinflammatory cytokines IL-17 and IL-23 in patients with alzheimer’s disease, mild cognitive impairment and vascular dementia. Wiad. Lek. 2021, 74, 68–71. [Google Scholar] [CrossRef]
- Jin, W.; Dong, C. IL-17 cytokines in immunity and inflammation. Emerg. Microbes Infect. 2013, 2, e60. [Google Scholar] [CrossRef]
- Cristiano, C.; Volpicelli, F.; Lippiello, P.; Buono, B.; Raucci, F.; Piccolo, M.; Iqbal, A.J.; Irace, C.; Miniaci, M.C.; Perrone Capano, C.; et al. Neutralization of IL-17 rescues amyloid-β-induced neuroinflammation and memory impairment. Br. J. Pharmacol. 2019, 176, 3544–3557. [Google Scholar] [CrossRef]
- Taylor, J.M.; Minter, M.R.; Newman, A.G.; Zhang, M.; Adlard, P.A.; Crack, P.J. Type-1 interferon signaling mediates neuro-inflammatory events in models of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Gorlé, N.; Vandenbroucke, R.E. Interferons: A molecular switch between damage and repair in ageing and Alzheimer’s disease. Mech. Ageing Dev. 2019, 183, 111148. [Google Scholar] [CrossRef]
- Figueira, A.J.; Moreira, G.G.; Saavedra, J.; Cardoso, I.; Gomes, C.M. Tetramerization of the S100B chaperone spawns a Ca2+ independent regulatory surface that enhances anti-aggregation activity and client specificity. J. Mol. Biol. 2022, 434, 167791. [Google Scholar] [CrossRef] [PubMed]
- Figueira, A.J.; Saavedra, J.; Cardoso, I.; Gomes, C.M. S100B chaperone multimers suppress the formation of oligomers during Aβ42 aggregation. Front. Neurosci. 2023, 17, 1162741. [Google Scholar] [CrossRef] [PubMed]
- Cristóvão, J.S.; Figueira, A.J.; Carapeto, A.P.; Rodrigues, M.S.; Cardoso, I.; Gomes, C.M. The S100B Alarmin Is a Dual-Function Chaperone Suppressing Amyloid-beta Oligomerization through Combined Zinc Chelation and Inhibition of Protein Aggregation. ACS Chem. Neurosci. 2020, 11, 2753–2760. [Google Scholar] [CrossRef] [PubMed]
- Cristóvão, J.S.; Moreira, G.G.; Rodrigues, F.E.P.; Carapeto, A.P.; Rodrigues, M.S.; Cardoso, I.; Ferreira, A.E.N.; Machuqueiro, M.; Fritz, G.; Gomes, C.M. Cu2+-binding to S100B triggers polymerization of disulfide cross-linked tetramers with enhanced chaperone activity against amyloid-β aggregation. Chem. Commun. 2021, 57, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-C.; Yu, W.-C.; Shih, Y.-H.; Chen, C.-Y.; Guo, Z.-H.; Huang, S.-J.; Chan, J.C.C.; Chen, Y.-R. Zinc ion rapidly induces toxic, off-pathway amyloid-β oligomers distinct from amyloid-β derived diffusible ligands in Alzheimer’s disease. Sci. Rep. 2018, 8, 4772. [Google Scholar] [CrossRef] [PubMed]
- Tonui, R.; John, R.O.; Edkins, A.L. Optimized Microscale Protein Aggregation Suppression Assay: A Method for Evaluating the Holdase Activity of Chaperones. Methods Mol. Biol. 2023, 2693, 113–123. [Google Scholar]
- Tittelmeier, J.; Nachman, E.; Nussbaum-Krammer, C. Molecular Chaperones: A Double-Edged Sword in Neurodegenerative Diseases. Front. Aging Neurosci. 2020, 12, 581374. [Google Scholar] [CrossRef]
- Muchowski, P.J.; Wacker, J.L. Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 2005, 6, 11–22. [Google Scholar] [CrossRef]
- Donato, R.; Sorci, G.; Riuzzi, F.; Arcuri, C.; Bianchi, R.; Brozzi, F.; Tubaro, C.; Giambanco, I. S100B's double life: Intracellular regulator and extracellular signal. Biochim. Biophys. Acta 2009, 1793, 1008–1022. [Google Scholar] [CrossRef]
- Chaplot, K.; Jarvela, T.S.; Lindberg, I. Secreted Chaperones in Neurodegeneration. Front. Aging Neurosci. 2020, 12, 268. [Google Scholar] [CrossRef] [PubMed]
- Österlund, N.; Frankel, R.; Carlsson, A.; Thacker, D.; Karlsson, M.; Matus, V.; Gräslund, A.; Emanuelsson, C.; Linse, S. The C-terminal domain of the antiamyloid chaperone DNAJB6 binds to amyloid-β peptide fibrils and inhibits secondary nucleation. J. Biol. Chem. 2023, 299, 105317. [Google Scholar] [CrossRef] [PubMed]
- Österlund, N.; Lundqvist, M.; Ilag, L.L.; Gräslund, A.; Emanuelsson, C. Amyloid-β oligomers are captured by the DNAJB6 chaperone: Direct detection of interactions that can prevent primary nucleation. J. Biol. Chem. 2020, 295, 8135–8144. [Google Scholar] [CrossRef]
- Cohen, S.I.A.; Arosio, P.; Presto, J.; Kurudenkandy, F.R.; Biverstal, H.; Dolfe, L.; Dunning, C.; Yang, X.; Frohm, B.; Vendruscolo, M.; et al. A molecular chaperone breaks the catalytic cycle that generates toxic Abeta oligomers. Nat. Struct. Mol. Biol. 2015, 22, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Andrade-Talavera, Y.; Tambaro, S.; Leppert, A.; Nilsson, H.E.; Zhong, X.; Landreh, M.; Nilsson, P.; Hebert, H.; Biverstål, H.; et al. Augmentation of Bri2 molecular chaperone activity against amyloid-β reduces neurotoxicity in mouse hippocampus in vitro. Commun. Biol. 2020, 3, 32. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Abelein, A.; Nilsson, H.E.; Leppert, A.; Andrade-Talavera, Y.; Tambaro, S.; Hemmingsson, L.; Roshan, F.; Landreh, M.; Biverstål, H.; et al. Bri2 BRICHOS client specificity and chaperone activity are governed by assembly state. Nat. Commun. 2017, 8, 2081. [Google Scholar] [CrossRef] [PubMed]
- Abelein, A.; Johansson, J. Amyloid inhibition by molecular chaperones in vitro can be translated to Alzheimer’s pathology in vivo. RSC Med. Chem. 2023, 14, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-beta Oligomer Hypothesis: Beginning of the Third Decade. J Alzheimers Dis 2018, 64 (Suppl. S1), S567–S610. [Google Scholar] [CrossRef]
- Linse, S. Monomer-dependent secondary nucleation in amyloid formation. Biophys. Rev. 2017, 9, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Selinfreund, R.H.; Barger, S.W.; Pledger, W.J.; Van Eldik, L.J. Neurotrophic protein S100 beta stimulates glial cell proliferation. Proc. Natl. Acad. Sci. USA 1991, 88, 3554–3558. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, A.; Seoane, R.; González Torres, A.; Rosciszewski, G.; Angelo, M.F.; Rossi, A.; Barker, P.A.; Ramos, A.J. S100B protein activates a RAGE-dependent autocrine loop in astrocytes: Implications for its role in the propagation of reactive gliosis. J. Neurochem. 2014, 131, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Dalskov, L.; Narita, R.; Andersen, L.L.; Jensen, N.; Assil, S.; Kristensen, K.H.; Mikkelsen, J.G.; Fujita, T.; Mogensen, T.H.; Paludan, S.R.; et al. Characterization of distinct molecular interactions responsible for IRF3 and IRF7 phosphorylation and subsequent dimerization. Nucleic Acids Res. 2020, 48, 11421–11433. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Yang, Q.; Wilder, P.T.; Carrier, F.; Weber, D.J. The calcium-binding protein S100B down-regulates p53 and apoptosis in malignant melanoma. J. Biol. Chem. 2010, 285, 27487–27498. [Google Scholar] [CrossRef] [PubMed]
- Jebelli, J.D.; Hooper, C.; Garden, G.A.; Pocock, J.M. Emerging roles of p53 in glial cell function in health and disease. Glia 2012, 60, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Botelho, H.M.; Fritz, G.; Gomes, C.M. Analysis of S100 oligomers and amyloids. Methods Mol. Biol. 2012, 849, 373–386. [Google Scholar] [PubMed]
- Walsh, D.M.; Thulin, E.; Minogue, A.M.; Gustavsson, N.; Pang, E.; Teplow, D.B.; Linse, S. A facile method for expression and purification of the Alzheimer’s disease-associated amyloid beta-peptide. FEBS J. 2009, 276, 1266–1281. [Google Scholar] [CrossRef]
- Zhang, Z.; Marshall, A.G. A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. J. Am. Soc. Mass Spectrom. 1998, 9, 225–233. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coelho, R.; De Benedictis, C.A.; Sauer, A.K.; Figueira, A.J.; Faustino, H.; Grabrucker, A.M.; Gomes, C.M. Secondary Modification of S100B Influences Anti Amyloid-β Aggregation Activity and Alzheimer’s Disease Pathology. Int. J. Mol. Sci. 2024, 25, 1787. https://doi.org/10.3390/ijms25031787
Coelho R, De Benedictis CA, Sauer AK, Figueira AJ, Faustino H, Grabrucker AM, Gomes CM. Secondary Modification of S100B Influences Anti Amyloid-β Aggregation Activity and Alzheimer’s Disease Pathology. International Journal of Molecular Sciences. 2024; 25(3):1787. https://doi.org/10.3390/ijms25031787
Chicago/Turabian StyleCoelho, Romina, Chiara A. De Benedictis, Ann Katrin Sauer, António J. Figueira, Hélio Faustino, Andreas M. Grabrucker, and Cláudio M. Gomes. 2024. "Secondary Modification of S100B Influences Anti Amyloid-β Aggregation Activity and Alzheimer’s Disease Pathology" International Journal of Molecular Sciences 25, no. 3: 1787. https://doi.org/10.3390/ijms25031787
APA StyleCoelho, R., De Benedictis, C. A., Sauer, A. K., Figueira, A. J., Faustino, H., Grabrucker, A. M., & Gomes, C. M. (2024). Secondary Modification of S100B Influences Anti Amyloid-β Aggregation Activity and Alzheimer’s Disease Pathology. International Journal of Molecular Sciences, 25(3), 1787. https://doi.org/10.3390/ijms25031787