
Experimental Evidences on Magnetism-Covalent Bonding Interplay in Structural Properties of Solids and during Chemisorption



Abstract
:1. Introduction
2. Discussion
2.1. On the Interplay between Magnetism and Covalent Bonding
2.2. Evidence of the Interplay between Magnetism and Covalent Bonding in Materials
2.3. Experimental Evidence on Magnetism–Covalent Bonding Interplay during Chemisorption
- (1)
- The orbital hybridization between the sp-electrons of the adsorbate and the d-electrons of the FM catalyst;
- (2)
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dowden, D.A. 56. Heterogeneous catalysis. Part I. Theoretical basis. J. Chem. Soc. 1950, 242–265. [Google Scholar] [CrossRef]
- Hagen, J. Industrial Catalysis: A Practical Approach, 2nd ed.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005. [Google Scholar]
- Blundell, S. Magnetism in Condensed Matter. In Oxford Master Series in Condensed Matter Physics; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Getzlaff, M. Fundamentals of Magnetism; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Wijn, H.J. Magnetic Properties of Metals. d-Elements, Alloys and Compounds. In Data in Science and Technology; Springer-Verlag: Berlin/Heidelberg, Germany, 1991. [Google Scholar]
- Ryden, W.D.; Lawson, A.W. Magnetic Susceptibility of IrO2 and RuO2. J. Chem. Phys. 1970, 52, 6058–6061. [Google Scholar] [CrossRef]
- Mabbs, F.E.; Machin, D.J. Magnetism and Transition Metal Complexes; Dover Publications, Inc.: Mineola, NY, USA, 2008. [Google Scholar]
- Biz, C.; Fianchini, M.; Gracia, J. Strongly Correlated Electrons in Catalysis: Focus on Quantum Exchange. ACS Catal. 2021, 11, 14249–14261. [Google Scholar] [CrossRef]
- Biz, C.; Gracia, J.; Fianchini, M. Review on Magnetism in Catalysis: From Theory to PEMFC Applications of 3d Metal Pt-Based Alloys. Int. J. Mol. Sci. 2022, 23, 14768. [Google Scholar] [CrossRef]
- Ishikawa, Y.M.N. Physics and Engineering Applications of Magnetism. In Springer Series in Solid-State Science 92; Springer-Verlag: Berlin/Heidelberg, Germany, 1991. [Google Scholar]
- Goodenough, J.B. Magnetism and the Chemical Bond; John Wiley & Sons: Hoboken, NJ, USA, 1963. [Google Scholar]
- Selwood, W. Magnetism and Catalysis. Chem. Rev. 1946, 38, 41–82. [Google Scholar] [CrossRef] [PubMed]
- Voorhoeve, R.J.H. Experimental Relationships between Catalysis and Magnetism. AIP Conf. Proc. 1974, 18, 19–32. [Google Scholar]
- Richardson, J.T. Magnetism and catalysis. J. Appl. Phys. 1978, 49, 1781–1786. [Google Scholar] [CrossRef]
- Cullen, D.A.; Neyerlin, K.C.; Ahluwalia, R.K.; Mukundan, R.; More, K.L.; Borup, R.L.; Weber, A.Z.; Myers, D.J.; Kusoglu, A. New roads and challenges for fuel cells in heavy-duty transportation. Nat. Energy 2021, 6, 462–474. [Google Scholar] [CrossRef]
- Sapountzi, F.M.; Gracia, J.M.; Weststrate, C.J.; Fredriksson, H.O.A.; Niemantsverdriet, J.W. Electrocatalysts for the generation of hydrogen, oxygen and synthesis gas. Prog. Energy Combust. Sci. 2017, 58, 1–35. [Google Scholar] [CrossRef]
- Anantharaj, S.; Kundu, S.; Noda, S. “The Fe Effect”: A review unveiling the critical roles of Fe in enhancing OER activity of Ni and Co based catalysts. Nano Energy 2020, 80, 105514. [Google Scholar] [CrossRef]
- Luo, S.; Elouarzaki, K.; Xu, Z. Electrochemistry in Magnetic Fields. Angew. Chem. Int. Ed. 2022, 61, e202203564. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Wu, T.; Gong, Z.; Pan, L.; Meng, J.; Yang, H.; Dagbjartsdottir, F.B.; Fisher, A.; Gao, H.-J.; Xu, Z.J. The origin of magnetization-caused increment in water oxidation. Nat. Commun. 2023, 14, 2482. [Google Scholar] [CrossRef] [PubMed]
- Kittel, C. Introduction to Solid State Physics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Rossler, U. Solid State Theory: An Introduction; Physica-Verlag: Heidelberg, Germany, 2009. [Google Scholar]
- Miller, G.J.; Zhang, Y.; Wagner, F. Chemical Bonding in Solids. In Handbook of Solid State Chemistry; Verlag GmbH & Co.: Weinheim, Germany, 2017; pp. 405–489. [Google Scholar]
- David, R.L. (Ed.) The Madelung Constant and Crystal Lattice Energy. In CRC Handbook of Chemistry and Physics, Internet Version; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules. A quantum theory. In The International Series of Monographs on Chemistry; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Darling, A.S. Cobalt-Platinum Alloys. A Critical Review of their Constitution and Properties. Platin. Met. Rev. 1963, 7, 96. [Google Scholar]
- Shi, Q.; Zhu, C.; Du, D.; Lin, Y. Robust noble metal-based electrocatalysts for oxygen evolution reaction. Chem. Soc. Rev. 2019, 48, 3181–3192. [Google Scholar] [CrossRef]
- Berlijn, T.; Snijders, P.C.; Delaire, O.; Zhou, H.D.; Maier, T.A.; Cao, H.B.; Chi, S.X.; Matsuda, M.; Wang, Y.; Koehler, M.R.; et al. Itinerant Antiferromagnetism in RuO2. Phys. Rev. Lett. 2017, 118, 077201. [Google Scholar] [CrossRef]
- Gracia, J.; Biz, C.; Fianchini, M.; Amthor, S. How advances in theoretical chemistry meet industrial expectations in electrocatalysts for water splitting. Catal. Sci. Technol. 2023, 13, 6751–6763. [Google Scholar] [CrossRef]
- Alaswad, A.; Omran, A.; Sodre, J.R.; Wilberforce, T.; Pignatelli, G.; Dassisti, M.; Baroutaji, A.; Olabi, A.G. Technical and Commercial Challenges of Proton-Exchange Membrane (PEM) Fuel Cells. Energies 2021, 14, 144. [Google Scholar] [CrossRef]
- Wang, X.X.; Swihart, M.T.; Wu, G. Achievements, challenges and perspectives on cathode catalysts in proton exchange membrane fuel cells for transportation. Nat. Catal. 2019, 2, 578–589. [Google Scholar] [CrossRef]
- Grigoriev, S.A.; Fateev, V.N.; Bessarabov, D.G.; Millet, P. Current status, research trends, and challenges in water electrolysis science and technology. Int. J. Hydrogen Energy 2020, 45, 26036–26058. [Google Scholar] [CrossRef]
- Ponikvar, Ž.; Likozar, B.; Gyergyek, S. Electrification of Catalytic Ammonia Production and Decomposition Reactions: From Resistance, Induction, and Dielectric Reactor Heating to Electrolysis. ACS Appl. Energy Mater. 2022, 5, 5457–5472. [Google Scholar] [CrossRef]
- Teržan, J.; Huš, M.; Likozar, B.; Djinović, P. Propylene Epoxidation using Molecular Oxygen over Copper- and Silver-Based Catalysts: A Review. ACS Catal. 2020, 10, 13415–13436. [Google Scholar] [CrossRef]
- David., R.L. (Ed.) Magnetic susceptibility of the elements and inorganic compounds. In CRC Handbook of Chemistry and Physics; Internet Version 2005; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Coey, J.M.D.; Venkatesan, M.; Xu, H. Introduction to Magnetic Oxides. In Functional Metal Oxides; Ogale, S., Venkatesan, T., Blamire, M., Eds.; Verlag GmbH & Co.: Weinheim, Germany, 2013; pp. 1–49. [Google Scholar]
- Liu, G.; Hagelin-Weaver, H.; Welt, B. A Concise Review of Catalytic Synthesis of Methanol from Synthesis Gas. Waste 2023, 1, 228–248. [Google Scholar] [CrossRef]
- Twigg, M.V.; Spencer, M.S. Deactivation of supported copper metal catalysts for hydrogenation reactions. Appl. Catal. A Gen. 2001, 212, 161–174. [Google Scholar] [CrossRef]
- Millar, G.J.; Collins, M. Industrial Production of Formaldehyde Using Polycrystalline Silver Catalyst. Ind. Eng. Chem. Res. 2017, 56, 9247–9265. [Google Scholar] [CrossRef]
- Pu, T.; Tian, H.; Ford, M.E.; Rangarajan, S.; Wachs, I.E. Overview of Selective Oxidation of Ethylene to Ethylene Oxide by Ag Catalysts. ACS Catal. 2019, 9, 10727–10750. [Google Scholar] [CrossRef]
- Mugiraneza, S.; Hallas, A.M. Tutorial: A beginner’s guide to interpreting magnetic susceptibility data with the Curie-Weiss law. Commun. Phys. 2022, 5, 95. [Google Scholar] [CrossRef]
- Hatscher, S.; Schilder, H.; Lueken, H.; Urland, W. Practical guide to measurement and interpretation of magnetic properties. IUPAC Tech. Rep. 2005, 77, 497–511. [Google Scholar]
- Gothe, M.L.; Silva, K.L.C.; Figueredo, A.L.; Fiorio, J.L.; Rozendo, J.; Manduca, B.; Simizu, V.; Freire, R.S.; Garcia, M.A.S.; Vidinha, P. Rhenium—A Tuneable Player in Tailored Hydrogenation Catalysis. Eur. J. Inorg. Chem. 2021, 2021, 4043–4065. [Google Scholar] [CrossRef]
- Luo, L.; Li, H.; Peng, Y.; Feng, C.; Zeng, J. Rh-Based Nanocatalysts for Heterogeneous Reactions. ChemNanoMat 2018, 4, 451–466. [Google Scholar] [CrossRef]
- Krajczewski, J.; Ambroziak, R.; Kudelski, A. Formation and selected catalytic properties of ruthenium, rhodium, osmium and iridium nanoparticles. RSC Adv. 2022, 12, 2123–2144. [Google Scholar] [CrossRef]
- Tofield, B.C. Covalency effects in magnetic interactions. J. Phys. Colloq. 1976, 37, 539–570. [Google Scholar] [CrossRef]
- Su, Y.; Wang, Z.; Gao, R.; Wu, Q.; Zhao, J.; Zhu, G.; Li, Q.; Xu, H.; Pan, Y.; Gu, K.; et al. Ferromagnetic L12-Pt3Co Nanowires with Spin-Polarized Orbitals for Fast and Selective Oxygen Reduction Electrocatalysis. Adv. Funct. Mater. 2023, 2311618. [Google Scholar] [CrossRef]
- Pauling, L. The nature of the chemical bond. Application of results obtained from the quantum mechanics and from a theory of paramagnetic susceptibility to the structure of molecules. J. Am. Chem. Soc. 1931, 53, 1367–1400. [Google Scholar] [CrossRef]
- Hubbard, J.; Marshall, W. Covalency effects in neutron diffraction from ferromagnetic and antiferromagnetic salts. Proc. Phys. Soc. 1965, 86, 561. [Google Scholar] [CrossRef]
- Owen, J.; Thornley, J.H.M. Covalent bonding and magnetic properties of transition metal ions. Rep. Prog. Phys. 1966, 29, 675. [Google Scholar] [CrossRef]
- Anderson, W. Theory of Magnetic Exchange Interactions:Exchange in Insulators and Semiconductors. In Solid State Physics; Seitz, F., Turnbull, D., Eds.; Academic Press: Cambridge, MA, USA, 1963; pp. 99–214. [Google Scholar]
- Tofield, B.C. The study of covalency by magnetic neutron scattering. In Recent Impact of Physics on Inorganic Chemistry; Springer: Berlin/Heidelberg, Germany, 1975. [Google Scholar]
- Tofield, B.C.; Fender, B.E.F. Covalency parameters for Cr3+, Fe3+ and Mn4+ in an oxide environment. J. Phys. Chem. Solids 1970, 31, 2741–2749. [Google Scholar] [CrossRef]
- Plakhty, V.P.; Gukasov, A.G.; Papoular, R.J.; Smirnov, O.P. Spin density on ligands O2− and covalency of Fe3+ ions in octahedral sites of the Ca3Fe2Ge3O12 garnet: A polarised neutron diffraction study. Europhys. Lett. 1999, 48, 233. [Google Scholar] [CrossRef]
- Sawatzky, G.A.; Van Der Woude, F. Covalency effects in hyperfine interactions. J. Phys. Colloq. 1974, 35, 47–60. [Google Scholar] [CrossRef]
- Dirac, A.M.; Fowler, R. On the theory of quantum mechanics. Proc. R. Soc. London. Ser. A Contain. Pap. A Math. Phys. Character 1997, 112, 661–677. [Google Scholar]
- Heisenberg, W. Mehrkörperproblem und Resonanz in der Quantenmechanik. Z. Phys. 1926, 38, 411–426. [Google Scholar] [CrossRef]
- Szabo, A.O.; Ostlund, N.S. Modern Quantum Chemistry. In Introduction to Advanced Electronic Structure Theory; Dover Publications, Inc.: Mineola, NY, USA, 1996. [Google Scholar]
- Goodenough, J.B.; Loeb, A.L. Theory of Ionic Ordering, Crystal Distortion, and Magnetic Exchange Due to Covalent Forces in Spinels. Phys. Rev. B 1955, 98, 391–408. [Google Scholar] [CrossRef]
- Bluhm, H.B.M.M.; von Plessen, G.; Stampfer, C. Electrons in solids. In Graduate Texts in Condensed Matter; Walter De Gruyter GmbH: Berlin,Germany; Boston, MA, USA, 2019. [Google Scholar]
- Candy, J. On the use of magnetic methods for studying the adsorption of H2, O2, CO, and hydrocarbons on supported palladium. J. Chem. Soc. Chem. Commun. 1983, 19, 1105–1106. [Google Scholar] [CrossRef]
- Candy, J.; Perrichon, V. Magnetic study of CO and C2 hydrocarbons adsorption on PdSiO2 catalyst. J. Catal. 1984, 89, 93–99. [Google Scholar] [CrossRef]
- Cotton, J.D.; Fensham, P.J. Magnetic Susceptibility Changes during the Adsorption of Oxygen and Carbon Monoxide on Cuprous Oxide. J. Phys. Chem. 1964, 68, 1052–1055. [Google Scholar] [CrossRef]
- Selwood, W. Magnetism and the Structure of Catalytically Active Solids. In Advances in Catalysis; Frankenburg, W.G., Komarewsky, V.I., Rideal, E.K., Emmett, P.H., Taylor, H.S., Eds.; Academic Press: Cambridge, MA, USA, 1951; pp. 27–106. [Google Scholar]
- Oikawa, M. On the Relation between the Hydrogen Overpotential and the Magnetic Susceptibility of Copper-Nickel Alloys. Bull. Chem. Soc. Jpn. 1955, 28, 626–629. [Google Scholar] [CrossRef]
- Mulay, L.N.; Mulay, I.L. Magnetic Susceptibility: Recent Aspects of Instrumentation and Applications. Anal. Chem. 1966, 38, 501–513. [Google Scholar] [CrossRef]
- Chernavskii, P.A.; Dalmon, J.A.; Perov, N.S.; Khodakov, A.Y. Magnetic Characterization of Fischer-Tropsch Catalysts. Oil Gas Sci. Technol.-Rev. IFP 2009, 64, 25–48. [Google Scholar] [CrossRef]
- Morris, H.; Selwood, P.W. Magnetic Measurements on Some Catalytically Active Substances. J. Am. Chem. Soc. 1943, 65, 2245–2252. [Google Scholar] [CrossRef]
- Kadyk, T.; Eikerling, M. Magnetic susceptibility as a direct measure of oxidation state in LiFePO4 batteries and cyclic water gas shift reactors. Phys. Chem. Chem. Phys. 2015, 17, 19834–19843. [Google Scholar] [CrossRef]
- Rodulfo-Baechler, S.M.; González-Cortés, S.L.; Orozco, J.; Sagredo, V.; Fontal, B.; Mora, A.J.; Delgado, G. Characterization of modified iron catalysts by X-ray diffraction, infrared spectroscopy, magnetic susceptibility and thermogravimetric analysis. Mater. Lett. 2004, 58, 2447–2450. [Google Scholar] [CrossRef]
- Dilke, M.H.; Eley, D.D.; Maxted, E.B. Catalytic Poisons and Magnetic Susceptibility. Nature 1948, 161, 804. [Google Scholar] [CrossRef]
- Stradella, L.; Genova, E. The influence of chemisorbed gases on the magnetization of transition metals. J. Mater. Sci. Lett. 1995, 14, 44–45. [Google Scholar] [CrossRef]
- Selwood, W. Chemisorption and Magnetization; Academic Press: Cambridge, MA, USA, 1975. [Google Scholar]
- Gradmann, U. Surface magnetism. J. Magn. Magn. Mater. 1991, 100, 481–496. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Lee, S.-C. Cooperation and competition between magnetism and chemisorption. Phys. Chem. Chem. Phys. 2021, 23, 3802–3809. [Google Scholar] [CrossRef] [PubMed]
- Silvent, J.A.; Selwood, P.W. The Mechanism of Benzene Chemisorption on a Supported Nickel Catalyst. J. Am. Chem. Soc. 1961, 83, 1033–1035. [Google Scholar] [CrossRef]
- Leak, R.J.; Selwood, W. The Chemisorption of Oxygen on Nickel. J. Phys. Chem. 1960, 64, 1114–1120. [Google Scholar] [CrossRef]
- Besten, I.E.D.; Fox, P.G.; Selwood, P.W. The mechanism of chemisorption: Carbon Monoxide and Carbon Dioxide on Nickel. J. Phys. Chem. 1962, 66, 450–453. [Google Scholar] [CrossRef]
- Schönhense, G.; Donath, M.; Kolac, U.; Dose, V. Exchange splitting of an oxygen 2p-derived band at Ni(100). Surf. Sci. 1988, 206, L888–L894. [Google Scholar] [CrossRef]
- Schmitt, W.; Hopster, H.; Güntherodt, G. Influence of adsorbates on surface magnetism studied by spin-resolved photoemission spectroscopy. Phys. Rev. B 1985, 31, 4035–4038. [Google Scholar] [CrossRef] [PubMed]
- Sinković, B.; Johnson, P.D.; Brookes, N.B.; Clarke, A.; Smith, N.V. Study of local magnetic properties of an adsorbate by spin-polarized Auger-electron spectroscopy. Phys. Rev. Lett. 1989, 62, 2740–2743. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.D.; Clarke, A.; Brookes, N.B.; Hulbert, S.L.; Sinkovic, B.; Smith, N.V. Exchange-Split Adsorbate Bands: The Role of Substrate Hybridization. Phys. Rev. Lett. 1988, 61, 2257–2260. [Google Scholar] [CrossRef] [PubMed]
- Peter, D.J. Spin-polarized photoemission. Rep. Prog. Phys. 1997, 60, 1217. [Google Scholar]
- Seiler, A.; Feigerle, C.S.; Pena, J.L.; Celotta, R.J.; Pierce, D.T. Connection between surface magnetism and electronic structure of oxygen on Ni(110). J. Appl. Phys. 1985, 57, 3638–3640. [Google Scholar] [CrossRef]
- Nishihara, K.; Nomitsu, T.; Nakagawa, T.; Mizuno, S. Structural investigation and magnetic properties of oxygen adsorption on ultrathin Fe(110) film. Surf. Sci. 2019, 685, 34–39. [Google Scholar] [CrossRef]
- Johnson, D. Magnetism and Chemisorption. J. Electron Spectrosc. Relat. Phenom. 1990, 51, 249–262. [Google Scholar] [CrossRef]
- Abeledo, C.R.; Selwood, W. Chemisorption of Hydrogen on Cobalt. J. Chem. Phys. 1962, 37, 2709–2712. [Google Scholar] [CrossRef]
- Göpel, W. “Magnetic dead layers” on chemisorption at ferromagnetic surfaces. Surf. Sci. 1979, 85, 400–412. [Google Scholar] [CrossRef]
- Feder, R.; Hopster, H. Chemisorption and surface ferromagnetism: Theoretical analysis of spin-resolved photoemission data from Ni(1 1 0)(2×1)−O. Solid State Commun. 1985, 55, 1043–1047. [Google Scholar] [CrossRef]
- Dietz, R.E.; Selwood, P.W. Effect of Chemisorbed Hydrogen on the Magnetization of Nickel. J. Chem. Phys. 1961, 35, 270–281. [Google Scholar] [CrossRef]
- Donath, M. Spin-resolved inverse photoemission of ferromagnetic surfaces. Appl. Phys. A 1989, 49, 351–364. [Google Scholar] [CrossRef]
- Forzatti, P.; Lietti, L. Catalyst deactivation. Catal. Today 1999, 52, 165–181. [Google Scholar] [CrossRef]
- Martin, G.A. How Far Can Magnetic Methods Help in the Understanding of Metal Catalyst Deactivation? In Studies in Surface Science and Catalysis; Bartholomew, C.H., Butt, J.B., Eds.; Elsevier: Amsterdam, The Netherlands, 1991; pp. 659–666. [Google Scholar]
- Ng, C.; Chang, Y. Arsine poisoning of nickel/silica catalysts : Hydrogen chemisorption study by magnetic method. Appl. Catal. 1991, 70, 213–224. [Google Scholar] [CrossRef]
- Brandt, S.; Knöll, J.; Jiang, H.; Saar, J.; Fougret, C. The Role of Magnetic Susceptibility in Detecting Iron Poisoning in FCC Equilibrium Catalyst Samples and Its Combination with Other Macroscopic Bulk Analysis Techniques. Ind. Eng. Chem. Res. 2019, 58, 20528–20535. [Google Scholar] [CrossRef]
- Schmitt, W.; Kämper, K.-P.; Güntherodt, G. Effect of adsorbates on the spin-polarized photoemission of itinerant ferromagnets. Phys. Rev. B 1987, 36, 3763–3768. [Google Scholar] [CrossRef] [PubMed]
- Landolt, M.; Campagna, M. Demagnetization of the Ni(100) Surface by Hydrogen Adsorption. Phys. Rev. Lett. 1977, 39, 568–570. [Google Scholar] [CrossRef]
- Feigerle, C.S.; Seiler, A.; Peña, J.L.; Celotta, R.J.; Pierce, D.T. Chemisorbed oxygen on Ni(110) studied by spin polarized inverse photoemission. J. Vac. Sci. Technol. A 1985, 3, 1487–1490. [Google Scholar] [CrossRef]
- Elmers, H.; Gradmann, U. Magnetometric analysis of the interaction of Ni(111) with oxygen. Surf. Sci. 1988, 193, 94–108. [Google Scholar] [CrossRef]
- Allenspach, R.; Taborelli, M.; Landolt, M. Oxygen on Fe(100): An Initial-Oxidation Study by Spin-Polarized Auger Spectroscopy. Phys. Rev. Lett. 1985, 55, 2599–2602. [Google Scholar] [CrossRef] [PubMed]
- Rau, C. Electron spin polarization esp at surfaces of ferromagnetic metals. J. Magn. Magn. Mater. 1982, 30, 141–174. [Google Scholar] [CrossRef]
- Feigerle, C.S.; Seiler, A.; Peña, J.L.; Celotta, R.J.; Pierce, D.T. CO Chemisorption on Ni(110): Effect on Surface Magnetism. Phys. Rev. Lett. 1986, 56, 2207–2210. [Google Scholar] [CrossRef]
- Elmers, H.J.; Gradmann, U. Surface magnetism of oxygen and hydrogen adsorption on Ni(111). J. Appl. Phys. 1988, 63, 3664–3666. [Google Scholar] [CrossRef]
- Quesada, A.; Chen, G.; N'Diaye, A.T.; Wang, P.; Wu, Y.Z.; Schmid, A.K. Non-monotonic magnetic anisotropy behavior as a function of adsorbate coverage in Fe ultrathin films near the spin reorientation transition. J. Mater. Chem. C 2021, 9, 2801–2805. [Google Scholar] [CrossRef]
- Ma, X.-D.; Nakagawa, T.; Yokoyama, T. Effect of surface chemisorption on the spin reorientation transition in magnetic ultrathin Fe film on Ag(001). Surf. Sci. 2006, 600, 4605–4612. [Google Scholar] [CrossRef]
- Chen, G.; Ophus, C.; Quintana, A.; Kwon, H.; Won, C.; Ding, H.; Wu, Y.; Schmid, A.K.; Liu, K. Reversible writing/deleting of magnetic skyrmions through hydrogen adsorption/desorption. Nat. Commun. 2022, 13, 1350. [Google Scholar] [CrossRef] [PubMed]
- Meron, S.; Shenberger, Y.; Ruthstein, S. The Advantages of EPR Spectroscopy in Exploring Diamagnetic Metal Ion Binding and Transfer Mechanisms in Biological Systems. Magnetochemistry 2022, 8, 3. [Google Scholar] [CrossRef]
- Bonke, S.A.; Risse, T.; Schnegg, A.; Brückner, A. In situ electron paramagnetic resonance spectroscopy for catalysis. Nat. Rev. Methods Prim. 2021, 1, 33. [Google Scholar] [CrossRef]
- Winkler, R.; Ciria, M.; Ahmad, M.; Plank, H.; Marcuello, C. A Review of the Current State of Magnetic Force Microscopy to Unravel the Magnetic Properties of Nanomaterials Applied in Biological Systems and Future Directions for Quantum Technologies. Nanomaterials 2023, 13, 2585. [Google Scholar] [CrossRef] [PubMed]
- Min, S.; Baek, J.; Kim, J.; Jeong, H.J.; Chung, J.; Jeong, K. Water-Compatible and Recyclable Heterogeneous SABRE Catalyst for NMR Signal Amplification. JACS Au 2023, 3, 2912–2917. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Mascaraque, A.; Jia, H.; Zimmermann, B.; Robertson, M.; Conte, R.L.; Hoffmann, M.; González Barrio, M.A.; Ding, H.; Wiesendanger, R.; et al. Large Dzyaloshinskii-Moriya interaction induced by chemisorbed oxygen on a ferromagnet surface. Sci. Adv. 2020, 6, eaba4924. [Google Scholar] [CrossRef]
- Janas, D.M.; Droghetti, A.; Ponzoni, S.; Cojocariu, I.; Jugovac, M.; Feyer, V.; Radonjić, M.M.; Rungger, I.; Chioncel, L.; Zamborlini, G.; et al. Enhancing Electron Correlation at a 3d Ferromagnetic Surface. Adv. Mater. 2023, 35, 2205698. [Google Scholar] [CrossRef]
- Kurahashi, M.; Yamauchi, Y. Spin Correlation in O2 Chemisorption on Ni(111). Phys. Rev. Lett. 2015, 114, 016101. [Google Scholar] [CrossRef] [PubMed]
- Kurahashi, M. Oxygen adsorption on surfaces studied by a spin- and alignment-controlled O2 beam. Prog. Surf. Sci. 2016, 91, 29–55. [Google Scholar] [CrossRef]
- Yu, X.; Cheng, Y.; Li, Y.; Polo-Garzon, F.; Liu, J.; Mamontov, E.; Li, M.; Lennon, D.; Parker, S.F.; Ramirez-Cuesta, A.J.; et al. Neutron Scattering Studies of Heterogeneous Catalysis. Chem. Rev. 2023, 123, 8638–8700. [Google Scholar] [CrossRef] [PubMed]
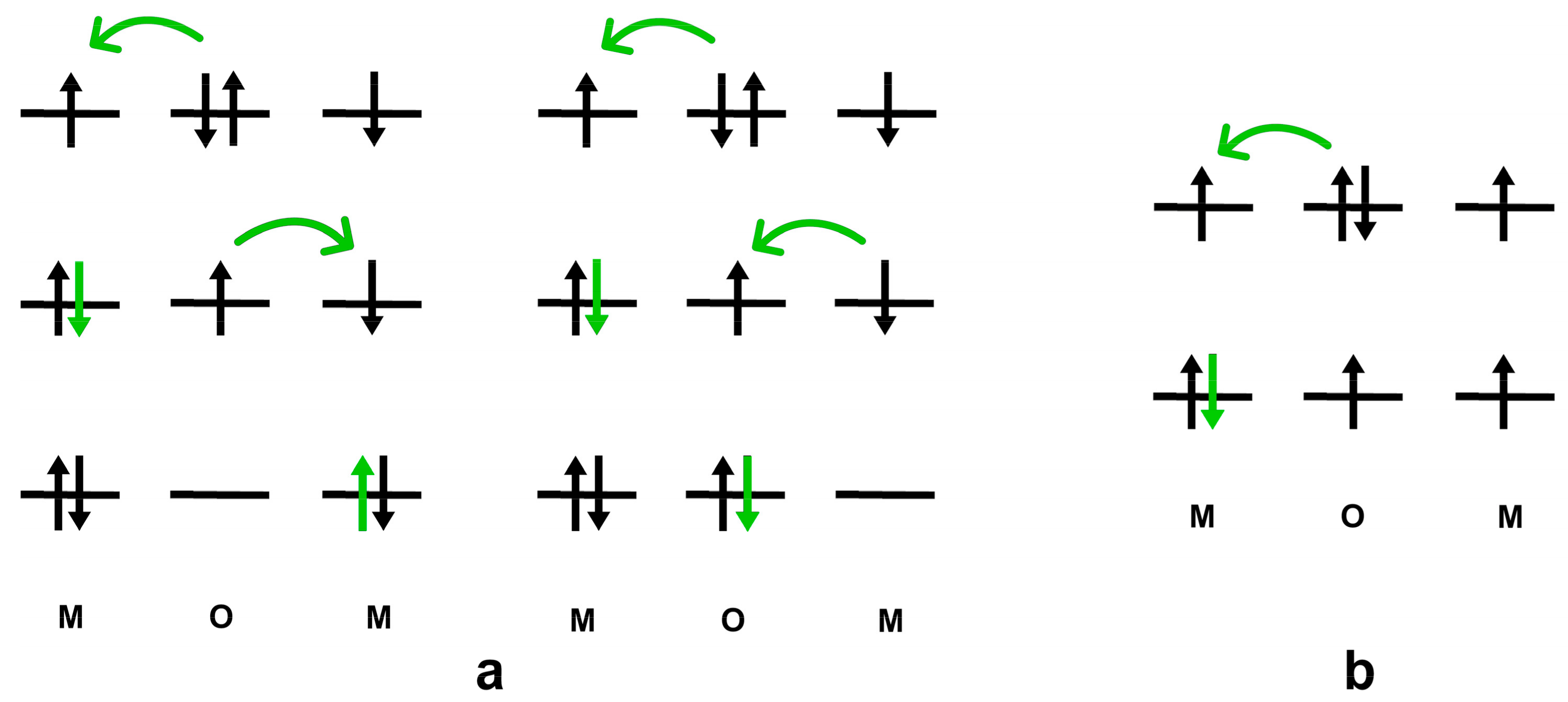



{kind=link}
{kind=link}
{kind=link}
| Name | Chemical Bond (Strength) | Features | Examples |
|---|---|---|---|
| Ionic solids | Ionic (strong) |
| NaCl, CsCl |
| Covalent solids | Covalent (strong) |
| diamond, Si, Ge |
| Metals | Metallic (medium) |
| Na, Nb, Pt, Ir |
| Molecular solids | Hydrogen bonds, van der Waals interactions, hydrophobic interactions, London dispersion forces, dipole–dipole interactions (weak individually, but may be strong in numbers) |
| crystalline Ar, solid HF, crystalline H2O |
| Catalytic Reaction | Catalyst | Operating Conditions | Ref. |
|---|---|---|---|
| Oxygen reduction reaction (ORR) | Platinum (Pt) | 334–394 K (PEMFC) a | [29] |
| L10 Platinum-Cobalt (PtCo) | [15,30] | ||
| Hydrogen evolution reaction (HER) | Platinum (Pt) | 298–393 K, 1–200 bar | [31] |
| Nickel-Molybdenum (Ni-Mo) alloys | |||
| Nickel (Ni), Nickel (Ni) foam | |||
| Oxygen evolution reaction (OER) | Iridium dioxide (IrO2) | 298–393 K, 1–200 bar | [31] |
| Ruthenium dioxide (RuO2) | |||
| Cobalt nickel oxide (CoNi2O4) | |||
| Ammonia (NH3) synthesis | Iron (Fe)-based catalysts | 674–774 K, 100–450 bar | [32] |
| Magnetite (Fe3O4), Potassium oxide (K2O), Alumina (Al2O3)) | 724–774 K, 250–450 bar | [2] | |
| Hydrogenation of ketones/aldehydes to alcohols | Nickel (Ni) | 374–424 K, 30 bar | [2] |
| Platinum (Pt) | |||
| Copper (Cu) | |||
| Methanol (CH3OH) synthesis | Zinc oxide-Chromic oxide (ZnO-Cr2O3) | 524–674 K, 200–300 bar | [2] |
| Oxidation of ethene to epoxyethane (H2C=CH2 → C2H4O) | Silver (Ag)/support | 474–524 K, 10–22 bar | [2] |
| Oxidation of methanol (CH3OH) to formaldehyde (CH2O) | Silver (Ag) crystalline | ~874 K | [2] |
| Polymerization of ethene (H2C=CH2) | Chromic oxide/Silica (Cr2O3/SiO2) | 324–424 K, 20–80 bar | [2] |
| Chromic oxide/Molybdenum trioxide (Cr2O3/MoO3) | |||
| Production of gasoline from cracking of kerosene | Alumina/Silica (Al2O3/SiO2) zeolites | 774–824 K, 1–20 bar | [2] |
| Propene (H2C=CHCH3) epoxidation by dioxygen (O2(g)) | Copper (Cu)-based catalysts | 474–673 K 0.01–0.5 bar | [33] |
| Silver (Ag)-based catalysts | |||
| Production of vinyl chloride from ethene (H2C=CH2) and hydrochloric acid/dioxygen (HCl/O2) | Cupric chloride/Alumina (CuCl2/Al2O3) | 474–514 K, 2–5 bar | [2] |
| Hamiltonian Operator | |||||
|---|---|---|---|---|---|
| Energy contributions | |||||
| kinetic energy of the electrons | |||||
| Coulomb attraction between nucleus and electrons | |||||
| electron–electron Coulomb repulsions | |||||
| crystal field potential | |||||
| energy contribution of the spin–orbit interaction | |||||
| Strength of the Energy contributions (eV) | n | Order of Energy contributions | |||
| nd | 3d > 4d > 5d ≈ 1.23 () = 1–10 (3d) | 3d < 4d < 5d ≈ 2.48 ≈1.5 (3d) | 3d < 4d < 5d ≈ 0.12 ≈0.1 for 3d | 3d N | ≈ > > > (for N = 6 (LS)) |
| 4d N | > > | ||||
| 5d N | > | ||||
| Catalytic Reaction | Catalyst Poisons | Refs. |
|---|---|---|
| Ammonia (NH3) synthesis | Strong: sulphur (S), carbon monoxide (CO), selenium (Se), tellurium (Te), phosphorus (P), halogens, arsenic (As)-containing compounds Weak: water (H2O), dioxygen (O2), nitric oxide (NO) | [2,91] |
| Hydrogenation of ketones/aldehydes to alcohols | Sulphur (S), selenium (Se), tellurium (Te), phosphorous (P), arsenic (As)-containing compounds, halogens, dioxygen (O2), carbon monoxide (CO) | [2] |
| Oxidation of ethene to epoxyethane (H2C=CH2 → C2H4O) | Acetylene (HC≡CH) | [91] |
| Oxidation of methanol (CH3OH) to formaldehyde (CH2O) | Iron (Fe), nickel (Ni), carbonyls | [91] |
| Production of gasoline from cracking of kerosene | Ammonia (NH3), sodium (Na), organic bases, heavy metals, coke | [2,91] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biz, C.; Gracia, J.; Fianchini, M. Experimental Evidences on Magnetism-Covalent Bonding Interplay in Structural Properties of Solids and during Chemisorption. Int. J. Mol. Sci. 2024, 25, 1793. https://doi.org/10.3390/ijms25031793
Biz C, Gracia J, Fianchini M. Experimental Evidences on Magnetism-Covalent Bonding Interplay in Structural Properties of Solids and during Chemisorption. International Journal of Molecular Sciences. 2024; 25(3):1793. https://doi.org/10.3390/ijms25031793
Chicago/Turabian StyleBiz, Chiara, Jose Gracia, and Mauro Fianchini. 2024. "Experimental Evidences on Magnetism-Covalent Bonding Interplay in Structural Properties of Solids and during Chemisorption" International Journal of Molecular Sciences 25, no. 3: 1793. https://doi.org/10.3390/ijms25031793
APA StyleBiz, C., Gracia, J., & Fianchini, M. (2024). Experimental Evidences on Magnetism-Covalent Bonding Interplay in Structural Properties of Solids and during Chemisorption. International Journal of Molecular Sciences, 25(3), 1793. https://doi.org/10.3390/ijms25031793