
Fibroblasts in Diabetic Foot Ulcers
 , ,
, ,
Abstract
:1. Introduction
2. Fibroblasts: A Heterogenous Cell Population with High Cellular Plasticity and Broad Functionality
2.1. Tissue Specificity and Heterogeneity
2.2. High Plasticity and Adaptation
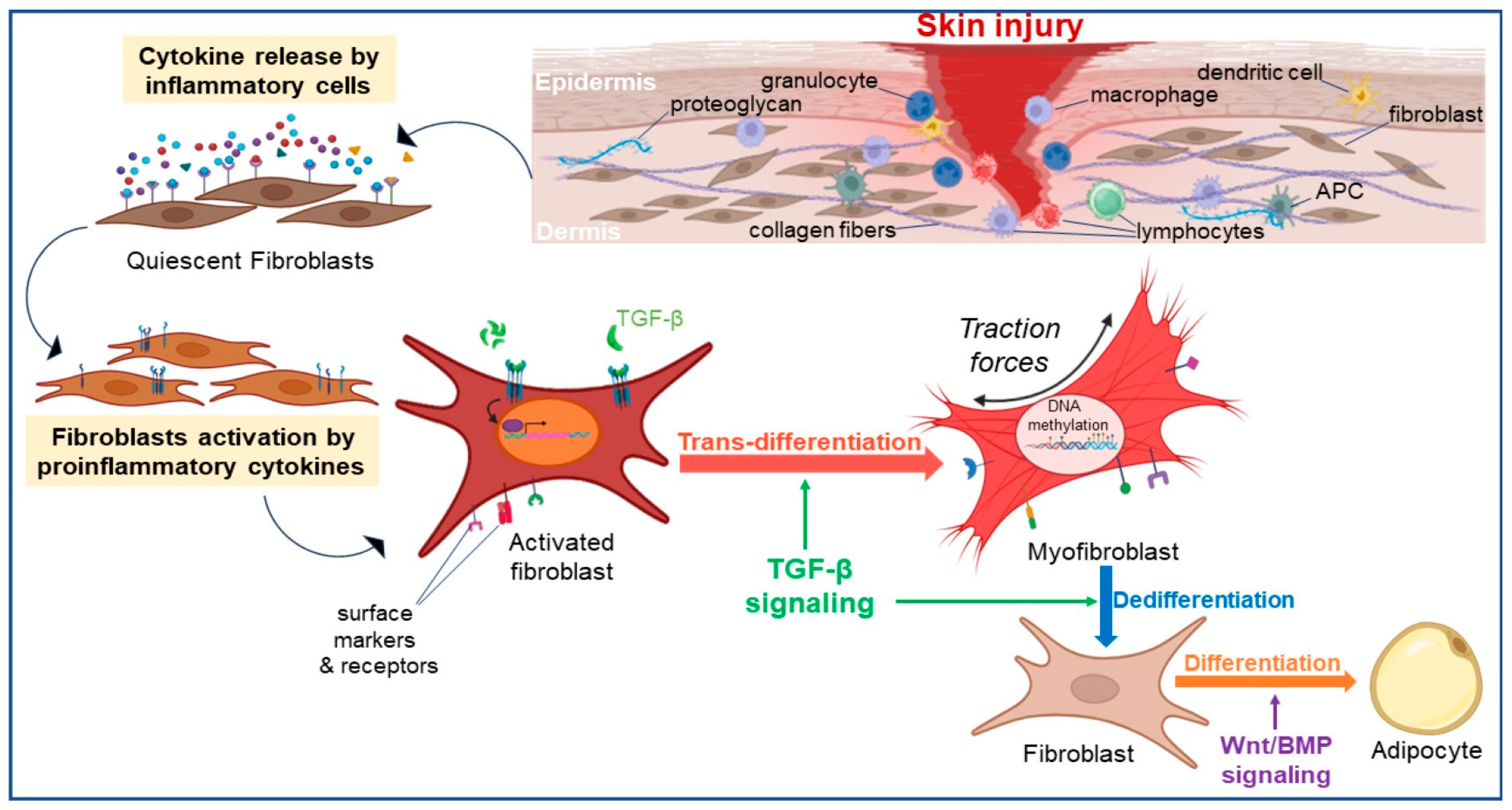
2.2.1. Fibroblast Trans-Differentiation
2.2.2. Fibroblast Dedifferentiation
2.3. Immune Response Modulation
3. Fibroblasts’ Key Role in Wound Healing
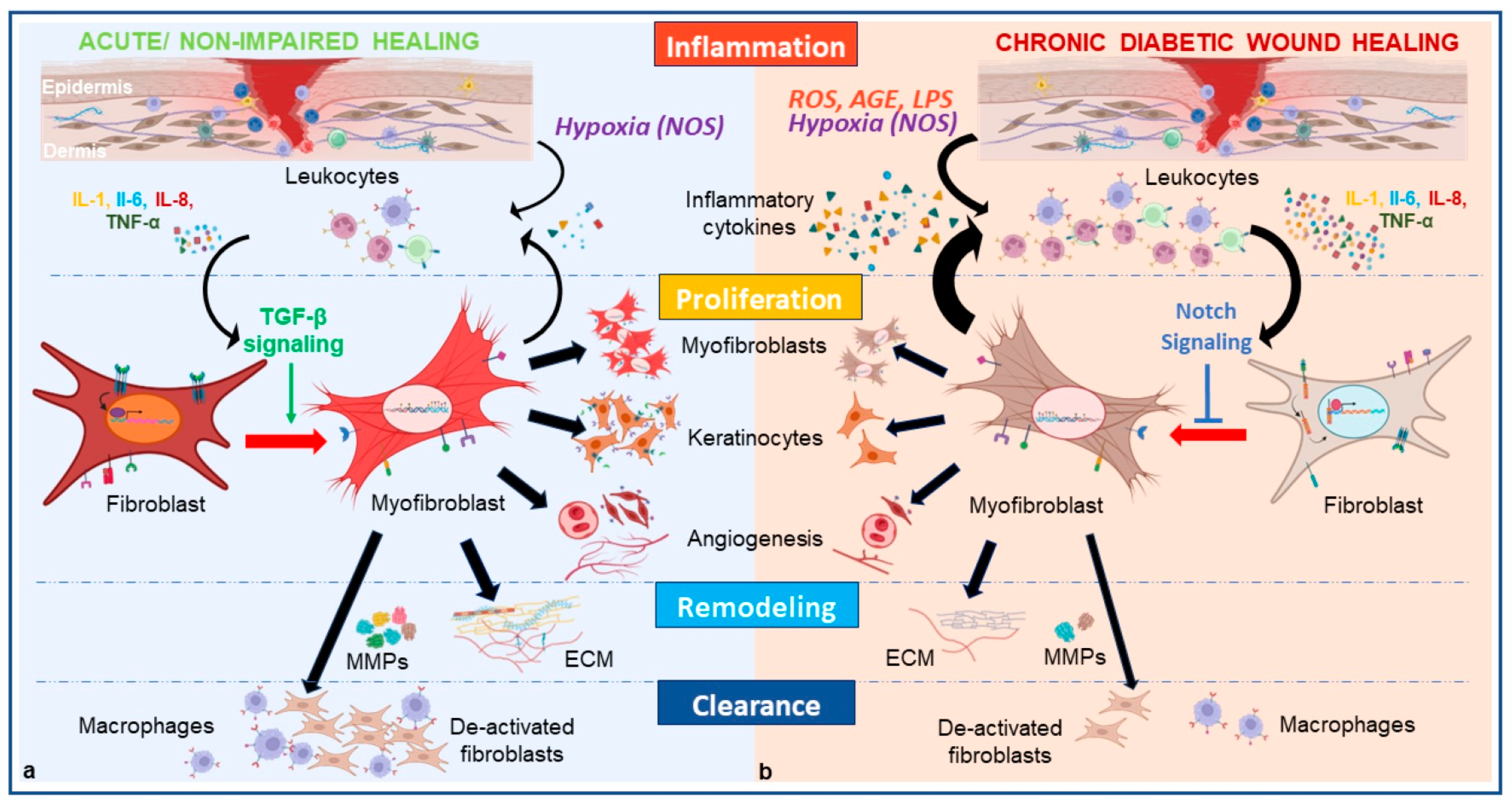
4. Fibroblasts Dysregulation Inherent to the Pathophysiology of DFU
4.1. Hyperglycemia Leads to Phenotypic Switch Alteration via Immune Cell Dysregulation Which Prolongs the Inflammatory Phase
4.2. Hyperglycemia Leads to Phenotypic Switch Alteration via Dermal Cell Dysregulation and Prevents Angiogenesis and ECM Production
4.3. Hyperglycemia Leads to Phenotypic Switch Alteration via Endothelial Cell Dysregulation
5. Preclinical Approaches to Treating Diabetic Wounds Using Fibroblasts and Fibroblast-Derived Products
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| α-SMA | α-smooth muscle actin |
| AGE | Advanced Glycation End |
| ALK | Activin-like kinase |
| APC | Adematous Polyposis Coli |
| APCs | Antigen Presenting Cells |
| BMP | Bone Morphogenic Protein |
| CAFs | Cancer-Associated Fibroblasts |
| Cav1 | Caveolin 1 |
| CDK | Cyclin-dependent kinase |
| CSL | C-promoter binding factor 1 (CBF1), suppressor of hairless (Su(H)), lin-12 and glp1 (Lag1) |
| COL1A1 | Collagen type I alpha 1 |
| CTGF | Connective Tissue Growth Factor |
| DCCT | Diabetes Control and Complications Trial |
| DDL | Deep dermal layer |
| DFF | Diabetic Foot Fibroblasts |
| DFU | Diabetic Foot Ulcer |
| DFUF | Diabetic Foot Ulcer Fibroblasts |
| Dlk1 | Delta-like non-canonical Notch ligand 1 |
| DM | Diabetes Mellitus |
| DNMT | DNA methyltransferase |
| DW | Diabetic Wound |
| ECM | Extracellular Matrix |
| EDIC | Epidemiology of Diabetes Interventions and Complications |
| EGF | Epidermal Growth Factor |
| ERK | Extracellular signal-regulated kinase |
| FAP | Fibroblast Activation Protein |
| Fbn1 | Fibrillin |
| FGF | Fibroblast Growth Factor |
| FSP1 | Fibroblast-specific protein 1 |
| FOXO1 | Forkhead box protein O1 |
| GSK | Glycogen synthase kinase |
| HGF | Hepatocyte Growth Factor |
| IFNγ | Interferon γ |
| IL | Interleukin |
| Itga8 | Alpha-8 integrin |
| ITG-β1 | Integrin-β1 |
| LEA | Lower extremity amputation |
| LPS | Lipopolysaccharides |
| LRIG1 | Leucine-rich repeats and immunoglobulin-like domains 1 |
| MAPK | Mitogen-activated protein kinase |
| MEK | Mitogen-activated extracellular signal-regulated kinase |
| MMPs | Matrix Metalloproteinases |
| MyoD | Myogenic Differentiation |
| NET | Neutrophil Extracellular Trap |
| NFF | Non-diabetic Foot Fibroblasts |
| NG2 | Neural/Glial antigen 2 |
| NLRP3 | Nod-Like Receptor Protein 3 |
| NOS | Nitric oxide synthase |
| PDGF | Platelet-Derived Growth Factor |
| PDGFR | Platelet Derived Growth Factor Receptor |
| Pdpn | Podoplanin |
| PGE2 | Prostaglandin E2 |
| ROS | Reactive Oxygen Species |
| SDL | Deep dermal layer |
| T2DM | Type 2 Diabetes Mellitus |
| TCF/LEF | T-cell factor/lymphoid enhancer factor |
| TGF-β | Transforming Growth Factor-β |
| TIMPs | Tissue Inhibitors of Metalloproteinases |
| TLR | Toll-Like Receptor |
| TNF-α | Tumor Necrosis Factor- α |
| VEGF | Vascular Endothelial Growth Factor |
| Wnt | Wingless-related integration site |
References
- Eming, S.A.; Martin, P.; Tomic-Canic, M. Wound repair and regeneration: Mechanisms, signaling, and translation. Sci. Transl. Med. 2014, 6, 265sr6. [Google Scholar] [CrossRef] [PubMed]
- Zimmet, P.; Alberti, K.G.; Magliano, D.J.; Bennett, P.H. Diabetes mellitus statistics on prevalence and mortality: Facts and fallacies. Nat. Rev. Endocrinol. 2016, 12, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, Y.; Deng, J.; Li, W.; Nie, X. Fibroblast Growth Factor in Diabetic Foot Ulcer: Progress and Therapeutic Prospects. Front. Endocrinol. 2021, 12, 744868. [Google Scholar] [CrossRef] [PubMed]
- McDermott, K.; Fang, M.; Boulton, A.J.; Selvin, E.; Hicks, C.W. Etiology, Epidemiology, and Disparities in the Burden of Diabetic Foot Ulcers. Diabetes Care 2022, 46, 209–221. [Google Scholar] [CrossRef]
- Jaacks, L.M.; Siegel, K.R.; Gujral, U.P.; Narayan, K.V. Type 2 diabetes: A 21st century epidemic. Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 331–343. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Neilson, E.G. Origin and functional heterogeneity of fibroblasts. FASEB J. 2020, 34, 3519–3536. [Google Scholar] [CrossRef]
- Lendahl, U.; Muhl, L.; Betsholtz, C. Identification, discrimination and heterogeneity of fibroblasts. Nat. Commun. 2022, 13, 1–14. [Google Scholar] [CrossRef]
- Griffin, M.F.; Desjardins-Park, H.E.; Mascharak, S.; Borrelli, M.R.; Longaker, M.T. Understanding the impact of fibroblast heterogeneity on skin fibrosis. Dis. Models Mech. 2020, 13, 44164. [Google Scholar] [CrossRef]
- Talbott, H.E.; Mascharak, S.; Griffin, M.; Wan, D.C.; Longaker, M.T. Wound healing, fibroblast heterogeneity, and fibrosis. Cell Stem Cell 2022, 29, 1161–1180. [Google Scholar] [CrossRef]
- Martin, P.; Nunan, R. Cellular and molecular mechanisms of repair in acute and chronic wound healing. Br. J. Dermatol. 2015, 173, 370–378. [Google Scholar] [CrossRef]
- Plikus, M.V.; Guerrero-Juarez, C.F.; Ito, M.; Li, Y.R.; Dedhia, P.H.; Zheng, Y.; Shao, M.; Gay, D.L.; Ramos, R.; Hsi, T.-C.; et al. Regeneration of fat cells from myofibroblasts during wound healing. Science 2017, 355, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Stunova, A.; Vistejnova, L. Dermal fibroblasts—A heterogeneous population with regulatory function in wound healing. Cytokine Growth Factor Rev. 2018, 39, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, M.; Kannan, S. Fibroblasts and mesenchymal stem cells: Two sides of the same coin? J. Cell Physiol. 2018, 233, 9099–9109. [Google Scholar] [CrossRef] [PubMed]
- Kashpur, O.; Smith, A.; Gerami-Naini, B.; Maione, A.G.; Calabrese, R.; Tellechea, A.; Theocharidis, G.; Liang, L.; Pastar, I.; Tomic-Canic, M.; et al. Differentiation of diabetic foot ulcer–derived induced pluripotent stem cells reveals distinct cellular and tissue phenotypes. FASEB J. 2019, 33, 1262–1277. [Google Scholar] [CrossRef] [PubMed]
- Saraswati, S.; Marrow, S.M.W.; Watch, L.A.; Young, P.P. Identification of a pro-angiogenic functional role for FSP1-positive fibroblast subtype in wound healing. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Huang, W.; Wang, J.; Li, G.; Xin, Q.; Lin, Z.; Chen, X.; Wang, X. Single-cell analysis reveals distinct functional heterogeneity of CD34+ cells in anagen wound and diabetic wound. Biochem. Biophys. Res. Commun. 2023, 639, 9–19. [Google Scholar] [CrossRef]
- Guerrero-Juarez, C.F.; Dedhia, P.H.; Jin, S.; Ruiz-Vega, R.; Ma, D.; Liu, Y.; Yamaga, K.; Shestova, O.; Gay, D.L.; Yang, Z.; et al. Single-cell analysis reveals fibroblast heterogeneity and myeloid-derived adipocyte progenitors in murine skin wounds. Nat. Commun. 2019, 10, 650. [Google Scholar] [CrossRef]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef]
- des Jardins-Park, H.E.; Foster, D.S.; Longaker, M.T. Fibroblasts and wound healing: An update. Regen. Med. 2018, 13, 491–495. [Google Scholar] [CrossRef]
- Muhl, L.; Genové, G.; Leptidis, S.; Liu, J.; He, L.; Mocci, G.; Sun, Y.; Gustafsson, S.; Buyandelger, B.; Chivukula, I.V.; et al. Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat. Commun. 2020, 11, 3953. [Google Scholar] [CrossRef]
- Rai, V.; Moellmer, R.; Agrawal, D.K. Role of fibroblast plasticity and heterogeneity in modulating angiogenesis and healing in the diabetic foot ulcer. Mol. Biol. Rep. 2023, 50, 1913–1929. [Google Scholar] [CrossRef]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef]
- Louault, K.; Li, R.-R.; DeClerck, Y.A. Cancer-Associated Fibroblasts: Understanding Their Heterogeneity. Cancers 2020, 12, 3108. [Google Scholar] [CrossRef]
- Davidson, S.; Coles, M.; Thomas, T.; Kollias, G.; Ludewig, B.; Turley, S.; Brenner, M.; Buckley, C.D. Fibroblasts as immune regulators in infection, inflammation and cancer. Nat. Rev. Immunol. 2021, 21, 704–717. [Google Scholar] [CrossRef]
- Pelon, F.; Bourachot, B.; Kieffer, Y.; Magagna, I.; Mermet-Meillon, F.; Bonnet, I.; Costa, A.; Givel, A.-M.; Attieh, Y.; Barbazan, J.; et al. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat. Commun. 2020, 11, 404. [Google Scholar] [CrossRef]
- Driskell, R.R.; Lichtenberger, B.M.; Hoste, E.; Kretzschmar, K.; Simons, B.D.; Charalambous, M.; Ferron, S.R.; Herault, Y.; Pavlovic, G.; Ferguson-Smith, A.C.; et al. Distinct fibroblast lineages de-termine dermal architecture in skin development and repair. Nature 2013, 504, 277–281. [Google Scholar] [CrossRef]
- Wu, S.; Rietveld, M.; Hogervorst, M.; de Gruijl, F.; van der Burg, S.; Vermeer, M.; van Doorn, R.; Welters, M.; El Ghalbzouri, A. Human Papillary and Reticular Fibroblasts Show Distinct Functions on Tumor Behavior in 3D-Organotypic Cultures Mimicking Melanoma and HNSCC. Int. J. Mol. Sci. 2022, 23, 11651. [Google Scholar] [CrossRef]
- Sorrell, J.M.; Baber, M.; Caplan, A. Site-matched papillary and reticular human dermal fibroblasts differ in their release of specific growth factors/cytokines and in their interaction with keratinocytes. J. Cell. Physiol. 2004, 200, 134–145. [Google Scholar] [CrossRef]
- Ghetti, M.; Topouzi, H.; Theocharidis, G.; Papa, V.; Williams, G.; Bondioli, E.; Cenacchi, G.; Connelly, J.; Higgins, C. Subpopulations of dermal skin fibroblasts secrete distinct extracellular matrix: Implications for using skin substitutes in the clinic. Br. J. Dermatol. 2017, 179, 381–393. [Google Scholar] [CrossRef]
- Philippeos, C.; Telerman, S.B.; Oules, B.; Pisco, A.O.; Shaw, T.J.; Elgueta, R.; Lombardi, G.; Driskell, R.R.; Soldin, M.; Lynch, M.D.; et al. Spatial and Single-Cell Transcriptional Profiling Identifies Functionally Distinct Human Dermal Fibroblast Subpopulations. J. Investig. Dermatol. 2018, 138, 811–825. [Google Scholar] [CrossRef]
- Jiang, D.; Rinkevich, Y. Scars or Regeneration?—Dermal Fibroblasts as Drivers of Diverse Skin Wound Responses. Int. J. Mol. Sci. 2020, 21, 617. [Google Scholar] [CrossRef]
- Chen, D.; Jarrell, A.; Guo, C.; Lang, R.; Atit, R. Dermal β-catenin activity in response to epidermal Wnt ligands is required for fi-broblast proliferation and hair follicle initiation. Development 2012, 139, 1522–1533. [Google Scholar] [CrossRef]
- Lichtenberger, B.M.; Mastrogiannaki, M.; Watt, F.M. Epidermal β-catenin activation remodels the dermis via paracrine signalling to distinct fibroblast lineages. Nat. Commun. 2017, 7, 10537. [Google Scholar] [CrossRef]
- Ito, M.; Yang, Z.; Andl, T.; Cui, C.; Kim, N.; Millar, S.E.; Cotsarelis, G. Wnt-dependent de novo hair follicle regeneration in adult mouse skin after wounding. Nature 2007, 447, 316–320. [Google Scholar] [CrossRef]
- Kennard, S.; Liu, H.; Lilly, B. Transforming growth factor-beta (TGF- 1) down-regulates Notch3 in fibroblasts to promote smooth muscle gene expression. J. Biol. Chem. 2008, 283, 1324–1333. [Google Scholar] [CrossRef]
- Li, B.; Gao, C.; Diao, J.; Wang, D.; Chu, F.; Li, Y.; Wang, G.; Guo, S.; Xia, W. Aberrant Notch signalling contributes to hypertrophic scar formation by modulating the phenotype of keratinocytes. Exp. Dermatol. 2016, 25, 137–142. [Google Scholar] [CrossRef]
- Foster, D.S.; Januszyk, M.; Yost, K.E.; Chinta, M.S.; Gulati, G.S.; Nguyen, A.T.; Burcham, A.R.; Salhotra, A.; Ransom, R.C.; Henn, D.; et al. Integrated spatial multiomics reveals fibroblast fate during tissue repair. Proc. Natl. Acad. Sci. USA 2021, 118, e2110025118. [Google Scholar] [CrossRef]
- Deng, J.-Y.; Wu, X.-Q.; He, W.-J.; Liao, X.; Tang, M.; Nie, X.-Q. Targeting DNA methylation and demethylation in diabetic foot ulcers. J. Adv. Res. 2023, 54, 119–131. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’His, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Vertino, P.M.; Issa, J.P.; Pereira-Smith, O.M.; Baylin, S.B. Stabilization of DNA methyltransferase levels and CpG island hypermeth-ylation precede SV40-induced immortalization of human fibroblasts. Cell Growth Differ. 1994, 5, 1395–1402. [Google Scholar] [PubMed]
- Zou, M.-L.; Teng, Y.-Y.; Wu, J.-J.; Liu, S.-Y.; Tang, X.-Y.; Jia, Y.; Chen, Z.-H.; Zhang, K.-W.; Sun, Z.-L.; Li, X.; et al. Fibroblasts: Heterogeneous Cells with Potential in Regenerative Therapy for Scarless Wound Healing. Front. Cell Dev. Biol. 2021, 9, 713605. [Google Scholar] [CrossRef] [PubMed]
- Korosec, A.; Frech, S.; Gesslbauer, B.; Vierhapper, M.; Radtke, C.; Petzelbauer, P.; Lichtenberger, B.M. Lineage Identity and Location within the Dermis Determine the Function of Papillary and Reticular Fibroblasts in Human Skin. J. Investig. Dermatol. 2019, 139, 342–351. [Google Scholar] [CrossRef]
- Buechler, M.B.; Pradhan, R.N.; Krishnamurty, A.T.; Cox, C.; Calviello, A.K.; Wang, A.W.; Yang, Y.A.; Tam, L.; Caothien, R.; Roose-Girma, M.; et al. Cross-tissue organization of the fibroblast lineage. Nature 2021, 593, 575–579. [Google Scholar] [CrossRef]
- Jopling, C.; Boue, S.; Izpisua Belmonte, J.C. Dedifferentiation, transdifferentiation and reprogramming: Three routes to regenera-tion. Nat. Rev. Mol. Cell Biol. 2011, 12, 79–89. [Google Scholar] [CrossRef]
- Martin, P. Wound Healing--Aiming for Perfect Skin Regeneration. Science 1997, 276, 75–81. [Google Scholar] [CrossRef]
- Hinz, B. The role of myofibroblasts in wound healing. Curr. Res. Transl. Med. 2016, 64, 171–177. [Google Scholar] [CrossRef]
- Shao, H.; Li, Y.; Pastar, I.; Xiao, M.; Prokupets, R.; Liu, S.; Yu, K.; Vazquez-Padron, R.I.; Tomic-Canic, M.; Velazquez, O.C.; et al. Notch1 signaling determines the plasticity and function of fibroblasts in diabetic wounds. Life Sci. Alliance 2020, 3, e202000769. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue re-modelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- Liu, Z.-J.; Velazquez, O.C. Hyperoxia, endothelial progenitor cell mobilization, and diabetic wound healing. Antioxid. Redox Signal. 2008, 10, 1869–1882. [Google Scholar] [CrossRef]
- Gallagher, K.A.; Liu, Z.-J.; Xiao, M.; Chen, H.; Goldstein, L.J.; Buerk, D.G.; Nedeau, A.; Thom, S.R.; Velazquez, O.C. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1α. J. Clin. Investig. 2007, 117, 1249–1259. [Google Scholar] [CrossRef]
- Desmoulière, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef]
- Goldberg, M.T.; Han, Y.-P.; Yan, C.; Shaw, M.C.; Garner, W.L. TNF-α Suppresses α-Smooth Muscle Actin Expression in Human Dermal Fibroblasts: An Implication for Abnormal Wound Healing. J. Investig. Dermatol. 2007, 127, 2645–2655. [Google Scholar] [CrossRef] [PubMed]
- Dees, C.; Tomcik, M.; Zerr, P.; Akhmetshina, A.; Horn, A.; Palumbo, K.; Beyer, C.; Zwerina, J.; Distler, O.; Schett, G.; et al. Notch signalling regulates fibroblast activation and collagen release in systemic sclerosis. Ann. Rheum. Dis. 2011, 70, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Fortier, S.M.; Penke, L.R.; King, D.; Pham, T.X.; Ligresti, G.; Peters-Golden, M. Myofibroblast dedifferentiation proceeds via distinct transcriptomic and phenotypic transitions. J. Clin. Investig. 2021, 6, 144799. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2020, 16, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Gerber, T.; Murawala, P.; Knapp, D.; Masselink, W.; Schuez, M.; Hermann, S.; Gac-Santel, M.; Nowoshilow, S.; Kageyama, J.; Khattak, S.; et al. Single-cell analysis uncovers convergence of cell identities during axolotl limb regeneration. Science 2018, 362, 421. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-Y.; Gerber, T.; Taniguchi-Sugiura, Y.; Murawala, P.; Hermann, S.; Grosser, L.; Shibata, E.; Treutlein, B.; Tanaka, E.M. Fibroblast dedifferentiation as a determinant of successful regeneration. Dev. Cell 2021, 56, 1541–1551.e6. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Jagirdar, R.; Jin, T.; Thannickal, V.J. Reversible differentiation of myofibroblasts by MyoD. Exp. Cell Res. 2011, 317, 1914–1921. [Google Scholar] [CrossRef]
- Kosla, J.; Dvorakova, M.; Dvorak, M.; Cermak, V. Effective myofibroblast dedifferentiation by concomitant inhibition of TGF-β signaling and perturbation of MAPK signaling. Eur. J. Cell Biol. 2013, 92, 363–373. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Pahwa, R.; Goyal, A.; Jialal, I. Chronic Inflammation; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Liu, Y.; Liu, Y.; He, W.; Mu, X.; Wu, X.; Deng, J.; Nie, X. Fibroblasts: Immunomodulatory factors in refractory diabetic wound healing. Front. Immunol. 2022, 13, 918223. [Google Scholar] [CrossRef]
- Guo, S.; DiPietro, L. Factors Affecting Wound Healing. J. Dent. Res. 2010, 89, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cai, S.; Shu, X.Z.; Shelby, J.; Prestwich, G.D. Release of basic fibroblast growth factor from a crosslinked glycosaminoglycan hydrogel promotes wound healing. Wound Repair Regen. 2007, 15, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Zhang, Y.; Cheng, S.; Zhou, X.; Zhang, H.; Gao, J.; Zhao, F. Inhibition of TGFβ1/Smad pathway by NF-κB induces inflammation leading to poor wound healing in high glucose. Cells Dev. 2022, 172, 203814. [Google Scholar] [CrossRef]
- Acosta, J.B.; del Barco, D.G.; Vera, D.C.; Savigne, W.; Lopez-Saura, P.; Nieto, G.G.; Schultz, G.S. The pro-inflammatory environment in recalcitrant diabetic foot wounds. Int. Wound J. 2008, 5, 530–539. [Google Scholar] [CrossRef]
- Henrich, D.; Zimmer, S.; Seebach, C.; Frank, J.; Barker, J.; Marzi, I. Trauma-activated polymorphonucleated leukocytes damage en-dothelial progenitor cells: Probable role of CD11b/CD18-CD54 interaction and release of reactive oxygen species. Shock 2011, 36, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Wilgus, T.A.; Roy, S.; McDaniel, J.C. Neutrophils and Wound Repair: Positive Actions and Negative Reactions. Adv. Wound Care 2013, 2, 379–388. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.-L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef]
- Ogle, M.E.; Segar, C.E.; Sridhar, S.; Botchwey, E.A. Monocytes and macrophages in tissue repair: Implications for immunoregener-ative biomaterial design. Exp. Biol. Me. 2016, 241, 1084–1097. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Meszaros, A.J.; Reichner, J.S.; Albina, J.E. Macrophage-Induced Neutrophil Apoptosis. J. Immunol. 2000, 165, 435–441. [Google Scholar] [CrossRef]
- Wang, X.; Chen, H.; Tian, R.; Zhang, Y.; Drutskaya, M.S.; Wang, C.; Ge, J.; Fan, Z.; Kong, D.; Wang, X.; et al. Macrophages induce AKT/β-catenin-dependent Lgr5+ stem cell activation and hair follicle regeneration through TNF. Nat. Commun. 2017, 8, 14091. [Google Scholar] [CrossRef]
- Rahmani, W.; Liu, Y.; Rosin, N.L.; Kline, A.; Raharjo, E.; Yoon, J.; Biernaskie, J. Macrophages Promote Wound-Induced Hair Follicle Regener-ation in a CXCR1- and TGF-β1-Dependent Manner. J. Investig. Dermatol. 2018, 138, 2111–2122. [Google Scholar] [CrossRef]
- Portou, M.; Baker, D.; Abraham, D.; Tsui, J. The innate immune system, toll-like receptors and dermal wound healing: A review. Vasc. Pharmacol. 2015, 71, 31–36. [Google Scholar] [CrossRef]
- Velazquez, O.C.; Snyder, R.; Liu, Z.-J.; Fairman, R.M.; Herlyn, M. Fibroblast-dependent differentiation of human microvascular en-dothelial cells into capillary-like 3-dimensional networks. FASEB J. 2002, 16, 1316–1318. [Google Scholar] [CrossRef]
- Gharbia, F.Z.; Abouhashem, A.S.; Moqidem, Y.A.; Elbaz, A.A.; Abdellatif, A.; Singh, K.; Sen, C.K.; Azzazy, H.M.E. Adult skin fibroblast state change in murine wound healing. Sci. Rep. 2023, 13, 886. [Google Scholar] [CrossRef]
- Johnson, K.E.; Wilgus, T.A. Vascular Endothelial Growth Factor and Angiogenesis in the Regulation of Cutaneous Wound Repair. Adv. Wound Care 2014, 3, 647–661. [Google Scholar] [CrossRef]
- Huerta, C.T.; Voza, F.A.; Ortiz, Y.Y.; Liu, Z.-J.; Velazquez, O.C. Mesenchymal stem cell-based therapy for non-healing wounds due to chronic limb-threatening ischemia: A review of preclinical and clinical studies. Front. Cardiovasc. Med. 2023, 10, 1113982. [Google Scholar] [CrossRef]
- Liu, Z.-J.; Tian, R.; Li, Y.; Zhang, L.; Shao, H.; Yang, C.; Velazquez, O.C. SDF-1α-induced dual pairs of E-selectin/ligand mediate endothelial progenitor cell homing to critical ischemia. Sci. Rep. 2016, 6, srep34416. [Google Scholar] [CrossRef]
- Mathew-Steiner, S.S.; Roy, S.; Sen, C.K. Collagen in Wound Healing. Bioengineering 2021, 8, 63. [Google Scholar] [CrossRef]
- Wall, I.B.; Moseley, R.; Baird, D.M.; Kipling, D.; Giles, P.; Laffafian, I.; Price, P.E.; Thomas, D.W.; Stephens, P. Fibroblast Dysfunction Is a Key Factor in the Non-Healing of Chronic Venous Leg Ulcers. J. Investig. Dermatol. 2008, 128, 2526–2540. [Google Scholar] [CrossRef]
- Zhao, R.; Liang, H.; Clarke, E.; Jackson, C.; Xue, M. Inflammation in Chronic Wounds. Int. J. Mol. Sci. 2016, 17, 2085. [Google Scholar] [CrossRef]
- Lévigne, D.; Tobalem, M.; Modarressi, A.; Pittet-Cuénod, B. Hyperglycemia Increases Susceptibility to Ischemic Necrosis. BioMed Res. Int. 2012, 2013, 1–5. [Google Scholar] [CrossRef]
- Blakytny, R.; Jude, E.B. Altered Molecular Mechanisms of Diabetic Foot Ulcers. Int. J. Low. Extremity Wounds 2009, 8, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Catrina, S.; Zheng, X. Disturbed hypoxic responses as a pathogenic mechanism of diabetic foot ulcers. Diabetes Metab. Res. Rev. 2016, 32 (Supp. 1), 179–185. [Google Scholar] [CrossRef]
- Pradhan, L.; Nabzdyk, C.; Andersen, N.D.; LoGerfo, F.W.; Veves, A. Inflammation and neuropeptides: The connection in diabetic wound healing. Expert Rev. Mol. Med. 2009, 11, e2. [Google Scholar] [CrossRef]
- Adler, A.I.; Erqou, S.; Lima, T.A.S.; Robinson, A.H.N. Association between glycated haemoglobin and the risk of lower extremity amputation in patients with diabetes mellitus—Review and meta-analysis. Diabetologia 2010, 53, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Al-Rikabi, A.H.A.; Tobin, D.J.; Riches-Suman, K.; Thornton, M.J. Dermal fibroblasts cultured from donors with type 2 diabetes mellitus retain an epigenetic memory associated with poor wound healing responses. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Intine, R.V.; Sarras, M.P. Metabolic Memory and Chronic Diabetes Complications: Potential Role for Epigenetic Mechanisms. Curr. Diabetes Rep. 2012, 12, 551–559. [Google Scholar] [CrossRef]
- Park, L.K.; Maione, A.G.; Smith, A.; Gerami-Naini, B.; Iyer, L.K.; Mooney, D.J.; Veves, A.; Garlick, J.A. Genome-wide DNA methylation analysis identifies a metabolic memory profile in patient-derived diabetic foot ulcer fibroblasts. Epigenetics 2014, 9, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Evangelatov, A.; Georgiev, G.; Arabadjiev, B.; Pankov, S.; Krastev, P.; Momchilova, A.; Pankov, R. Hyperglycemia attenuates fibroblast contractility via suppression of TβRII receptor modulated α-smooth muscle actin expression. Biotechnol. Biotechnol. Equip. 2022, 36, 35–44. [Google Scholar] [CrossRef]
- Zubair, M.; Ahmad, J. Role of growth factors and cytokines in diabetic foot ulcer healing: A detailed review. Rev. Endocr. Metab. Disord. 2019, 20, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Ceilley, R. Chronic Wound Healing: A Review of Current Management and Treatments. Adv. Ther. 2017, 34, 599–610. [Google Scholar] [CrossRef]
- Mendoza-Marí, Y.; García-Ojalvo, A.; Fernández-Mayola, M.; Rodríguez-Rodríguez, N.; Martinez-Jimenez, I.; Berlanga-Acosta, J. Epidermal growth factor effect on lipopolysaccharide-induced inflammation in fibroblasts derived from diabetic foot ulcer. Scars, Burn. Heal. 2022, 8, 20595131211067380. [Google Scholar] [CrossRef] [PubMed]
- Maione, A.G.; Smith, A.; Kashpur, O.; Yanez, V.; Knight, E.; Mooney, D.J.; Veves, A.; Tomic-Canic, M.; Garlick, J.A. Altered ECM deposition by diabetic foot ulcer-derived fibroblasts implicates fibronectin in chronic wound repair. Wound Repair Regen. 2016, 24, 630–643. [Google Scholar] [CrossRef]
- Liu, D.; Yang, P.; Gao, M.; Yu, T.; Shi, Y.; Zhang, M.; Zhang, X. NLRP3 activation induced by neutrophil extracellular traps sustains in-flammatory response in the diabetic wound. Clin. Sci. 2019, 133, 565–582. [Google Scholar] [CrossRef]
- Sawaya, A.P.; Stone, R.C.; Brooks, S.R.; Pastar, I.; Jozic, I.; Hasneen, K.; O’Neill, K.; Mehdizadeh, S.; Head, C.R.; Strbo, N.; et al. Deregulated immune cell recruitment orchestrated by FOXM1 impairs human diabetic wound healing. Nat. Commun. 2020, 11, 4678. [Google Scholar] [CrossRef]
- Mirza, R.E.; Fang, M.M.; Ennis, W.J.; Koh, T.J. Blocking Interleukin-1β Induces a Healing-Associated Wound Macrophage Phenotype and Improves Healing in Type 2 Diabetes. Diabetes 2013, 62, 2579–2587. [Google Scholar] [CrossRef]
- Theocharidis, G.; Baltzis, D.; Roustit, M.; Tellechea, A.; Dangwal, S.; Khetani, R.S.; Shu, B.; Zhao, W.; Fu, J.; Bhasin, S.; et al. Integrated Skin Transcriptomics and Serum Multiplex Assays Reveal Novel Mechanisms of Wound Healing in Diabetic Foot Ulcers. Diabetes 2020, 69, 2157–2169. [Google Scholar] [CrossRef]
- Nassiri, S.; Zakeri, I.; Weingarten, M.S.; Spiller, K.L. Relative Expression of Proinflammatory and Antiinflammatory Genes Reveals Differences between Healing and Nonhealing Human Chronic Diabetic Foot Ulcers. J. Investig. Dermatol. 2015, 135, 1700–1703. [Google Scholar] [CrossRef]
- Rai, V.; Moellmer, R.; Agrawal, D.K. The role of CXCL8 in chronic nonhealing diabetic foot ulcers and phenotypic changes in fibroblasts: A molecular perspective. Mol. Biol. Rep. 2022, 49, 1565–1572. [Google Scholar] [CrossRef]
- Littig, J.P.B.; Moellmer, R.; Estes, A.M.; Agrawal, D.K.; Rai, V. Increased Population of CD40+ Fibroblasts Is Associated with Impaired Wound Healing and Chronic Inflammation in Diabetic Foot Ulcers. J. Clin. Med. 2022, 11, 6335. [Google Scholar] [CrossRef]
- Werner, S.; Krieg, T.; Smola, H. Keratinocyte–Fibroblast Interactions in Wound Healing. J. Investig. Dermatol. 2007, 127, 998–1008. [Google Scholar] [CrossRef]
- Wang, Y.; Graves, D.T. Keratinocyte Function in Normal and Diabetic Wounds and Modulation by FOXO1. J. Diabetes Res. 2020, 2020, 1–9. [Google Scholar] [CrossRef]
- Ponugoti, B.; Xu, F.; Zhang, C.; Tian, C.; Pacios, S.; Graves, D.T. FOXO1 promotes wound healing through the up-regulation of TGF-β1 and prevention of oxidative stress. J. Cell Biol. 2013, 203, 327–343. [Google Scholar] [CrossRef]
- Lan, C.-C.E.; Wu, C.-S.; Huang, S.-M.; Wu, I.-H.; Chen, G.-S. High-glucose environment enhanced oxidative stress and increased in-terleukin-8 secretion from keratinocytes: New insights into impaired diabetic wound healing. Diabetes 2013, 62, 2530–2538. [Google Scholar] [CrossRef]
- Rangarajan, A.; Talora, C.; Okuyama, R.; Nicolas, M.; Mammucari, C.; Oh, H.; Aster, J.C.; Krishna, S.; Metzger, D.; Chambon, P.; et al. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 2001, 20, 3427–3436. [Google Scholar] [CrossRef]
- Siekmann, A.F.; Lawson, N.D. Notch Signalling and the Regulation of Angiogenesis. Cell Adhes. Migr. 2007, 1, 104–105. [Google Scholar] [CrossRef]
- Pardali, E.; Goumans, M.-J.; ten Dijke, P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol. 2010, 20, 556–567. [Google Scholar] [CrossRef]
- Itoh, F.; Itoh, S.; Goumans, M.-J.; Valdimarsdottir, G.; Iso, T.; Dotto, G.P.; Hamamori, Y.; Kedes, L.; Kato, M.; ten Dijke, P. Synergy and antagonism between Notch and BMP re-ceptor signaling pathways in endothelial cells. EMBO J. 2004, 23, 541–551. [Google Scholar] [CrossRef]
- Larrivée, B.; Prahst, C.; Gordon, E.; del Toro, R.; Mathivet, T.; Duarte, A.; Simons, M.; Eichmann, A. ALK1 Signaling Inhibits Angiogenesis by Cooperating with the Notch Pathway. Dev. Cell 2012, 22, 489–500. [Google Scholar] [CrossRef]
- Ziyadeh, N.; Fife, D.; Walker, A.M.; Wilkinson, G.S.; Seeger, J.D. A Matched Cohort Study of the Risk of Cancer in Users of Becaplermin. Adv. Ski. Wound Care 2011, 24, 31–39. [Google Scholar] [CrossRef]
- Leal, E.C.; Carvalho, E.; Tellechea, A.; Kafanas, A.; Tecilazich, F.; Kearney, C.; Kuchibhotla, S.; Auster, M.E.; Kokkotou, E.; Mooney, D.J.; et al. Substance P Promotes Wound Healing in Diabetes by Modulating Inflammation and Macrophage Phenotype. Am. J. Pathol. 2015, 185, 1638–1648. [Google Scholar] [CrossRef]
- Freedman, B.R.; Hwang, C.; Talbot, S.; Hibler, B.; Matoori, S.; Mooney, D.J. Breakthrough treatments for accelerated wound healing. Sci. Adv. 2023, 9, eade7007. [Google Scholar] [CrossRef]
- Han, J.; Lin, K.; Choo, H.; He, J.; Wang, X.; Wu, Y.; Chen, X. β-Catenin Signaling Evokes Hair Follicle Senescence by Accelerating the Differentiation of Hair Follicle Mesenchymal Progenitors. Front. Cell Dev. Biol. 2022, 10, 839519. [Google Scholar] [CrossRef]
- Whyte, J.L.; Smith, A.A.; Helms, J.A. Wnt Signaling and Injury Repair. Cold Spring Harb. Perspect. Biol. 2012, 4, a008078. [Google Scholar] [CrossRef]
- Han, X.; Wu, P.; Li, L.; Sahal, H.M.; Ji, C.; Zhang, J.; Wang, Y.; Wang, Q.; Qian, H.; Shi, H.; et al. Exosomes derived from autologous dermal fibroblasts promote diabetic cutaneous wound healing through the Akt/β-catenin pathway. Cell Cycle 2021, 20, 616–629. [Google Scholar] [CrossRef]
- Hart, C.E.; Loewen-Rodriguez, A.; Lessem, J. Dermagraft: Use in the Treatment of Chronic Wounds. Adv. Wound Care 2012, 1, 138–141. [Google Scholar] [CrossRef]
- Zaulyanov, L.; Kirsner, R.S. A review of a bi-layered living cell treatment (Apligraf) in the treatment of venous leg ulcers and diabetic foot ulcers. Clin. Interv. Aging 2007, 2, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, G.W. Grafix®, a Cryopreserved Placental Membrane, for the Treatment of Chronic/Stalled Wounds. Adv. Wound Care 2015, 4, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Albanna, M.; Binder, K.W.; Murphy, S.V.; Kim, J.; Qasem, S.A.; Zhao, W.; Tan, J.; El-Amin, I.B.; Dice, D.D.; Marco, J.; et al. In Situ Bioprinting of Autologous Skin Cells Accelerates Wound Healing of Extensive Excisional Full-Thickness Wounds. Sci. Rep. 2019, 9, 1856. [Google Scholar] [CrossRef]
- Ishihara, J.; Ishihara, A.; Fukunaga, K.; Sasaki, K.; White, M.J.V.; Briquez, P.S.; Hubbell, J.A. Laminin heparin-binding peptides bind to several growth factors and enhance diabetic wound healing. Nat. Commun. 2018, 9, 2163. [Google Scholar] [CrossRef]
- Zheng, Z.; Jian, J.; Zhang, X.; Zara, J.N.; Yin, W.; Chiang, M.; Liu, Y.; Wang, J.; Pang, S.; Ting, K.; et al. Reprogramming of human fibroblasts into multipotent cells with a single ECM proteoglycan, fibromodulin. Biomaterials 2012, 33, 5821–5831. [Google Scholar] [CrossRef]
- Jiang, W.; Ting, K.; Lee, S.; Zara, J.N.; Song, R.; Li, C.; Chen, E.; Zhang, X.; Zhao, Z.; Soo, C.; et al. Fibromodulin reduces scar size and increases scar tensile strength in normal and excessive-mechanical-loading porcine cutaneous wounds. J. Cell. Mol. Med. 2018, 22, 2510–2513. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Characteristics | Fibroblast Subpopulations | |
|---|---|---|
| Morphology | Papillary | Reticular |
| Spindle-Shape | Stellate-Shape | |
| Dermal distribution | SDL | DDL |
| CD26+ | CD26- | |
| DLK1- | DLK1+ | |
| Surface markers | FSP1+ | FSP1+ |
| LRIG1+ | LRIG 1- | |
| PDGFR-α+ | PDGFR-α+ | |
| Genetic signature | fgf-7 | Col1A1 |
| Itga8 | Fbn1 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voza, F.A.; Huerta, C.T.; Le, N.; Shao, H.; Ribieras, A.; Ortiz, Y.; Atkinson, C.; Machuca, T.; Liu, Z.-J.; Velazquez, O.C. Fibroblasts in Diabetic Foot Ulcers. Int. J. Mol. Sci. 2024, 25, 2172. https://doi.org/10.3390/ijms25042172
Voza FA, Huerta CT, Le N, Shao H, Ribieras A, Ortiz Y, Atkinson C, Machuca T, Liu Z-J, Velazquez OC. Fibroblasts in Diabetic Foot Ulcers. International Journal of Molecular Sciences. 2024; 25(4):2172. https://doi.org/10.3390/ijms25042172
Chicago/Turabian StyleVoza, Francesca A., Carlos Theodore Huerta, Nga Le, Hongwei Shao, Antoine Ribieras, Yulexi Ortiz, Carl Atkinson, Tiago Machuca, Zhao-Jun Liu, and Omaida C. Velazquez. 2024. "Fibroblasts in Diabetic Foot Ulcers" International Journal of Molecular Sciences 25, no. 4: 2172. https://doi.org/10.3390/ijms25042172
APA StyleVoza, F. A., Huerta, C. T., Le, N., Shao, H., Ribieras, A., Ortiz, Y., Atkinson, C., Machuca, T., Liu, Z.-J., & Velazquez, O. C. (2024). Fibroblasts in Diabetic Foot Ulcers. International Journal of Molecular Sciences, 25(4), 2172. https://doi.org/10.3390/ijms25042172