
Chemical Hypoxia Induces Pyroptosis in Neuronal Cells by Caspase-Dependent Gasdermin Activation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
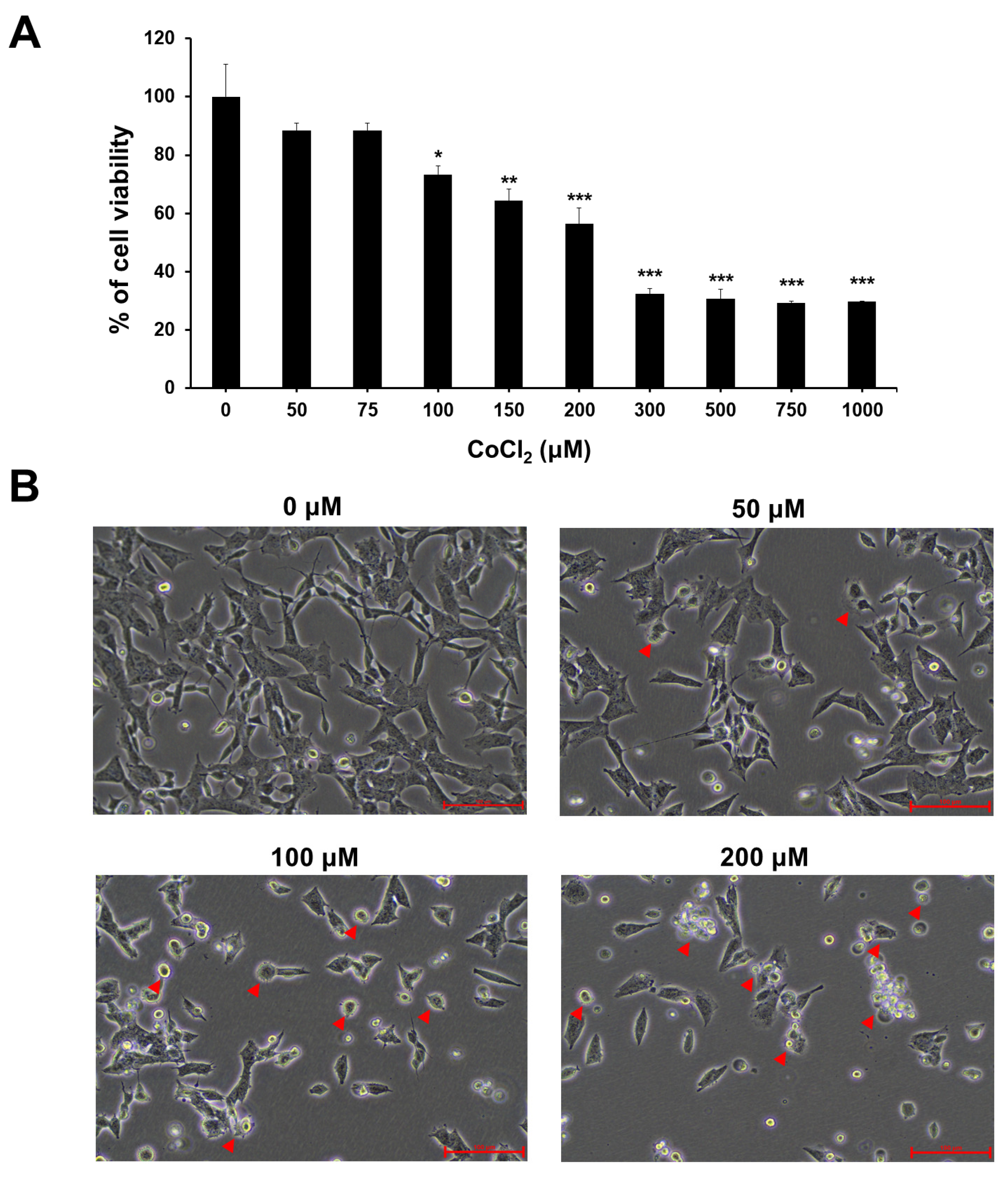
2.1. Effect of CoCl2-Induced Hypoxia on Cell Viability and Morphological Changes
2.2. Chemical Hypoxia Induces Accumulation of HIF-1α in SH-SY5Y Cells
2.3. Chemical Hypoxia Enhances Cytosolic and Mitochondrial ROS
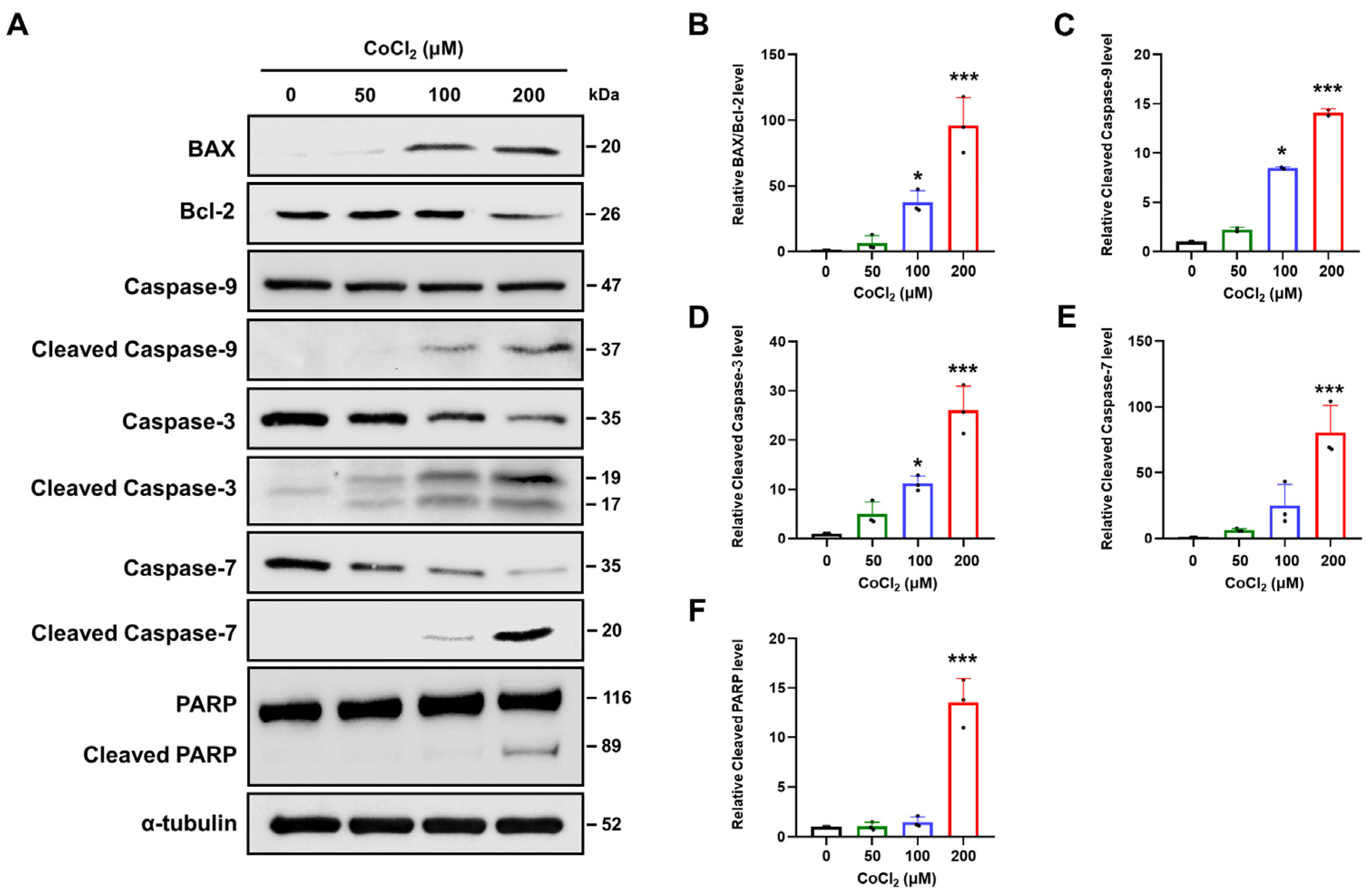
2.4. Chemical Hypoxia Triggers Intrinsic Apoptotic Pathways in SH-SY5Y Cells
2.5. Chemical Hypoxia Activates Caspase-3/GSDME-Mediated Pyroptosis
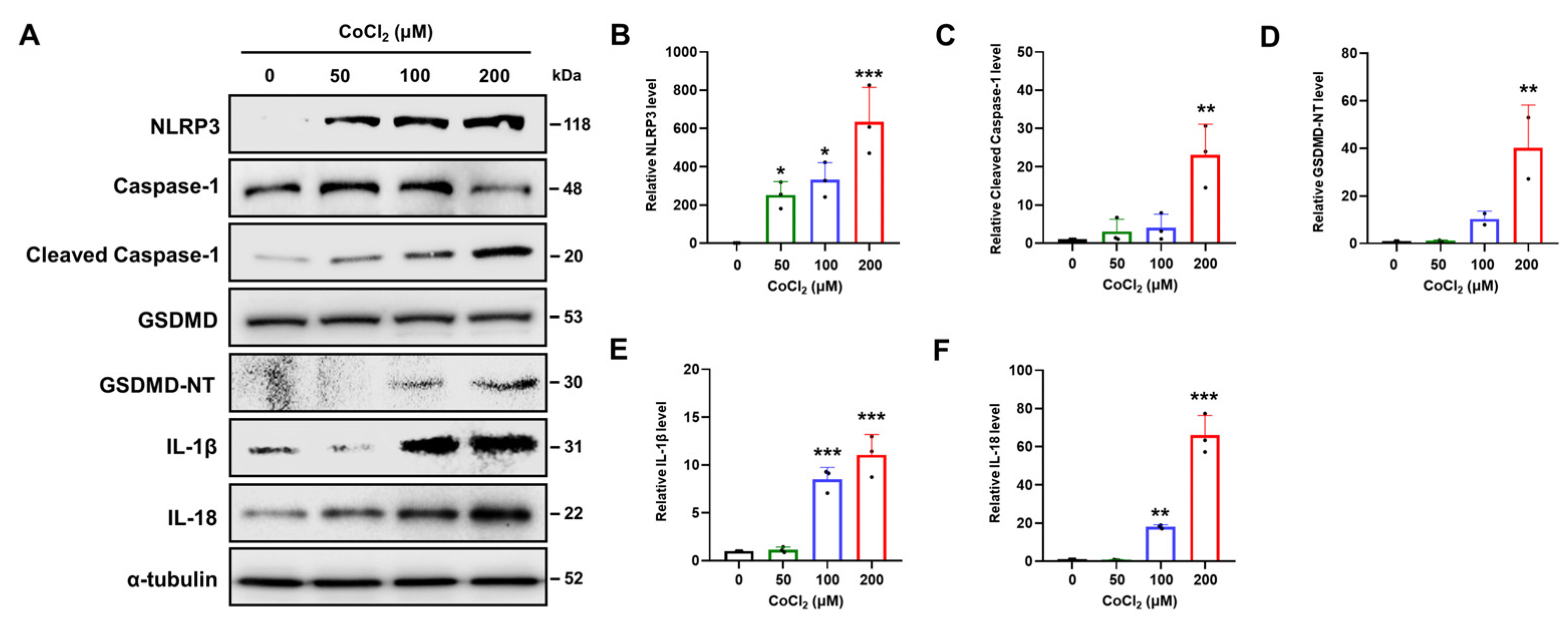
2.6. Chemical Hypoxia Causes NLRP3/Caspase-1/GSDMD-Mediated Pyroptosis
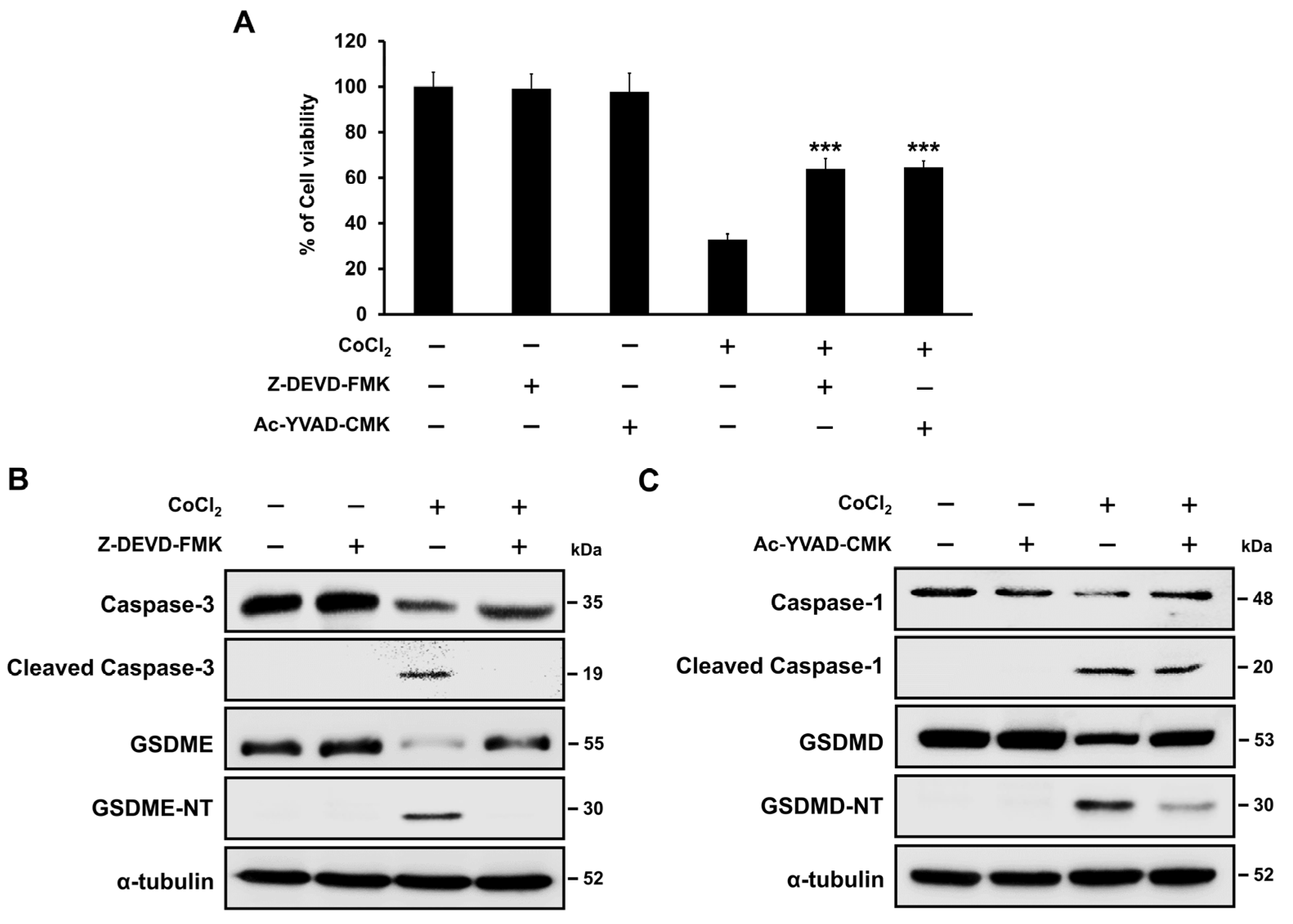
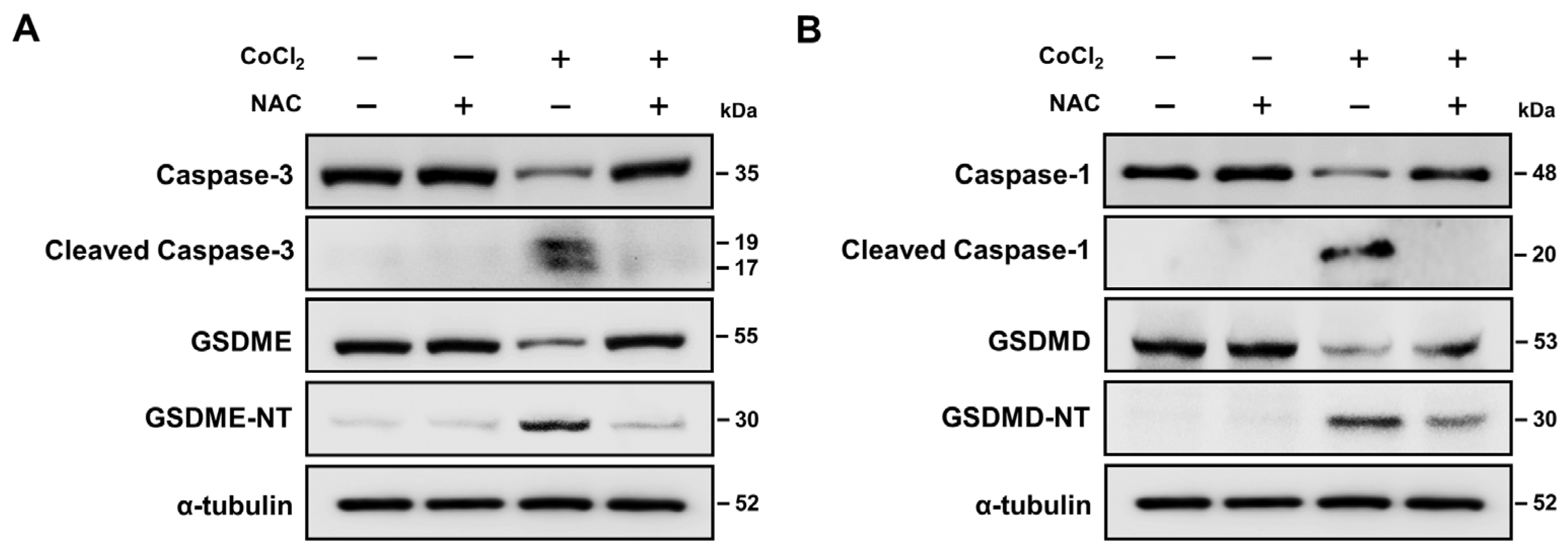
2.7. Hypoxia-Mediated Pyroptosis Depends on the Caspase Activation
2.8. ROS Activation Contributed to Hypoxia-Mediated Pyroptosis
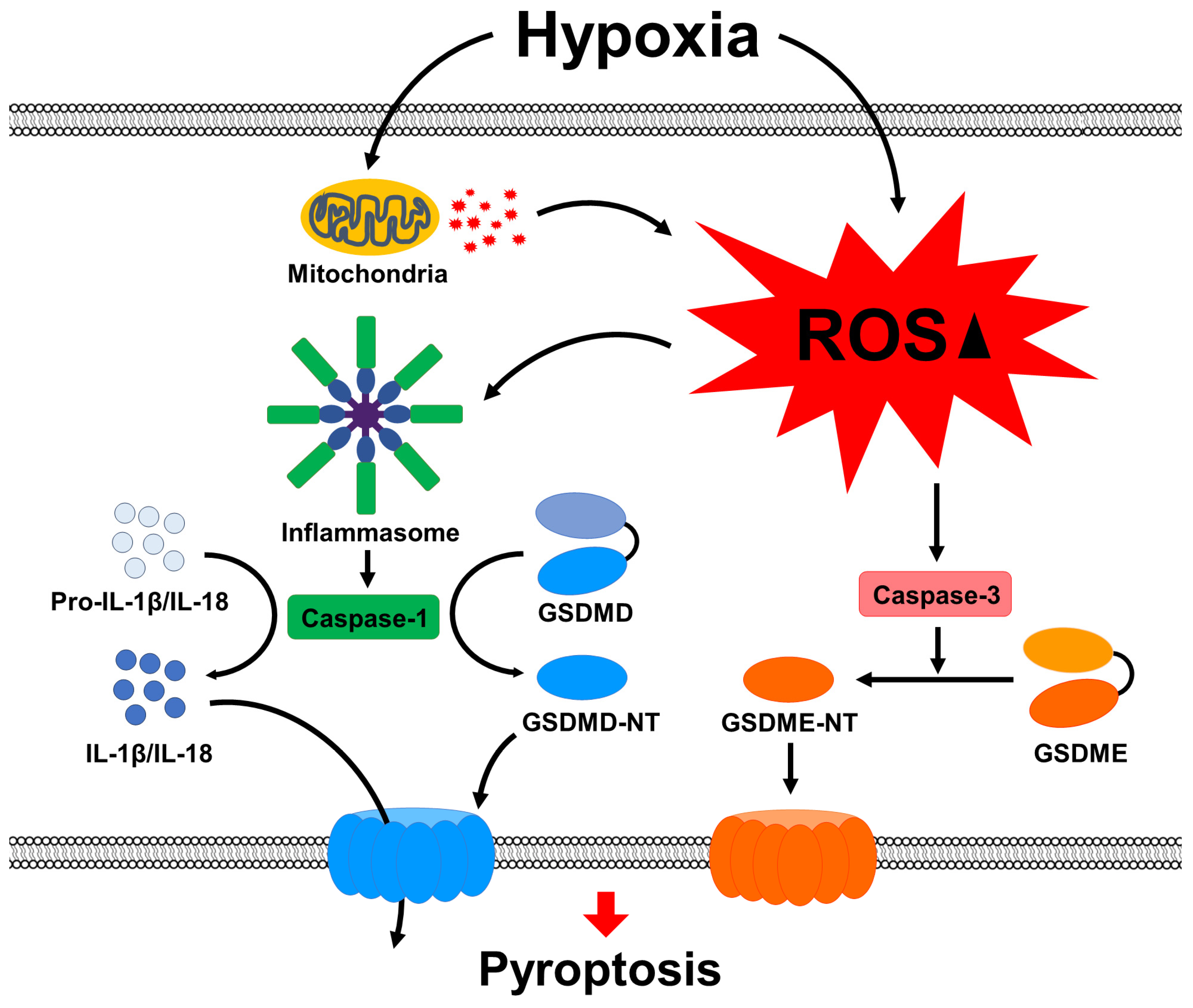
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Line and Culture
4.3. Cell Viability Assay
4.4. Cell Morphology
4.5. ROS Production Assay
4.6. Western Blot Analysis
4.7. Treatment of Cells with Caspase Inhibitors and ROS Scavenger
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, P.S.; Chiu, W.T.; Hsu, P.L.; Lin, S.C.; Peng, I.C.; Wang, C.Y.; Tsai, S.J. Pathophysiological implications of hypoxia in human diseases. J. Biomed. Sci. 2020, 27, 63. [Google Scholar] [CrossRef]
- Zhu, X.H.; Lu, M.; Chen, W. Quantitative imaging of brain energy metabolisms and neuroenergetics using in vivo X-nuclear 2H, 17O and 31P MRS at ultra-high field. J. Magn. Reson. 2018, 292, 155–170. [Google Scholar] [CrossRef]
- Neupane, P.; Bhuju, S.; Thapa, N.; Bhattarai, H.K. ATP synthase: Structure, function and inhibition. Biomol. Concepts 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Minhas, G.; Mathur, D.; Ragavendrasamy, B.; Sharma, N.K.; Paanu, V.; Anand, A. Hypoxia in CNS pathologies: Emerging role of miRNA-based neurotherapeutics and yoga based alternative therapies. Front. Neurosci. 2017, 11, 386. [Google Scholar] [CrossRef] [PubMed]
- Solaini, G.; Baracca, A.; Lenaz, G.; Sgarbi, G. Hypoxia and mitochondrial oxidative metabolism. Biochim. Biophys. Acta 2010, 1797, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Lall, R.; Mohammed, R.; Ojha, U. What are the links between hypoxia and Alzheimer’s disease? Neuropsychiatr. Dis. Treat. 2019, 15, 1343–1354. [Google Scholar] [CrossRef]
- Lestón Pinilla, L.; Ugun-Klusek, A.; Rutella, S.; De Girolamo, L.A. Hypoxia signaling in parkinson’s disease: There is use in asking “What HIF?”. Biology 2021, 10, 723. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Milner, R. Hypoxia in multiple sclerosis; is it the chicken or the egg? Brain 2021, 144, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, J.; Maglione, V.; Di Pardo, A.; Millet, G.P.; Schwarzer, C.; Zangrandi, L. A rationale for hypoxic and chemical conditioning in Huntington’s disease. Int. J. Mol. Sci. 2021, 22, 582. [Google Scholar] [CrossRef]
- Sandrelli, F.; Bisaglia, M. Molecular and physiological determinants of amyotrophic lateral sclerosis: What the DJ-1 protein teaches us. Int. J. Mol. Sci. 2023, 24, 7674. [Google Scholar] [CrossRef]
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial reactive oxygen species and their contribution in chronic kidney disease progression through oxidative stress. Front. Physiol. 2021, 12, 627837. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Zhou, T.; Prather, E.R.; Garrison, D.E.; Zuo, L. Interplay between ROS and antioxidants during ischemia-reperfusion injuries in cardiac and skeletal muscle. Int. J. Mol. Sci. 2018, 19, 417. [Google Scholar] [CrossRef]
- Kung-Chun Chiu, D.; Pui-Wah Tse, A.; Law, C.T.; Ming-Jing Xu, I.; Lee, D.; Chen, M.; Kit-Ho Lai, R.; Wai-Hin Yuen, V.; Wing-Sum Cheu, J.; Wai-Hung Ho, D.; et al. Hypoxia regulates the mitochondrial activity of hepatocellular carcinoma cells through HIF/HEY1/PINK1 pathway. Cell Death Dis. 2019, 12, 934. [Google Scholar] [CrossRef]
- Hambali, A.; Kumar, J.; Hashim, N.F.M.; Maniam, S.; Mehat, M.Z.; Cheema, M.S.; Mustapha, M.; Adenan, M.I.; Stanslas, J.; Hamid, H.A. Hypoxia-induced neuroinflammation in Alzheimer’s disease: Potential neuroprotective effects of centella asiatica. Front. Physiol. 2021, 12, 712317. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS generation in microglia: Understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Jiang, B.H.; Rue, E.; Wang, G.L.; Roe, R.; Semenza, G.L. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 17771–17778. [Google Scholar] [CrossRef] [PubMed]
- Lisy, K.; Peet, D.J. Turn me on: Regulating HIF transcriptional activity. Cell Death Differ. 2008, 15, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Hypoxia/ischemia activate processing of amyloid precursor protein: Impact of vascular dysfunction in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2017, 140, 536–549. [Google Scholar] [CrossRef]
- Lee, E.; Song, C.H.; Bae, S.J.; Ha, K.T.; Karki, R. Regulated cell death pathways and their roles in homeostasis, infection, inflammation, and tumorigenesis. Exp. Mol. Med. 2023, 55, 1632–1643. [Google Scholar] [CrossRef]
- Peng, F.; Liao, M.; Qin, R.; Zhu, S.; Peng, C.; Fu, L.; Chen, Y.; Han, B. Regulated cell death (RCD) in cancer: Key pathways and targeted therapies. Signal Transduct. Target Ther. 2022, 7, 286. [Google Scholar] [CrossRef]
- Weir, A.; Vince, J.E. No longer married to inflammasome signaling: The diverse interacting pathways leading to pyroptotic cell death. Biochem. J. 2022, 479, 1083–1102. [Google Scholar] [CrossRef]
- Zou, J.; Zheng, Y.; Huang, Y.; Tang, D.; Kang, R.; Chen, R. The versatile gasdermin family: Their function and roles in diseases. Front. Immunol. 2021, 12, 751533. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.B.; Nakashima, A.; Huber, W.J.; Davis, S.; Banerjee, S.; Huang, Z.; Saito, S.; Sadovsky, Y.; Sharma, S. Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis. 2019, 10, 927. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Wu, X.; Chen, D.; Cao, Z.; Li, Z.; Liu, Y.; Zhao, Q. The hypoxia-related signature predicts prognosis, pyroptosis and drug sensitivity of osteosarcoma. Front. Cell Dev. Biol. 2022, 10, 814722. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Zhao, R.; Xia, W.; Chang, C.W.; You, Y.; Hsu, J.M.; Nie, L.; Chen, Y.; Wang, Y.C.; Liu, C.; et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 2020, 22, 1264–1275. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.M.; Zhang, W.H.; Han, X.X.; Li, Y.Y.; Lu, Y.; Pan, J.; Mao, J.Q.; Zhu, L.Y.; Deng, J.J.; Huang, W.; et al. Hypoxia-Induced ROS Contribute to Myoblast Pyroptosis during Obstructive Sleep Apnea via the NF-κB/HIF-1α Signaling Pathway. Oxid Med. Cell Longev. 2019, 2019, 4596368. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.; Zhu, Y.; Dong, Q.; Zhang, Y. Chronic intermittent hypoxia induces the pyroptosis of renal tubular epithelial cells by activating the NLRP3 inflammasome. Bioengineered 2022, 13, 7528–7540. [Google Scholar] [CrossRef] [PubMed]
- Hirsilä, M.; Koivunen, P.; Xu, L.; Seeley, T.; Kivirikko, K.I.; Myllyharju, J. Effect of desferrioxamine and metals on the hydroxylases in the oxygen sensing pathway. FASEB J. 2005, 19, 1308–1310. [Google Scholar] [CrossRef] [PubMed]
- McGarry, T.; Biniecka, M.; Veale, D.J.; Fearon, U. Hypoxia, oxidative stress and inflammation. Free Radic. Biol. Med. 2018, 125, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Klimova, T.; Chandel, N.S. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008, 15, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Park, J.H. ROS-dependent caspase-9 activation in hypoxic cell death. FEBS Lett. 2003, 549, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Brentnall, M.; Rodriguez-Menocal, L.; De Guevara, R.L.; Cepero, E.; Boise, L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Qi, L.; Li, L.; Li, Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov. 2020, 6, 112. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Wang, C.; Ruan, J. Mechanistic Insights into Gasdermin Pore Formation and Regulation in Pyroptosis. J. Mol. Biol. 2022, 434, 167297. [Google Scholar] [CrossRef]
- Watanabe, S.; Usui-Kawanishi, F.; Karasawa, T.; Kimura, H.; Kamata, R.; Komada, T.; Inoue, Y.; Mise, N.; Kasahara, T.; Takahashi, M. Glucose regulates hypoxia-induced NLRP3 inflammasome activation in macrophages. J. Cell. Physiol. 2020, 235, 7554–7566. [Google Scholar] [CrossRef]
- Tian, K.; Yang, Y.; Zhou, K.; Deng, N.; Tian, Z.; Wu, Z.; Liu, X.; Zhang, F.; Jiang, Z. The role of ROS-induced pyroptosis in CVD. Front. Cardiovasc. Med. 2023, 10, 1116509. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, P.; Chen, Q.; Huang, Z.; Zou, D.; Zhang, J.; Gao, X.; Lin, Z. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J. Mol. Cell. Biol. 2019, 11, 1069–1082. [Google Scholar] [CrossRef]
- Zimna, A.; Kurpisz, M. Hypoxia-inducible factor-1 in physiological and pathophysiological angiogenesis: Applications and therapies. Biomed. Res. Int. 2015, 2015, 549412. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7987–7992. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Hilliard, G.; Ferguson, T.; Millhorn, D.E. Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-alpha. J. Biol. Chem. 2003, 278, 15911–15916. [Google Scholar] [CrossRef] [PubMed]
- Fleury, C.; Mignotte, B.; Vayssière, J.L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie 2002, 84, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Marsden, V.S.; Ekert, P.G.; Van Delft, M.; Vaux, D.L.; Adams, J.M.; Strasser, A. Bcl-2-regulated apoptosis and cytochrome c release can occur independently of both caspase-2 and caspase-9. J. Cell. Biol. 2004, 165, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Bratton, S.B. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid. Redox Signal. 2013, 19, 546–558. [Google Scholar] [CrossRef]
- Xia, W.; Li, Y.; Wu, M.; Jin, Q.; Wang, Q.; Li, S.; Huang, S.; Zhang, A.; Zhang, Y.; Jia, Z. Gasdermin E deficiency attenuates acute kidney injury by inhibiting pyroptosis and inflammation. Cell Death Dis. 2021, 12, 139. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sun, J.; Zhou, X.; Lu, Y.; Cui, W.; Miao, L. Mini-Review: GSDME-mediated pyroptosis in diabetic nephropathy. Front. Pharmacol. 2021, 12, 780790. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, J.; Zeng, Y.; Chen, K.; Wang, C.; Yang, S.; Sun, N.; Chen, H.; Duan, K.; Zeng, G. Pyroptosis: A pro-inflammatory type of cell death in cardiovascular disease. Clin. Chim. Acta 2020, 510, 62–72. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 inflammasome activation and cell death. Cell Mol. Immunol. 2021, 18, 2114–2127. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, C.H.; Park, J.Y.; Cho, W.G. Chemical Hypoxia Induces Pyroptosis in Neuronal Cells by Caspase-Dependent Gasdermin Activation. Int. J. Mol. Sci. 2024, 25, 2185. https://doi.org/10.3390/ijms25042185
Park CH, Park JY, Cho WG. Chemical Hypoxia Induces Pyroptosis in Neuronal Cells by Caspase-Dependent Gasdermin Activation. International Journal of Molecular Sciences. 2024; 25(4):2185. https://doi.org/10.3390/ijms25042185
Chicago/Turabian StylePark, Chan Ho, Jun Young Park, and Won Gil Cho. 2024. "Chemical Hypoxia Induces Pyroptosis in Neuronal Cells by Caspase-Dependent Gasdermin Activation" International Journal of Molecular Sciences 25, no. 4: 2185. https://doi.org/10.3390/ijms25042185
APA StylePark, C. H., Park, J. Y., & Cho, W. G. (2024). Chemical Hypoxia Induces Pyroptosis in Neuronal Cells by Caspase-Dependent Gasdermin Activation. International Journal of Molecular Sciences, 25(4), 2185. https://doi.org/10.3390/ijms25042185