
Aldehyde Dehydrogenase 2 (ALDH2) Deficiency, Obesity, and Atrial Fibrillation Susceptibility: Unraveling the Connection
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
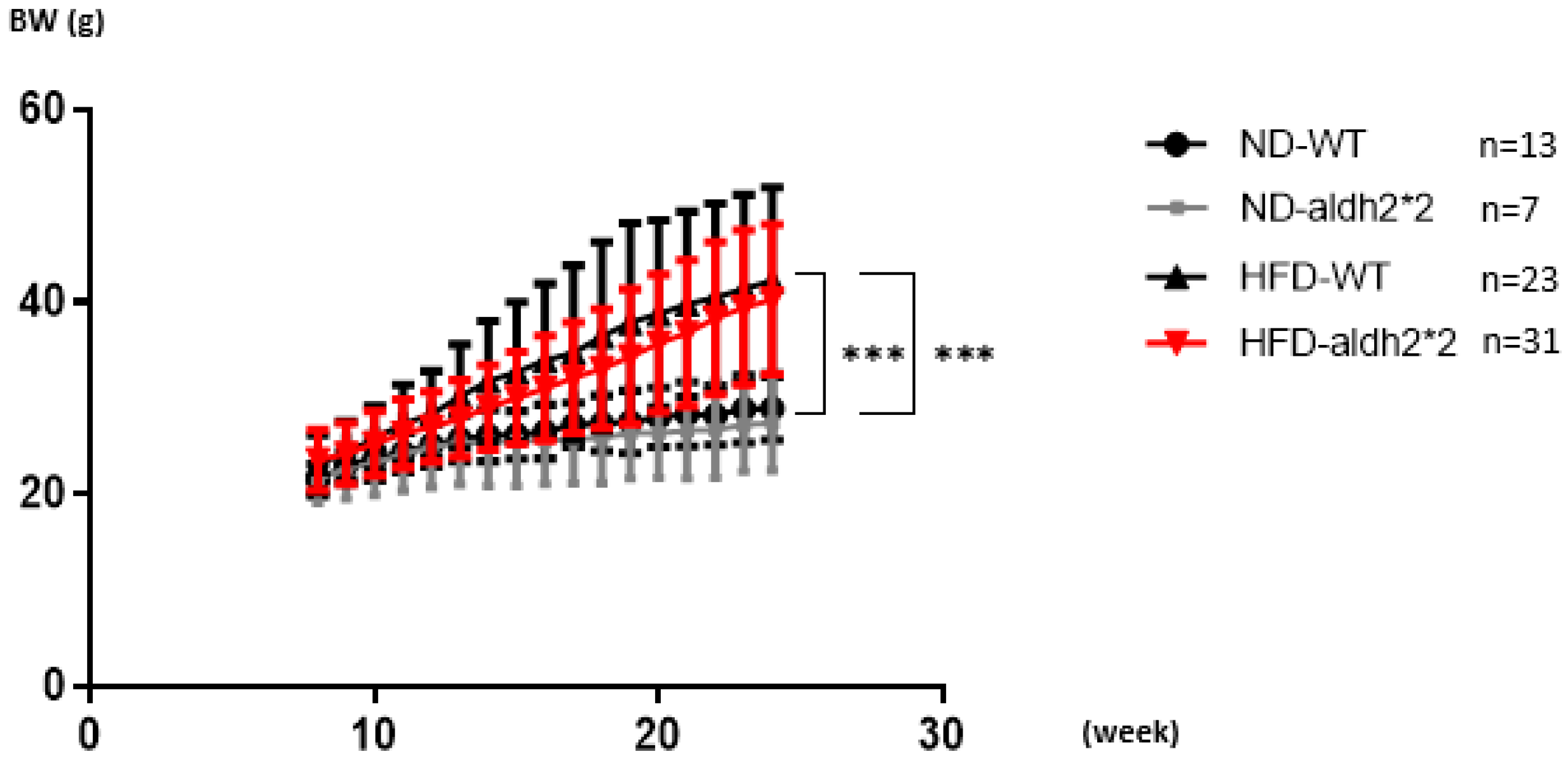
2.1. High-Fat Diet Inducing Obesity
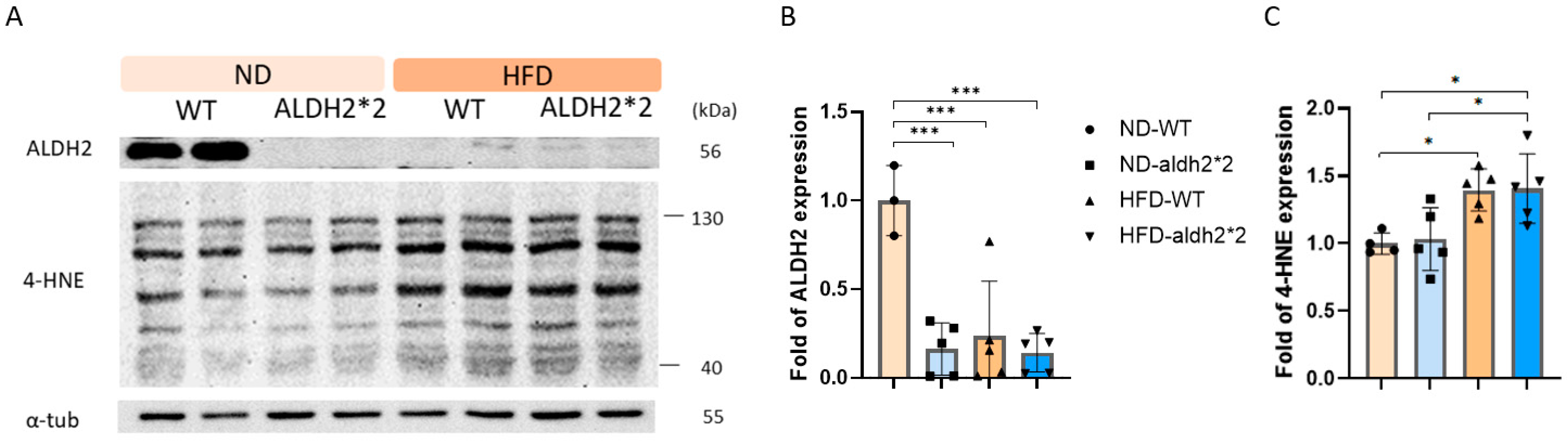
2.2. 4-HNE and ALDH2 Production in Wild-Type and ALDH2*2 KI Mice with High-Fat Diets
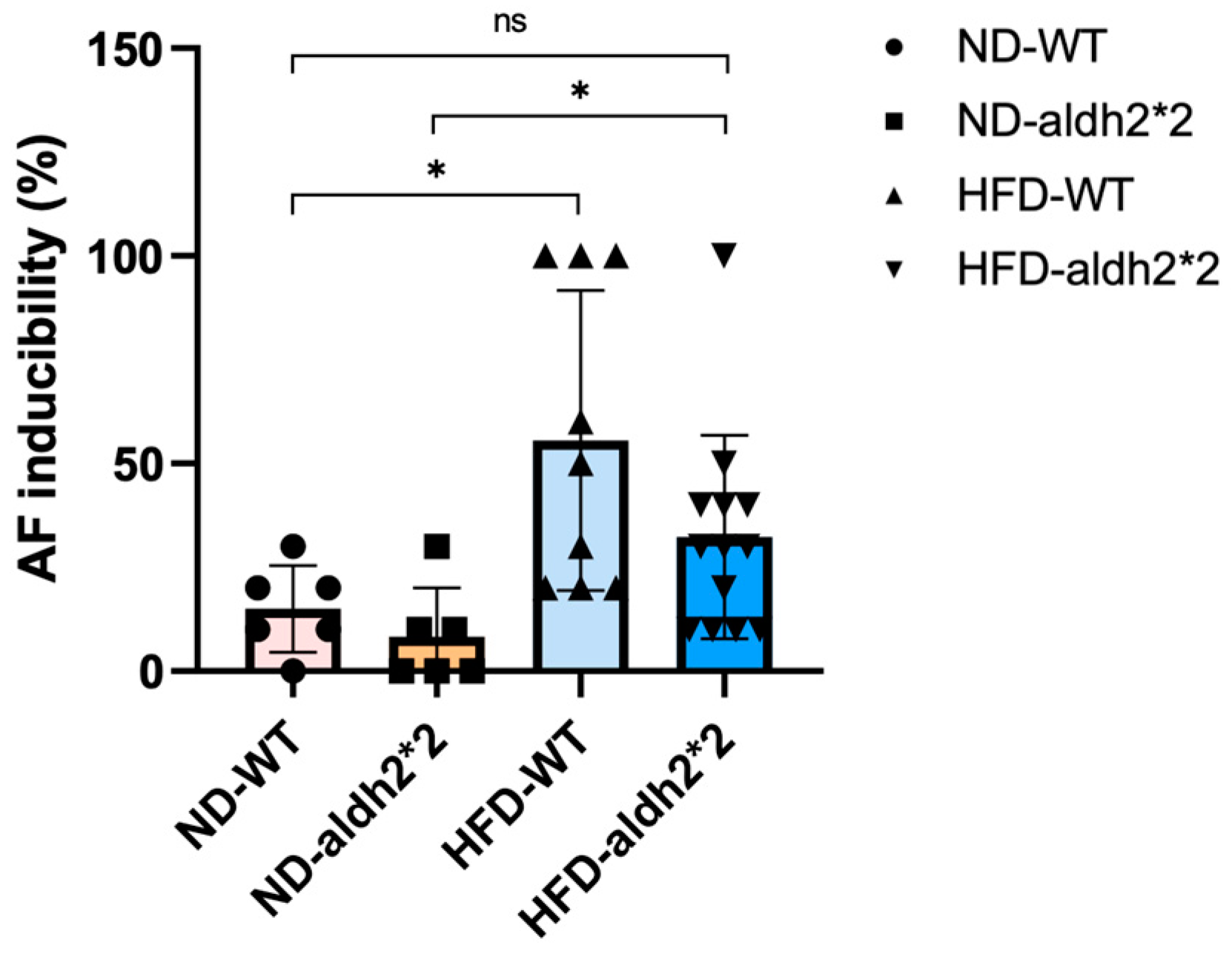
2.3. AF Inducibility in ALDH2*2 KI Mice with Chronic Diet-Induced Obesity
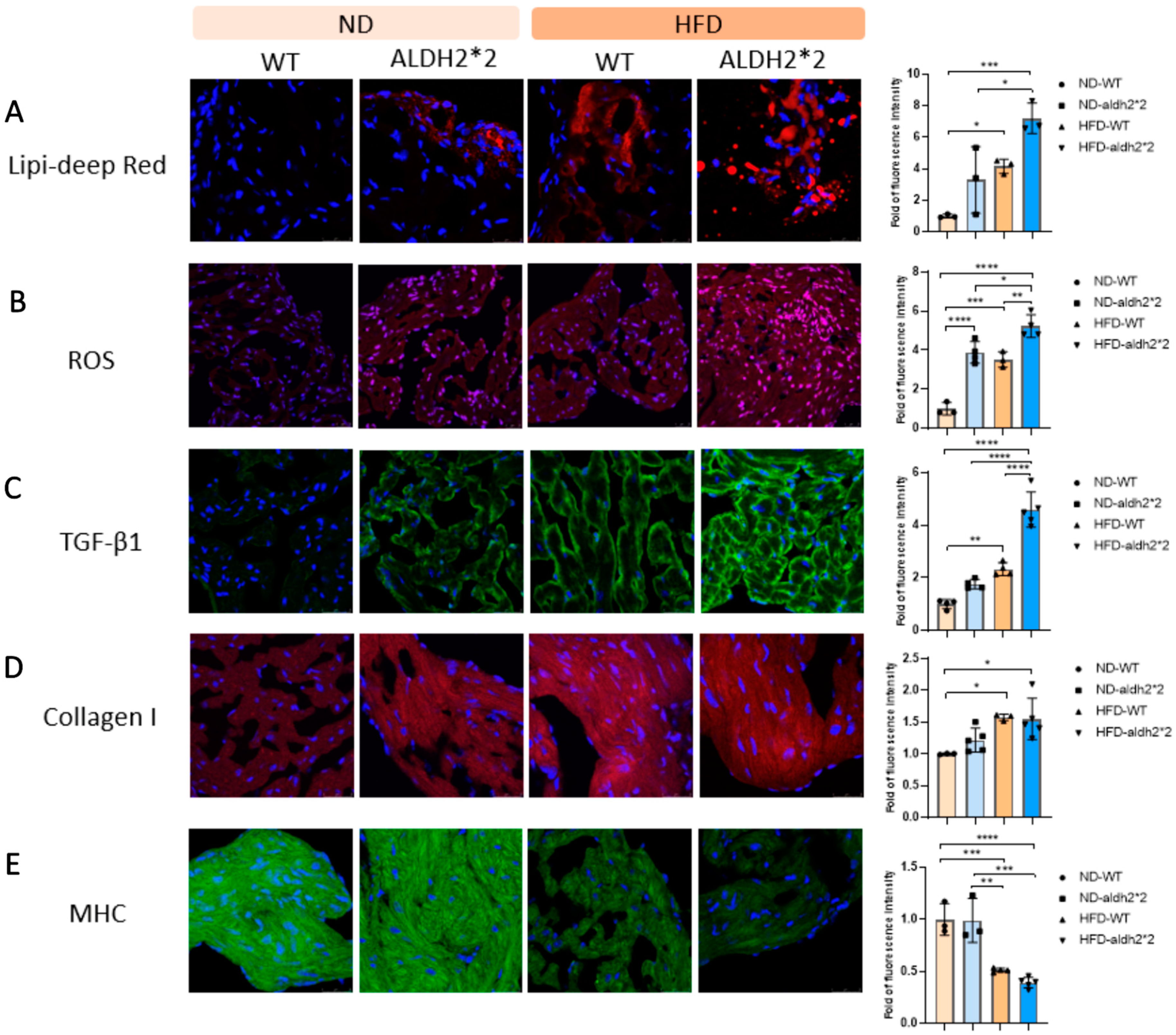
2.4. Increased Fat Accumulation and Oxidative Stress in ALDH2*2 KI Mice with Chronic HFD Consumption
2.5. Atrial Fibrosis and Structure Remodeling in ALDH2*2 KI Mice with Chronic HFD Consumption
2.6. Role of HO-1 in the Differential Effect of Chronic HFD Consumption on ALDH2*2 KI Mice
3. Discussion
3.1. Role of ALDH2 in Obesity-Induced Cardiac Dysfunction
3.2. Impact of ALDH2*2 on Body Weight and Metabolic Parameters in Obesity
3.3. Expanding beyond Alcohol Metabolism: ALDH2*2 in Obesity-Related Cardiovascular Complications
3.4. Limitations
4. Materials and Methods
4.1. Ethics Statement
4.2. Generation of ALDH2*2 KI Mice Using CRISPR/CAS9 to Mimic Humans with ALDH2*2
4.3. Mice Maintenance and Diet-Induced Obesity Model
4.4. Programmed Electrical Stimulation to Evaluate AF Vulnerability
4.5. Western Blot Analysis to Study ALDH2-Related Oxidative Stress and Atrial Remodeling
4.6. Histology and Immunohistochemistry Analyses to Examine Atrial ALDH2-Related Oxidative Stress and Atrial Remodeling
4.7. Real-Time Quantitative Reverse Transcription–PCR (RT-PCR)
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chugh, S.S.; Havmoeller, R.; Narayanan, K.; Singh, D.; Rienstra, M.; Benjamin, E.J.; Gillum, R.F.; Kim, Y.H.; McAnulty, J.H., Jr.; Zheng, Z.J.; et al. Worldwide epidemiology of atrial fibrillation: A global burden of disease 2010 study. Circulation 2014, 129, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Schotten, U.; Verheule, S.; Kirchhof, P.; Goette, A. Pathophysiological mechanisms of atrial fibrillation: A translational appraisal. Physiol. Rev. 2011, 91, 265–325. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J.; Parise, H.; Levy, D.; D’Agostino, R.B., Sr.; Wolf, P.A.; Vasan, R.S.; Benjamin, E.J. Obesity and the risk of new-onset atrial fibrillation. JAMA 2004, 292, 2471–2477. [Google Scholar] [CrossRef] [PubMed]
- Abed, H.S.; Wittert, G.A.; Leong, D.P.; Shirazi, M.G.; Bahrami, B.; Middeldorp, M.E.; Lorimer, M.F.; Lau, D.H.; Antic, N.A.; Brooks, A.G.; et al. Effect of weight reduction and cardiometabolic risk factor management on symptom burden and severity in patients with atrial fibrillation: A randomized clinical trial. JAMA 2013, 310, 2050–2060. [Google Scholar] [CrossRef]
- Jamaly, S.; Carlsson, L.; Peltonen, M.; Jacobson, P.; Sjostrom, L.; Karason, K. Bariatric surgery and the risk of new-onset atrial fibrillation in Swedish obese subjects. J. Am. Coll. Cardiol. 2016, 68, 2497–2504. [Google Scholar] [CrossRef]
- Mahajan, R.; Lau, D.H.; Brooks, A.G.; Shipp, N.J.; Manavis, J.; Wood, J.P.; Finnie, J.W.; Samuel, C.S.; Royce, S.G.; Twomey, D.J.; et al. Electrophysiological, electroanatomical, and structural remodeling of the atria as consequences of sustained obesity. J. Am. Coll. Cardiol. 2015, 66, 1–11. [Google Scholar] [CrossRef]
- Hohl, M.; Lau, D.H.; Muller, A.; Elliott, A.D.; Linz, B.; Mahajan, R.; Hendriks, J.M.L.; Bohm, M.; Schotten, U.; Sanders, P.; et al. Concomitant obesity and metabolic syndrome add to the atrial arrhythmogenic phenotype in male hypertensive rats. J. Am. Heart. Assoc. 2017, 6, e006717. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Abe, I.; Gotoh, K.; Fukui, A.; Takanari, H.; Ishii, Y.; Ikebe, Y.; Kira, S.; Oniki, T.; Saito, S.; et al. Interleukin 10 treatment ameliorates high-fat diet induced inflammatory atrial remodeling and fibrillation. Circ. Arrhythm. Electrophysiol. 2018, 11, e006040. [Google Scholar] [CrossRef]
- Takahashi, K.; Sasano, T.; Sugiyama, K.; Kurokawa, J.; Tamura, N.; Soejima, Y.; Sawabe, M.; Isobe, M.; Furukawa, T. High-fat diet increases vulnerability to atrial arrhythmia by conduction disturbance via miR-27b. J. Mol. Cell Cardiol. 2016, 90, 38–46. [Google Scholar] [CrossRef]
- Fang, G.; Cao, W.; Chen, L.; Song, S.; Li, Y.; Yuan, J.; Fei, Y.; Ge, Z.; Chen, Y.; Zhou, L.; et al. Cadherin 11 deficiency mitigates high fat diet induced inflammatory atrial remodeling and vulnerability to atrial fibrillation. J. Cell Physiol. 2021, 236, 5725–5741. [Google Scholar] [CrossRef]
- Lubbers, E.R.; Price, M.V.; Mohler, P.J. Arrhythmogenic Substrates for Atrial Fibrillation in Obesity. Front. Physiol. 2018, 9, 1482. [Google Scholar] [CrossRef]
- McCauley, M.D.; Hong, L.; Sridhar, A.; Menon, A.; Perike, S.; Zhang, M.; da Silva, I.B.; Yan, J.; Bonini, M.G.; Ai, X.; et al. Ion Channel and Structural Remodeling in Obesity Mediated Atrial Fibrillation. Circ. Arrhythm. Electrophysiol. 2020, 13, e008296. [Google Scholar] [CrossRef]
- Dewal, R.S.; Greer, S.A.; Lane, C.; Nirengi, S.; Manzano, P.A.; Hernández-Saavedra, D.; Wright, K.R.; Nassal, D.; Baer, L.A.; Mohler, P.J.; et al. Phospho ablation of cardiac sodium channel Na v 1.5 mitigates susceptibility to atrial fibrillation and improves glucose homeostasis under conditions of diet induced obesity. Int. J. Obes. 2021, 45, 795–807. [Google Scholar] [CrossRef]
- Aromolaran, A.S.; Colecraft, H.M.; Boutjdir, M. High-fat diet-dependent modulation of the delayed rectifier K(+) current in adult guinea pig atrial myocytes. Biochem. Biophys. Res. Commun. 2016, 474, 554–559. [Google Scholar] [CrossRef]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Mizuno, Y.; Harada, E.; Morita, S.; Kinoshita, K.; Hayashida, M.; Shono, M.; Morikawa, Y.; Murohara, T.; Nakayama, M.; Yoshimura, M.; et al. East Asian Variant of Aldehyde Dehydrogenase 2 Is Associated with Coronary Spastic Angina: Possible Roles of Reactive Aldehydes and Implications of Alcohol Flushing Syndrome. Circulation 2015, 131, 1665–1673. [Google Scholar] [CrossRef]
- Jo, S.A.; Kim, E.K.; Park, M.H.; Han, C.; Park, H.Y.; Jang, Y.; Song, B.J.; Jo, I.A. Glu487Lys polymorphism in the gene for mitochondrial aldehyde dehydrogenase 2 is associated with myocardial infarction in elderly Korean men. Clin. Chim. Acta. 2007, 382, 43–47. [Google Scholar] [CrossRef]
- Takagi, S.; Iwai, N.; Yamauchi, R.; Kojima, S.; Yasuno, S.; Baba, T.; Terashima, M.; Tsutsumi, Y.; Suzuki, S.; Morii, I.; et al. Aldehyde dehydrogenase 2 gene is a risk factor for myocardial infarction in Japanese men. Hypertens. Res. 2002, 25, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.F.; Lu, C.C.; Lee, J.T.; Hung, Y.J.; Hu, C.J.; Jeng, J.S.; Chiou, H.Y.; Peng, G.S. Homozygous ALDH2*2 Isan Independent Risk Factor for Ischemic Stroke in Taiwanese Men. Stroke 2016, 47, 2174–2179. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Ochi, H.; Onohara, Y.; Sairaku, A.; Tokuyama, T.; Matsumura, H.; Tomomori, S.; Amioka, M.; Hironomobe, N.; Motoda, C.; et al. Genetic Variations of Aldehyde Dehydrogenase 2 and Alcohol Dehydrogenase 1B Are Associated with the Etiology of Atrial Fibrillation in Japanese. J. Biomed. Sci. 2016, 23, 89. [Google Scholar] [CrossRef] [PubMed]
- Sakaue, S.; Kanai, M.; Tanigawa, Y.; Karjalainen, J.; Kurki, M.; Koshiba, S.; Narita, A.; Konuma, T.; Yamamoto, K.; Akiyama, M.; et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet. 2021, 53, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.A.; Tsai, F.C.; Yeh, Y.H.; Chang, C.J.; Kuo, C.T.; Chen, W.J.; Tsai, H.Y.; Chang, G.J. Aldehyde dehydrogenase 2 ameliorates chronic alcohol consumption-induced atrial fibrillation through detoxification of 4-hne. Int. J. Mol. Sci. 2020, 21, 6678. [Google Scholar] [CrossRef] [PubMed]
- Purohit, V.; Brenner, D.A. Mechanisms of alcohol-induced hepatic fibrosis: A summary of the Ron Thurman Symposium. Hepatology 2006, 43, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.H.; Hsu, L.A.; Chen, Y.H.; Kuo, C.T.; Chang, G.J.; Chen, W.J. Protective Role of Heme oxygenase-1 in Atrial Remodeling. Basic Res. Cardiol. 2016, 111, 58. [Google Scholar] [CrossRef] [PubMed]
- Korantzopoulos, P.; Kolettis, T.M.; Galaris, D.; Goudevenos, J.A. The role of oxidative stress in the pathogenesis and perpetuation of atrial fibrillation. Int. J. Cardiol. 2007, 115, 135–143. [Google Scholar] [CrossRef]
- Srivastava, S.; Chandra, A.; Wang, L.F.; Seifert, W.E., Jr.; DaGue, B.B.; Ansari, N.H.; Srivastava, S.K.; Bhatnagar, A. Metabolism of the lipid peroxidation product, 4-hydroxy-trans-2-nonenal, in isolated perfused rat heart. J. Biol. Chem. 1998, 273, 10893–10900. [Google Scholar] [CrossRef]
- Chen, C.H.; Budas, G.R.; Churchill, E.N.; Disatnik, M.H.; Hurley, T.D.; Mochly-Rosen, D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 2008, 321, 1493–1495. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Mali, V.R.; Ning, R.; Chen, J.; Yang, X.P.; Xu, J.; Palaniyandi, S.S. Impairment of aldehyde dehydrogenase-2 by 4-hydroxy-2-nonenal adduct formation and cardiomyocyte hypertrophy in mice fed a high-fat diet and injected with low-dose streptozotocin. Exp. Biol. Med. 2014, 239, 610–618. [Google Scholar] [CrossRef]
- Li, C.; Li, X.; Chang, Y.; Zhao, L.; Liu, B.; Wei, S.; Xu, F.; Zhang, Y.; Chen, Y. Aldehyde Dehydrogenase-2 Attenuates Myocardial Remodeling and Contractile Dysfunction Induced by a High-Fat Diet. Cell Physiol. Biochem. 2018, 48, 1843–1853. [Google Scholar] [CrossRef]
- Wang, S.; Wang, C.; Turdi, S.; Richmond, K.L.; Zhang, Y.; Ren, J. ALDH2 protects against high fat diet-induced obesity cardiomyopathy and defective autophagy: Role of CaM kinase II, histone H3K9 methyltransferase SUV39H, Sirt1, and PGC-1α deacetylation. Int. J. Obes. 2018, 42, 1073–1087. [Google Scholar] [CrossRef]
- Chang, Y.C.; Lee, H.L.; Yang, W.; Hsieh, M.L.; Liu, C.C.; Lee, T.Y.; Huang, J.Y.; Nong, J.Y.; Li, F.A.; Chuang, H.L.; et al. A common East-Asian ALDH2 mutation causes metabolic disorders and the therapeutic effect of ALDH2 activators. Nat. Commun. 2023, 14, 5971. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, D.; Zhou, D.; Li, Z.; Li, Z.; Fang, L.; Yang, M.; Shan, Z.; Li, H.; Chen, J.; et al. Susceptibility loci for metabolic syndrome and metabolic components identified in Han Chinese: A multi-stage genome-wide association study. J. Cell Mol. Med. 2017, 21, 1106–1116. [Google Scholar] [CrossRef]
- Wang, D.; Zou, Y.; Yu, S.; Lin, S.; Li, H.; Yin, Y.; Qiu, L.; Xu, T.; Wu, J. The effect of ALDH2 rs671 gene mutation on clustering of cardiovascular risk factors in a big data study of Chinese population: Associations differ between the sexes. BMC Cardiovasc. Disord. 2020, 20, 509. [Google Scholar] [CrossRef]
- Huang, L.O.; Rauch, A.; Mazzaferro, E.; Preuss, M.; Carobbio, S.; Bayrak, C.S.; Chami, N.; Wang, Z.; Schick, U.M.; Yang, N.; et al. Genome-wide discovery of genetic loci that uncouple excess adiposity from its comorbidities. Nat. Metab. 2021, 3, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Tan, A.; Yu, L.; Hou, C.; Kuang, H.; Wu, Q.; Su, J.; Zhou, Q.; Zhu, Y.; Zhang, C.; et al. A phenomics-based approach for the detection and interpretation of shared genetic influences on 29 biochemical indices in southern Chinese men. BMC Genomics 2019, 20, 983. [Google Scholar] [CrossRef]
- Hu, J.; Yang, L.; Peng, X.; Mao, M.; Liu, X.; Song, J.; Li, H. ALDH2 Hampers Immune Escape in Liver Hepatocellular Carcinoma through ROS/Nrf2-mediated Autophagy. Inflammation 2022, 45, 2309–2324. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.M.; Borges, J.I.; Suster, M.S.; Sizova, A.; Cora, N.; Desimine, V.L.; Lymperopoulos, A. Regulator of G-Protein Signaling-4 Attenuates Cardiac Adverse Remodeling and Neuronal Norepinephrine Release-Promoting Free Fatty Acid Receptor FFAR3 Signaling. Int. J. Mol. Sci. 2022, 23, 5803. [Google Scholar] [CrossRef]
- Del Calvo, G.; Baggio Lopez, T.; Lymperopoulos, A. The therapeutic potential of targeting cardiac RGS4. Ther. Adv. Cardiovasc. Dis. 2023, 17, 17539447231199350. [Google Scholar] [CrossRef]
- Verheule, S.; Sato, T.; Everett, T., 4th; Engle, S.K.; Otten, D.; Rubart-von der Lohe, M.; Nakajima, H.O.; Nakajima, H.; Field, L.J.; Olgin, J.E. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta1. Circ. Res. 2004, 94, 1458–1465. [Google Scholar] [CrossRef]
- Quiles, J.M.; Pepin, M.E.; Sunny, S.; Shelar, S.B.; Challa, A.K.; Dalley, B.; Hoidal, J.R.; Pogwizd, S.M.; Wende, A.R.; Rajasekaran, N.S. Identification of Nrf2-responsive microRNA networks as putative mediators of myocardial reductive stress. Sci. Rep. 2021, 11, 11977. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, L.-A.; Yeh, Y.-H.; Chang, C.-J.; Chen, W.-J.; Tsai, H.-Y.; Chang, G.-J. Aldehyde Dehydrogenase 2 (ALDH2) Deficiency, Obesity, and Atrial Fibrillation Susceptibility: Unraveling the Connection. Int. J. Mol. Sci. 2024, 25, 2186. https://doi.org/10.3390/ijms25042186
Hsu L-A, Yeh Y-H, Chang C-J, Chen W-J, Tsai H-Y, Chang G-J. Aldehyde Dehydrogenase 2 (ALDH2) Deficiency, Obesity, and Atrial Fibrillation Susceptibility: Unraveling the Connection. International Journal of Molecular Sciences. 2024; 25(4):2186. https://doi.org/10.3390/ijms25042186
Chicago/Turabian StyleHsu, Lung-An, Yung-Hsin Yeh, Chi-Jen Chang, Wei-Jan Chen, Hsin-Yi Tsai, and Gwo-Jyh Chang. 2024. "Aldehyde Dehydrogenase 2 (ALDH2) Deficiency, Obesity, and Atrial Fibrillation Susceptibility: Unraveling the Connection" International Journal of Molecular Sciences 25, no. 4: 2186. https://doi.org/10.3390/ijms25042186
APA StyleHsu, L.-A., Yeh, Y.-H., Chang, C.-J., Chen, W.-J., Tsai, H.-Y., & Chang, G.-J. (2024). Aldehyde Dehydrogenase 2 (ALDH2) Deficiency, Obesity, and Atrial Fibrillation Susceptibility: Unraveling the Connection. International Journal of Molecular Sciences, 25(4), 2186. https://doi.org/10.3390/ijms25042186