
Extreme Reconfiguration of Plastid Genomes in Papaveraceae: Rearrangements, Gene Loss, Pseudogenization, IR Expansion, and Repeats

Abstract
1. Introduction
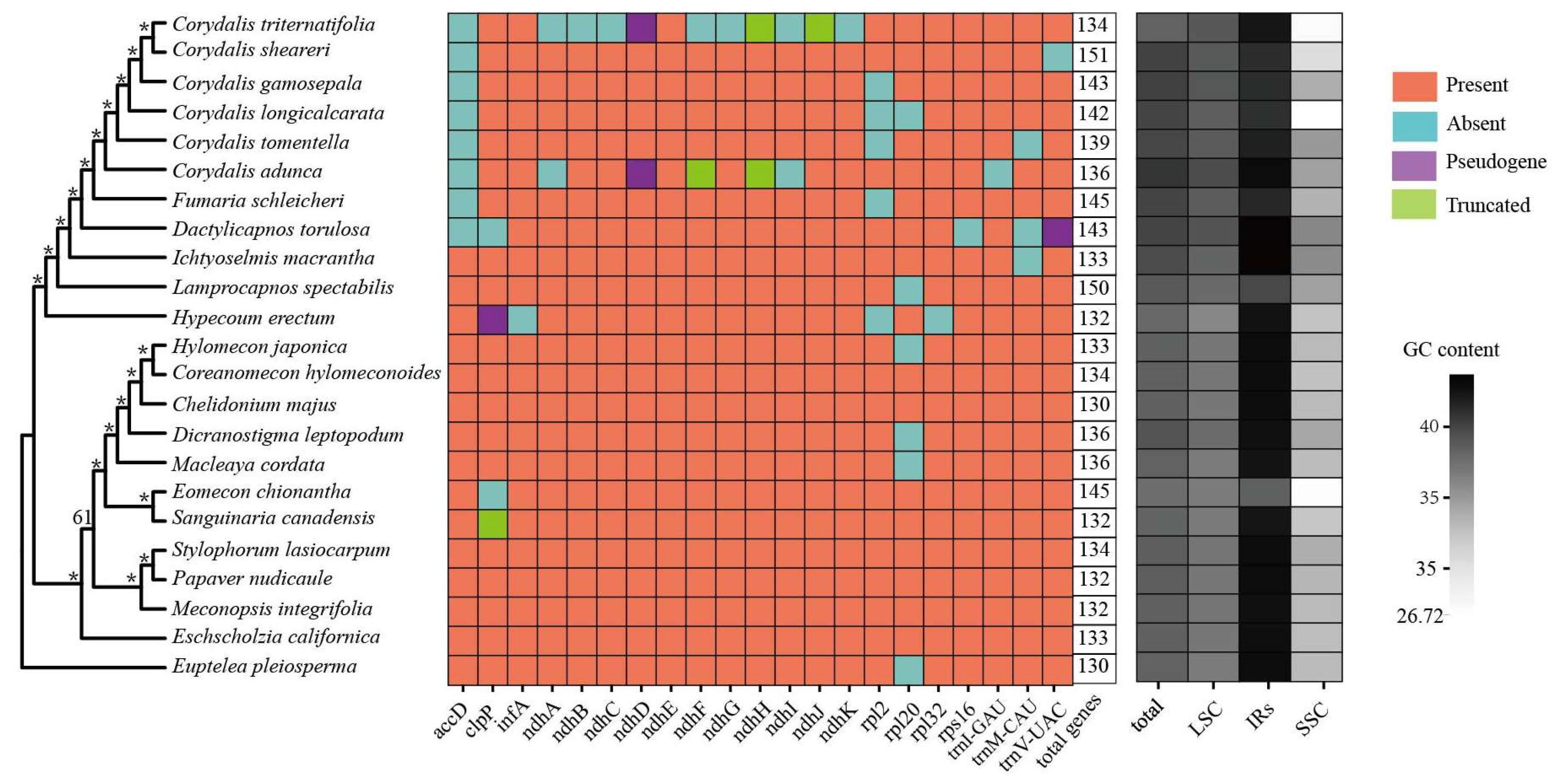
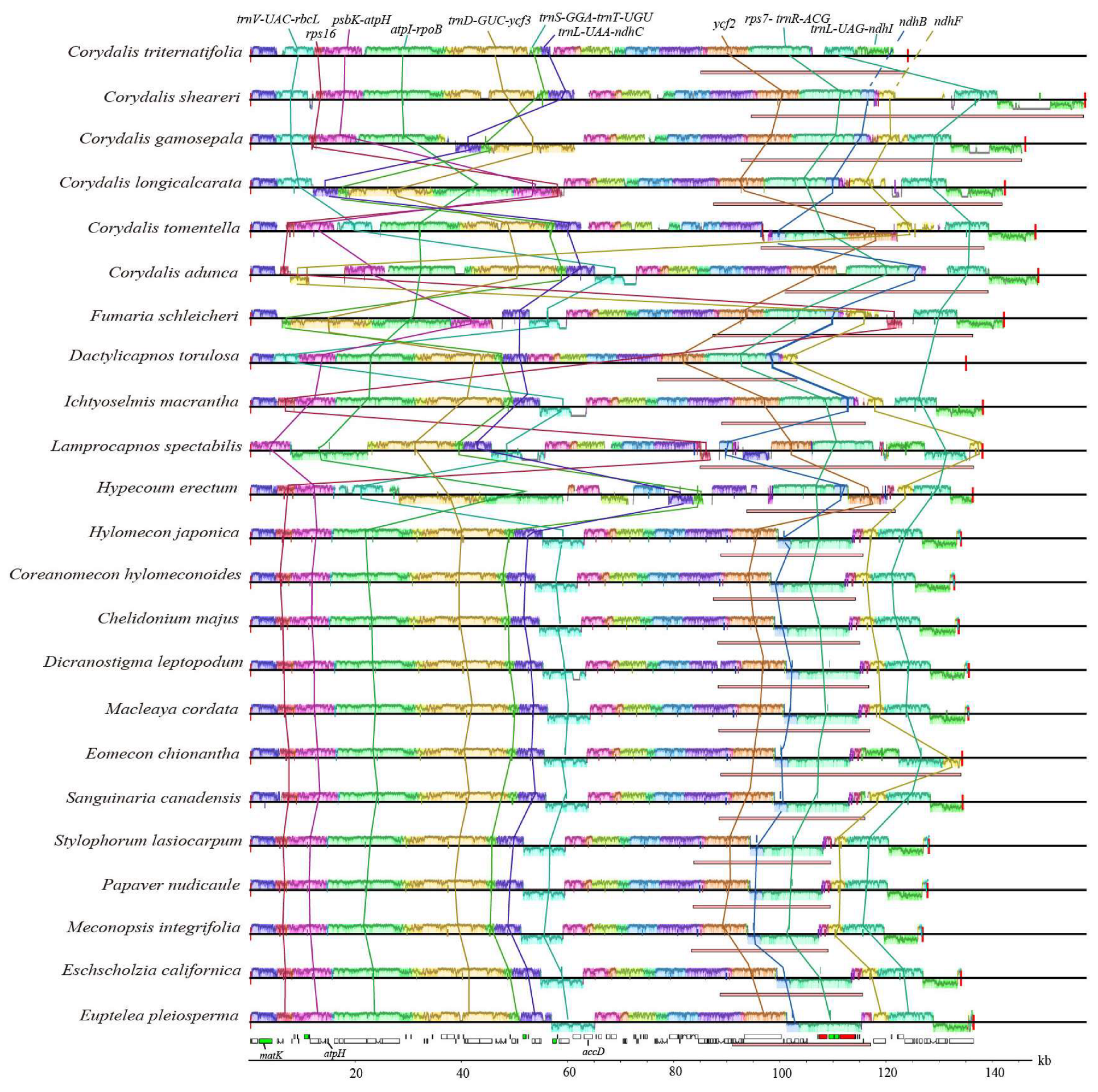
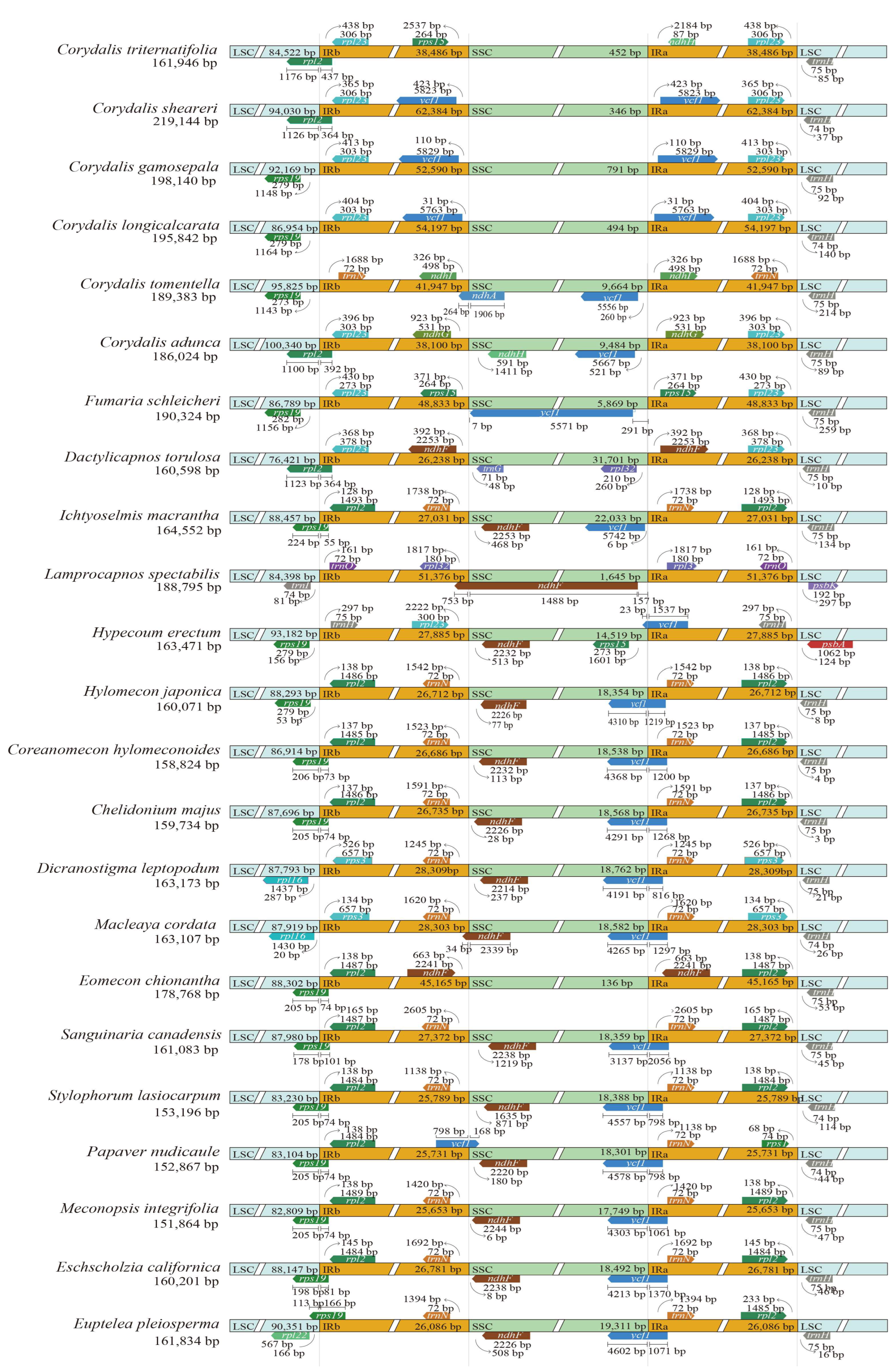
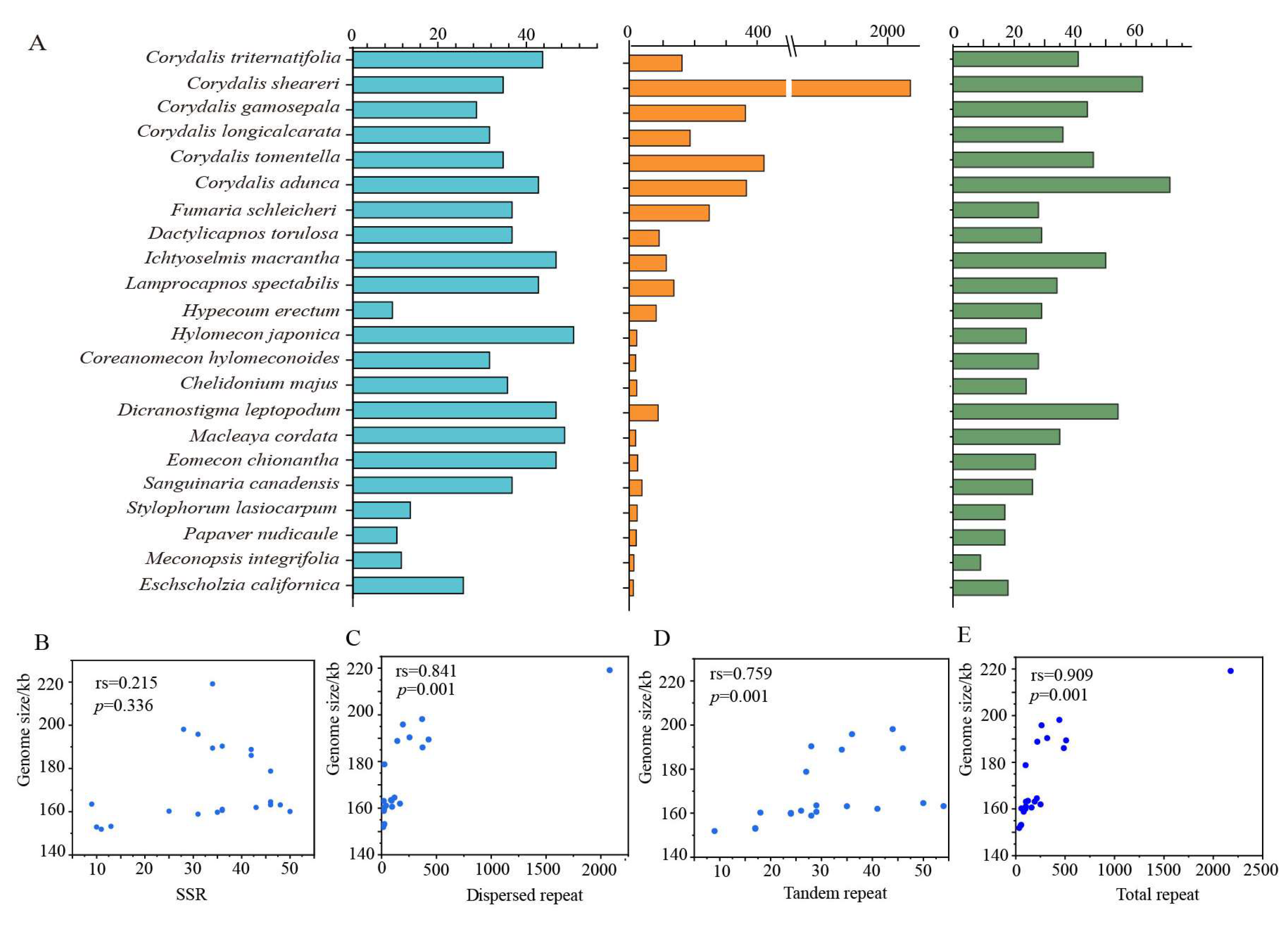
2. Results
2.1. Chloroplast Genome Characterization
2.2. Phylogenetic Analyses
2.3. Genome Structure Variations
2.4. Codon Usage and Repeat Sequence Analysis
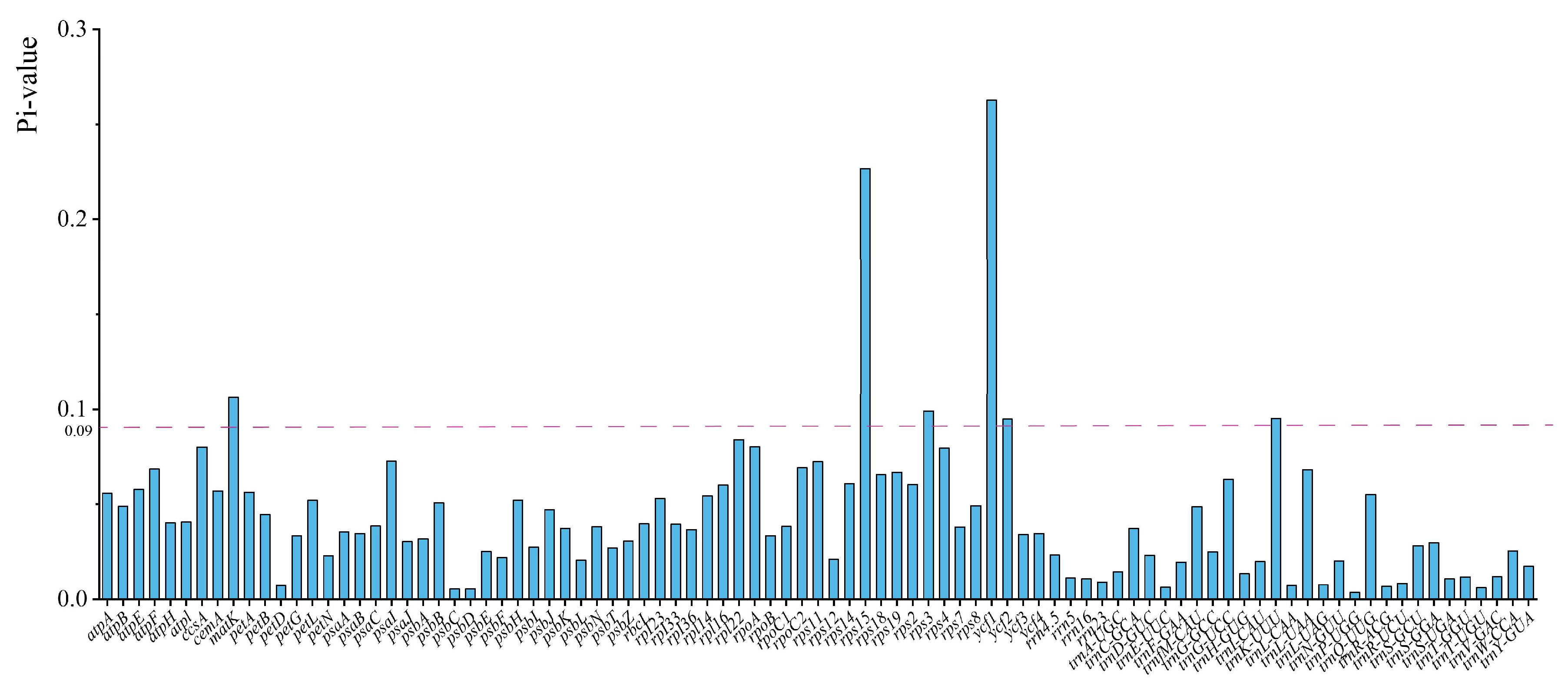
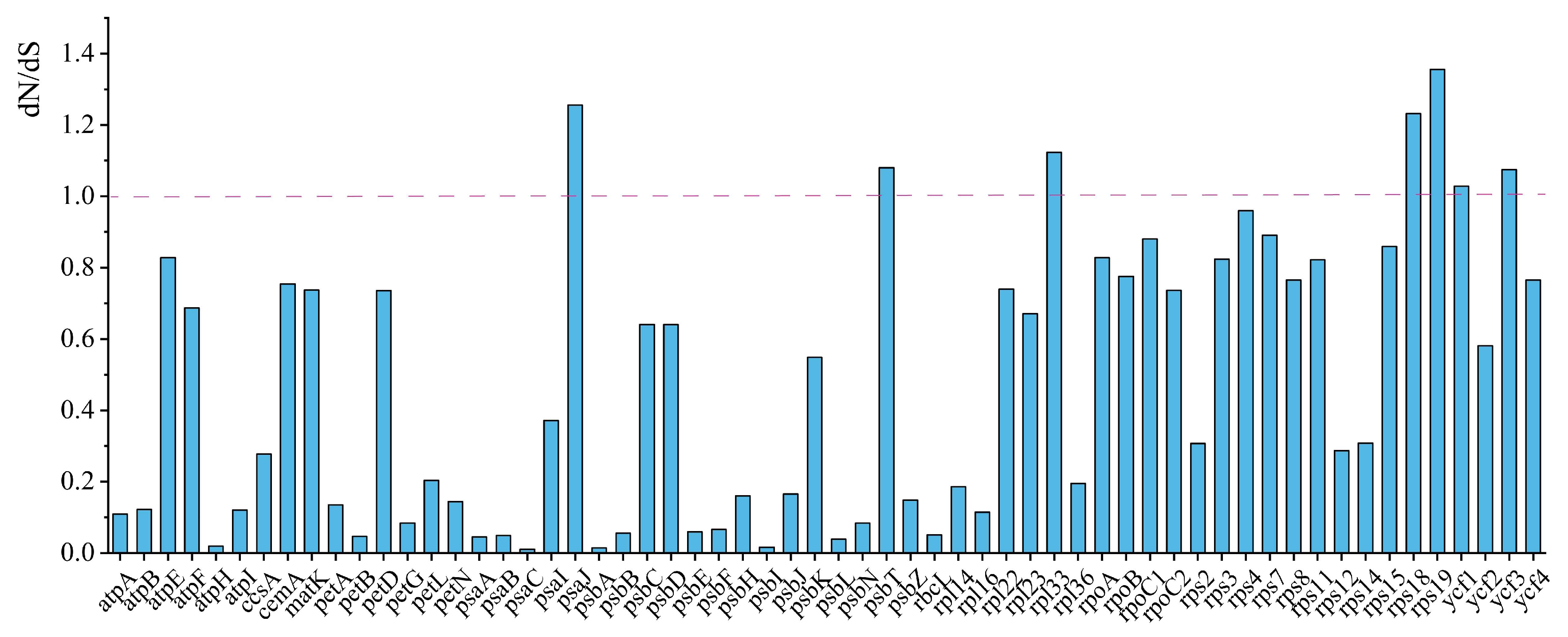
2.5. Nucleotide Diversity and Positive Selection Analyses
3. Discussion
4. Materials and Methods
4.1. Plant Materials, Taxon Sampling, DNA Extraction, and Sequencing
4.2. Plastome Assembly, Annotation, and Plastid Gene Extraction
4.3. Phylogenetic Analyses
4.4. Genome Structure Analyses
4.5. Codon Usage and Repeat Sequence Analysis
4.6. Nucleotide Diversity and Positive Selection Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebens-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef]
- Mower, J.P.; Vickrey, T.L. Structural diversity among plastid genomes of land plants. Adv. Bot. Res. 2018, 85, 263–292. [Google Scholar]
- Maier, R.M.; Neckermann, K.; Igloi, G.L.; Kössel, H. Complete sequence of the maize chloroplast genome: Gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J. Mol. Biol. 1995, 251, 614–628. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kan, S.L.; Liao, X.Z.; Zhou, J.W.; Tembrock, L.R.; Daniell, H.; Jin, S.X.; Wu, Z.Q. Plant organellar genomes: Much done, much more to do. Trends Plant Sci. 2024. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.-R.; Oyebanji, O.; Zhang, R.; Yi, T.S. Plastid phylogenomic insights into the evolution of subfamily Dialioideae (Leguminosae). Plant Divers. 2021, 43, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Guisinger, M.; Kim, H.-G.; Ruck, E.; Blazier, J.C.; McMurtry, V.; Kuehl, J.V.; Boore, J.; Jansen, R.K. Extensive reorganization of the plastid genome of Trifolium subterraneum (Fabaceae) is associated with numerous repeated sequences and novel DNA insertions. J. Mol. Evol. 2008, 67, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Kolodner, R.; Tewari, K.K. Inverted repeats in chloroplast DNA from higher plants. Proc. Natl. Acad. Sci. USA 1979, 76, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.D.; Osorio, B.; Thompson, W.F. Evolutionary significance of inversions in legume chloroplast DNAs. Curr. Genet. 1988, 14, 65–74. [Google Scholar] [CrossRef]
- Tangphatsornruang, S.; Sangsrakru, D.; Chanprasert, J.; Uthaipaisanwong, P.; Yoocha, T.; Jomchai, N.; Tragoonrung, S. The chloroplast genome sequence of mungbean (Vigna radiata) determined by high-throughput pyrosequencing: Structural organization and phylogenetic relationships. DNA Res. 2010, 17, 11–22. [Google Scholar] [CrossRef]
- Blazier, J.C.; Jansen, R.K.; Mower, J.P.; Govindu, M.; Zhang, J.; Weng, M.-L.; Ruhlman, T.A. Variable presence of the inverted repeat and plastome stability in Erodium. Ann. Bot. 2016, 117, 1209–1220. [Google Scholar] [CrossRef]
- Guisinger, M.; Kuehl, J.; Boore, J.; Jansen, R. Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: Rearrangements, repeats, and codon usage. Mol. Biol. Evol. 2011, 28, 583–600. [Google Scholar] [CrossRef] [PubMed]
- Ruhlman, T.A.; Zhang, J.; Blazier, J.C.; Sabir, J.S.M.; Jansen, R.K. Recombination-dependent replication and gene conversion homogenize repeat sequences and diversify plastid genome structure. Am. J. Bot. 2017, 104, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.L.; Blazier, J.C.; Govindu, M.; Jansen, R.K. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol. Biol. Evol. 2013, 31, 645–659. [Google Scholar] [CrossRef]
- Amaral, D.T.; Bombonato, J.R.; da Silva Andrade, S.C.; Moraes, E.M.; Franco, F.F. The genome of a thorny species: Comparative genomic analysis among South and North American Cactaceae. Planta 2021, 254, 44. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, E.M.; Sader, M.A.; Rodriguez, P.E.; Loeuille, B.; Felix, L.P.; Pedrosa-Harand, A. Assembling the puzzle: Complete chloroplast genome sequences of Discocactus bahiensis Britton & Rose and Melocactus ernestii Vaupel (Cactaceae) and their evolutionary significance. Braz. J. Bot. 2021, 44, 877–888. [Google Scholar]
- Cosner, M.E.; Raubeson, L.A.; Jansen, R.K. Chloroplast DNA rearrangements in Campanulaceae: Phylogenetic utility of highly rearranged genomes. BMC Evol. Biol. 2004, 4, 27. [Google Scholar] [CrossRef]
- Haberle, R.C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Extensive rearrangements in the chloroplast genome of Trachelium caeruleum are associated with repeats and tRNA Genes. J. Mol. Evol. 2008, 66, 350–361. [Google Scholar] [CrossRef]
- Cauz dos Santos, L.; Munhoz, C.; Rodde, N.; Cauet, S.; Santos, A.; Penha, H.; Dornelas, M.; Varani, A.; Oliveira, G.; Bergès, H.; et al. The chloroplast genome of Passiflora edulis (Passifloraceae) assembled from long sequence reads: Structural organization and phylogenomic studies in Malpighiales. Front. Plant Sci. 2017, 8, 334. [Google Scholar] [CrossRef]
- Cauz dos Santos, L.; Portugal, Z.; Callot, C.; Cauet, S.; Zucchi, M.I.; Bergès, H.; van den Berg, C.; Vieira, M.-L. A repertory of rearrangements and the loss of an inverted repeat region in Passiflora chloroplast genomes. Genome Biol. Evol. 2020, 12, 1841–1857. [Google Scholar] [CrossRef]
- Rabah, S.O.; Shrestha, B.; Hajrah, H.N.; Sabir, M.J.; Alharby, H.F.; Sabir, M.J.; Alhebshi, A.M.; Sabir, L.S.M.; Gilbert, L.E.; Ruhlman, T.A.; et al. Passiflora plastome sequencing reveals widespread genomic rearrangements. J. Syst. Evol. 2019, 57, 1–14. [Google Scholar] [CrossRef]
- Shrestha, B.; Weng, M.-L.; Theriot, E.C.; Gilbert, L.E.; Ruhlman, T.A.; Krosnick, S.E.; Jansen, R.K. Highly accelerated rates of genomic rearrangements and nucleotide substitutions in plastid genomes of Passiflora subgenus Decaloba. Mol. Phylogenet. Evol. 2019, 138, 53–64. [Google Scholar] [CrossRef]
- Li, B.; Zheng, Y. Dynamic evolution and phylogenomic analysis of the chloroplast genome in Schisandraceae. Sci. Rep. 2018, 8, 9285. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Yu, W.B.; Tan, Y.; Liu, B.; Yao, X.; Jin, J.; Padmanaba, M.; Yang, J.B.; Corlett, R. Evolutionary comparisons of the chloroplast genome in Lauraceae and insights into loss events in the Magnoliids. Genome Biol. Evol. 2017, 9, 2354–2364. [Google Scholar] [CrossRef]
- Palmer, J.D.; Thompson, W.F.J.C. Chloroplast DNA rearrangements are more frequent when a large inverted repeat sequence is lost. Cell 1982, 29, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.W.; Xiang, K.L.; Erst, A.S.; Lian, L.; Ortiz, R.D.C.; Jabbour, F.; Chen, Z.D.; Wang, W. A complete genus-level phylogeny reveals the Cretaceous biogeographic diversification of the poppy family. Mol. Phylogenet. Evol. 2023, 181, 107712. [Google Scholar] [CrossRef]
- Jacomet, S. Plant economy and village life in Neolithic lake dwellings at the time of the Alpine Iceman. Veg. Hist. Archaeobot. 2009, 18, 47–59. [Google Scholar] [CrossRef]
- Chen, N.; Qi, Y.; Ma, X.; Xiao, X.; Liu, Q.; Xia, T.; Xiang, J.; Zeng, J.; Tang, J. Rediscovery of traditional plant medicine: An underestimated anticancer drug of chelerythrine. Front. Pharmacol. 2022, 13, 906301. [Google Scholar] [CrossRef]
- Inada, M.; Shindo, M.; Kobayashi, K.; Sato, A.; Yamamoto, Y.; Akasaki, Y.; Ichimura, K.; Tanuma, S.I. Anticancer effects of a non-narcotic opium alkaloid medicine, papaverine, in human glioblastoma cells. PLoS ONE 2019, 14, e0216358. [Google Scholar] [CrossRef] [PubMed]
- Kilic, M.; Sener, B.; Kaya, E. Anti-cholinergic activities of Turkish Corydalis DC. species. Phytochem. Lett. 2021, 45, 142–156. [Google Scholar] [CrossRef]
- Lou, G.; Wang, J.; Hu, J.; Gan, Q.; Peng, C.; Xiong, H.; Huang, Q. Sanguinarine: A double-edged sword of anticancer and carcinogenesis and its future application prospect. Anti-Cancer Agent. Med. Chem. 2021, 21, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Huang, R.; Hua, J.; Liang, H.; Pan, Y.; Dai, L.; Liang, D.; Wang, H. Antitumor lignanamides from the aerial parts of Corydalis saxicola. Phytomedicine 2016, 23, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Arreola, E.; Hernandez, L.; Sanchez-Salas, J.; Martínez-Espino, G. Alkaloids from Bocconia frutescens and biological activity of their extracts. Pharm. Biol. 2008, 44, 540–543. [Google Scholar] [CrossRef]
- Qin, F.; Chen, Y.; Wang, F.F.; Tang, S.Q.; Fang, Y.L. Corydalis saxicola Bunting: A review of its traditional uses, phytochemistry, pharmacology, and clinical applications. Int. J. Mol. Sci. 2023, 24, 1626. [Google Scholar] [CrossRef] [PubMed]
- Hoot, S.B.; Kadereit, J.W.; Blattner, F.R.; Jork, K.B.; Schwarzbach, A.E.; Crane, P.R. Data congruence and phylogeny of the Papaveraceae s.l. based on four data sets: atpB and rbcL sequences, trnK restriction sites, and morphological characters. Syst. Bot. 1997, 22, 575–590. [Google Scholar] [CrossRef]
- Wang, W.; Lu, A.M.; Ren, Y.; Endress, M.E.; Chen, Z.D. Phylogeny and classification of Ranunculales: Evidence from four molecular loci and morphological data. Perspect. Plant Ecol. Evol. Syst. 2009, 11, 81–110. [Google Scholar] [CrossRef]
- Hoot, S.; Wefferling, K.; Wulff, J. Phylogeny and character evolution of Papaveraceae s. l. (Ranunculales). Syst. Bot. 2015, 40, 474–488. [Google Scholar] [CrossRef]
- Park, S.; An, B.; Park, S. Reconfiguration of the plastid genome in Lamprocapnos spectabilis: IR boundary shifting, inversion, and intraspecific variation. Sci. Rep. 2018, 8, 13568. [Google Scholar] [CrossRef]
- Xu, X.; Wang, D. Comparative chloroplast genomics of Corydalis species (Papaveraceae): Evolutionary perspectives on their unusual large-scale rearrangements. Front. Plant Sci. 2021, 11, 600354. [Google Scholar] [CrossRef]
- Raman, G.; Nam, G.H.; Park, S. Extensive reorganization of the chloroplast genome of Corydalis platycarpa: A comparative analysis of their organization and evolution with other Corydalis plastomes. Front. Plant Sci. 2022, 13, 1043740. [Google Scholar] [CrossRef]
- Kim, S.C.; Ha, Y.H.; Park, B.K.; Jang, J.E.; Kang, E.S.; Kim, Y.S.; Kimspe, T.H.; Kim, H.J. Comparative analysis of the complete chloroplast genome of Papaveraceae to identify rearrangements within the Corydalis chloroplast genome. PLoS ONE 2023, 18, e0289625. [Google Scholar] [CrossRef]
- Wang, L.; Li, F.; Wang, N.; Gao, Y.; Liu, K.; Zhang, G.; Sun, J. Characterization of the Dicranostigma leptopodum chloroplast genome and comparative analysis within subfamily Papaveroideae. BMC Genom. 2022, 23, 794. [Google Scholar] [CrossRef]
- Sun, Y.; Moore, M.J.; Lin, N.; Adelalu, K.F.; Meng, A.; Jian, S.; Yang, L.; Li, J.; Wang, H. Complete plastome sequencing of both living species of Circaeasteraceae (Ranunculales) reveals unusual rearrangements and the loss of the ndh gene family. BMC Genom. 2017, 18, 592. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Luo, Y.; Pei, L.; Li, M.; Xiao, J.; Li, W.; Wu, H.; Luo, Y.; He, J.; Cheng, J.; et al. Complete plastid genomes of nine species of Ranunculeae (Ranunculaceae) and their phylogenetic inferences. Genes 2023, 14, 2140. [Google Scholar] [CrossRef] [PubMed]
- Szczecińska, M.; Sawicki, J. Genomic resources of three Pulsatilla species reveal evolutionary hotspots, species-specific sites and variable plastid structure in the family Ranunculaceae. Int. J. Mol. Sci. 2015, 16, 22258–22279. [Google Scholar] [CrossRef] [PubMed]
- Nyamgerel, N.; Baasanmunkh, S.; Oyuntsetseg, B.; Bayarmaa, G.A.; Erst, A.S.; Park, I.; Choi, H.J.J. Insight into chloroplast genome structural variation of the Mongolian endemic species Adonis mongolica (Ranunculaceae) in the Adonideae tribe. Sci. Rep. 2023, 13, 22014. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Yao, M.; Lyu, R.D.; Lin, L.L.; Liu, H.J.; Pei, L.Y.; Yan, S.X.; Xie, L.; Cheng, J. Structural variation of the complete chloroplast genome and plastid phylogenomics of the genus Asteropyrum (Ranunculaceae). Sci. Rep. 2019, 9, 15285. [Google Scholar] [CrossRef] [PubMed]
- Tong, R.; Gui, C.; Zhang, Y.; Su, N.; Hou, X.; Liu, M.; Yang, Z.; Kang, B.; Chang, Z.; Jabbour, F.; et al. Phylogenomics, plastome structure and species identification in Mahonia (Berberidaceae). BMC Genom. 2022, 23, 766. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.S.; Wang, Y.N.; Hsu, C.Y.; Lin, C.P.; Chaw, S.M. Loss of different inverted repeat copies from the chloroplast genomes of Pinaceae and cupressophytes and influence of heterotachy on the evaluation of gymnosperm phylogeny. Genome Biol. Evol. 2011, 3, 1284–1295. [Google Scholar] [CrossRef]
- Chen, J.T.; Lidén, M.; Huang, X.H.; Zhang, L.; Zhang, X.J.; Kuang, T.H.; Landis, J.B.; Wang, D.; Deng, T.; Sun, H. An updated classification for the hyper-diverse genus Corydalis (Papaveraceae: Fumarioideae) based on phylogenomic and morphological evidence. J. Integr. Plant Biol. 2023, 65, 2138–2156. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Cao, J.L.; Kan, S.L.; Wang, P.H.; Wang, J.L.; Cao, Y.N.; Wang, H.W.; Li, J.M. Phylogenomic analyses sheds new light on the phylogeny and diversification of Corydalis DC in Himalaya–Hengduan Mountains and adjacent regions. Mol. Phylogenet. Evol. 2024, in press. [Google Scholar] [CrossRef]
- Peng, L.; Yamamoto, H.; Shikanai, T. Structure and biogenesis of the chloroplast NAD(P)H dehydrogenase complex. Biochim. Biophys. Acta 2011, 1807, 945–953. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; dePamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef]
- Chris Blazier, J.; Guisinger, M.M.; Jansen, R.K. Recent loss of plastid-encoded ndh genes within Erodium (Geraniaceae). Plant Mol. Biol. 2011, 76, 263–272. [Google Scholar] [CrossRef]
- Guo, Y.Y.; Yang, J.X.; Bai, M.-Z.; Zhang, G.Q.; Liu, Z.J. The chloroplast genome evolution of Venus slipper (Paphiopedilum): IR expansion, SSC contraction, and highly rearranged SSC regions. BMC Plant Biol. 2021, 21, 248. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.S.; Wang, Y.N.; Liu, S.M.; Chaw, S.M. Chloroplast genome (cpDNA) of Cycas taitungensis and 56 cp protein-coding genes of Gnetum parvifolium: Insights into cpDNA evolution and phylogeny of extant seed plants. Mol. Biol. Evol. 2007, 24, 1366–1379. [Google Scholar] [CrossRef]
- Kim, H.T.; Kim, J.S.; Moore, M.J.; Neubig, K.M.; Williams, N.H.; Whitten, W.M.; Kim, J.H. Seven new complete plastome sequences reveal rampant independent loss of the ndh gene family across Orchids and associated instability of the inverted repeat/small single-copy region boundaries. PLoS ONE 2015, 10, e0142215. [Google Scholar] [CrossRef] [PubMed]
- Caroca, R.; Howell, K.A.; Malinova, I.; Burgos, A.; Tiller, N.; Pellizzer, T.; Annunziata, M.G.; Hasse, C.; Ruf, S.; Karcher, D.; et al. Knockdown of the plastid-encoded acetyl-CoA carboxylase gene uncovers functions in metabolism and development. Plant Physiol. 2021, 185, 1091–1110. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.; Stanne, T.; Sjögren, L. The ATP-dependent Clp protease in chloroplasts of higher plants. Physiol. Plant 2005, 123, 406–412. [Google Scholar] [CrossRef]
- Shikanai, T.; Shimizu, K.; Ueda, K.; Nishimura, Y.; Kuroiwa, T.; Hashimoto, T. The chloroplast clpP gene, encoding a proteolytic subunit of ATP-dependent protease, is indispensable for chloroplast development in tobacco. Plant Cell Physiol. 2001, 42, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Olinares, P.D.; Oh, S.H.; Ghisaura, S.; Poliakov, A.; Ponnala, L.; van Wijk, K.J. Modified Clp protease complex in the ClpP3 null mutant and consequences for chloroplast development and function in Arabidopsis. Plant Physiol. 2013, 162, 157–179. [Google Scholar] [CrossRef]
- Claude, S.J.; Park, S.; Park, S. Gene loss, genome rearrangement, and accelerated substitution rates in plastid genome of Hypericum ascyron (Hypericaceae). BMC Plant Biol. 2022, 22, 135. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.S.; Yun, B.K.; Yoon, Y.H.; Hong, S.Y.; Mekapogu, M.; Kim, K.H.; Yang, T.J. Complete chloroplast genome sequence of tartary buckwheat (Fagopyrum tataricum) and comparative analysis with common buckwheat (F. esculentum). PLoS ONE 2015, 10, e0125332. [Google Scholar] [CrossRef] [PubMed]
- Yamane, K.; Yasui, Y.; Ohnishi, O. Intraspecific cpDNA variations of diploid and tetraploid perennial buckwheat, Fagopyrum cymosum (Polygonaceae). Am. J. Bot. 2003, 90, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Erixon, P.; Oxelman, B. Whole-gene positive selection, elevated synonymous substitution rates, duplication, and indel evolution of the chloroplast clpP1 gene. PLoS ONE 2008, 3, e1386. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Jun, M.; Park, S.; Park, S. Lineage-specific variation in IR boundary shift events, inversions, and substitution rates among Caprifoliaceae s.l. (Dipsacales) plastomes. Int. J. Mol. Sci. 2021, 22, 10485. [Google Scholar] [CrossRef]
- Rogers, S.O.; Bendich, A.J. Extraction of DNA from milligram amounts of fresh, herbarium and mummified plant tissues. Plant Mol. Biol. 1985, 5, 69–76. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 2017, 7, 1–6. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2014. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 December 2023).
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Qu, X.J.; Moore, M.J.; Li, D.Z.; Yi, T.S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Guo, Q.; Xu, L.; Gao, H.; Liu, L.; Zhou, X. CPJSdraw: Analysis and visualization of junction sites of chloroplast genomes. PeerJ 2023, 11, e15326. [Google Scholar] [CrossRef]
- Grantham, R. Working of the genetic code. Trends Biochem. Sci. 1980, 5, 327–331. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- IBM Corp. IBM SPSS Statistics for Windows, Version 22.0; IBMCorp: Armonk, NY, USA, 2013. [Google Scholar]
- Akoglu, H. User’s guide to correlation coefficients. Turkish J. Emerg. Med. 2018, 18, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Subfamily | Genome Size (bp) | LSC (bp) | IR (bp) | SSC (bp) | GC (bp) | PCG | tRNA | rRNA |
|---|---|---|---|---|---|---|---|---|---|
| Corydalis triternatifolia | Fumarioideae | 161,946 | 84,522 | 38,486 | 452 | 38.6 | 85 | 37 | 8 |
| Corydalis sheareri | Fumarioideae | 219,144 | 94,030 | 62,384 | 346 | 40.4 | 99 | 44 | 8 |
| Corydalis gamosepala | Fumarioideae | 198,140 | 92,169 | 52,590 | 791 | 40.5 | 95 | 40 | 8 |
| Corydalis longicalcarata | Fumarioideae | 195,842 | 86,954 | 54,197 | 494 | 40.3 | 96 | 38 | 8 |
| Corydalis tomentella | Fumarioideae | 189,383 | 95,825 | 41,947 | 9664 | 40.2 | 94 | 38 | 8 |
| Corydalis adunca | Fumarioideae | 186,049 | 100,365 | 38,100 | 9484 | 41.1 | 92 | 36 | 8 |
| Fumaria schleicheri | Fumarioideae | 190,324 | 86,789 | 48,833 | 5869 | 40.3 | 97 | 40 | 8 |
| Dactylicapnos torulosa | Fumarioideae | 160,598 | 76,421 | 26,238 | 31,701 | 40.3 | 96 | 40 | 8 |
| Ichtyoselmis macrantha | Fumarioideae | 164,552 | 88,457 | 27,031 | 22,033 | 39.9 | 89 | 37 | 8 |
| Lamprocapnos spectabilis | Fumarioideae | 188,795 | 84,398 | 51,376 | 1645 | 39.2 | 100 | 42 | 8 |
| Hypecoum erectum | Hypecoideae | 163,471 | 93,182 | 27,885 | 14,519 | 38.2 | 86 | 38 | 8 |
| Hylomecon japonica | Papaveroideae | 160,071 | 88,293 | 26,712 | 18,354 | 38.8 | 88 | 37 | 8 |
| Coreanomecon hylomeconoides | Papaveroideae | 158,824 | 86,914 | 26,686 | 18,538 | 38.7 | 89 | 37 | 8 |
| Chelidonium majus | Papaveroideae | 159,734 | 87,696 | 26,735 | 18,568 | 38.7 | 89 | 37 | 8 |
| Dicranostigma leptopodum | Papaveroideae | 163,173 | 87,793 | 28,309 | 18,762 | 39.4 | 91 | 37 | 8 |
| Macleaya cordata | Papaveroideae | 163,107 | 87,919 | 28,303 | 18,582 | 38.6 | 91 | 37 | 8 |
| Eomecon chionantha | Papaveroideae | 178,768 | 88,302 | 45,165 | 136 | 37.9 | 99 | 38 | 8 |
| Sanguinaria canadensis | Papaveroideae | 161,083 | 87,980 | 27,372 | 18,359 | 38.5 | 87 | 37 | 8 |
| Stylophorum lasiocarpum | Papaveroideae | 153,196 | 83,230 | 25,789 | 18,388 | 38.9 | 89 | 37 | 8 |
| Papaver nudicaule | Papaveroideae | 152,867 | 83,104 | 25,731 | 18,301 | 38.9 | 85 | 37 | 8 |
| Meconopsis integrifolia | Papaveroideae | 151,864 | 82,809 | 25,653 | 17,749 | 38.8 | 87 | 37 | 8 |
| Eschscholzia californica | Papaveroideae | 160,201 | 88,147 | 26,781 | 18,492 | 38.7 | 88 | 37 | 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, J.; Wang, H.; Cao, Y.; Kan, S.; Li, J.; Liu, Y. Extreme Reconfiguration of Plastid Genomes in Papaveraceae: Rearrangements, Gene Loss, Pseudogenization, IR Expansion, and Repeats. Int. J. Mol. Sci. 2024, 25, 2278. https://doi.org/10.3390/ijms25042278
Cao J, Wang H, Cao Y, Kan S, Li J, Liu Y. Extreme Reconfiguration of Plastid Genomes in Papaveraceae: Rearrangements, Gene Loss, Pseudogenization, IR Expansion, and Repeats. International Journal of Molecular Sciences. 2024; 25(4):2278. https://doi.org/10.3390/ijms25042278
Chicago/Turabian StyleCao, Jialiang, Hongwei Wang, Yanan Cao, Shenglong Kan, Jiamei Li, and Yanyan Liu. 2024. "Extreme Reconfiguration of Plastid Genomes in Papaveraceae: Rearrangements, Gene Loss, Pseudogenization, IR Expansion, and Repeats" International Journal of Molecular Sciences 25, no. 4: 2278. https://doi.org/10.3390/ijms25042278
APA StyleCao, J., Wang, H., Cao, Y., Kan, S., Li, J., & Liu, Y. (2024). Extreme Reconfiguration of Plastid Genomes in Papaveraceae: Rearrangements, Gene Loss, Pseudogenization, IR Expansion, and Repeats. International Journal of Molecular Sciences, 25(4), 2278. https://doi.org/10.3390/ijms25042278