1. Introduction
Cutaneous melanoma (CM) is a malignant melanocytic tumor, representing less than 5% of all skin cancers. Despite its low prevalence, it is the most severe skin neoplasia, accounting for 80% of skin cancer-associated deaths, especially among the young adult population [
1]. In Europe and the USA, the incidence of CM has increased four-fold in the last 30 years [
2]. Established CM risk factors include ultraviolet (UV) radiation exposure, phenotypic traits with a solid genetic component, such as fair skin complexion and red or blonde hair, and an increased number of common and atypical melanocytic nevi, alongside a positive family history for melanoma [
3]. All these conditions subside to chronic inflammation events that favor tumorigenesis [
4]. Death from CM is mainly due to distant metastasis or drug resistance [
5]. The 5-year relative survival rate for advanced CM is 25% and falls to 10% surviving at ten years [
6,
7]. Notably, more than 95% of CM cases can be successfully treated if they are discovered in the early stages, suggesting that it is critical to understand this disease’s biology to improve its clinical management [
7]. CM arises from the neoplastic transformation of neural crest stem cell-derived melanocytes [
8]. Recent studies evaluating the mutational status of human melanomas have revealed an increased mutation load associated with the molecular signatures of UV damage [
9,
10,
11]. In line with these observations, the latest World Health Organization (WHO) classification of skin tumors highlights the existence of two major melanoma subtypes—the low cumulative sun damage (low CSD) subtype and the high cumulative sun damage (high CSD) subtype, emphasizing solar exposure as the main pro-inflammatory carcinogen involved in CM formation [
12]. Melanomas that arise against a background of low-CSD typically harbor
BRAF V600E mutations (and rarely
NRAS Q61 mutations) and originate from benign nevi. Most of these melanomas occur on the trunk and extremities of adults between the ages of 20 and 60 [
13]. The conversion of
BRAF V600E positive nevi to full-blown melanomas is achieved through a plethora of genetic and epigenetic alterations [
14,
15]. Additional mutations, such as in cyclin-dependent kinase inhibitor 2A (
CDKN2A) or the promoter of the human telomerase reverse transcriptase (
hTERT) and chromosomal copy number aberrations have been frequently reported in low-CSD melanoma [
16]. By contrast, high-CSD melanomas do not harbor
BRAF V600E mutations but present other mitogen-activated protein kinase (
MAPK) pathway mutations, such as
BRAF V600K,
NRAS G12/G13, or
KIT mutations, or inactivation of the negative regulators of
Ras, such as
NF1 or
RASA2. Melanoma in situ is considered the main precursor lesion of high-CSD tumors, as pre-existing nevi are not commonly observed in those tumors. Finally, high-CSD melanomas encompass the lentigo maligna and desmoplastic melanoma subtypes, whereas superficial spreading melanomas mainly represent low-CSD tumors [
13]. The 2018 WHO classification of melanomas intends to incorporate all the causal mechanisms and clinical/histopathological parameters of cutaneous tumors to improve the diagnosis and surveillance in the clinical setting [
17].
The genomic profiling of melanoma tumors revealed several recurrent mutations involved in their pathogenesis and evolution, such as
BRAF,
NRAS, and
KIT, contributing to these tumors’ genomic subtyping [
18]. At the molecular level, it is supposed that somatic mutations in
NRAS and
BRAF, which occur mutually exclusive, are critical in this multistep development of melanoma. These mutations cause the constitutive activation of the serine-threonine kinases in the
ERK–MAPK pathway, which results in augmented tumor proliferation and growth [
19]. The assessment of
BRAF mutational status is critical, as it indicates those tumors amenable to Food and Drug Administration (FDA)-approved targeted therapies, such as BRAF inhibitors (BRAFi: vemurafenib, dabrafenib) and MEK inhibitors (MEKi: trametinib, cobimetinib) [
20]. However, the exact role of
BRAF mutations in the initiation or progression of melanoma is still controversial. The
BRAF gene encodes a protein that plays a pivotal role in
MAPK pathway activation, contributing to cellular growth, differentiation, survival, and proliferation [
21].
BRAF mutations are usually identified in the codon 600 of the
BRAF gene and are reported in 40–60% of melanoma cases [
22,
23,
24,
25]. The
BRAF V600E mutation, which involves the substitution of glutamic acid with valine at codon 600 (
BRAF V600E: nucleotides 1799 T > A; codon GTG > GAG), is the most frequent mutation reported in CM. This mutation is present in over 90% of BRAF-positive melanomas [
26]. Less common modifications include substitutions of valine for lysine (V600K), arginine (V600R), leucine (V600M), or aspartic acid (V600D) [
22]. Many clinical studies have highlighted that the
BRAF V600E mutation is usually identified in younger patients and depicted in tumors on skin subjected to intermittent sun exposure, such as the upper limbs, trunk and, less frequently, the head and neck. Wild-type melanoma tumors are typically found on the lower limbs [
27,
28]. Nonetheless, melanomas with
BRAF V600 mutations respond well to current FDA-approved BRAFi, as well as combined BRAF/MEK inhibitor therapy [
29]. Moreover, the ratio of mutant/wild-type alleles of
BRAF was reported to be a potential biomarker for prognosis in this hard-to-treat disease, as an increased ratio was associated with superior clinical outcomes and reasonable response to BRAFi therapy [
30].
The
RAS gene family includes
KRAS,
NRAS, and
HRAS proto-oncogenes, which encode several small GTPases involved in cell growth and proliferation. Mutated
NRAS is the second most common genetic alteration in CM, reported in 15–20% of cases. Hotspot mutations in the
NRAS gene occur mainly in exon 2 (codons 12 and 13) and exon 3 (codon 61) [
31]. Current evidence reveals prominent
PI3K/AKT signaling in
NRAS G12 mutant cells and augmented
MAPK signaling in
NRAS Q61 variants, suggesting that
NRAS G12 and
NRAS Q61-mutant melanoma tumors differ in biology. Therefore,
NRAS-mutant tumors might be approached with more specific and effective therapeutic strategies in the future [
32]. Moreover, compared to
BRAF-mutant tumors,
NRAS-mutant melanomas tend to be thicker, with increased mitotic rates, usually occurring in older patients (>55 years) with a history of chronic UV exposure [
33,
34,
35,
36]. Several recent studies have also shown that
NRAS mutations might result in inferior clinical outcomes with a lower survival rate [
37]. Despite the general opinion, which assumes that
BRAF mutations cannot coexist with
NRAS mutations in melanocytic tumors, several research groups have also reported their synchronous occurrence [
24,
38]. Finally,
KRAS is the rarest mutated proto-oncogene in melanoma (~2% of cases).
KRAS genetic alterations also imply the alteration of the G12, G13, and Q61 residues of the protein [
33,
39]. It is also worth mentioning that
KRAS mutations have been recently linked with an increased propensity of melanoma tumors to metastasize to the brain and, therefore, envisioned as valuable biomarkers of patient risk stratification in clinical settings [
40].
The activation of the RAS signaling pathway by
EGFR in cancer is well known [
41]; however, in CM, there is limited information available on the role of this tyrosine kinase receptor (TKR), apart from its expression in melanocytic nevi and some melanomas [
42,
43,
44,
45]. CM is a heterogeneous disease, and its treatment is challenging; therefore, new therapeutic options are needed. As targeted anti-
EGFR therapy has shown promising results in decreasing melanoma cell growth and hindering its invasive abilities [
46], we examined melanomas and their precursors for mutations that lead to the constitutive activation of kinase activity, such as exon 19 deletions (Del19) [
41]. Studies of melanoma cell lines that express
BRAF mutation indicate a mechanism related to
EGFR overexpression and activation of the
MAPK and
PI3K-AKT pathways after drug administration [
47]. Moreover, melanoma cell lines with higher
EGFR expression are more prone to becoming resistant to vemurafenib than cells with lower
EGFR expression [
48]. Additionally, melanomas treated with BRAF inhibitors tend to accumulate mutations in
NRAS or
MEK1/2, exhibiting excessive activation of
ERK1/2 or
SFK-STAT3 [
49]. Therefore, tumor cells may again be sensitized to vemurafenib in the therapeutical approach if the drug is administered simultaneously with EGFR and AKT inhibitors [
41]. Since there are very few studies investigating the mutations of this pathway in solid tumors (only 9 out of a total of 32 in the last five years), we have embarked on a pilot study on skin melanomas to assess the
RAS-RAF-MAPK pathway mutations. Therefore, in the present study, we evaluated the
RAS,
BRAF, and
EGFR mutational status of tissue specimens representing normal skin, benign nevi, and cutaneous melanomas to determine the genetic landscape of pre-malignant and malignant stages of melanoma development and to investigate the extent to which these mutational signatures overlap.
3. Discussion
Studies performed on individual healthy-skin melanocytes show that, as expected, sun-shielded melanocytes had fewer mutations related to UV signatures than sun-exposed melanocytes. In addition, melanocytes from chronically sun-exposed areas (e.g., the face, neck, and bald scalp) displayed a lower mutational burden than melanocytes on intermittently exposed skin (e.g., the back and the limbs) [
12]. This assertion mirrors the clinical observations that, compared to other forms of skin cancer, melanomas are more common in intermittently exposed skin than in chronically exposed skin [
54,
55,
56].
UVR induces erythema, edema, and dermal vasodilation, a clear portrait of inflammation events. As sunburn is a transient inflammatory response, chronic UVR contact activates prolonged inflammatory factors. Chromophores can absorb certain wavelength ranges that induce inflammation. The main chromophores for UVR in the human skin are nucleic acids, urocanic acid, amino acids with aromatic structures, and melanin [
57]. The action of UVR on these chromophores initiates events that trigger inflammatory and immunological processes that favor skin tumorigenesis [
58]. Furthermore, melanocytes located in the vicinity of a skin cancer were reported as having a similar mutational burden to melanomas, but a significantly higher mutational burden compared to melanocytes from donors without skin cancer, suggesting that the mutation burden of normal skin may be exploited as an indicator of UV-related DNA damage and skin cancer predisposition [
12]. The genes most frequently mutated in healthy melanocytes encode for suppressors of the
MAPK pathway, such as
NF1,
CBL, and
RASA2, but gain-of-function alterations in the
BRAF,
NRAS, and
MAP2K1 genes have also been reported [
12]. In line with these findings, we identified
NRAS G12/G13 mutations in a healthy skin specimen included in our study. In particular, for
NRAS G12/G13 mutations, the current literature highlights that they tend to be weakly oncogenic, giving rise to noticeable lesions only in conjunction with additional oncogenic mutations [
53,
59].
Melanomas often arise from typical precursor lesions, such as benign nevi, dysplastic lesions, or melanoma in situ, which allows for tracking their evolution [
60,
61,
62]. Somatic mutations in key melanoma oncogenes, such as
BRAF or
NRAS, are already present in benign nevi, indicating that they occur early during disease progression. However, little is known about the additional mutations that drive the neoplastic transformation of benign nevi and their sequential order [
63]. Interestingly, in our nevi samples, the majority have shown
BRAF mutations, and some of them also
NRAS, confirming previous studies. But although nevi are common, and they have such a high rate of mutations, they stop growing on their own [
64]. The arrested growth of nevi was shown to be the trigger for oncogene-induced senescence as an intrinsic cellular response hindering proliferation [
65]. However, as not all nevi have senescence markers, and some nevi can take up growing after decades or even transform into skin melanomas, a hypothesis has developed that there are also external signals that hinder nevi proliferation [
66,
67].
CDKN2A is a gene located at chromosome 9, band p21.3, and encodes p16INK4a and p14arf proteins [
68], p16 being an important negative cell cycle regulator [
69]. P16 is critical for melanocyte’s senescence, hindering tumorigenesis toward melanoma [
70]. As we previously published, nevi have a high expression of p16 [
71]. In our current work, the evaluated nevi had a high mutational burden but doubled by a high p16 expression, proving that the cellular cycle is arrested in the nevi.
According to the Clark model, which describes the histopathological changes occurring during normal melanocytes’ linear progression to melanoma via a benign naevus,
BRAF mutations have a crucial role in melanoma development and progression [
72].
BRAF mutations were first described in 2003 when Pollock and colleagues discovered them in 80% of their batch of nevi samples [
73]. Further studies aiming to assess the mutational rate of
BRAF in acquired melanocytic lesions have demonstrated similar prevalence rates; however,
NRAS mutations were identified at a much lower percentage (6%) in benign melanocytic nevi [
74]. Moreover, Shain et al. reported that
NRAS and
BRAF mutations may have a strict specificity regarding the type of nevi in which they occur, with
BRAF V600E mutations being more frequent in benign lesions compared to
NRAS mutations, which occur predominantly in intermediate precursor lesions, such as dysplastic nevi [
75]. Despite the claimed oncogenic role of
BRAF mutations, the mutant melanocytes within a benign nevus lose their proliferative activity, as the entire benign entity comprising them stops growing and temporarily stabilizes in size. These observations are supported by the fact that
BRAF V600E mutations can also be present in normal human melanocytes, triggering cell cycle arrest and p16INK4a overexpression, features also reported in early-stage melanomas [
70,
76]. Thus, acquired melanocytic nevi are likely benign clonal tumors that are initially stimulated to proliferate via oncogenic
BRAF signaling but which finally undergo growth arrest due to oncogene-induced senescence [
74]. It is unanimously accepted that
BRAF gene mutations are insufficient to drive the malignant transformation of normal melanocytes; thus, more research regarding the factors involved in this process is needed [
16,
77]. Finally, the almost perfect overlap of the
BRAF mutational profile between melanomas and their benign counterparts, together with the existence of some
BRAF wild-type melanomas originating from
BRAF-mutant nevi, suggests that melanomagenesis is a multifaceted process that involves numerous factors, many of them not yet elucidated [
78]. Here, we report in 85.7% of the cases the presence of
BRAF V600E mutations for the analyzed benign nevi. Moreover, we highlight that
NRAS G12/G13 mutations may also be present but at very low allelic frequencies in acquired melanocytic nevi. Usually, there is no overlap regarding mutations in both
NRAS and
BRAF in nevi or CM [
16], but surprisingly, we reported several cases of simultaneous mutations in both of those two genes. We consider that the detection of
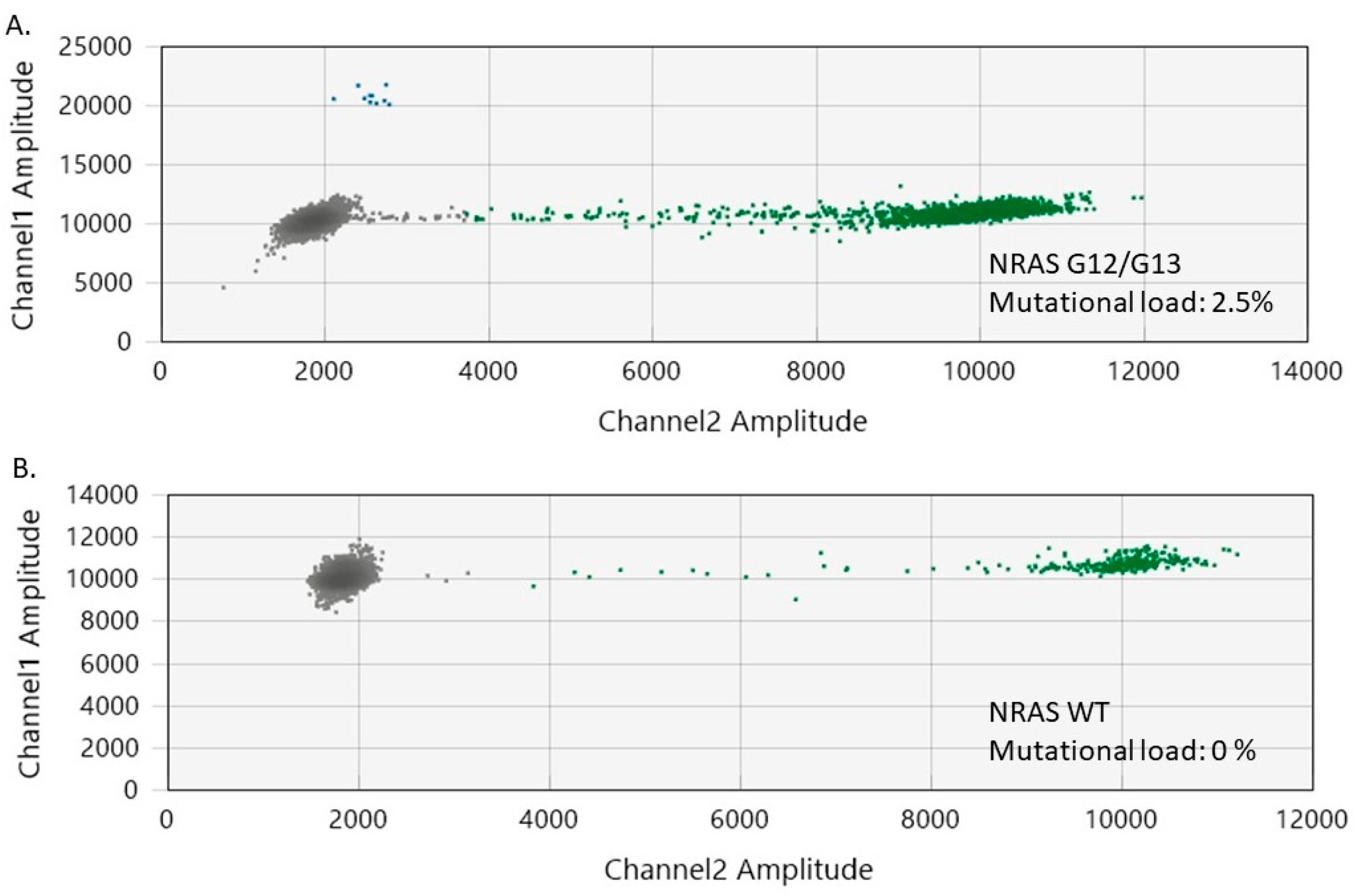
NRAS G12/G13 mutations was enabled by the ultrasensitive power of ddPCR, which has a limit of detection (LoD) of 0.005%. The unique
NRAS mutation obtained in the two cases of benign nevi sustains the fact that the investigated nevi were congenital. However, the precise role of
NRAS G12/G13 mutations in benign nevi is not yet well defined and remains to be explored in further studies. At least in our batch of investigated nevi, the
NRAS mutations accompanying
BRAF mutations were not associated with the nevi location, highly exposed versus non-exposed skin areas, as were detected on the scalp or trunk.
As mentioned, the most frequently mutated genes in cutaneous melanoma are
BRAF and
NRAS. The
BRAF V600 mutation has been linked to younger age at diagnosis, lack of chronic UV damage, a high total body nevus count, and localization of the primary tumor on the body extremities [
25,
26]. Some studies also suggest an association with the male gender [
79] or any gender association [
80]. The frequency of
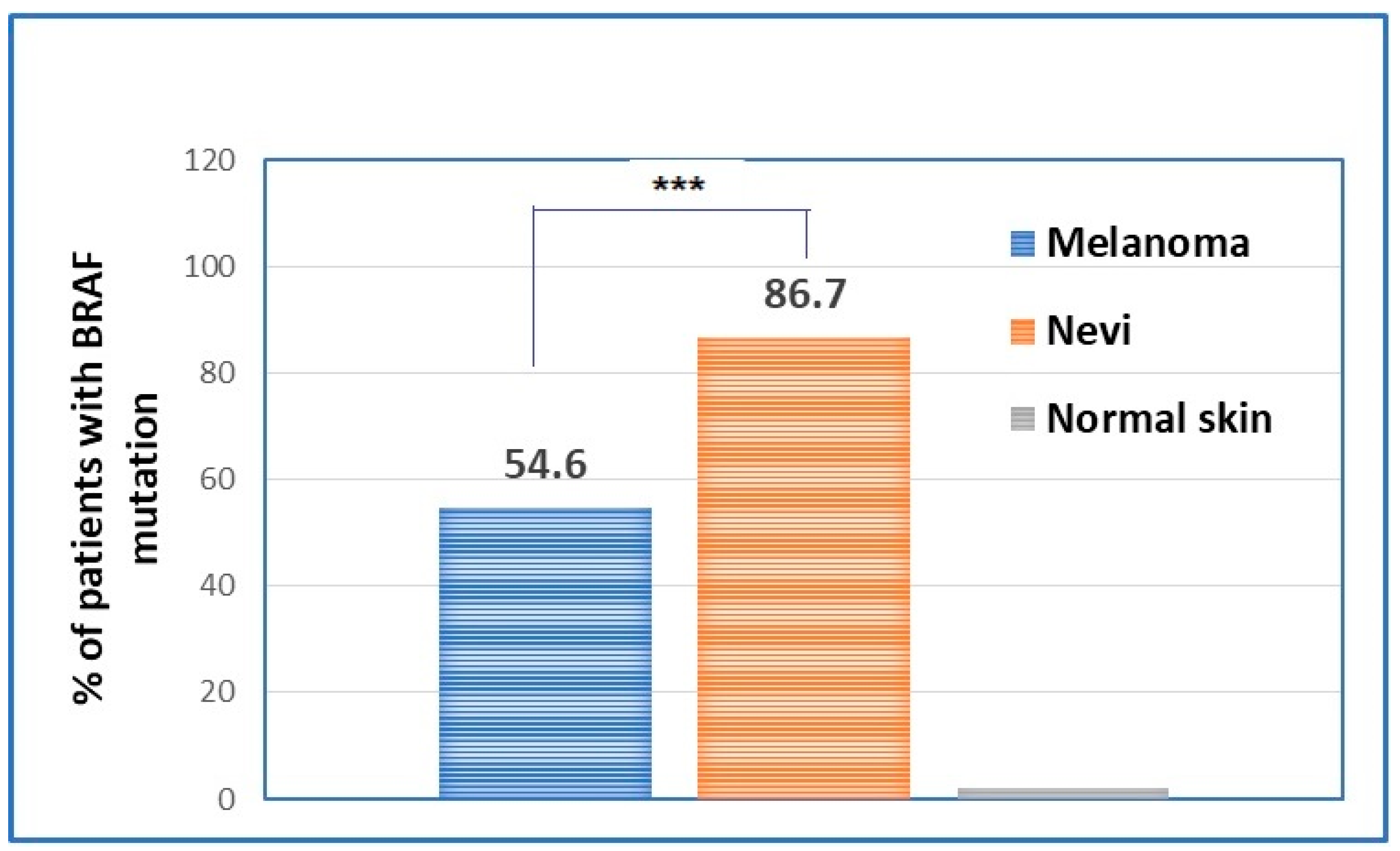
BRAF mutations in primary melanomas is commonly reported to be between 40 and 60% [
21,
22], with 54.6% seen in our study. Regarding tumor characteristics, at least three studies demonstrated an association with tumor thickness [
24,
36]. Furthermore, other investigations suggest a correlation between
BRAF mutation and aggressive tumor features, such as increased mitotic rates [
81] and the presence of ulceration [
82,
83]. However, our study found no associations between the patient age, gender, Clark level, mitotic index, tumor localization, histological type, ulceration, and the
BRAF V600 mutational status. The only significant associations were with Breslow thickness (
p = 0.029) and the presence of TILs (
p = 0.027). Thus, our study highlights a dual role of
BRAF V600 mutations in cutaneous melanoma: one related to sustaining tumor growth and another linked to the modulation of the immune response.
In accordance with previous evidence [
84,
85] and our findings, a potential correlation is emerging between the presence of TILs and
BRAF mutation in CM. The current literature also highlights that half of the primary
BRAF-mutated melanomas exhibit the non-brisk phenotype, while approximately 30% fall within the TIL brisk group [
86]. However, while the clinical significance of TILs in
BRAF-mutant melanomas remains largely unknown, the immunomodulatory effects linked to
BRAF-targeted therapy–leading to increased expression of melanoma antigens and a more favorable tumor microenvironment—indicate that this feature may hold particular importance in the therapeutic setting [
87]. Therefore, adopting TIL assessment in routine histopathological practice may be helpful for melanoma risk stratification and therapeutic guidance.
Mutated
NRAS is the second most frequent genetic alteration in CM, occurring in 15% to 20% of cases [
31]. In our study, the frequency of
NRAS mutations was higher than previously reported (50%), and we attribute this to the ultrasensitive power of the ddPCR technology. It has been demonstrated that patients with
NRAS mutant melanoma were older than 55 years and had a previous history of UV exposure, and thus, higher pro-inflammatory conditions than those with
BRAF mutant tumors [
31].
NRAS-mutant melanomas are typically located on the upper extremities [
37]. In addition,
NRAS mutations were also found to be associated with the nodular subtype of the primary tumor [
13] and with unfavorable prognostic factors, such as increased Breslow thickness, mitotic activity, and lymphovascular invasion [
35,
36,
73].
In our small batch of melanoma samples,
NRAS mutational status exhibited correlations with two of the most important histological predictors of clinical outcome: Breslow thickness (
p = 0.01) and the mitotic index (
p = 0.04). We observed no associations between
NRAS mutations and the patient age, gender, Clark level of invasion, tumor site, histological type, ulceration, or presence of TILs. Therefore, our study highlights the potential involvement of
NRAS mutations in modulating the aggressiveness of CM. These findings align with previous studies in the field [
24,
33,
36] and underscore the importance of
NRAS mutation molecular testing in clinical practice. Such testing may offer insights into tumor behavior and significantly contribute to patients’ risk stratification.
NRAS mutations are known to occur independently of
BRAF mutations in melanocytic tumors; however, recent research using advanced technologies revealed that these two mutations may co-exist in almost one-third of CM cases. Patients with both
BRAF and
NRAS mutations have a poorer prognosis compared to those bearing either
NRAS wild-type or
BRAF-mutant melanoma [
37]. Our study confirms the co-occurrence of
BRAF and
NRAS mutations in melanoma and emphasizes the metastatic potential of a melanoma tumor that displays both of the mentioned mutations. Moreover, we show the co-occurrence of
BRAF and
NRAS mutations for the first time even in acquired melanocytic nevi.
At the same time, we did not find any
EGFR exon 19 (Del19 EGFR) deletions or
KRAS Q61 mutations in the healthy skin, benign nevi, or melanoma FFPE specimens.
EGFR exon19 deletion characterizes non-small cell lung cancer and confers sensitivity to EGFR tyrosine kinase inhibitors (TKIs) [
88]. No reports were found regarding this deletion in melanomas. Few reports show
KRAS Q61 mutations in UV-inducible melanoma models [
89]. Using the same ddPCR technology (Bio-Rad Laboratories, Hercules, CA, USA), an
NRAS Q61 mutation was found in malignant peripheral nerve sheath tumors that were further identified as melanoma [
90].
However, it is still unknown for CM if the oncogenic BRAF gene is capable of maintaining oncogenic MAPK kinase signaling on its own.
The intensely debated role of
BRAF mutations in benign nevi outlines two distinct hypotheses regarding their functionality in benign and pre-malignant settings. Despite the notable changes reported in melanocytic nevi after exposure to BRAF inhibitor treatment, a significant percentage of nevi do not regress when exposed to targeted therapies such as vemurafenib, although they are underpinned by oncogenic
BRAF V600E mutations [
91]. This lack of sensitivity to therapy suggests that within the melanocytic nevi genome, there may be an internal inhibitor that attenuates the pro-oncogenic activity associated with
MAPK signaling, preventing nevi from behaving like a malignant tumor and regressing under targeted therapy (the tumor-suppressor hypothesis). In parallel, another hypothesis (the second oncogene hypothesis) claims that not the
BRAF gene but several cofactors that acquire oncogenic mutations are the key players that facilitate the neoplastic transformation of melanocytic nevi into CM [
91]. Therefore, additional studies are needed to unravel the genomic landscape of healthy skin and benign melanocytic nevi and determine the key genetic alterations triggering melanocyte neoplastic transformation.
Using The Cancer Genome Atlas (TCGA) for melanoma, it was shown that in all stages, there are genes significantly related to
MAPK, neurotrophin, the focal adhesion signaling pathway, and, moreover, immune and inflammation pathways. Recently, a six-lncRNA prognostic signature that can stratify risk patients was reported in relation to these genes [
92]. Although
MAPK pathway mutations can induce the proliferation of melanocytes within a nevus, it is obvious that not all nevi (bearing
BRAF mutations) will turn to melanoma. Thus, our results come in favor of the oncogene-induced senescence hypothesis [
64], as oncogene activation induces growth arrest.
As a limitation, our study has a low number of cases. We compensated for this by evaluating a high number of genetic parameters performing a thorough IHC assessment, with the results confirmed by recent publications as well. ddPCR technology has a high sensitivity [
93] and can detect lower levels of mutations in the FFPE tissue samples, therefore recommending this technology for further extended studies. Another limitation of our study is that mutational screening is performed on a diverse collection of skin cells, which generates an average signal of the tissue sample instead of depicting the mutational landscape of individual melanocytes. Moreover, although we report the coexistence of
BRAF and
NRAS mutations within the same tumor, this does not necessarily imply these mutations can co-occur within single cells, as each tumor cell has its transcriptional repertoire that dictates its fate. Thus, although the present study offers a comprehensive overview of the mutational landscape of healthy skin, nevi, and cutaneous melanomas, the most accurate information regarding the molecular events that lead to melanoma can be obtained through single-cell sequencing. While single-cell sequencing technologies are expensive and require sophisticated technology and trained personnel, they have the potential to provide a more exhaustive picture of CM intratumoral heterogeneity and the associated transcriptional programs that modulate disease progression, drug sensitivity, or tolerance, holding promise for more personalized approaches in the clinical setting.
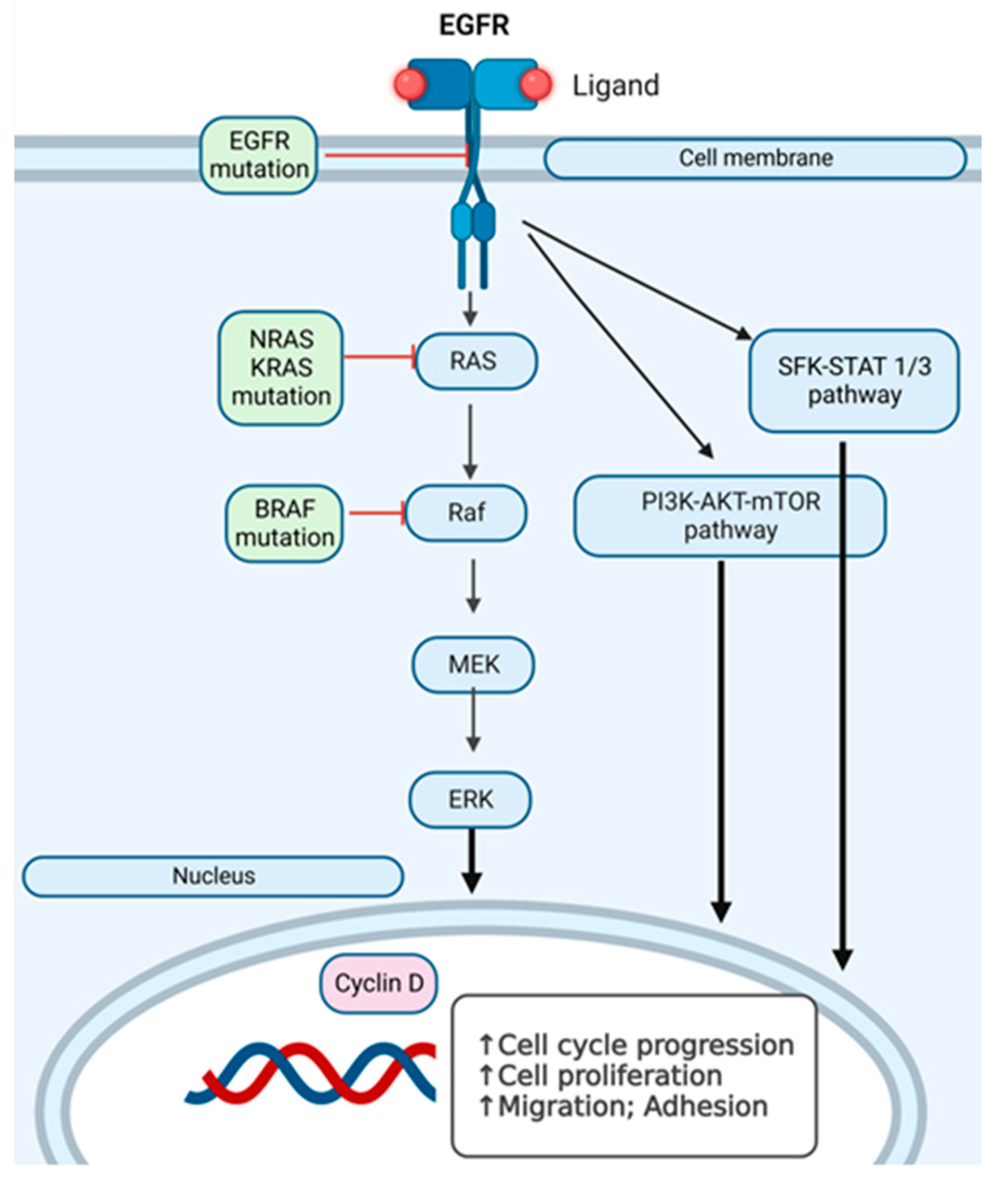
We have investigated
EGFR and
RAF-RAS-MAPK pathway mutations in the search for additional hit targets for further therapeutical approaches that can overcome immunotherapy and/or targeted therapy resistance, with the aim of personalizing the therapy. In
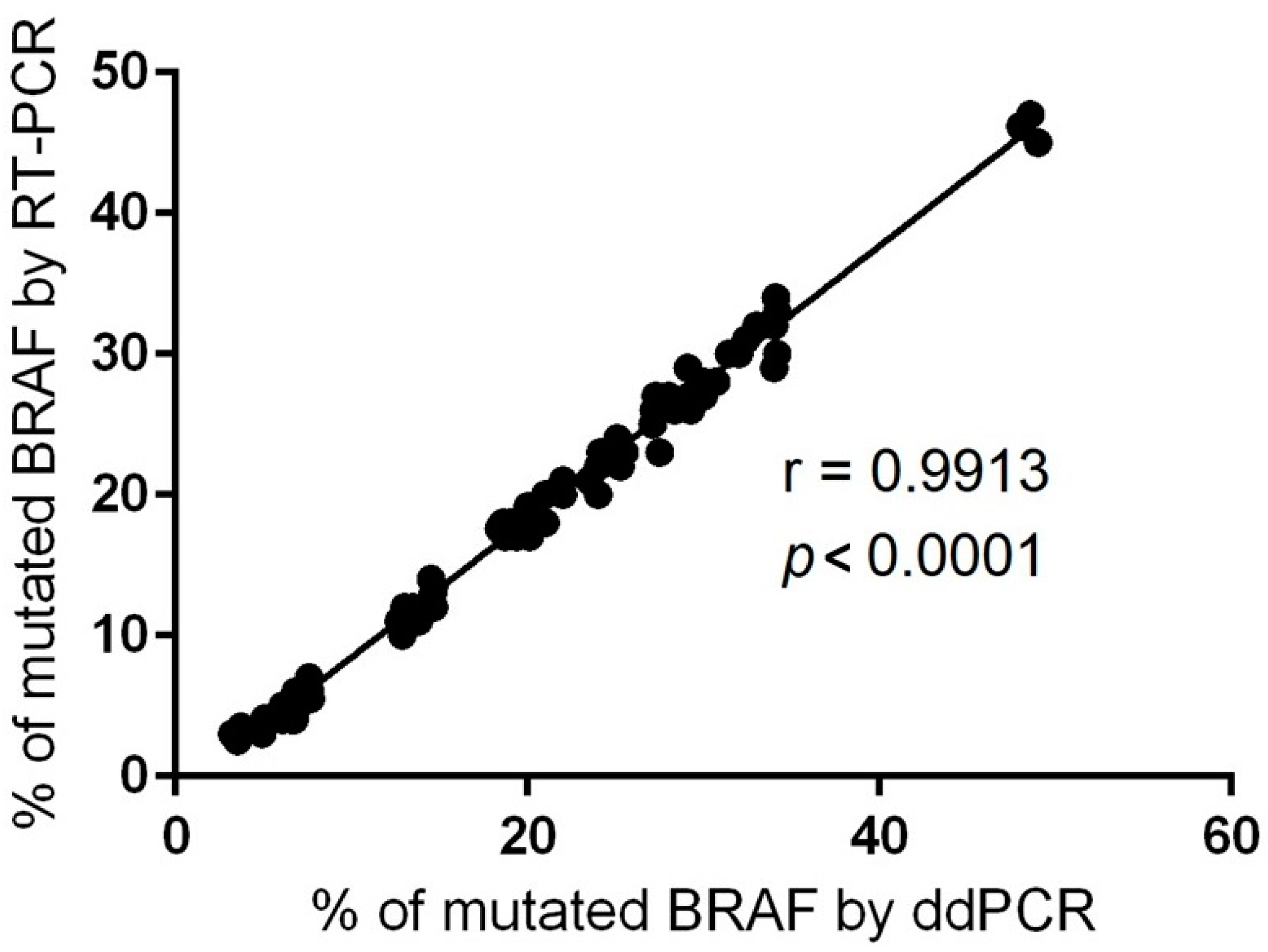
Figure 7, we present an overview of the pathways we have investigated at the mutational level in skin melanomas, nevi, and healthy skin.
Future perspectives of this complex genetic evaluation that are still in the research stages can drive new therapeutical approaches for CM, such as one-time autologous TIL cell therapy [
94], extracellular vesicles as drug transporters [
95], or new adjuvants like peptides [
96,
97].


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}