
Generating Potential RET-Specific Inhibitors Using a Novel LSTM Encoder–Decoder Model


Abstract
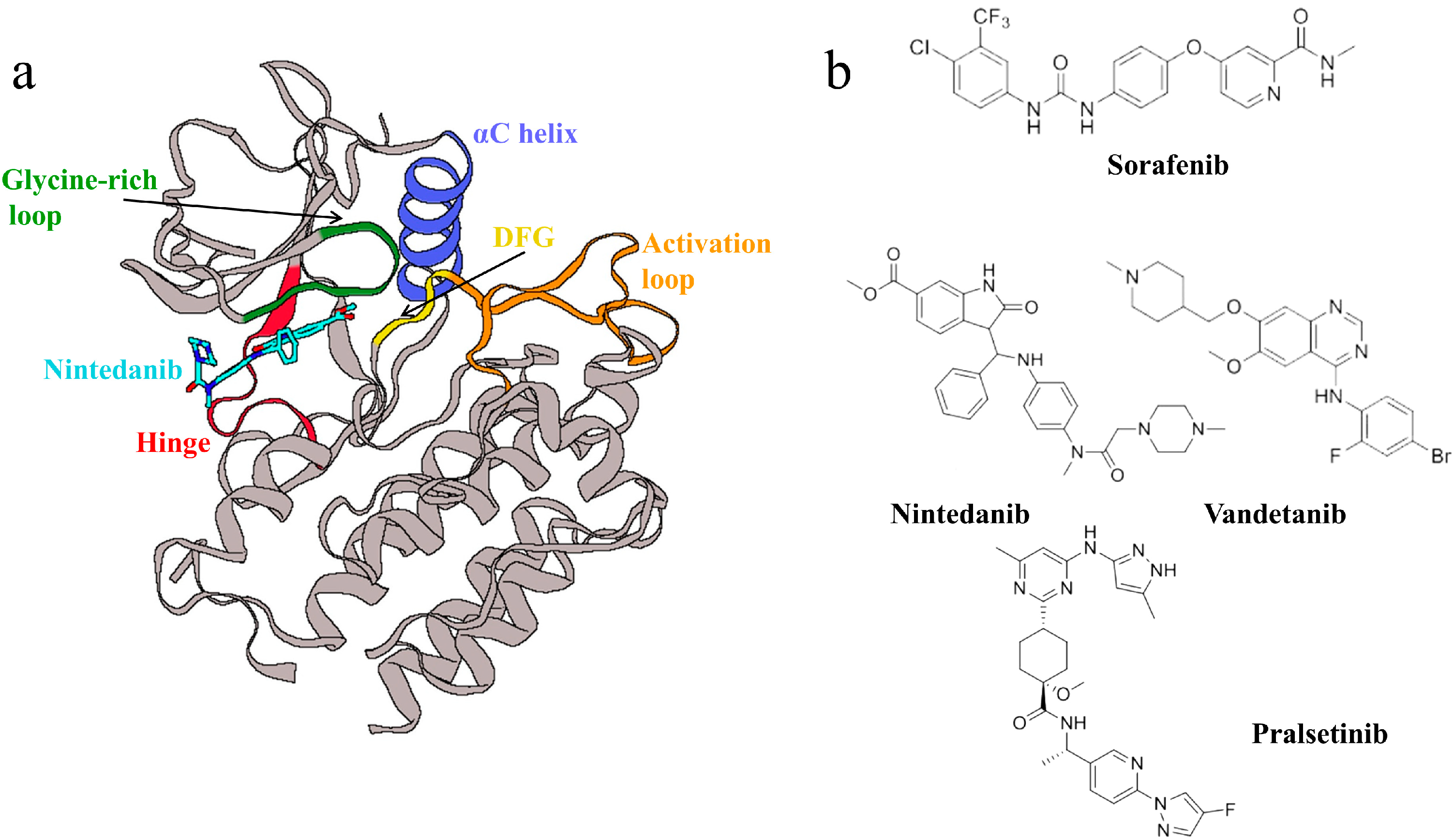
:1. Introduction
2. Results
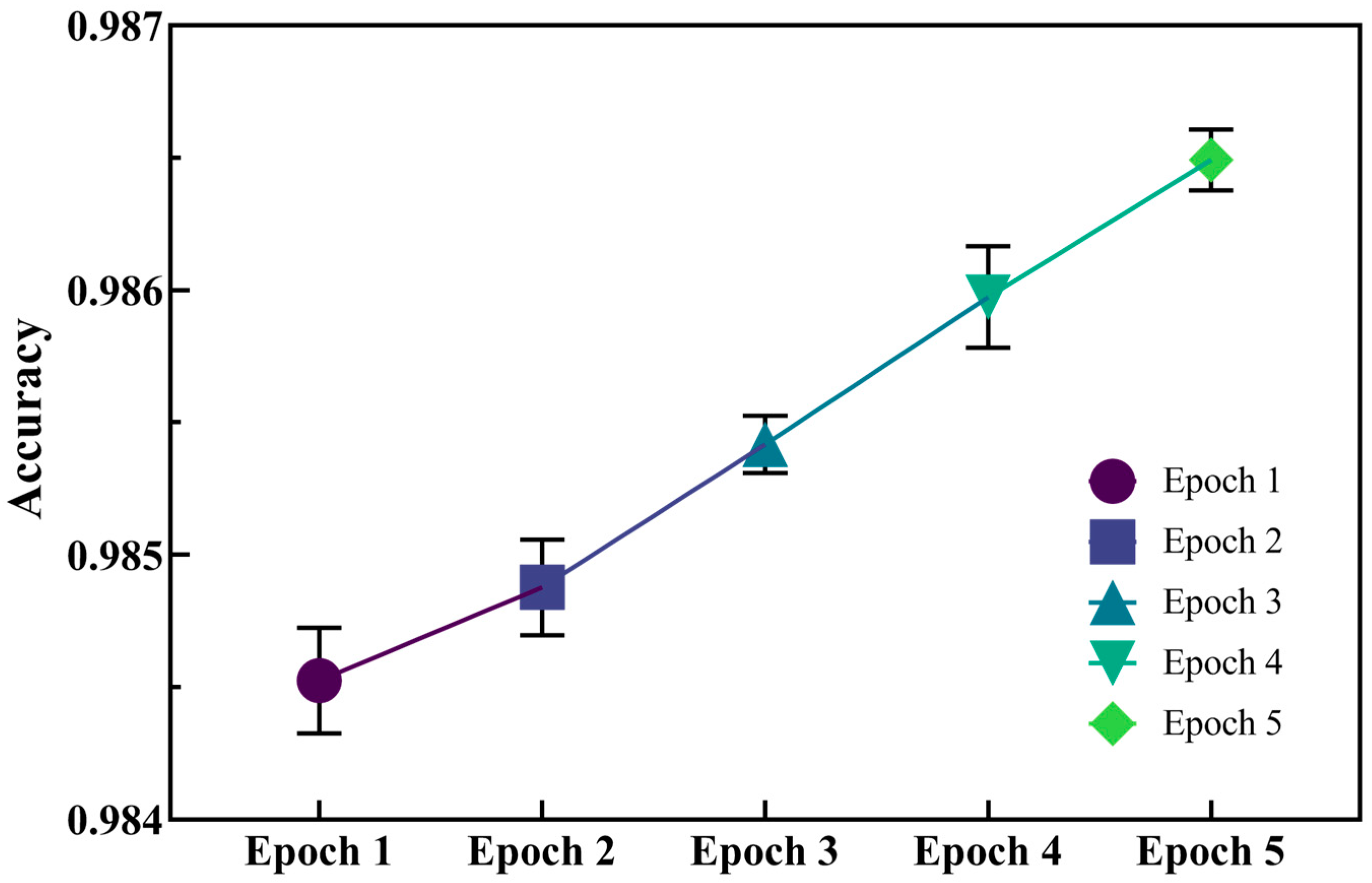
2.1. Implementation and Evaluation of the Model
2.2. Transfer Learning
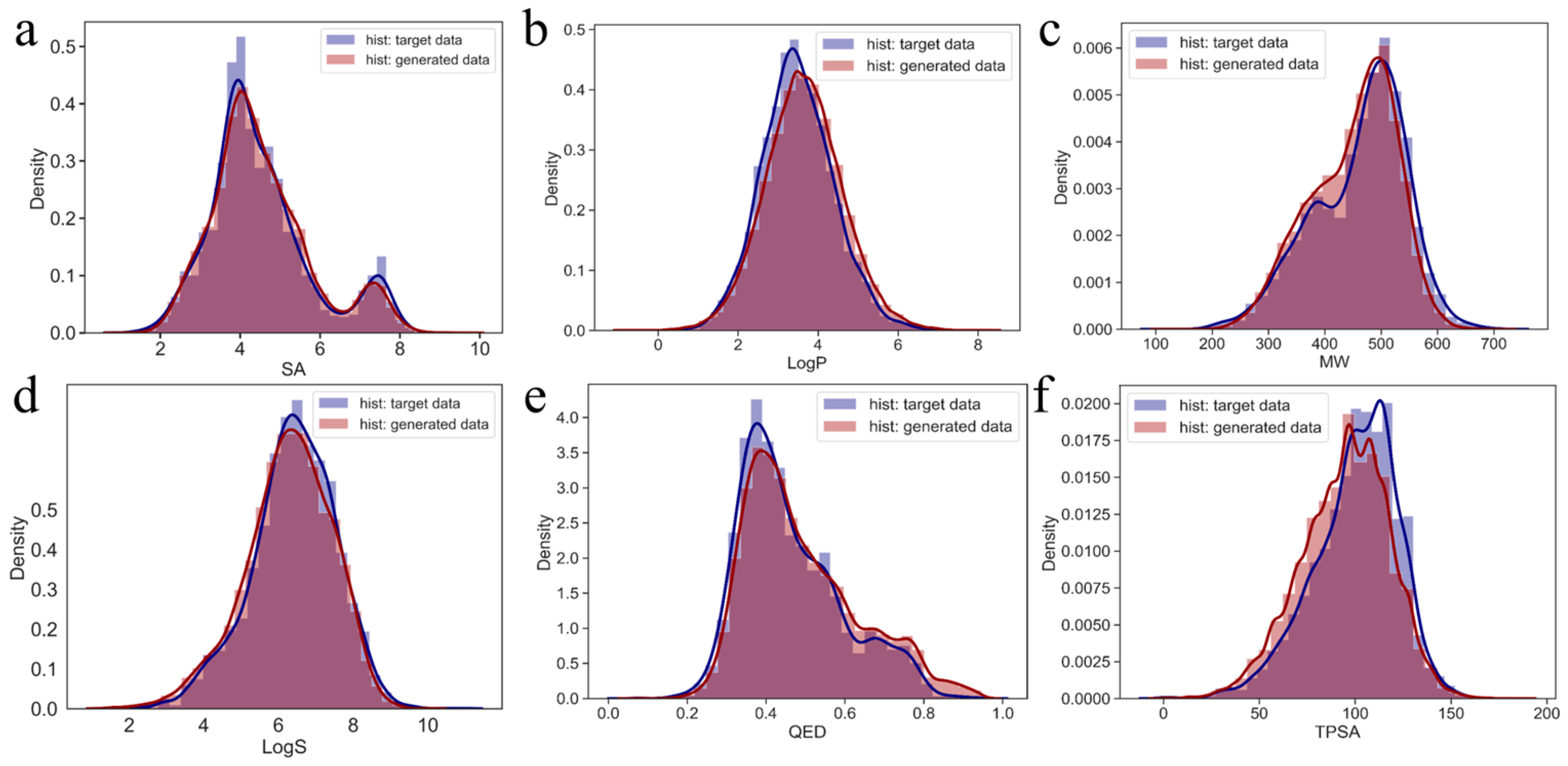
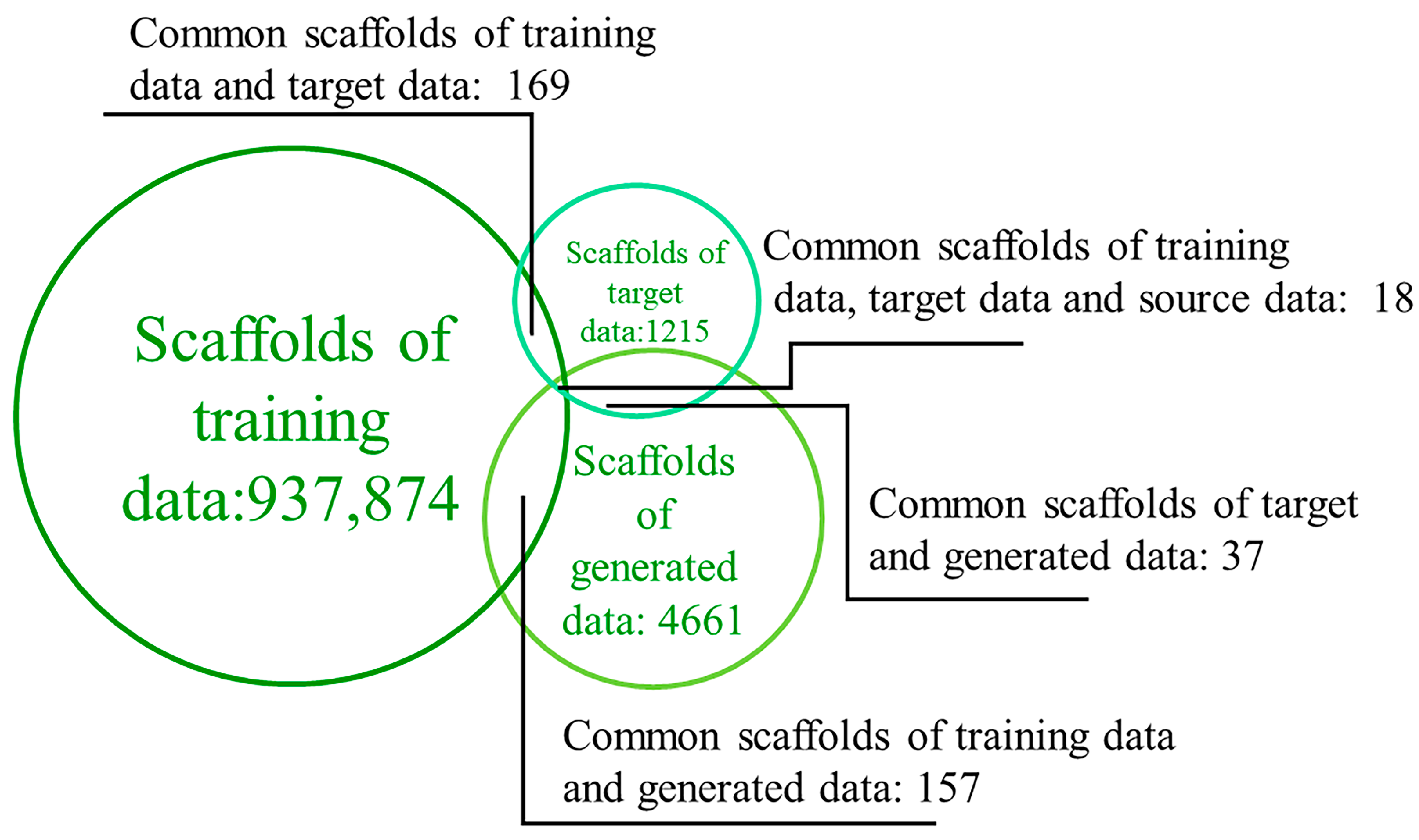
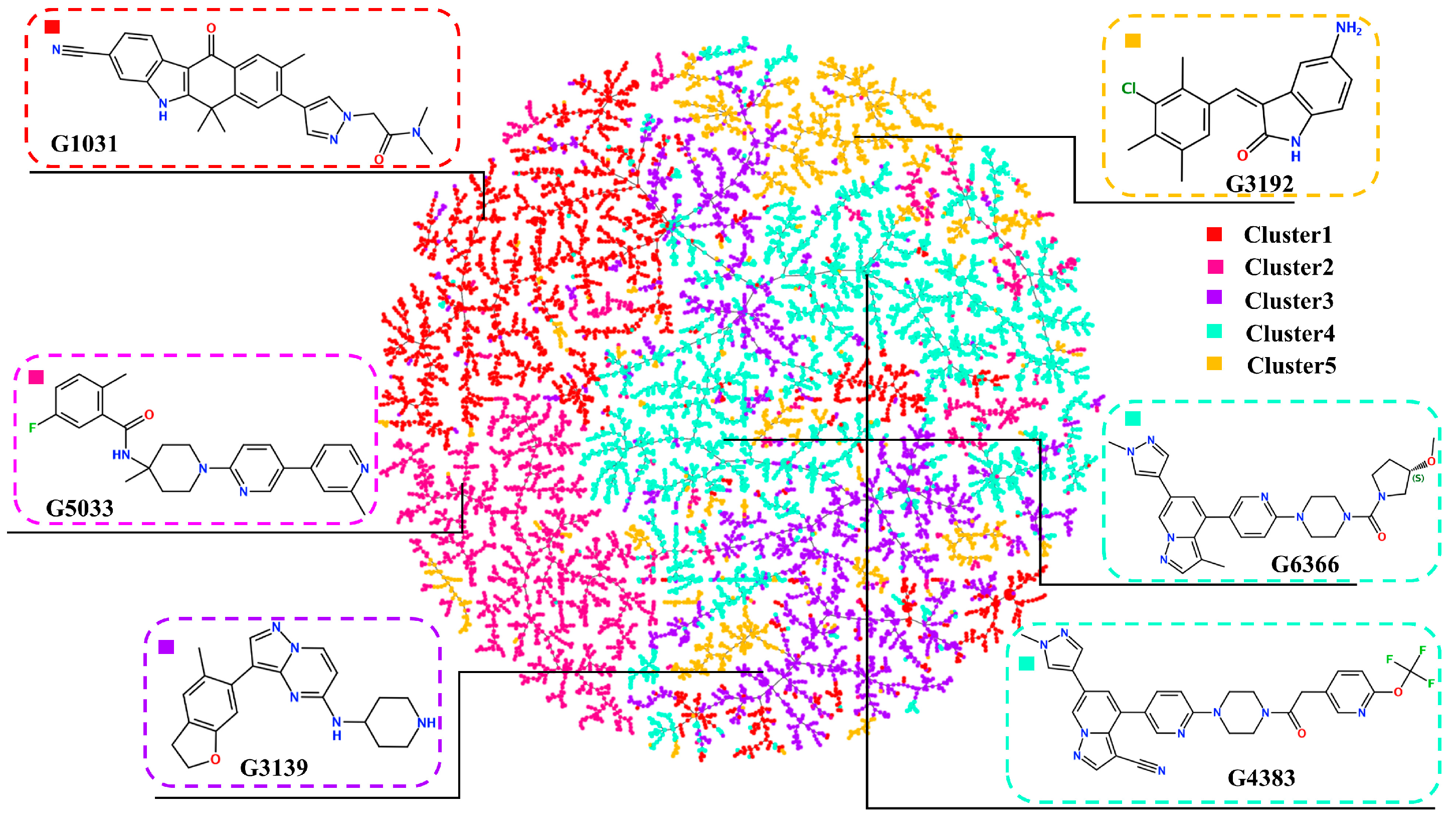
2.3. Diversity of Generated Molecules
2.4. Virtual Screening
2.5. ADMET Predictions
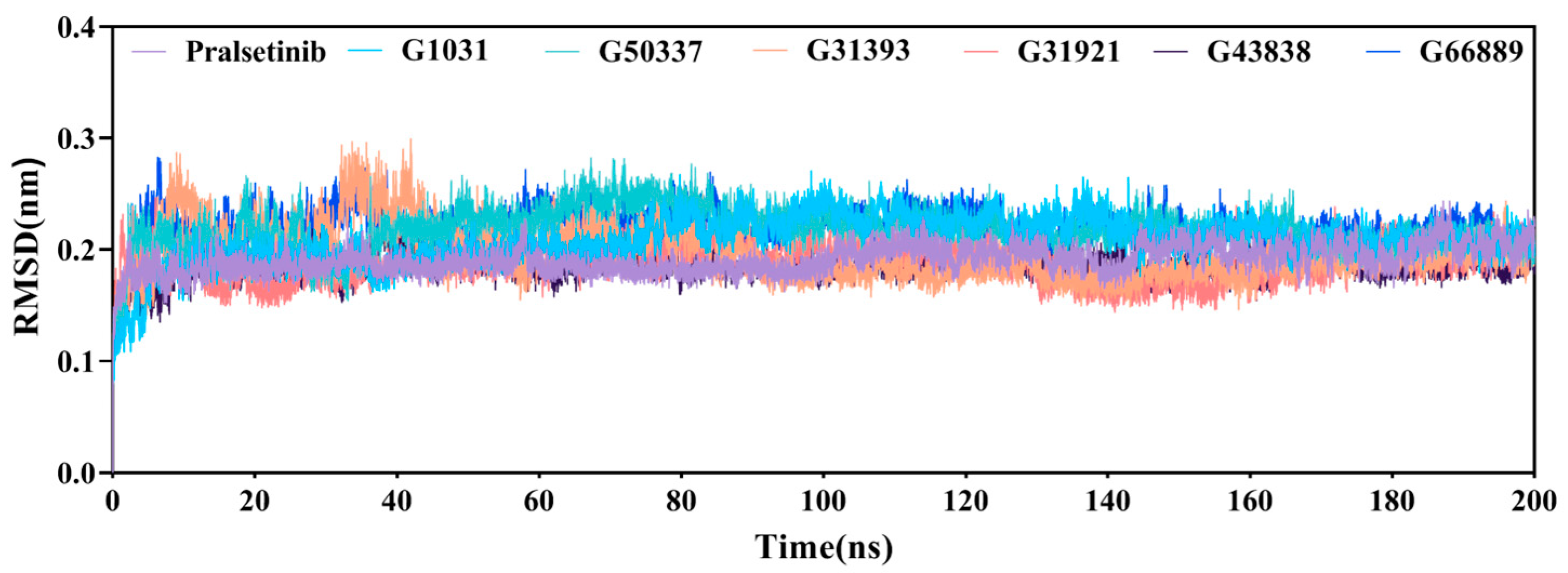
2.6. Molecular Dynamics Simulations
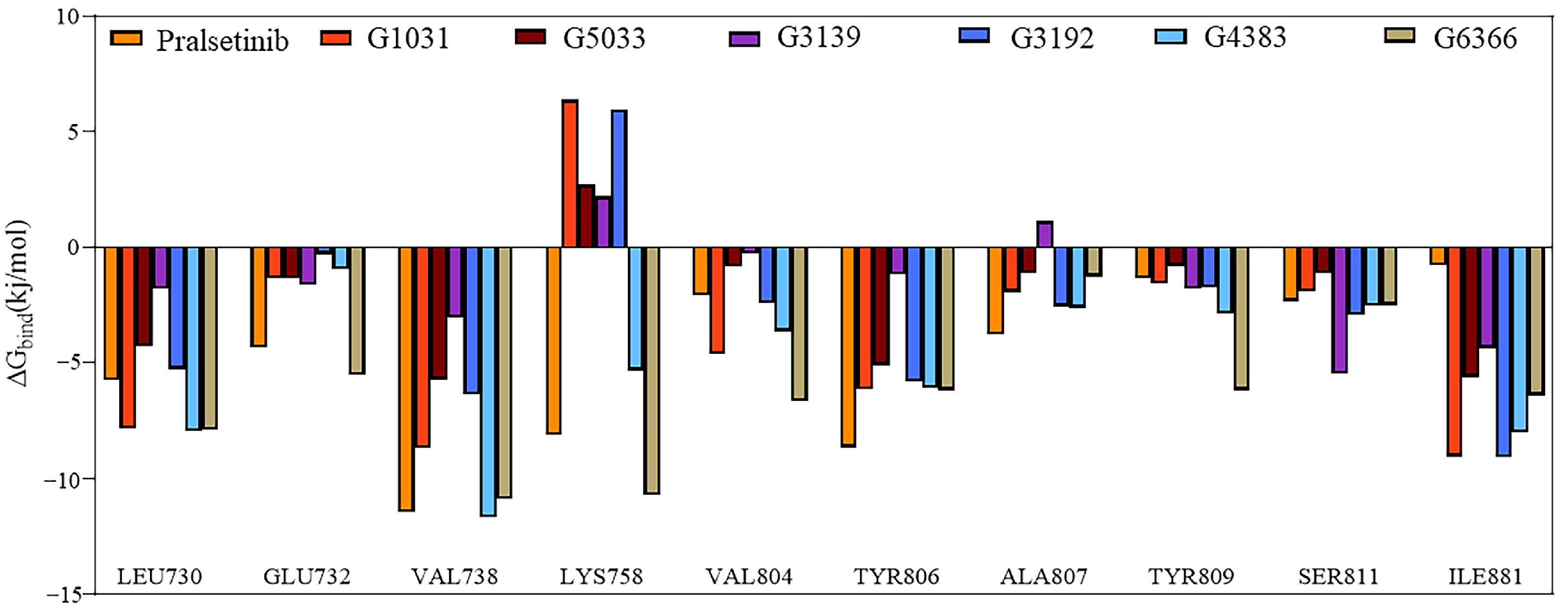
2.7. Binding Free Energy
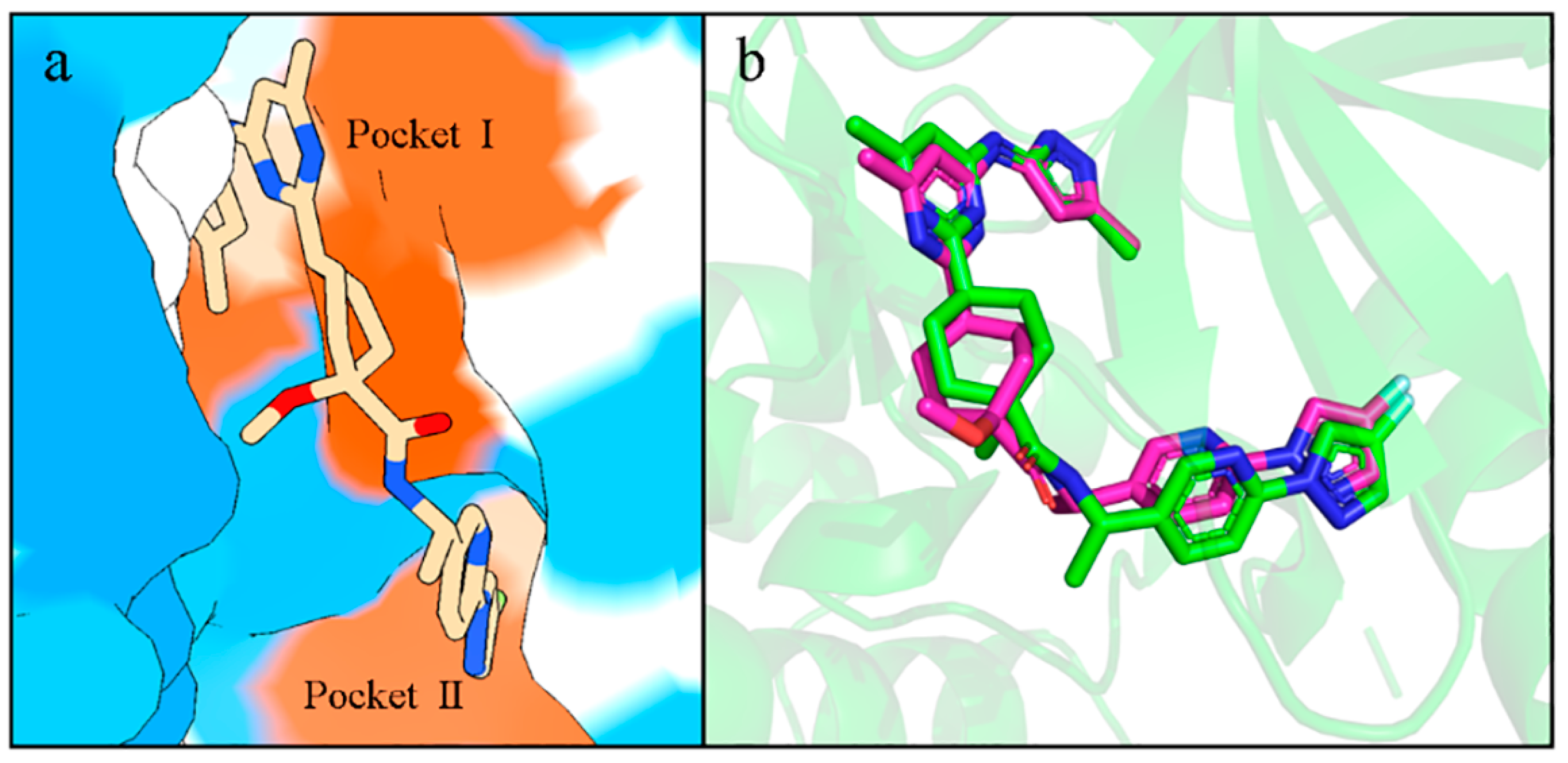
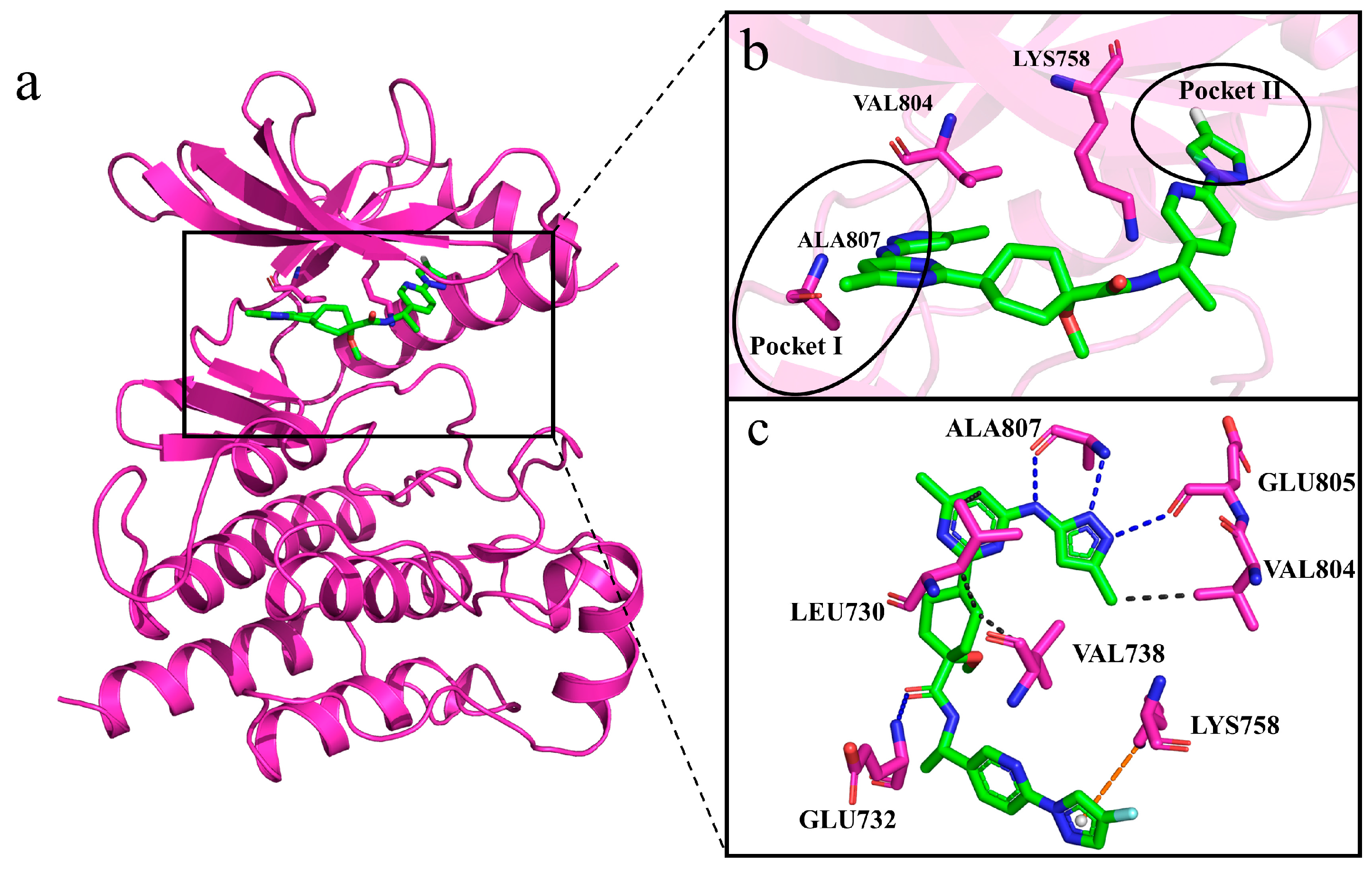
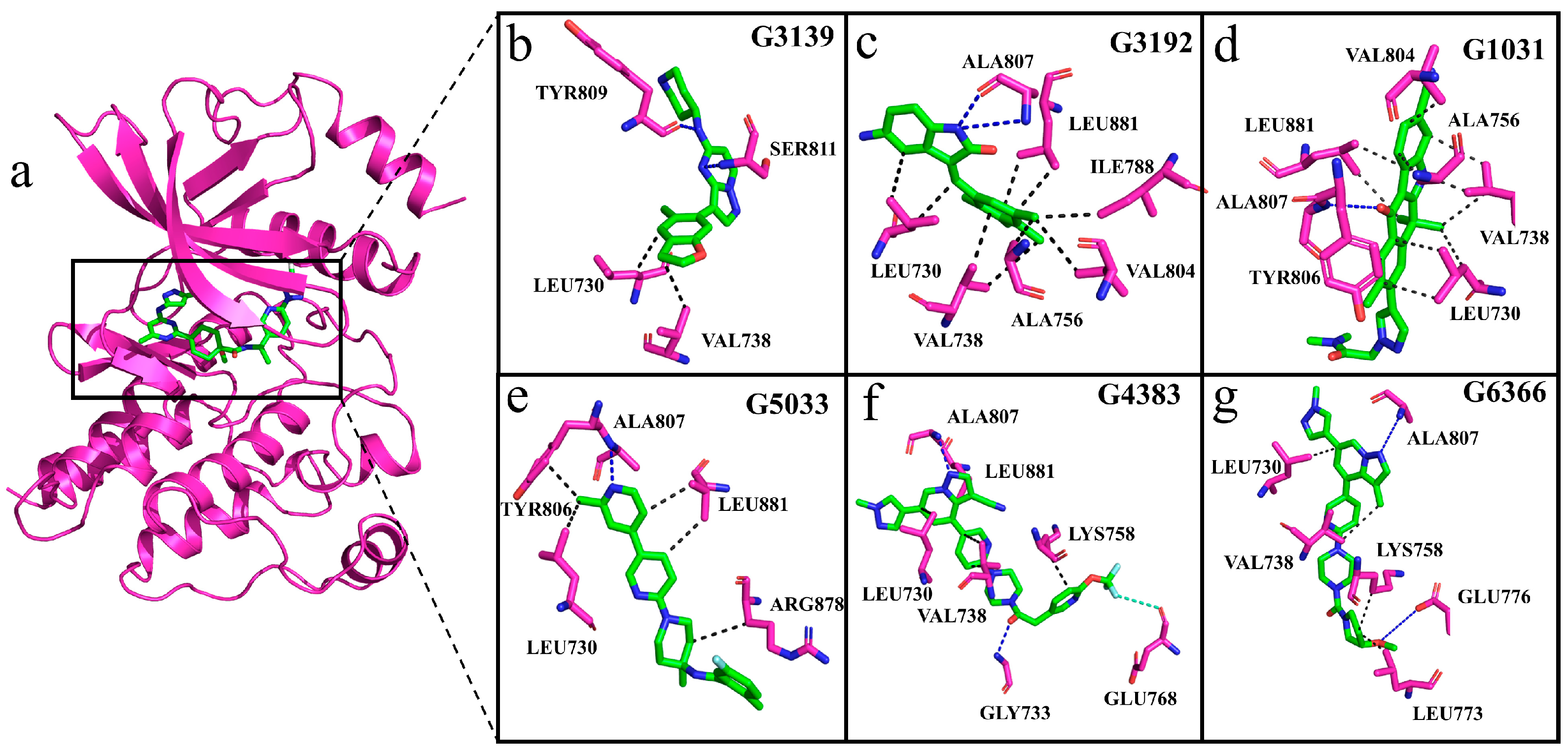
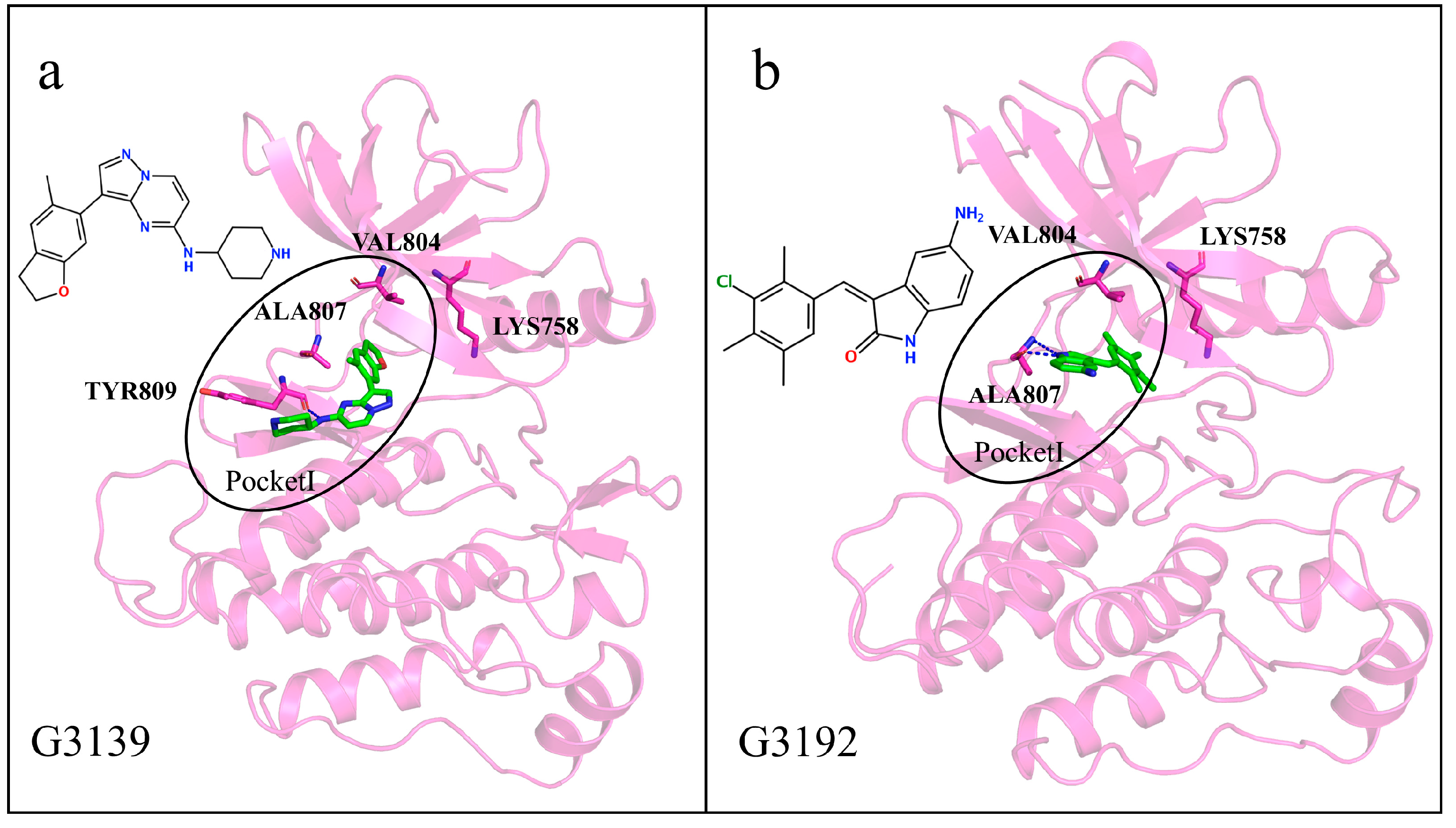
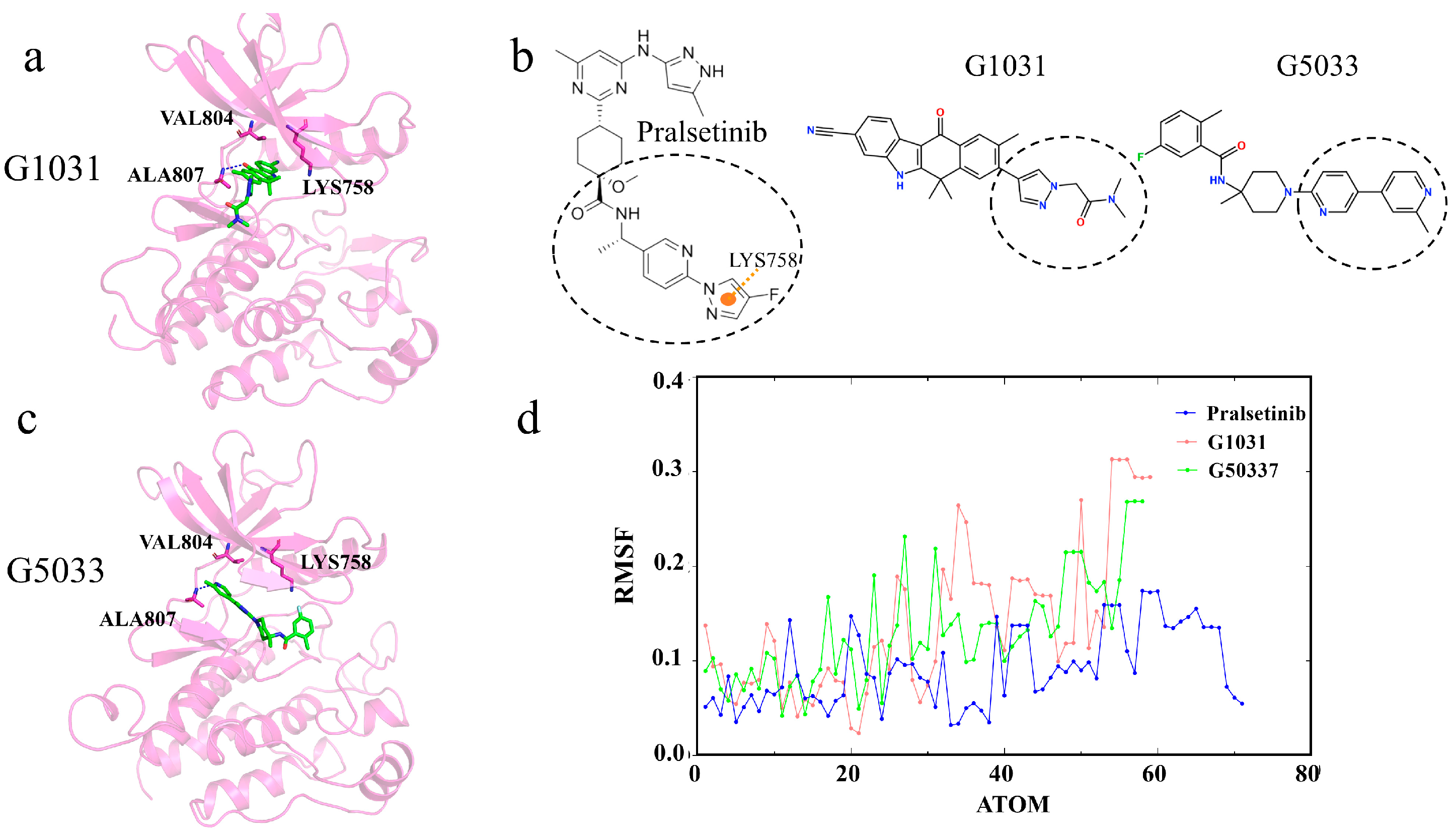
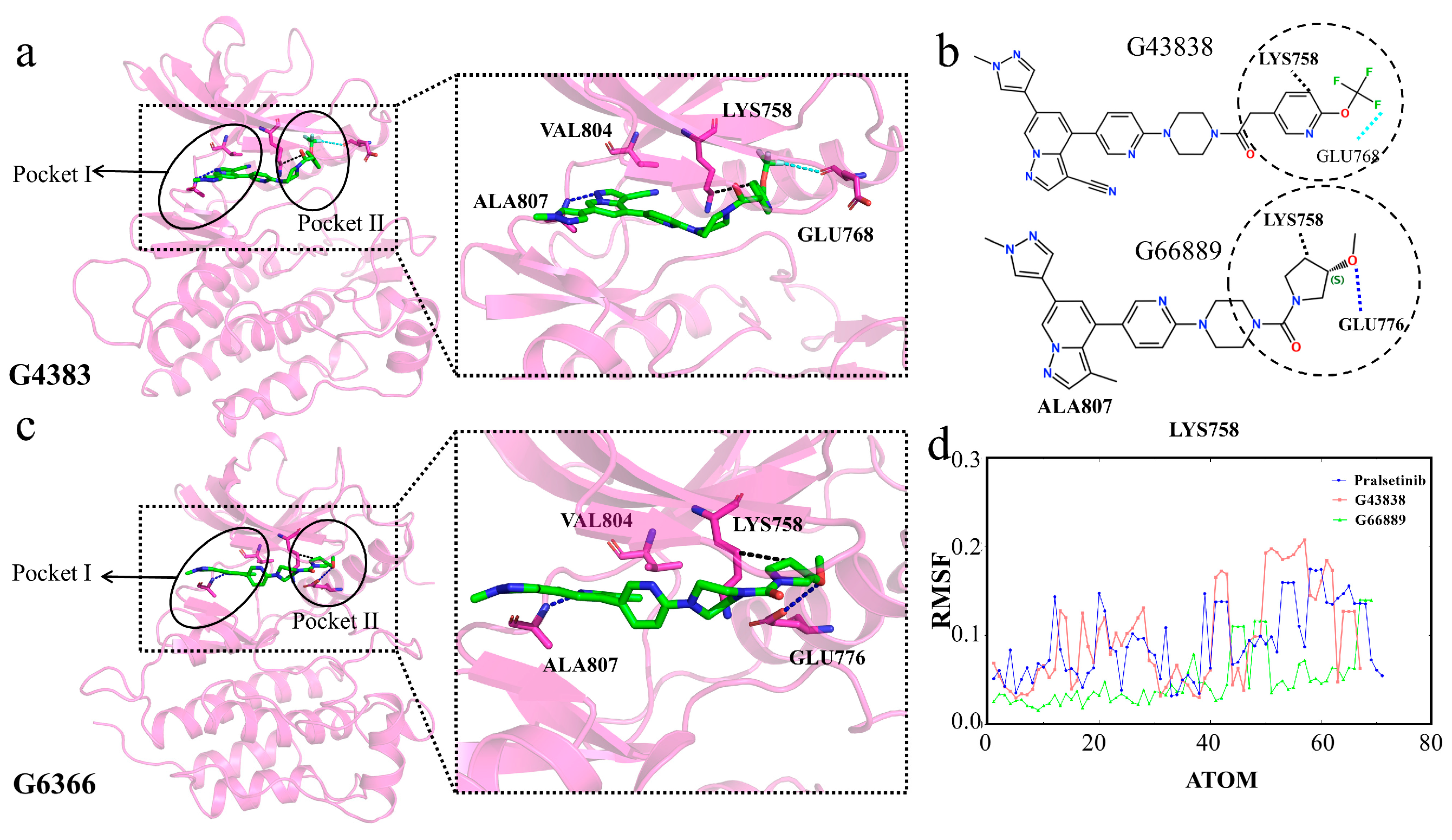
2.8. Interactions of Generated Molecules with RET
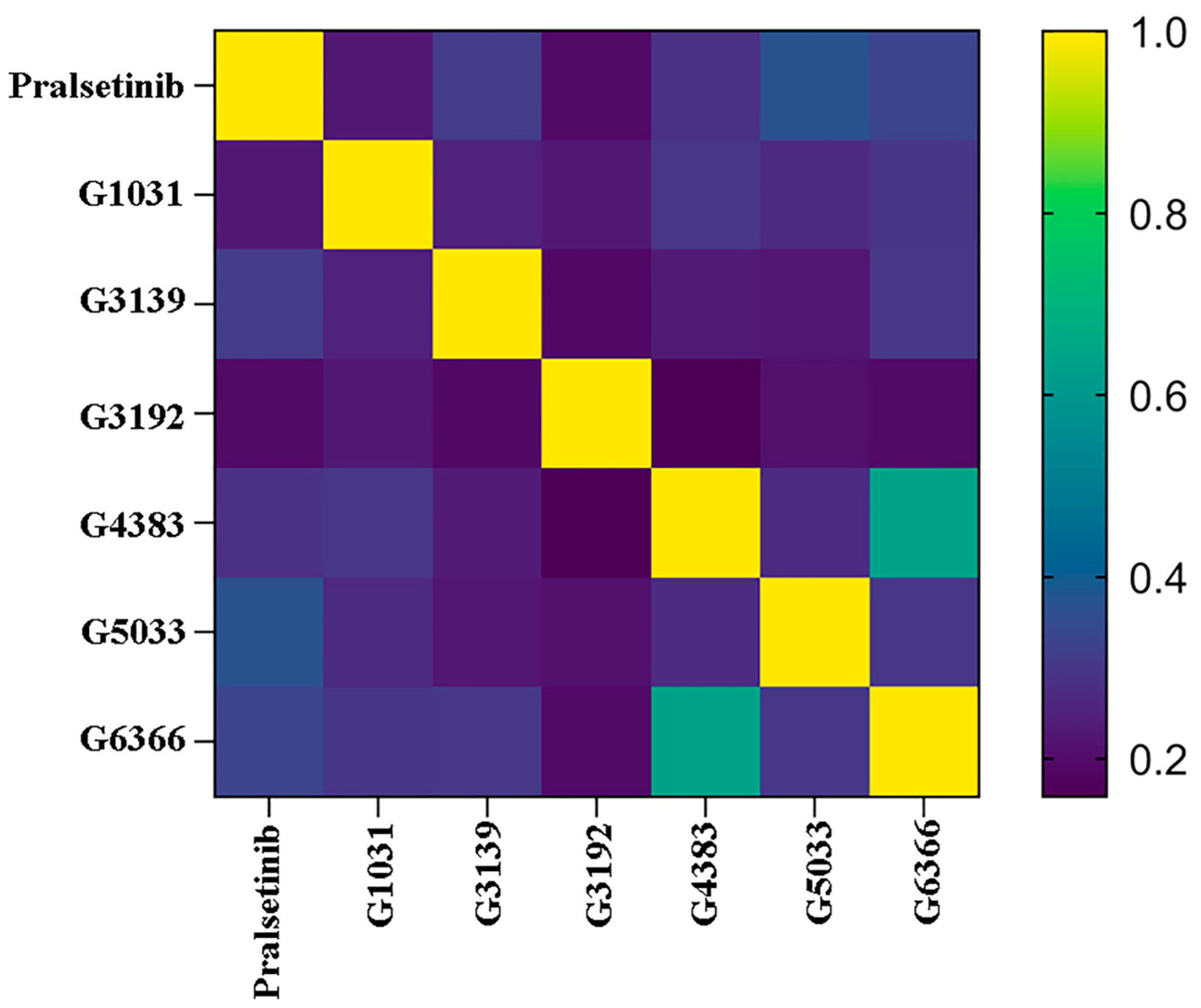
2.9. Selectivity between Generated Molecules with RET
3. Discussion
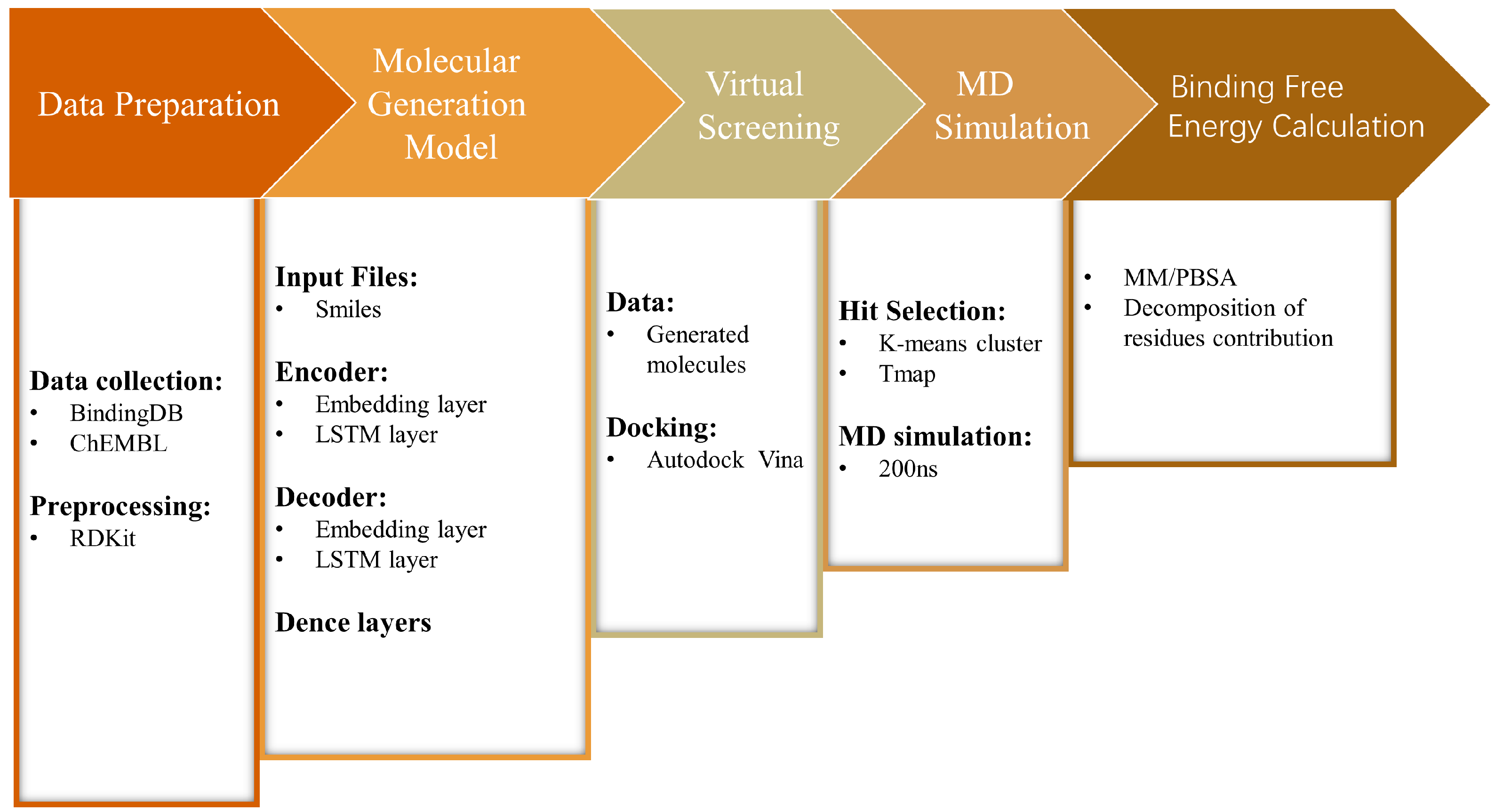
4. Materials and Methods
4.1. Preparation of the Dataset
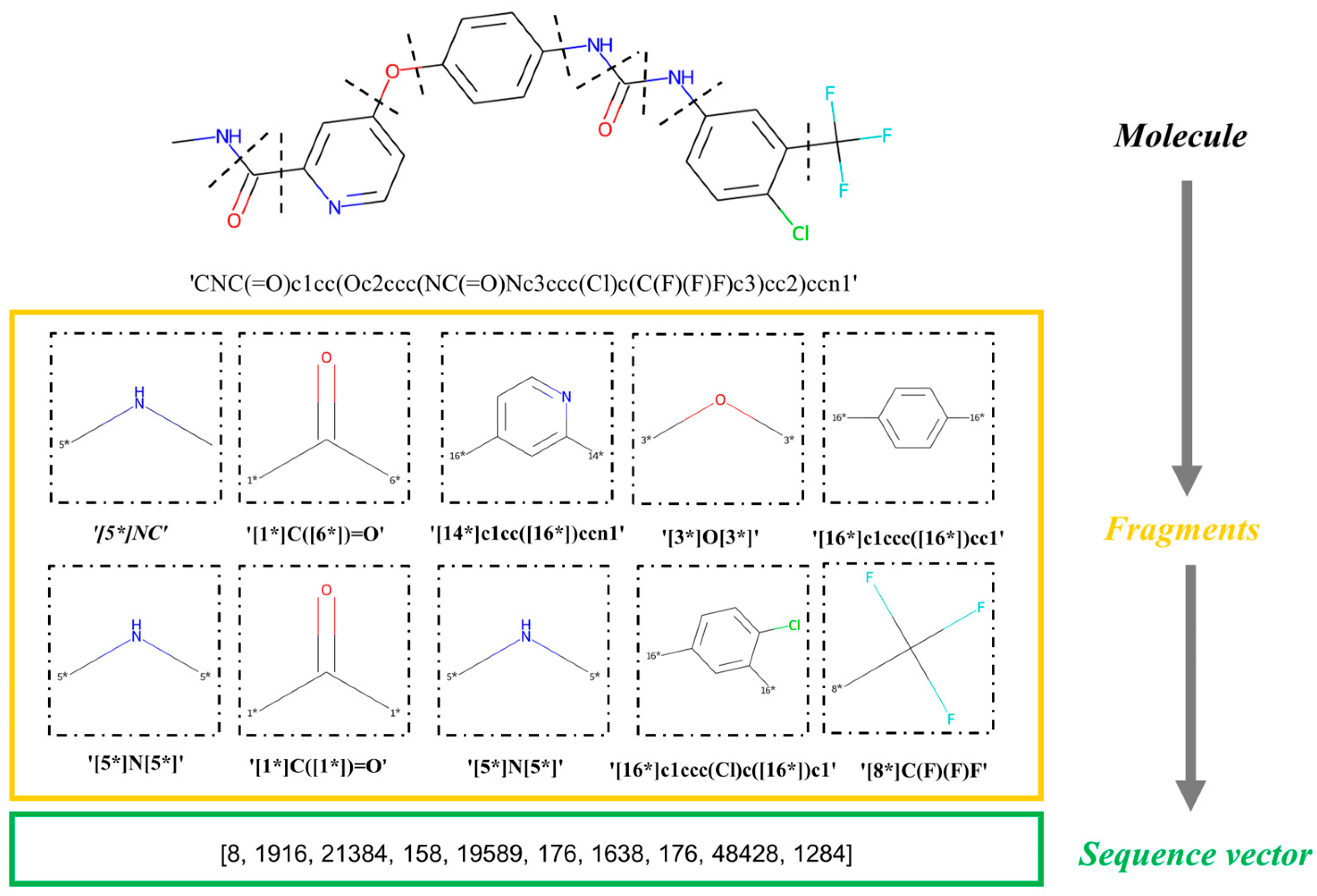
4.2. Molecular Fragmentation
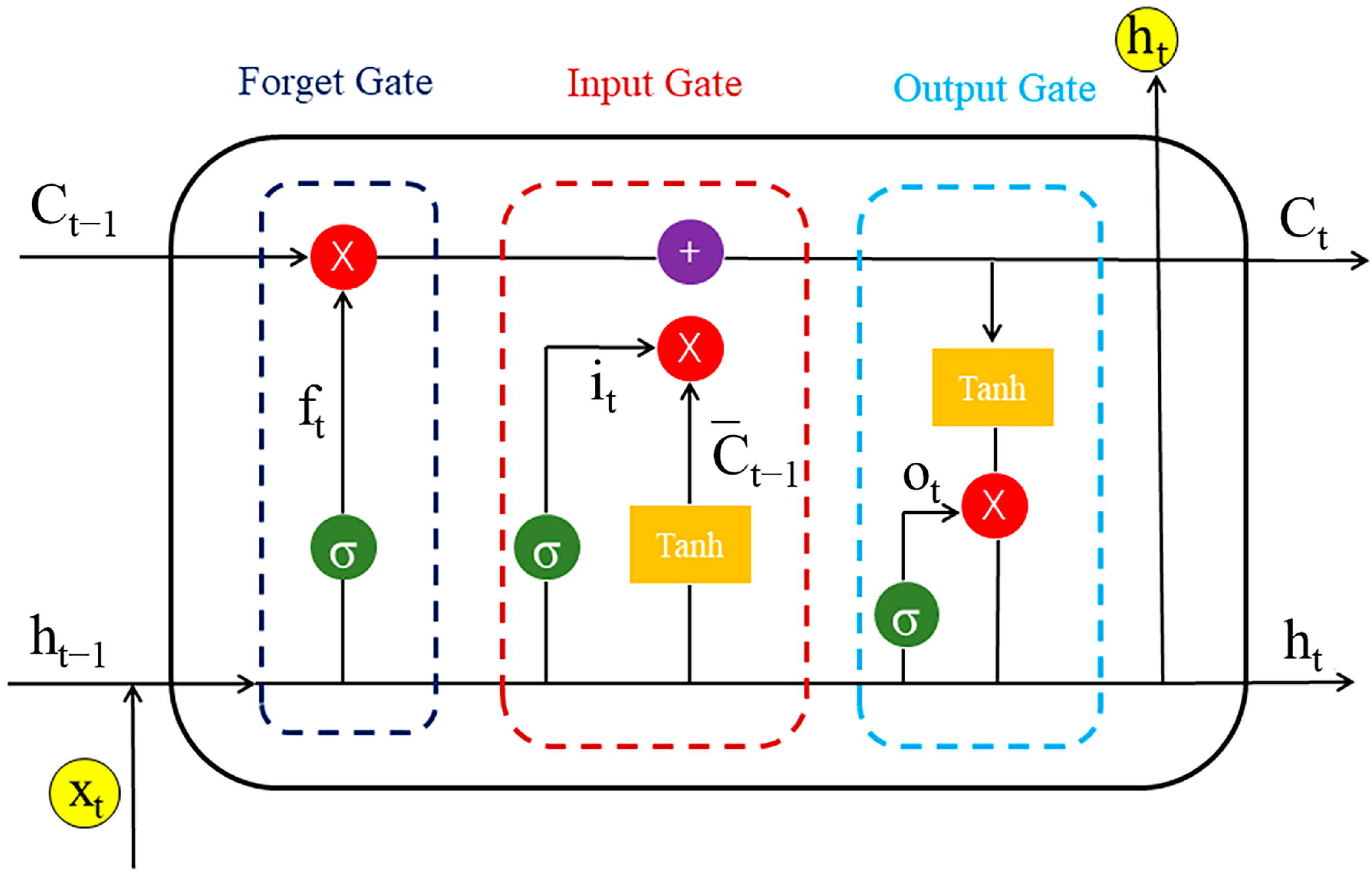
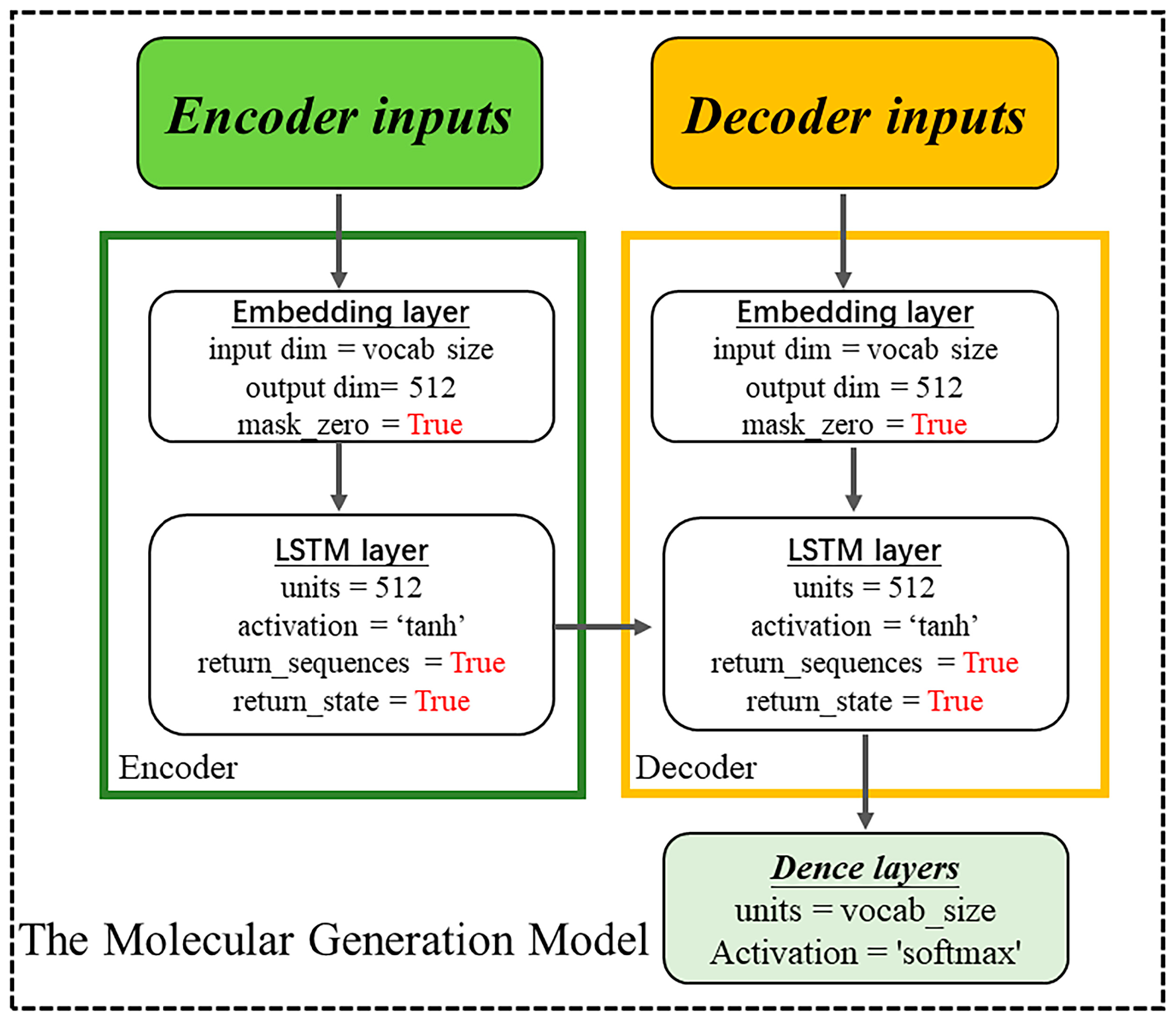
4.3. Construction of the Chemical Language Model
4.4. Model Evaluation
4.5. Molecular Property Calculation
4.6. Molecular Docking and Clustering
4.7. ADMET Prediction
4.8. Molecular Dynamics Simulation
4.9. Binding Free Energy Calculation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jhiang, S.M. The RET proto-oncogene in human cancers. Oncogene 2000, 19, 5590–5597. [Google Scholar] [CrossRef] [PubMed]
- Schuchardt, A.; D’Agati, V.; Larsson-Blomberg, L.; Costantini, F.; Pachnis, V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 1994, 367, 380–383. [Google Scholar] [CrossRef]
- Wang, X. Structural studies of GDNF family ligands with their receptors-Insights into ligand recognition and activation of receptor tyrosine kinase RET. Biochim. Biophys. Acta 2013, 1834, 2205–2212. [Google Scholar] [CrossRef] [PubMed]
- Worby, C.A.; Vega, Q.C.; Zhao, Y.; Chao, H.H.; Seasholtz, A.F.; Dixon, J.E. Glial cell line-derived neurotrophic factor signals through the RET receptor and activates mitogen-activated protein kinase. J. Biol. Chem. 1996, 271, 23619–23622. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef]
- Airaksinen, M.S.; Saarma, M. The GDNF family: Signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002, 3, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Knowles, P.; Murray-Rust, J.; Kjaer, S.; Scott, R.; Hanrahan, S.; Santoro, M.; Ibáñez, C.; McDonald, N. Structure and Chemical Inhibition of the RET Tyrosine Kinase Domain. J. Biol. Chem. 2006, 281, 33577–33587. [Google Scholar] [CrossRef]
- Sariola, H.; Saarma, M. Novel functions and signalling pathways for GDNF. J. Cell Sci. 2003, 116, 3855–3862. [Google Scholar] [CrossRef]
- Terzyan, S.S.; Shen, T.; Liu, X.; Huang, Q.L.; Teng, P.; Zhou, M.; Hilberg, F.; Cai, J.F.; Mooers, B.H.M.; Wu, J. Structural basis of resistance of mutant RET protein-tyrosine kinase to its inhibitors nintedanib and vandetanib. J. Biol. Chem. 2019, 294, 10428–10437. [Google Scholar] [CrossRef]
- Horiike, A.; Takeuchi, K.; Uenami, T.; Kawano, Y.; Tanimoto, A.; Kaburaki, K.; Tambo, Y.; Kudo, K.; Yanagitani, N.; Ohyanagi, F.; et al. Sorafenib treatment for patients with fusion-positive non-small cell lung cancer. Lung Cancer 2016, 93, 43–46. [Google Scholar] [CrossRef]
- Yoh, K.; Seto, T.; Satouchi, M.; Nishio, M.; Yamamoto, N.; Murakami, H.; Nogami, N.; Matsumoto, S.; Kohno, T.; Tsuta, K.; et al. Vandetanib in patients with previously treated RET-rearranged advanced non-small-cell lung cancer (LURET): An open-label, multicentre phase 2 trial. Lancet Respir. Med. 2017, 5, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Chougnet, C.N.; Schlumberger, M.; Leboulleux, S.; Baudin, E. Vandetanib, in the management of patients with locally advanced or metastatic medullary thyroid carcinomas. Bull. Cancer 2014, 101, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A.; Robinson, B.G.; Gagel, R.F. Vandetanib in Patients With Locally Advanced or Metastatic Medullary Thyroid Cancer: A Randomized, Double-Blind Phase III Trial. J. Clin. Oncol. 2013, 31, 3049. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Curigliano, G.; Kim, D.W.; Lee, D.H.; Besse, B.; Baik, C.S.; Doebele, R.C.; Cassier, P.A.; Lopes, G.; Tan, D.S.W.; et al. Pralsetinib for RET fusion-positive non-small-cell lung cancer (ARROW): A multi-cohort, open-label, phase 1/2 study. Lancet Oncol. 2021, 22, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Luna, J.; Grisoni, F.; Weskamp, N.; Schneider, G. Artificial intelligence in drug discovery: Recent advances and future perspectives. Expert. Opin. Drug Discov. 2021, 16, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Sumathi, S.; Suganya, K.; Swathi, K.; Sudha, B.; Poornima, A.; Varghese, C.A.; Aswathy, R. A Review on Deep Learning-driven Drug Discovery: Strategies, Tools and Applications. Curr. Pharm. Des. 2023, 29, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Merrienboer, B.; Gulcehre, C.; Bougares, F.; Schwenk, H.; Bengio, Y. Learning Phrase Representations using RNN Encoder-Decoder for Statistical Machine Translation. arXiv 2014, arXiv:1406.1078. [Google Scholar]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef]
- Grisoni, F.; Moret, M.; Lingwood, R.; Schneider, G. Bidirectional Molecule Generation with Recurrent Neural Networks. J. Chem. Inf. Model. 2020, 60, 1175–1183. [Google Scholar] [CrossRef]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Z.; Rao, J.; Yang, Y.; Dai, Z. Gene based message passing for drug repurposing. iScience 2023, 26, 107663. [Google Scholar] [CrossRef] [PubMed]
- Computational Intelligence and Neuroscience. Retracted: Learning Representations Using RNN Encoder-Decoder for Edge Security Control. Comput. Intell. Neurosci. 2023, 2023, 9871380. [Google Scholar] [CrossRef]
- Awale, M.; Sirockin, F.; Stiefl, N.; Reymond, J.L. Drug Analogs from Fragment-Based Long Short-Term Memory Generative Neural Networks. J. Chem. Inf. Model. 2019, 59, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Farmer, B.T.; Reitz, A.B. Chapter 11—Fragment-Based Drug Discovery. In The Practice of Medicinal Chemistry, 3rd ed.; Wermuth, C.G., Ed.; Academic Press: New York, NY, USA, 2008; pp. 228–243. [Google Scholar]
- Kumar, A.; Voet, A.; Zhang, K.Y. Fragment based drug design: From experimental to computational approaches. Curr. Med. Chem. 2012, 19, 5128–5147. [Google Scholar] [CrossRef]
- Podda, M.; Bacciu, D.; Micheli, A. A Deep Generative Model for Fragment-Based Molecule Generation. In Proceedings of the International Conference on Artificial Intelligence and Statistics, Virtual, 26–28 August 2020. [Google Scholar]
- Degen, J.; Wegscheid-Gerlach, C.; Zaliani, A.; Rarey, M. On the art of compiling and using ‘drug-like’ chemical fragment spaces. ChemMedChem 2008, 3, 1503–1507. [Google Scholar] [CrossRef]
- Chen, Z.; Min, M.R.; Parthasarathy, S.; Ning, X. A deep generative model for molecule optimization via one fragment modification. Nat. Mach. Intell. 2021, 3, 1040–1049. [Google Scholar] [CrossRef]
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.S.; Davis, A.; Dean, J.; Devin, M.; et al. TensorFlow: Large-Scale Machine Learning on Heterogeneous Distributed Systems. arXiv 2016, arXiv:1603.04467. [Google Scholar]
- Timmerman, M.E.; Ceulemans, E.; De Roover, K.; Van Leeuwen, K. Subspace K-means clustering. Behav. Res. Methods 2013, 45, 1011–1023. [Google Scholar] [CrossRef]
- Probst, D.; Reymond, J.-L. Visualization of very large high-dimensional data sets as minimum spanning trees. J. Cheminform. 2020, 12, 12. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP Density Functional Methods for a Large Set of Organic Molecules. J. Chem. Theory Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef]
- Cao, S.; Tan, C.; Fei, A.; Hu, G.; Fu, M.; Lv, J. Insights into pralsetinib resistance to the non-gatekeeper RET kinase G810C mutation through molecular dynamics simulations. J. Mol. Model. 2022, 29, 24. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Kholmurodov, K.; Smith, W.; Yasuoka, K.; Darden, T.; Ebisuzaki, T. A smooth-particle mesh Ewald method for DL_POLY molecular dynamics simulation package on the Fujitsu VPP700. J. Comput. Chem. 2000, 21, 1187–1191. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Kiel, C.; Case, D.A. Insights into Protein–Protein Binding by Binding Free Energy Calculation and Free Energy Decomposition for the Ras–Raf and Ras–RalGDS Complexes. J. Mol. Biol. 2003, 330, 891–913. [Google Scholar] [CrossRef] [PubMed]
- Keretsu, S.; Bhujbal, S.P.; Joo Cho, S. Computational study of paroxetine-like inhibitors reveals new molecular insight to inhibit GRK2 with selectivity over ROCK1. Sci. Rep. 2019, 9, 13053. [Google Scholar] [CrossRef] [PubMed]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef] [PubMed]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Generated Molecules | Physicochemical Properties | Pharmacokinetics | Toxicity | Drug-Likeness | SA | ||
|---|---|---|---|---|---|---|---|
| LogP | LogS | HIA | LogKp | AMES | Lipinski Rule | ||
| G3139 | 3.790 | −3.75 | 0.996 | −6.47 cm/s | 0.937 | Accepted | 2.577 |
| G3192 | 4.368 | −4.41 | 0.997 | −5.59 cm/s | 0.946 | Accepted | 2.402 |
| G1031 | 4.246 | −5.952 | 0.976 | −6.19 cm/s | 0.788 | Accepted | 3.001 |
| G5033 | 4.930 | −4.208 | 0.997 | −5.70 cm/s | 0.837 | Accepted | 2.577 |
| G4383 | 5.002 | −5.17 | 0.996 | −7.73 cm/s | 0.931 | Accepted | 3.214 |
| G6366 | 3.927 | −4.299 | 0.996 | −8.14 cm/s | 0.956 | Accepted | 3.449 |
| System | ΔEele | ΔEvdw | ΔGPB | ΔGNP | −TΔS | ΔGbind |
|---|---|---|---|---|---|---|
| Pralsetinib | −10.124 | −63.360 | 47.151 | −10.125 | 2.860 | −30.580 |
| G1031 | −5.648 | −55.550 | 27.913 | −6.125 | 3.443 | −35.568 |
| G5033 | −8.529 | −44.978 | 25.027 | −7.696 | 5.160 | −31.016 |
| G3139 | −6.012 | −39.480 | 21.128 | −4.610 | 2.439 | −26.535 |
| G3192 | −5.908 | −45.231 | 22.457 | −4.731 | 2.849 | −30.565 |
| G4383 | −5.553 | −65.031 | 32.635 | −7.667 | 6.177 | −39.441 |
| G6366 | −9.329 | −68.719 | 14.055 | −7.354 | 4.333 | −40.015 |
| Molecules | Residues | Occupancies |
|---|---|---|
| Pralsetinib | Ala807 | 94.2% |
| G1031 | Ala 807 | 53.81% |
| Asp892 | 22.23% | |
| G5033 | Ala807 | 62.8% |
| Glu733 | 12.4% | |
| G3139 | Tyr809 | 50.3% |
| G3192 | Ala807 | 80.8% |
| G4383 | Ala807 | 85.1% |
| Glu733 | 40.5% | |
| G6366 | Ala807 | 89.3% |
| Glu766 | 50.6% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Zhao, X.; Huang, X. Generating Potential RET-Specific Inhibitors Using a Novel LSTM Encoder–Decoder Model. Int. J. Mol. Sci. 2024, 25, 2357. https://doi.org/10.3390/ijms25042357
Liu L, Zhao X, Huang X. Generating Potential RET-Specific Inhibitors Using a Novel LSTM Encoder–Decoder Model. International Journal of Molecular Sciences. 2024; 25(4):2357. https://doi.org/10.3390/ijms25042357
Chicago/Turabian StyleLiu, Lu, Xi Zhao, and Xuri Huang. 2024. "Generating Potential RET-Specific Inhibitors Using a Novel LSTM Encoder–Decoder Model" International Journal of Molecular Sciences 25, no. 4: 2357. https://doi.org/10.3390/ijms25042357
APA StyleLiu, L., Zhao, X., & Huang, X. (2024). Generating Potential RET-Specific Inhibitors Using a Novel LSTM Encoder–Decoder Model. International Journal of Molecular Sciences, 25(4), 2357. https://doi.org/10.3390/ijms25042357